
Low-Dose Methylmercury-Induced Genes Regulate Mitochondrial Biogenesis via miR-25 in Immortalized Human Embryonic Neural Progenitor Cells

Abstract
:1. Introduction
2. Results
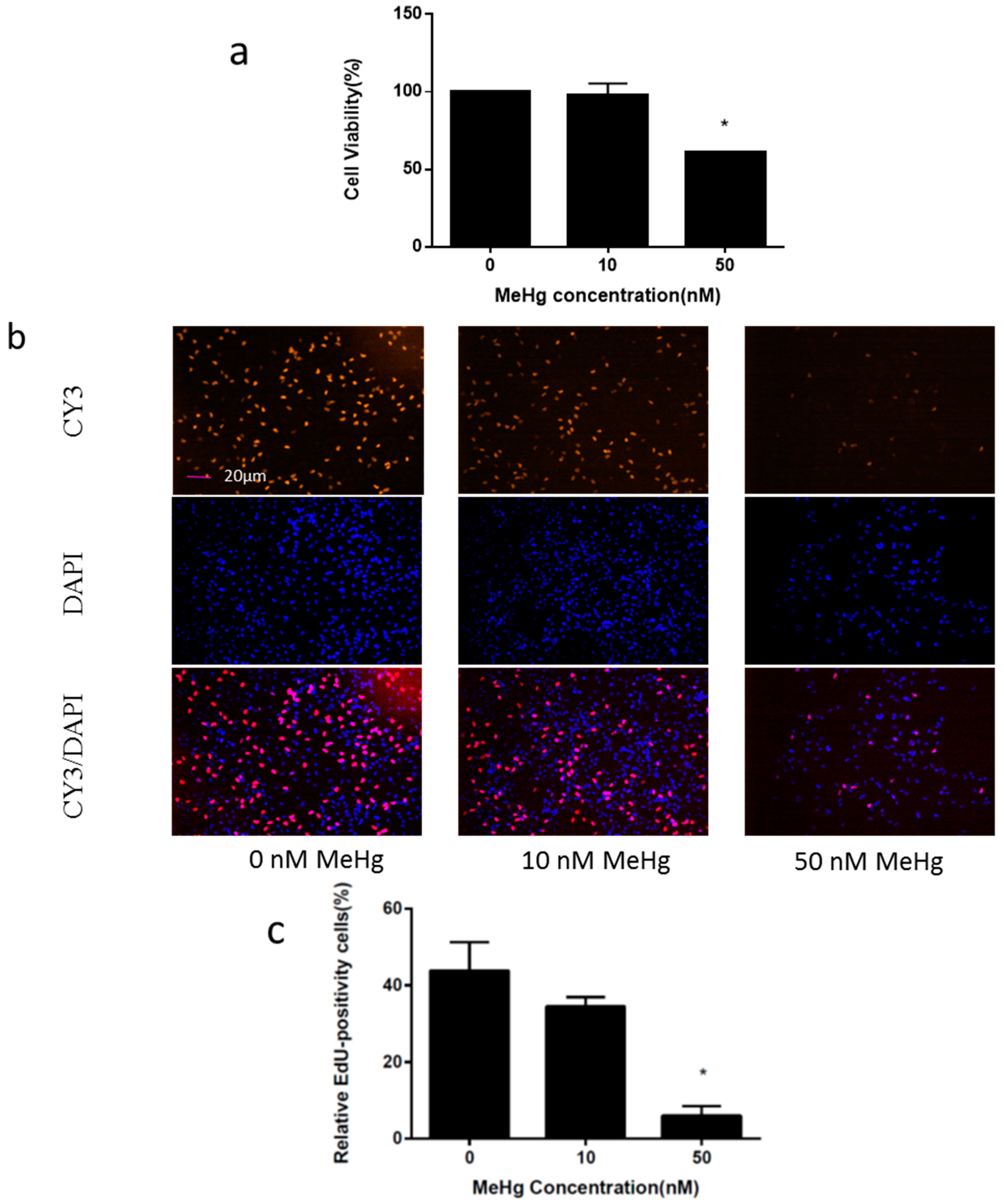
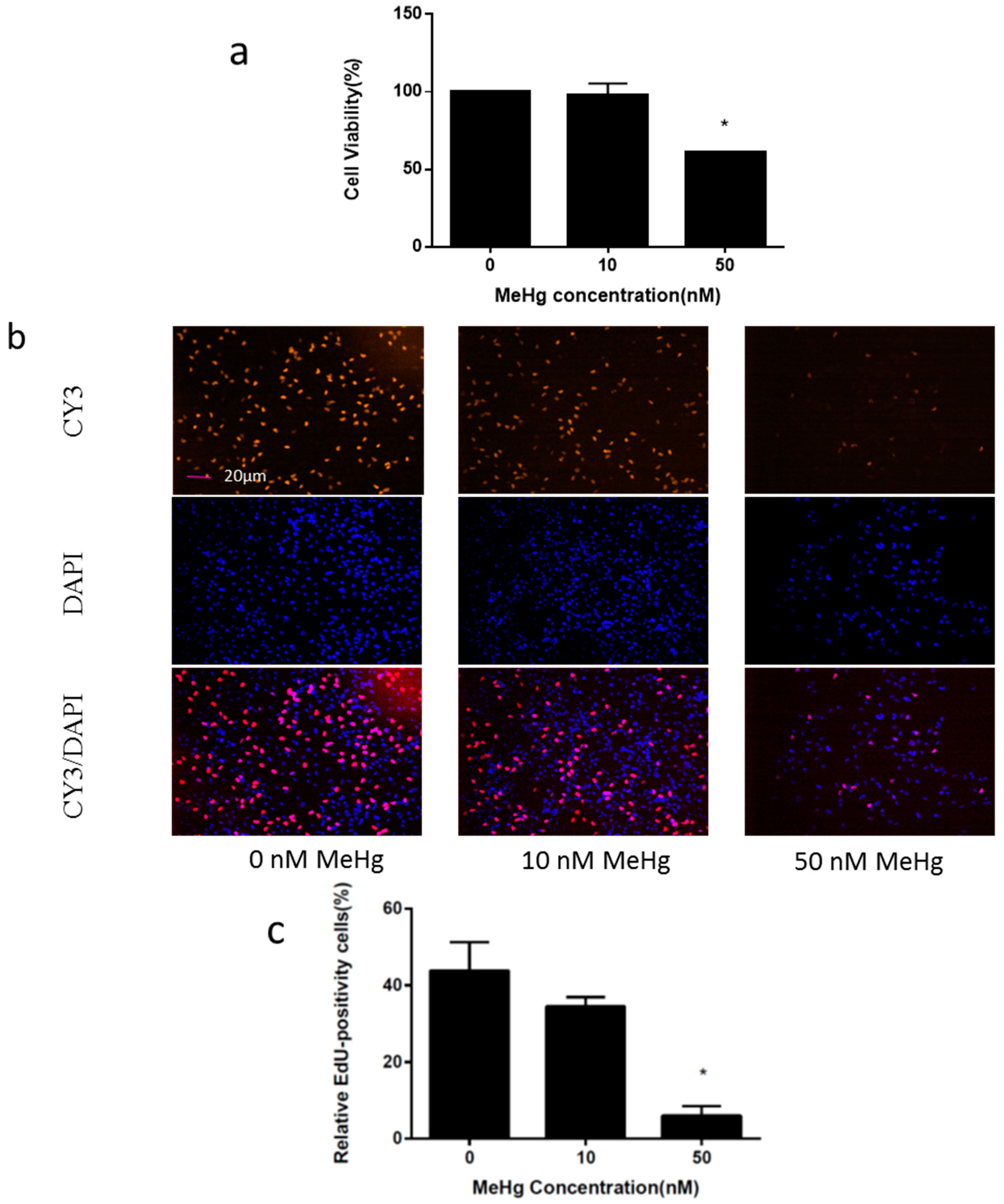
2.1. Immortalized Human Neural Progenitor Cells Are Highly Susceptible to MeHg Toxicity
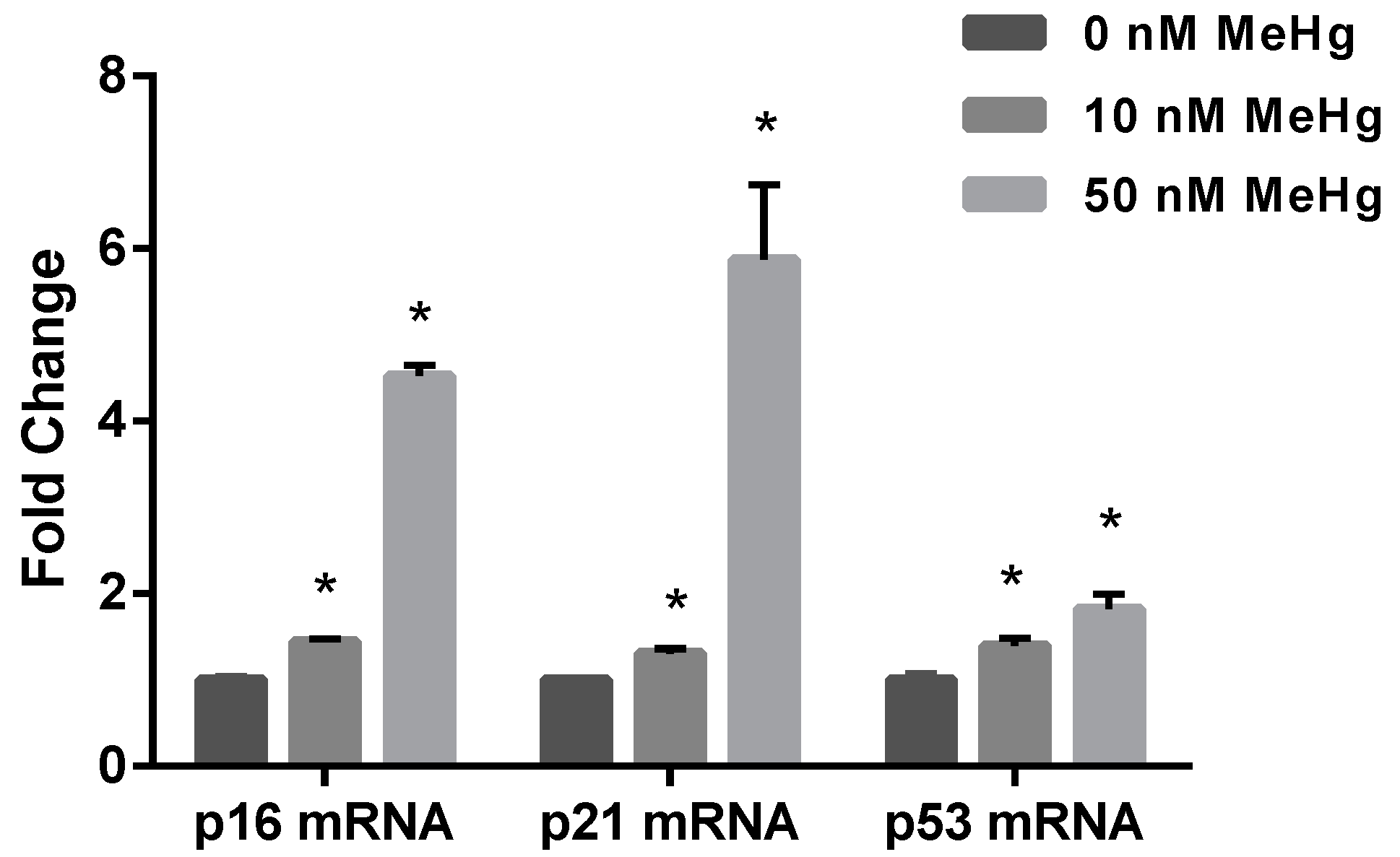
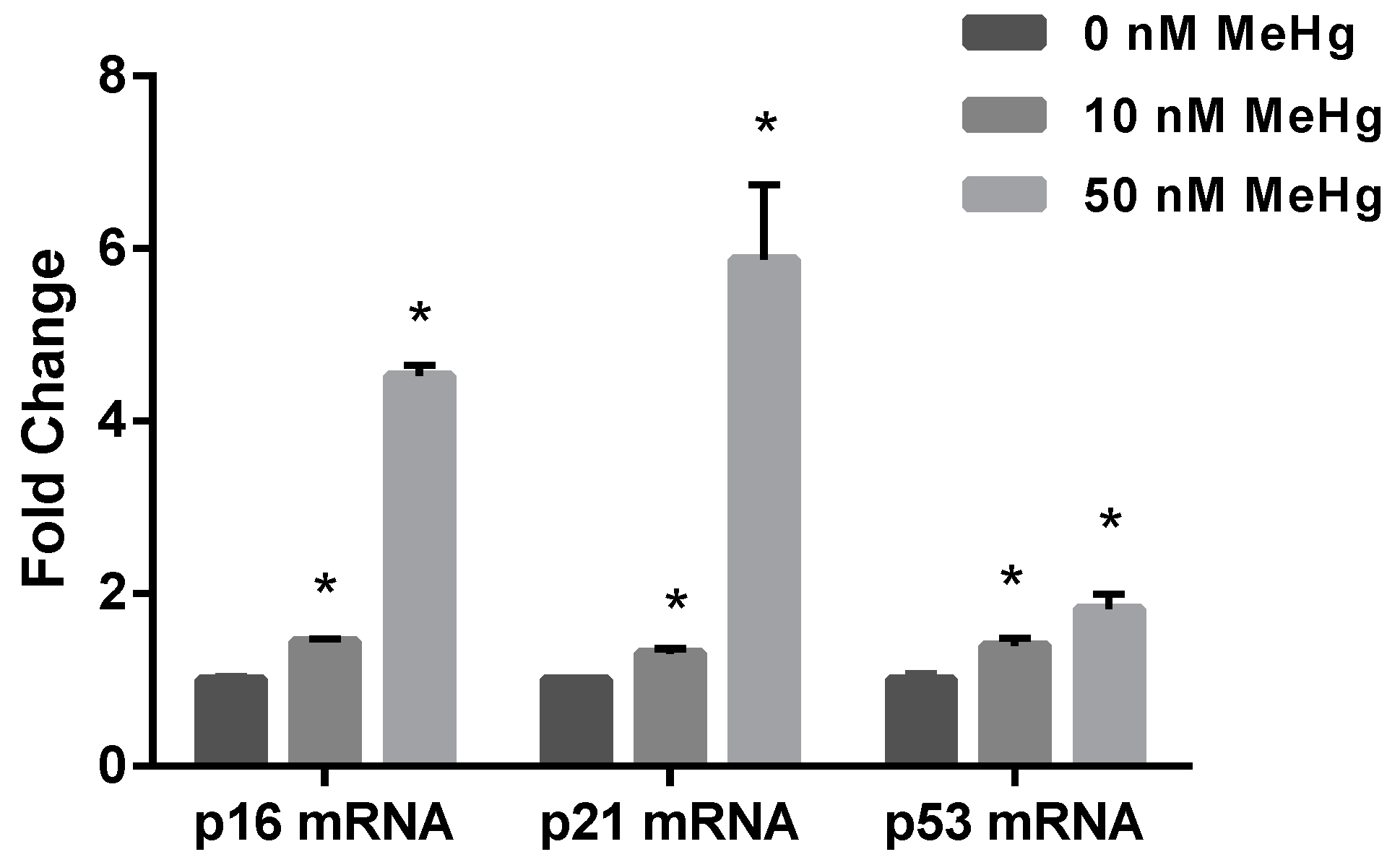
2.2. Effect of MeHg on Cell Cycle Regulatory Genes
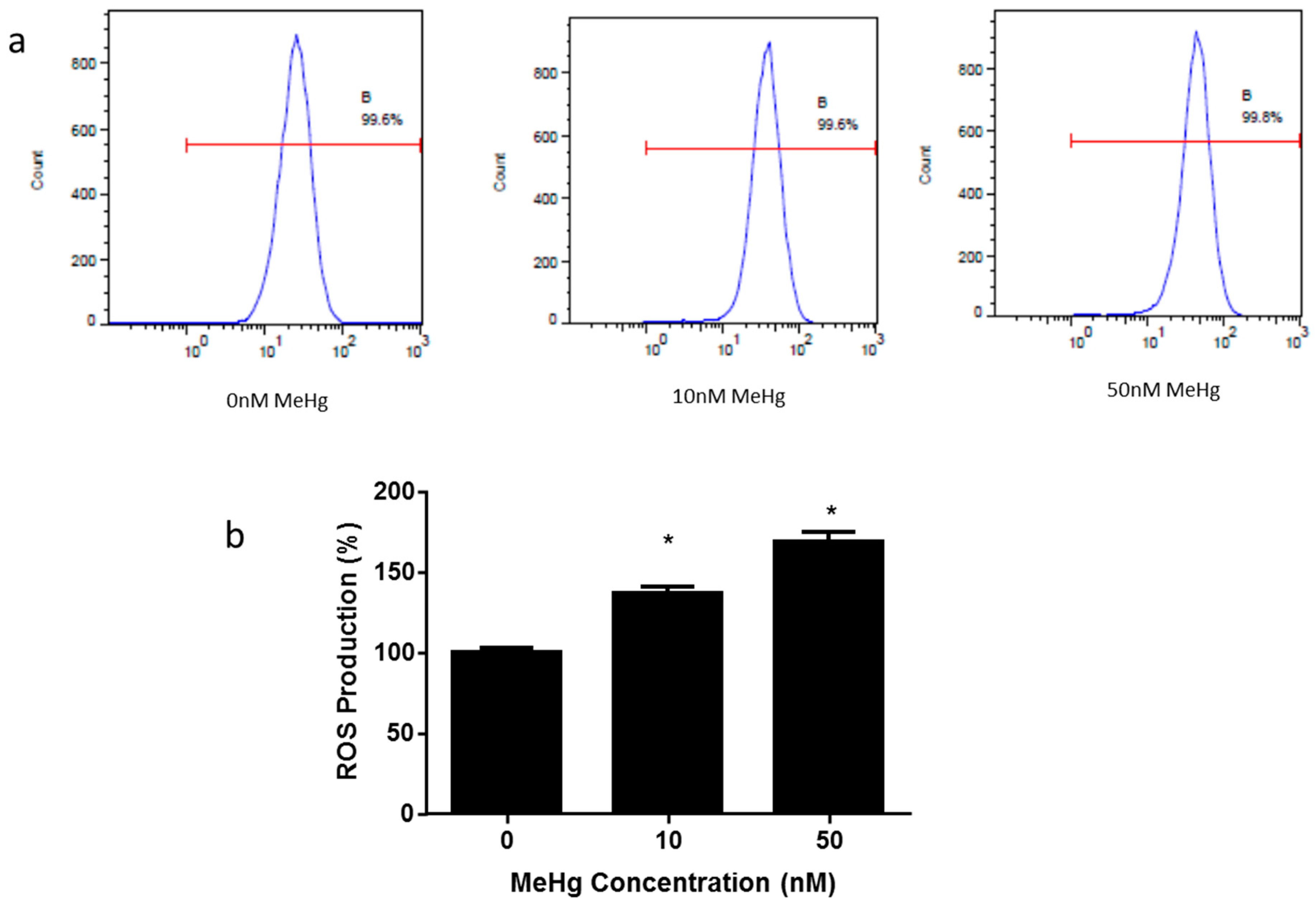
2.3. Effect of MeHg on Intracellular Reactive Oxygen Species Generation
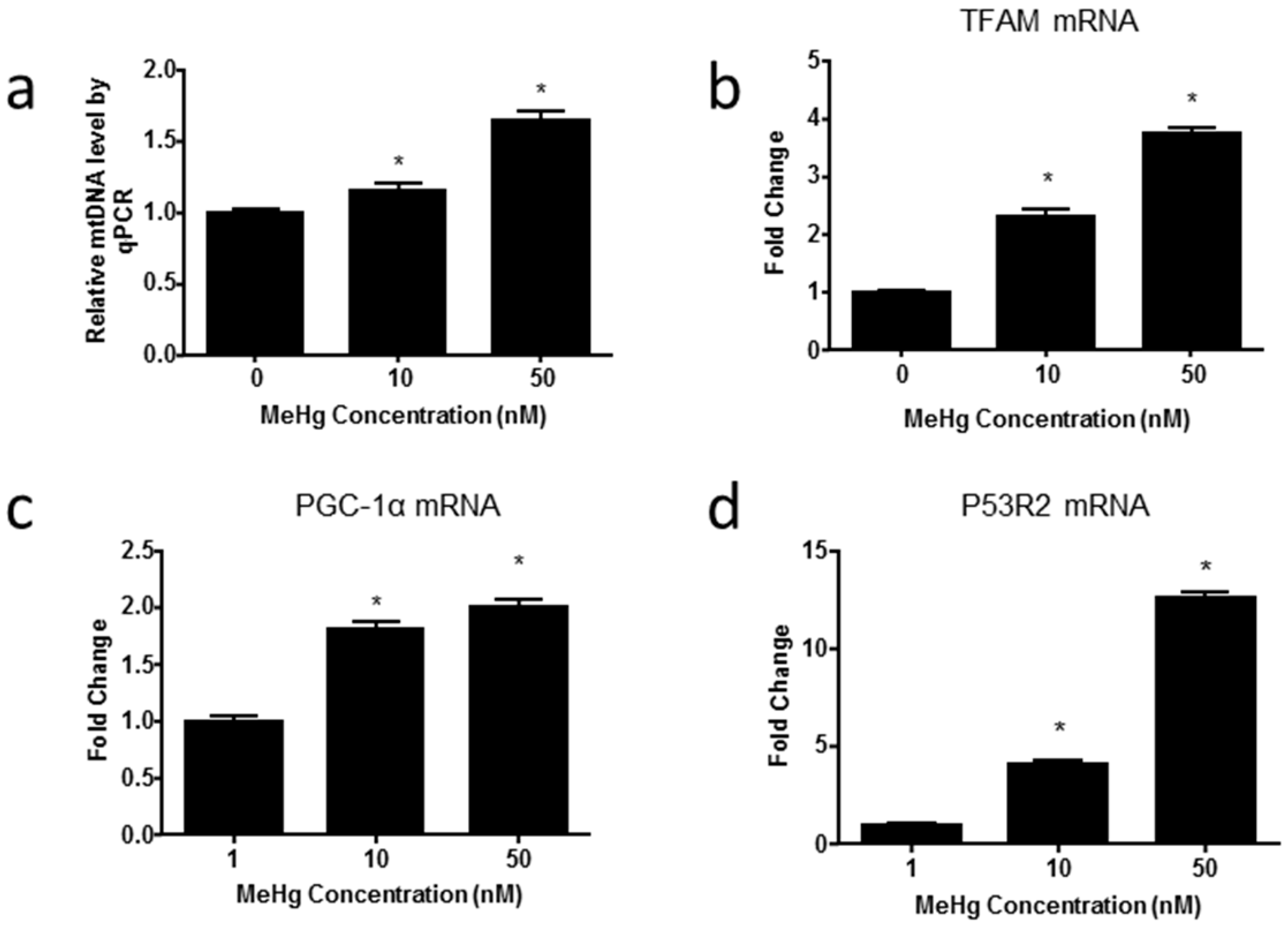
2.4. MeHg Alters Genes Regulating Mitochondrial Biogenesis
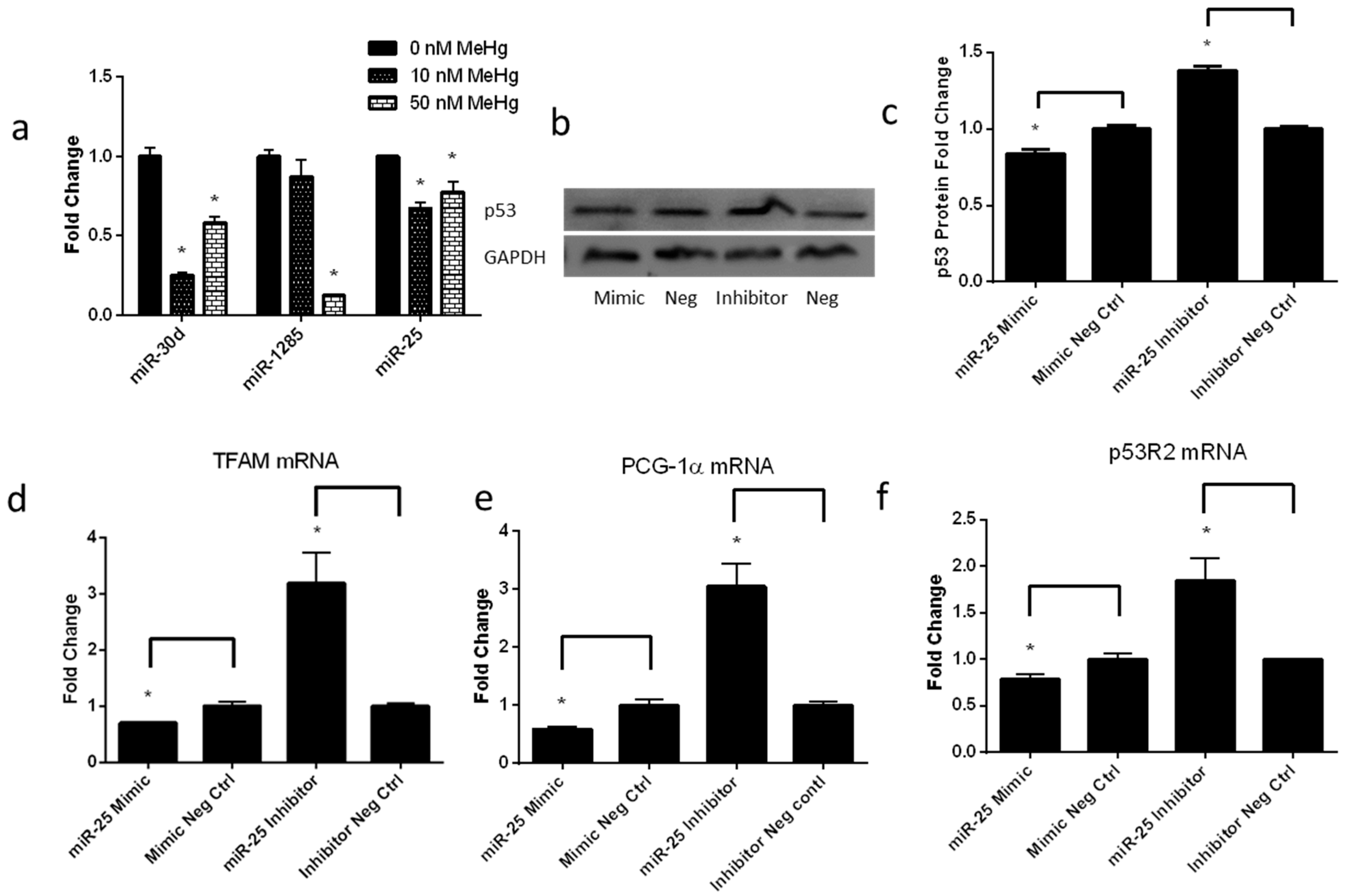
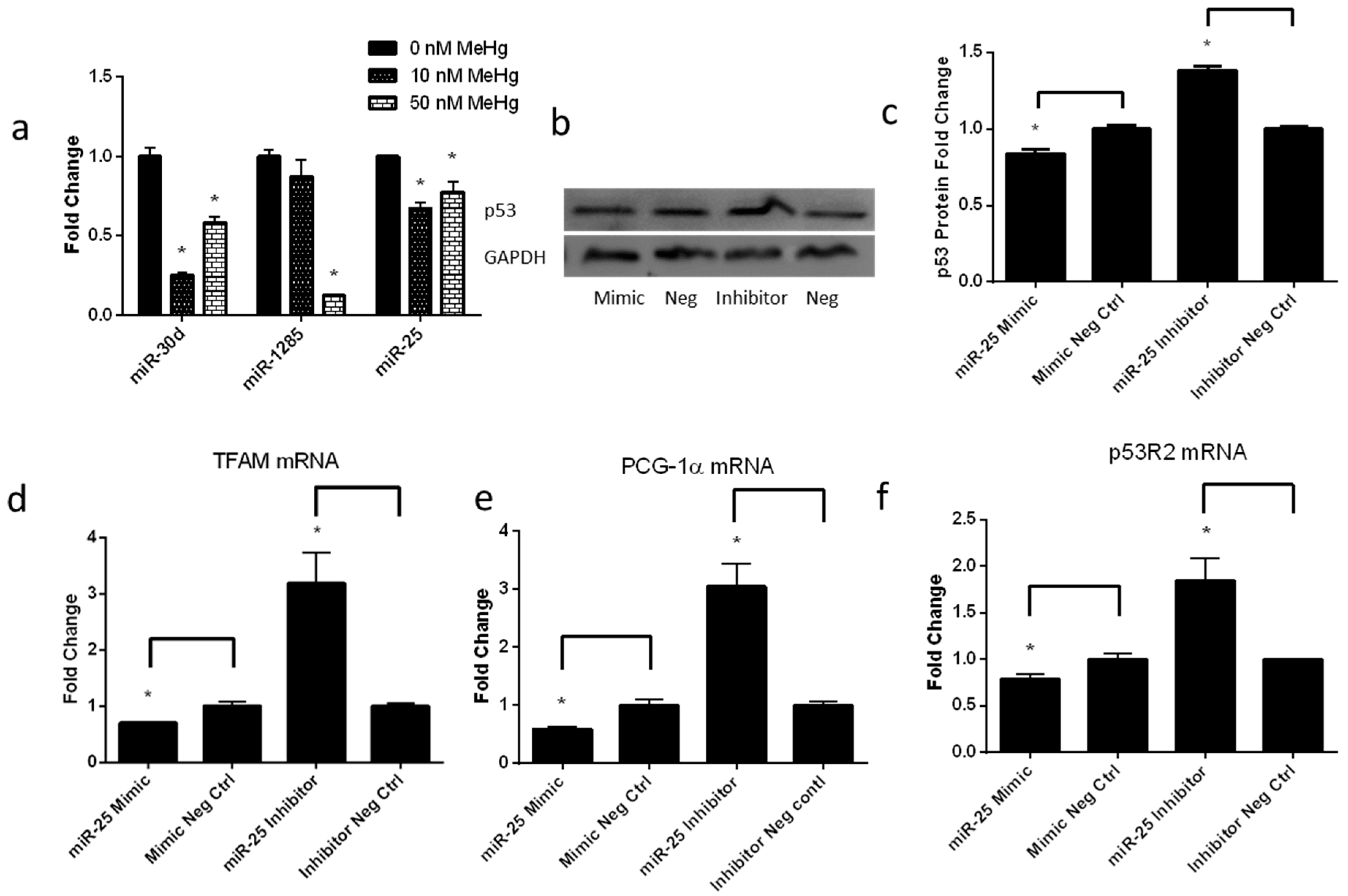
2.5. Low-Level MeHg Induces Genes Regulating Mitochondrial Biogenesis via miR-25
3. Discussion
4. Material and Methods
4.1. Cell Culture
4.2. MeHg Treatment
4.3. Assay of Cell Viability
4.4. 5-Ethynyl-2’-deoxyuridine Incorporation Assay
4.5. Reactive Oxygen Species Measurement
4.6. Analysis of mtDNA Copy Number by Quantitative Real-Time PCR
4.7. Quantitative Real Time PCR of mRNA and miRNAs
4.8. Cell Transfection
4.9. Western Blotting
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clarkson, T.W. The three modern faces of mercury. Environ. Health Perspect. 2002, 110 (Suppl. 1), 11–23. [Google Scholar] [CrossRef] [PubMed]
- Clarkson, T.W. The toxicology of mercury. Crit. Rev. Clin. Lab. Sci. 1997, 34, 369–403. [Google Scholar] [CrossRef] [PubMed]
- Ceccatelli, S.; Bose, R.; Edoff, K.; Onishchenko, N.; Spulber, S. Long-lasting neurotoxic effects of exposure to methylmercury during development. J. Intern. Med. 2013, 273, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, P.; Satoh, H.; Murata, K.; Eto, K. Adverse effects of methylmercury: Environmental health research implications. Environ. Health Perspect. 2010, 118, 1137–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandjean, P.; Weihe, P.; White, R.F. Milestone development in infants exposed to methylmercury from human milk. Neurotoxicology 1995, 16, 27–33. [Google Scholar] [PubMed]
- Grandjean, P.; Weihe, P.; White, R.F.; Debes, F.; Araki, S.; Yokoyama, K.; Murata, K.; Sorensen, N.; Dahl, R.; Jorgensen, P.J. Cognitive deficit in 7-year-old children with prenatal exposure to methylmercury. Neurotoxicol. Teratol. 1997, 19, 417–428. [Google Scholar] [CrossRef]
- Spurgeon, A. Prenatal methylmercury exposure and developmental outcomes: Review of the evidence and discussion of future directions. Environ. Health Perspect. 2006, 114, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.Q.; Yan, C.H.; Cai, S.Z.; Yuan, X.B.; Shen, X.M. Low level prenatal exposure to methylmercury disrupts neuronal migration in the developing rat cerebral cortex. Toxicology 2013, 304, 57–68. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Imanishi, S.; Sone, H.; Nagano, R.; Qin, X.Y.; Yoshinaga, J.; Akanuma, H.; Yamane, J.; Fujibuchi, W.; Ohsako, S. Effects of methylmercury exposure on neuronal differentiation of mouse and human embryonic stem cells. Toxicol. Lett. 2012, 212, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ceccatelli, S.; Dare, E.; Moors, M. Methylmercury-induced neurotoxicity and apoptosis. Chem. Biol. Interact. 2010, 188, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Falluel-Morel, A.; Sokolowski, K.; Sisti, H.M.; Zhou, X.; Shors, T.J.; Dicicco-Bloom, E. Developmental mercury exposure elicits acute hippocampal cell death, reductions in neurogenesis, and severe learning deficits during puberty. J. Neurochem. 2007, 103, 1968–1981. [Google Scholar] [CrossRef] [PubMed]
- Sokolowski, K.; Falluel-Morel, A.; Zhou, X.; DiCicco-Bloom, E. Methylmercury (MeHg) elicits mitochondrial-dependent apoptosis in developing hippocampus and acts at low exposures. Neurotoxicology 2011, 32, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Sokolowski, K.; Obiorah, M.; Robinson, K.; McCandlish, E.; Buckley, B.; DiCicco-Bloom, E. Neural stem cell apoptosis after low-methylmercury exposures in postnatal hippocampus produce persistent cell loss and adolescent memory deficits. Dev. Neurobiol. 2013, 73, 936–949. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, A.A.; Appel Hort, M.; Culbreth, M.; Lopez-Granero, C.; Farina, M.; Rocha, J.B.; Aschner, M. Methylmercury and brain development: A review of recent literature. J. Trace Elem. Med. Biol. 2016, 38, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.; Cheng, Y.; Li, B.; Petrov, A.; Joshi, P.; Berman, R.F.; Reuhl, K.R.; DiCicco-Bloom, E. Methylmercury elicits rapid inhibition of cell proliferation in the developing brain and decreases cell cycle regulator, cyclin E. Neurotoxicology 2006, 27, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Bose, R.; Onishchenko, N.; Edoff, K.; Janson Lang, A.M.; Ceccatelli, S. Inherited effects of low-dose exposure to methylmercury in neural stem cells. Toxicol. Sci. 2012, 130, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Farina, M.; Aschner, M.; Rocha, J.B. Oxidative stress in MeHg-induced neurotoxicity. Toxicol. Appl. Pharmacol. 2011, 256, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, J.; Post, J.A. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 2004, 337, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.H.; Hsieh, S.Y.; Yen, C.C.; Wu, H.C.; Chen, K.L.; Hung, D.Z.; Chen, C.H.; Wu, C.C.; Su, Y.C.; Chen, Y.W.; et al. Involvement of oxidative stress-mediated ERK1/2 and p38 activation regulated mitochondria-dependent apoptotic signals in methylmercury-induced neuronal cell injury. Toxicol. Lett. 2011, 204, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Kulawiec, M.; Ayyasamy, V.; Singh, K.K. p53 regulates mtDNA copy number and mitocheckpoint pathway. J. Carcinog. 2009, 8, 8. [Google Scholar] [PubMed]
- Wang, D.B.; Kinoshita, C.; Kinoshita, Y.; Morrison, R.S. p53 and mitochondrial function in neurons. Biochim. Biophys. Acta 2014, 1842, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Motti, D.; Bixby, J.L.; Lemmon, V.P. MicroRNAs and neuronal development. Semin. Fetal Neonatal Med. 2012, 17, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Pallocca, G.; Fabbri, M.; Sacco, M.G.; Gribaldo, L.; Pamies, D.; Laurenza, I.; Bal-Price, A. miRNA expression profiling in a human stem cell-based model as a tool for developmental neurotoxicity testing. Cell Biol. Toxicol. 2013, 29, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Breier, J.M.; Radio, N.M.; Mundy, W.R.; Shafer, T.J. Development of a high-throughput screening assay for chemical effects on proliferation and viability of immortalized human neural progenitor cells. Toxicol. Sci. 2008, 105, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Huang, S.; Wu, S.; Guo, W.; Li, J.; He, X. MicroRNA-1285 inhibits the expression of p53 by directly targeting its 3’ untranslated region. Biochem. Biophys. Res. Commun. 2010, 396, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Lupini, L.; Patergnani, S.; Rimessi, A.; Missiroli, S.; Bonora, M.; Bononi, A.; Corra, F.; Giorgi, C.; de Marchi, E.; et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr. Biol. CB 2013, 23, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Lu, Z.; Takwi, A.A.; Chen, W.; Callander, N.S.; Ramos, K.S.; Young, K.H.; Li, Y. Negative regulation of the tumor suppressor p53 gene by microRNAs. Oncogene 2011, 30, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Culbreth, M.E.; Harrill, J.A.; Freudenrich, T.M.; Mundy, W.R.; Shafer, T.J. Comparison of chemical-induced changes in proliferation and apoptosis in human and mouse neuroprogenitor cells. Neurotoxicology 2012, 33, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Takanezawa, Y.; Nakamura, R.; Sone, Y.; Uraguchi, S.; Kiyono, M. Atg5-dependent autophagy plays a protective role against methylmercury-induced cytotoxicity. Toxicol. Lett. 2016, 262, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, Y.; St Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.H.; Naito, A.; Mizumachi, T.; Evans, T.T.; Douglas, M.G.; Cooney, C.A.; Fan, C.Y.; Higuchi, M. Mitochondrial regulation of cancer associated nuclear DNA methylation. Biochem. Biophys. Res. Commun. 2007, 364, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Tamm, C.; Duckworth, J.; Hermanson, O.; Ceccatelli, S. High susceptibility of neural stem cells to methylmercury toxicity: Effects on cell survival and neuronal differentiation. J. Neurochem. 2006, 97, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Balansky, R.; Novikov, L.; Giannoni, P.; Izzotti, A.; de Flora, S. No effect of treatment with carcinogens on cytosine methylation of mitochondrial DNA isolated from rat organs by phenol-free alkaline extraction. Cancer Lett. 1995, 97, 17–23. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552 Pt 2, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Wei, Y.H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol. 2005, 37, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Czajka, A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 2013, 13, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef] [PubMed]
- Saleem, A.; Adhihetty, P.J.; Hood, D.A. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol. Genom. 2009, 37, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Castegna, A.; Infantino, V.; Andria, G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 2013, 110, 25–34. [Google Scholar] [CrossRef] [PubMed]
- miRWalk2.0: A Comprehensive Atlas of Predicted and Validated miRNA-Target Interactions. Available online: http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/ (accessed on 23 September 2016).
- Chang, X.; Lu, W.; Dou, T.; Wang, X.; Lou, D.; Sun, X.; Zhou, Z. Paraquat inhibits cell viability via enhanced oxidative stress and apoptosis in human neural progenitor cells. Chem. Biol. Interact. 2013, 206, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, M.; Miyazono, Y.; Sasamoto, K.; Ohkura, Y.; Ueno, K. A highly water-soluble disulfonated tetrazolium salt as a chromogenic indicator for NADH as well as cell viability. Talanta 1997, 44, 1299–1305. [Google Scholar] [CrossRef]
- Izawa, Y.; Takahashi, S.; Suzuki, N. Pioglitazone enhances pyruvate and lactate oxidation in cultured neurons but not in cultured astroglia. Brain Res. 2009, 1305, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.D.; Mahmud, A.; McKenzie, M.; Trounce, I.A.; St John, J.C. Mitochondrial DNA copy number is regulated in a tissue specific manner by DNA methylation of the nuclear-encoded DNA polymerase gamma A. Nucleic Acids Res. 2012, 40, 10124–10138. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Chauhan, V.; Kaur, K.; Brown, W.T.; LaFauci, G.; Wegiel, J.; Chauhan, A. Alterations in mitochondrial DNA copy number and the activities of electron transport chain complexes and pyruvate dehydrogenase in the frontal cortex from subjects with autism. Transl. Psychiatry 2013, 3, e299. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Park, S.G.; Song, S.Y.; Kim, J.K.; Sung, J.H. Reactive oxygen species-responsive miR-210 regulates proliferation and migration of adipose-derived stem cells via PTPN2. Cell Death Dis. 2013, 4, e588. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Forward | Reverse |
|---|---|---|
| p16 | 5′-CTCGTGCTGATGCTACTGAGGA-3′ | 5′-GGTCGGCGCAGTTGGGCTCC-3′ |
| p21 | 5′-AGGTGGACCTGGAGACTCTCAG-3′ | 5′-TCCTCTTGGAGAAGATCAGCCG-3′ |
| p53 | 5′-CCTCAGCATCTTATCCGAGTGG-3′ | 5′-TGGATGGTGGTACAGTCAGAGC-3′ |
| PGC-1α | 5′-CCAAAGGATGCGCTCTCGTTCA-3′ | 5′-CGGTGTCTGTAGTGGCTTGACT-3′ |
| TFAM | 5′-GTGGTTTTCATCTGTCTTGGCAAG-3′ | 5′-TTCCCTCCAACGCTGGGCAATT-3′ |
| p53R2 | 5′-ACTTCATCTCTCACATCTTAGCCT-3′ | 5′-AAACAGCGAGCCTCTGGAACCT-3′ |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Yan, M.; Zhao, L.; Wu, Q.; Wu, C.; Chang, X.; Zhou, Z. Low-Dose Methylmercury-Induced Genes Regulate Mitochondrial Biogenesis via miR-25 in Immortalized Human Embryonic Neural Progenitor Cells. Int. J. Mol. Sci. 2016, 17, 2058. https://doi.org/10.3390/ijms17122058
Wang X, Yan M, Zhao L, Wu Q, Wu C, Chang X, Zhou Z. Low-Dose Methylmercury-Induced Genes Regulate Mitochondrial Biogenesis via miR-25 in Immortalized Human Embryonic Neural Progenitor Cells. International Journal of Molecular Sciences. 2016; 17(12):2058. https://doi.org/10.3390/ijms17122058
Chicago/Turabian StyleWang, Xinjin, Mengling Yan, Lina Zhao, Qing Wu, Chunhua Wu, Xiuli Chang, and Zhijun Zhou. 2016. "Low-Dose Methylmercury-Induced Genes Regulate Mitochondrial Biogenesis via miR-25 in Immortalized Human Embryonic Neural Progenitor Cells" International Journal of Molecular Sciences 17, no. 12: 2058. https://doi.org/10.3390/ijms17122058