What Does This Mutation Mean? The Tools and Pitfalls of Variant Interpretation in Lymphoid Malignancies

 ,
,
Abstract
:1. Introduction
2. Is It a Somatic Variant?
2.1. Pre-Analytical and Bioinformatic Issues
2.1.1. Obtaining Germline DNA
2.1.2. Technical Considerations: FFPE Tissues
2.1.3. Bioinformatic Aspects
- -
- Some variant callers such as Freebayes or VarScan can be run in a multi-sample mode, allowing sequencing data to be obtained for all samples regarding a particular allele, if found to be mutated in one sample. From this, the minimal/mean/median frequency of reads supporting the altered sequence among all samples can be computed and inform about the background signal at this position; a VAF/median frequency ratio can be calculated, and a filtering threshold applied to select more probable true somatic mutations.
- -
- The distribution of reference allele and alternate allele between forward and reverse sequencing strand should be similar, i.e., the ratio (reference forward reads)/(reference reverse reads) should be comparable to the ratio (alternate forward reads)/(alternate reverse reads). An Allele Strand Ratio (ASR) can be calculated, (reference F/R reads)/(alternate F/R reads); for real mutations ASR should be close to 1, conversely an ASR very distant from 1 suggests possible artifact variation.
- -
- The occurrence of a variant in samples of the same run should be calculated, a too high recurrence would point out an artifact.
- -
- >Over time, a local database of recurrent artifacts can be built to help remove known false positive calls.
2.2. Databases for Germline and Somatic Variants
2.2.1. Constitutional Databases
- The Single Nucleotide Polymorphism Database (dbSNP) of nucleotide sequence variation (SNV) from the National Cancer Bioinformatics Institute (NCBI): This public domain catalogue, started in 1998 [32] and first released in 2000 by the NCBI as a part of the PubMed website includes only variants from non-tumor samples. The definition of small variations comprises SNPs, small (<50 nucleotides) insertions or deletions (InDels), and retroposable element insertions and microsatellite repeat variations. The catalogue is based on voluntary contributions as any public laboratory and private organizations can submit data that, after review, will be implemented as “first class data”. The “second-class data” available are computed from the original submitted data, automatically gathered from PubMed during the dbSNP build cycle. In 2004, its false positive rate was estimated at 15–17% by Mitchell et al. [34] mainly due to its conception. It accepts submissions from many sources that are difficult to verify regarding uncritical bioinformatic alignments of highly similar but distinct DNA sequences, or PCRs with primers that cannot discriminate between similar but distinct DNA sequences. In February 2017, the last build regarding Homo Sapiens was released, build 150 comprising 325,658,303 variants of which 135,967,291 were validated (as first-class data). In this database, MYD88 L265P is known as rs387907272, and was considered as a SNP until very recently. It has now been curated as a single nucleotide variant with a pathogenic clinical significance and a variant frequency <0.01%. This well-known pathogenic mutation highlights the difficulty to filter out polymorphisms due to false positives in such databases that are constantly evolving.
- 1000 Genomes Project: The goal of the 1000 Genomes Project was to identify genetic variants with frequencies of at least 1% in the populations studied. It ran between 2008 and 2015 and sequenced 2504 samples from 26 populations [30,31]. Whilst the samples for the 1000 Genomes Project had no associated medical or phenotype data, all participants had to declare themselves to be healthy with self-reported ethnicity and gender. MYD88 L265P is described with an overall allele frequency of 0.02%, from one non-Finnish European population. In this population, the MAF was still below a threshold that most studies would consider as rare (<0.01%) [35]. It is of note that the SNPs and short Indels of the 1000 Genomes Project are included in the dbSNP, making it redundant to consult both databases.
- The Exome Aggregation Consortium (ExAC): This database, compiled by the Broad Institute, tends to aggregate and harmonize exome sequencing data from a variety of large-scale sequencing projects. All of these projects provided their raw sequencing data (generated using various technologies), which were then reprocessed and variant called through one unique pipeline to increase consistency. Notably, the germline information obtained by The Cancer Genome Atlas (see below) is available in the ExAC database.Given the nature of the projects aggregated (such as “Inflammatory Bowel Disease”, “Jackson Heart Study”, or “Schizophrenia Trios from Taiwan”), not all of the patients sequenced were healthy. In ExAC, 60,706 unrelated individuals have been sequenced [29], where people with severe pediatric diseases and their first-degree relatives have been removed. This database is not intended to be further extended, and a new project called “The Genome Aggregation Database” (gnomAD; see below) includes all the data contained in the ExAC database. In this ExAC database, MYD88 p.L265P is present at a frequency of 0.01% in the general population and is considered as too common to plausibly cause disease.
- The Genome Aggregation Database (gnomAD): This database aims to aggregate the data from genome and exome studies into one database and is mainly driven by the Broad Institute. It uses the data from the ExAC database and from a consortium of more than 100 investigators and uses the same process as that used for ExAC (same pipeline and variant calling to re-process all data). It therefore contains data from the ExAC, 1000 Genomes Project, and the Cancer Genome Atlas (TCGA; see below) among others and spans 123,136 exomes and 15,496 genomes from unrelated individuals. In the gnomAD, MYD88 p.L265P is described with an allele frequency 0.0036% and count of 9.
- National Heart, Lung and Blood Institute Grand Opportunity (NHLBI GO) Exome Sequencing Project (ESP): This is an on-going project bringing together US investigators aiming to discover new genes and mechanisms leading to heart disorders, lung disorders, or benign blood disorders. No cancer sequencing data are included in this database comprising 6503 samples in the ESP6500SI-V2 release. The variant calling and analysis of the data are centralized. A subset of the data was published in 2012 [33] and is therefore present in the dbSNP.
2.2.2. Cancer Databases
- The Cancer Genome Atlas (TCGA): This project, that ended in 2017, was a collaboration between the National Cancer Institute (NCI) and National Human Genome Research Institute (NHGRI) [36]. More than 11,000 patients suffering from a total 33 types of cancer (in hematology, only acute myeloblastic leukaemia and diffuse large B cell lymphoma were targeted) were included. Cancer tissue and matched normal tissues were collected as well as clinical data, then sequenced, and registered.
- The International Cancer Genome Consortium (ICGC): The ICGC is a confederation of international working groups that aims to describe driver somatic mutations in more than 50 types or subtypes of cancers. Most working groups are required to sequence at least 500 samples by Whole Genome Shotgun analyses, with exceptions made for rare or very homogenous types of cancer. As this technique is not yet available everywhere for such large-scale projects, interim goals are accepted such as sequencing only the region of interest, analysis of low genome coverage of paired-end reads for rearrangements, or genotyping arrays. In addition to building this catalogue of somatic mutations, analyses of DNA methylation and RNA expression are planned. Processing the samples must be conducted according to the ICGC guidelines to ensure similar quality in the projects. Lymphoid malignancies are well represented with six different dedicated projects [37,38].
- The Catalogue of Somatic Mutation in Cancer (COSMIC): As is the case for dbSNP, two different types of data are present in the COSMIC. The first-class data is expert-curated, with manual input data after comprehensive review of selected genes after its submission by a group or laboratory. These genes are those presented in Census genes, a dynamic catalogue of genes that have been recognized as implicated in cancer that was initially published in Nature Reviews Cancer [40]. The second type is genome-wide screening data, uploaded from publications or imported from other databases such as the TCGA or ICGC. The uploaded data from publications imply that some false positives are included in this database through the lack of germline sample controls, simple laboratory errors, or poor-quality samples such as FFPE ones. Nevertheless, this catalogue is the most comprehensive resource for information on somatic mutations in human cancer and aims at providing somatic mutation frequencies [39].
2.3. Assessing the Functional Consequences and Clinical Impact of the Variants
2.3.1. In Silico Modelling
2.3.2. In Vitro Modelling
2.3.3. Limits of Current Prediction Tools and Models to Predict the Clinical Impact
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pastore, A.; Jurinovic, V.; Kridel, R.; Hoster, E.; Staiger, A.M.; Szczepanowski, M.; Pott, C.; Kopp, N.; Murakami, M.; Horn, H.; et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: A retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015, 16, 1111–1122. [Google Scholar] [CrossRef]
- Morschhauser, F.; Salles, G.; McKay, P.; Tilly, H.; Schmitt, A.; Gerecitano, J.; Johnson, P.; Le Gouill, S.; Dickinson, M.J.; Fruchart, C.; et al. Interim Report from a Phase 2 Multicenter Study of Tazemetostat, an Ezh2 Inhibitor, in Patients with Relapsed or Refractory B-Cell Non-Hodgkin Lymphomas. Hematol. Oncol. 2017, 35, 24–25. [Google Scholar] [CrossRef]
- Roy, S.; Coldren, C.; Karunamurthy, A.; Kip, N.S.; Klee, E.W.; Lincoln, S.E.; Leon, A.; Pullambhatla, M.; Temple-Smolkin, R.L.; Voelkerding, K.V.; et al. Standards and Guidelines for Validating Next-Generation Sequencing Bioinformatics Pipelines: A Joint Recommendation of the Association for Molecular Pathology and the College of American Pathologists. J. Mol. Diagn. 2018, 20, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [PubMed]
- Do, H.; Dobrovic, A. Sequence artifacts in DNA from formalin-fixed tissues: Causes and strategies for minimization. Clin. Chem. 2015, 61, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Choi, Y.-L.; Kwon, M.J.; Kim, R.N.; Kim, Y.J.; Song, J.-Y.; Jung, K.S.; Shin, Y.K. Comparison of Accuracy of Whole-Exome Sequencing with Formalin-Fixed Paraffin-Embedded and Fresh Frozen Tissue Samples. PLoS ONE 2015, 10, e0144162. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Li, J.; Tan, A.Y.-C.; Vedururu, R.; Pang, J.-M.B.; Do, H.; Ellul, J.; Doig, K.; Bell, A.; MacArthur, G.A.; et al. CANCER 2015 Cohort Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med. Genom. 2014, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute. Genome Analysis Toolkit. Available online: https://software.broadinstitute.org/gatk/ (accessed on 11 February 2018).
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv, 2012. Available online: https://arxiv.org/abs/1207.3907(accessed on 11 February 2018)arXiv:1207.3907.
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Krøigård, A.B.; Thomassen, M.; Lænkholm, A.-V.; Kruse, T.A.; Larsen, M.J. Evaluation of Nine Somatic Variant Callers for Detection of Somatic Mutations in Exome and Targeted Deep Sequencing Data. PLoS ONE 2016, 11, e0151664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Jia, P.; Li, F.; Chen, H.; Ji, H.; Hucks, D.; Dahlman, K.B.; Pao, W.; Zhao, Z. Detecting somatic point mutations in cancer genome sequencing data: A comparison of mutation callers. Genome Med. 2013, 5, 91. [Google Scholar] [CrossRef] [PubMed]
- Sandmann, S.; de Graaf, A.O.; Karimi, M.; van der Reijden, B.A.; Hellström-Lindberg, E.; Jansen, J.H.; Dugas, M. Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Sci. Rep. 2017, 7, 43169. [Google Scholar] [CrossRef] [PubMed]
- Kockan, C.; Hach, F.; Sarrafi, I.; Bell, R.H.; McConeghy, B.; Beja, K.; Haegert, A.; Wyatt, A.W.; Volik, S.V.; Chi, K.N.; et al. SiNVICT: Ultra-sensitive detection of single nucleotide variants and indels in circulating tumour DNA. Bioinformatics 2017, 33, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Xuei, X.; Li, L.; Nakshatri, H.; Edenberg, H.J.; Liu, Y. RareVar: A Framework for Detecting Low-Frequency Single-Nucleotide Variants. J. Comput. Biol. 2017, 24, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cuesta, L.; Perdomo, S.; Avogbe, P.H.; Leblay, N.; Delhomme, T.M.; Gaborieau, V.; Abedi-Ardekani, B.; Chanudet, E.; Olivier, M.; Zaridze, D.; et al. Identification of Circulating Tumor DNA for the Early Detection of Small-cell Lung Cancer. EBioMedicine 2016, 10, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Muller, E.; Goardon, N.; Brault, B.; Rousselin, A.; Paimparay, G.; Legros, A.; Fouillet, R.; Bruet, O.; Tranchant, A.; Domin, F.; et al. OutLyzer: Software for extracting low-allele-frequency tumor mutations from sequencing background noise in clinical practice. Oncotarget 2016, 7, 79485–79493. [Google Scholar] [CrossRef] [PubMed]
- Shlien, A.; Malkin, D. Copy number variations and cancer. Genome Med. 2009, 1, 62. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Mack, S.C.; Witt, H.; Piro, R.M.; Gu, L.; Zuyderduyn, S.; Stütz, A.M.; Wang, X.; Gallo, M.; Garzia, L.; Zayne, K.; et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014, 506, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.; Mohankumar, K.M.; Punchihewa, C.; Weinlich, R.; Dalton, J.D.; Li, Y.; Lee, R.; Tatevossian, R.G.; Phoenix, T.N.; Thiruvenkatam, R.; et al. C11orf95-RELA fusions drive oncogenic NF-κB signalling in ependymoma. Nature 2014, 506, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T. 1000 G. P. A global reference for human genetic variation. Nature 2015, 526, 68. [Google Scholar] [CrossRef] [Green Version]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.-Y.; et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015, 526, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999, 9, 677–679. [Google Scholar] [PubMed]
- Tennessen, J.A.; Bigham, A.W.; O’Connor, T.D.; Fu, W.; Kenny, E.E.; Gravel, S.; McGee, S.; Do, R.; Liu, X.; Jun, G.; et al. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012, 337, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.A.; Zwick, M.E.; Chakravarti, A.; Cutler, D.J. Discrepancies in dbSNP confirmation rates and allele frequency distributions from varying genotyping error rates and patterns. Bioinformatics 2004, 20, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Panoutsopoulou, K.; Tachmazidou, I.; Zeggini, E. In search of low-frequency and rare variants affecting complex traits. Hum. Mol. Genet. 2013, 22, R16–R21. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- The ICGC MMML-Seq Project. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [CrossRef]
- Ramsay, A.J.; Martínez-Trillos, A.; Jares, P.; Rodríguez, D.; Kwarciak, A.; Quesada, V. Next-generation sequencing reveals the secrets of the chronic lymphocytic leukemia genome. Clin. Transl. Oncol. 2013, 15, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Bhamra, G.; Bamford, S.; Dawson, E.; Kok, C.; Clements, J.; Menzies, A.; Teague, J.W.; Futreal, P.A.; Stratton, M.R. The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr. Protoc. Hum. Genet. 2008. [CrossRef]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- MSKCC. cBioPortal for Cancer Genomics. Available online: http://www.cbioportal.org/ (accessed on 11 February 2018).
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.-J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF Mutations in Hairy-Cell Leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [PubMed]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P Somatic Mutation in Waldenström’s Macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef] [PubMed]
- Odejide, O.; Weigert, O.; Lane, A.A.; Toscano, D.; Lunning, M.A.; Kopp, N.; Kim, S.; van Bodegom, D.; Bolla, S.; Schatz, J.H.; et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood 2014, 123, 1293–1296. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Couronné, L.; Khiabanian, H.; Kim, M.-Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Bogusz, A.M.; Bagg, A. Genetic aberrations in small B-cell lymphomas and leukemias: Molecular pathology, clinical relevance and therapeutic targets. Leuk. Lymphoma 2016, 57, 1991–2013. [Google Scholar] [CrossRef] [PubMed]
- Rosenquist, R.; Rosenwald, A.; Du, M.-Q.; Gaidano, G.; Groenen, P.; Wotherspoon, A.; Ghia, P.; Gaulard, P.; Campo, E.; Stamatopoulos, K. European Research Initiative on CLL (ERIC) and the European Association for Haematopathology (EAHP) Clinical impact of recurrently mutated genes on lymphoma diagnostics: State-of-the-art and beyond. Haematologica 2016, 101, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Tsang, H.; Addepalli, K.; Davis, S.R. Resources for Interpreting Variants in Precision Genomic Oncology Applications. Front. Oncol. 2017, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.H.; Brooks, A.N.; Wu, X.; Shrestha, Y.; Chouinard, C.; Piccioni, F.; Bagul, M.; Kamburov, A.; Imielinski, M.; Hogstrom, L.; et al. High-throughput Phenotyping of Lung Cancer Somatic Mutations. Cancer Cell 2016, 30, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011, 32, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.C.; Joyce, C.; Ritchie, G.R.S.; John, S.L.; Cunningham, F.; Asimit, J.; Zeggini, E. A combined functional annotation score for non-synonymous variants. Hum. Hered. 2012, 73, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Mathe, E.; Olivier, M.; Kato, S.; Ishioka, C.; Hainaut, P.; Tavtigian, S.V. Computational approaches for predicting the biological effect of p53 missense mutations: A comparison of three sequence analysis based methods. Nucleic Acids Res. 2006, 34, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Miñana, B.; Valcárcel, J.; Gabaldón, T.; Lehner, B. Synonymous mutations frequently act as driver mutations in human cancers. Cell 2014, 156, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Gotea, V.; Gartner, J.J.; Qutob, N.; Elnitski, L.; Samuels, Y. The functional relevance of somatic synonymous mutations in melanoma and other cancers. Pigment Cell Melanoma Res. 2015, 28, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.; Mardis, E.R.; et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012, 22, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Chen, S.; Isik, L.; Tyekucheva, S.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Cancer-specific high-throughput annotation of somatic mutations: Computational prediction of driver missense mutations. Cancer Res. 2009, 69, 6660–6667. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Lopez-Bigas, N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012, 40, e169. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 2013, 155, 948–962. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUST: Exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2013, 29, 2238–2244. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Kandoth, C.; Reimand, J.; Lawrence, M.S.; Getz, G.; Bader, G.D.; Ding, L.; et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci. Rep. 2013, 3, 2650. [Google Scholar] [CrossRef] [PubMed]
- Tokheim, C.J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; Karchin, R. Evaluating the evaluation of cancer driver genes. Proc. Natl. Acad. Sci. USA 2016, 113, 14330–14335. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Cancer Cell Line Encyclopedia Consortium. Genomics of Drug Sensitivity in Cancer Consortium Pharmacogenomic agreement between two cancer cell line data sets. Nature 2015, 528, 84–87. [CrossRef]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Home Page—Cancerrxgene—Genomics of Drug Sensitivity in Cancer. Available online: http://www.cancerrxgene.org/ (accessed on 11 February 2018).
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Cancer Therapeutics Response Portal. Available online: https://portals.broadinstitute.org/ctrp.v2.1/ (accessed on 11 February 2018).
- Peck, D.; Crawford, E.D.; Ross, K.N.; Stegmaier, K.; Golub, T.R.; Lamb, J. A method for high-throughput gene expression signature analysis. Genome Biol. 2006, 7, R61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Khiabanian, H.; Spina, V.; Ciardullo, C.; Bruscaggin, A.; Famà, R.; Rasi, S.; Monti, S.; Deambrogi, C.; De Paoli, L.; et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 2014, 123, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Malcikova, J.; Stano-Kozubik, K.; Tichy, B.; Kantorova, B.; Pavlova, S.; Tom, N.; Radova, L.; Smardova, J.; Pardy, F.; Doubek, M.; et al. Detailed analysis of therapy-driven clonal evolution of TP53 mutations in chronic lymphocytic leukemia. Leukemia 2015, 29, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Nadeu, F.; Delgado, J.; Royo, C.; Baumann, T.; Stankovic, T.; Pinyol, M.; Jares, P.; Navarro, A.; Martín-García, D.; Beà, S.; et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood 2016, 127, 2122–2130. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, S.; Clifford, R.; Antoniou, P.; Parker, H.; Robbe, P.; Larrayoz, M.; Davis, Z. The Contribution of Gene Mutations to Long-Term Clinical Outcomes: Data from the Randomised UK LRF CLL4 Trial. Am. Soc. Hematol. 2017, 130, 259. [Google Scholar]
- Cooper, J.N.; Young, N.S. Clonality in context: Hematopoietic clones in their marrow environment. Blood 2017, 130, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, X.; Munshi, M.; Xu, L.; Tsakmaklis, N.; Demos, M.; Kofides, A. BTK(Cys481Ser) Mutation Drives Ibrutinib Resistance through ERK1/2 Hyperactivation, and Can Confer a Protective Effect on Bystander Waldenstrom’s Macroglobulinemia and ABC DLBCL Cells through Paracrine Mediated Pro-Survival Signaling. Am. Soc. Hematol. 2017, 130, 803. [Google Scholar]
- Liu, M.; Watson, L.T.; Zhang, L. Predicting the combined effect of multiple genetic variants. Hum. Genom. 2015, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica 2015, 100, e438–e442. [Google Scholar] [CrossRef] [PubMed]
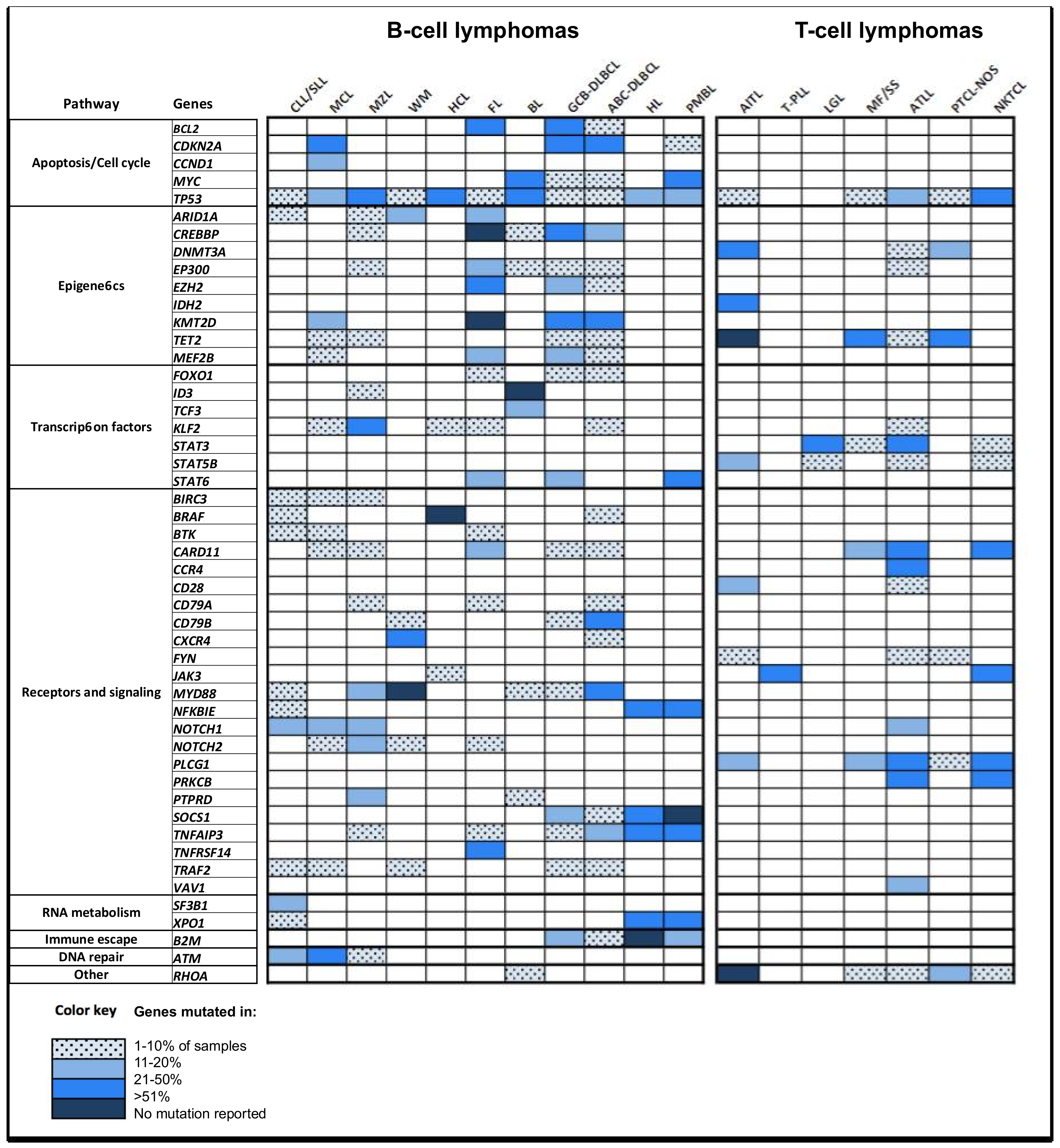

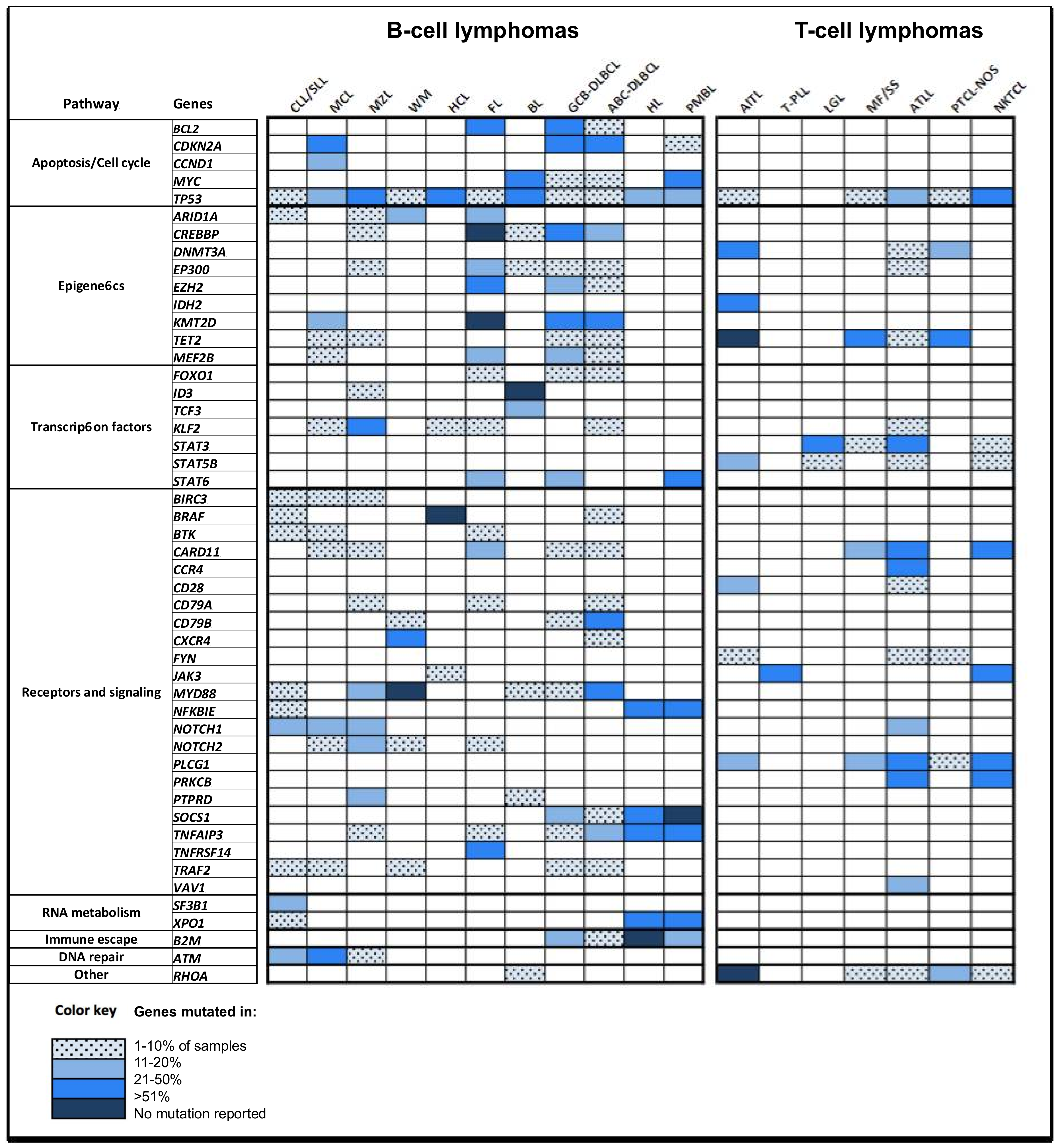{kind=link}
| Database | Cell of Origin | Healthy/Non Cancer Disease | Data | Number of Exome/Genome | URL |
|---|---|---|---|---|---|
| ExAC | Germline | both | Exome | 60,706 | http://exac.broadinstitute.org/ |
| gnomAD | Germline | both | Exome/Genome | 136,632 * | http://gnomad.broadinstitute.org/about |
| 1000 Genomes | Germline | Healthy | Exome/Genome | 2504 | http://www.internationalgenome.org/home |
| dbSNP | Germline | both | Exome/Genome | NA | https://www-ncbi-nlm-nih-gov.gate2.inist.fr/projects/SNP/ |
| ESP | Germline | both | Exome | 6503 | http://evs.gs.washington.edu/EVS/ |
| Database | % African/African American | % Latino/Mixed Americans | % East Asian | % Finnish | % Non-Finnish European | % South Asian | % Ashkenazi | % Other | MYD88 p.L265P Allele Frequency | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| ExAC | 8.57 | 9.53 | 7.13 | 5.45 | 54.97 | 13.6 | NA | 0.75 | 0.01% | [29] |
| gnomAD | 8.80 | 12.60 | 6.91 | 9.44 | 46.38 | 11.26 | 3.72 | 2.37 | 0.0036% | [29] |
| 1000 Genomes | 26.4 | 13.86 | 19.53 | 3.95 | 16.13 | 20.13 | NA | 0 | 0.02% | [30,31] |
| dbSNP | NA | NA | NA | NA | NA | NA | NA | NA | * | [32] |
| ESP | NA | NA | NA | NA | NA | NA | NA | NA | Not present | [33] |
| Database | Cell of Origin | Data | Number of Exome/Genome | Link | Reference |
|---|---|---|---|---|---|
| TCGA | Somatic | Exome/Genome | 11,077 | https://tcga-data.nci.nih.gov/docs/publications/tcga/ | [36] |
| ICGC | Somatic | Exome/Genome | 17,000 | http://icgc.org/ | [37,38] |
| COSMIC | Somatic | Exome/Genome | 32,000 genomes + 25,000 peer reviewed papers (genomes and/or exomes) | http://cancer.sanger.ac.uk/cosmic | [39,40] |
| Gene | Hotspot Mutation | Lymphoid Neoplasms (Frequency) | Commentary |
|---|---|---|---|
| BRAF | V600E | HCL (>90%), MM (5%) | targeted therapy available |
| EZH2 | Y646, A692 * | FL (30%), DLBCL (10%) | targeted therapy available;* Amino-acid numbering based on transcript NM_004456.4 (sometimes reported as Y641 and A687 with NM_001203247.1) |
| IDH2 | R172 | AITL (40%) | targeted therapy available |
| K/N/H-RAS | G12, G13, Q61 | MM (40%), DLBCL (10%) | targeted therapy available |
| MYD88 | L265P ** | LPL (95%), MGUS (50%), DLBCL (10%), CLL (5%), PCNSL (50%), EMZL/MALT (5%), NMZL (5%) | ** Amino-acid numbering based on transcript NM_002468.4 (sometimes reported as L273P with NM_001172567.1) |
| RHOA | G17V | AITL (60%), PTCL-NOS (20%) | - |
| SF3B1 | K700E, K666 | CLL (15%) | Prognostic impact in CLL |
| XPO1 | E571 | PMBL (25%), cHL (25%), CLL (5%) | targeted therapy available |
| Resource | URL | References |
|---|---|---|
| SIFTSorting Intolerant From Tolerant | http://sift.jcvi.org | [55,56] |
| PROVEANProtein Variation Effect Analyzer | http://provean.jcvi.org/index.php | [57] |
| PolyPhen-2Polymorphism Phenotyping v2 | http://genetics.bwh.harvard.edu/pph2 | [58] |
| MutationAssessor | http://mutationassessor.org | [59] |
| MutationTaster | http://www.mutationtaster.org/ | [60] |
| CAROL *Combined Annotation scoRing toOL | http://www.sanger.ac.uk/science/tools/carol | [61] |
| Align GCGD ** | http://agvgd.hci.utah.edu/ | [62] |
| dbNSFP v3.0 ***database for Nonsynonymous SNPs’ Functional Predictions | https://sites.google.com/site/jpopgen/dbNSFP | [54] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guillermin, Y.; Lopez, J.; Chabane, K.; Hayette, S.; Bardel, C.; Salles, G.; Sujobert, P.; Huet, S. What Does This Mutation Mean? The Tools and Pitfalls of Variant Interpretation in Lymphoid Malignancies. Int. J. Mol. Sci. 2018, 19, 1251. https://doi.org/10.3390/ijms19041251
Guillermin Y, Lopez J, Chabane K, Hayette S, Bardel C, Salles G, Sujobert P, Huet S. What Does This Mutation Mean? The Tools and Pitfalls of Variant Interpretation in Lymphoid Malignancies. International Journal of Molecular Sciences. 2018; 19(4):1251. https://doi.org/10.3390/ijms19041251
Chicago/Turabian StyleGuillermin, Yann, Jonathan Lopez, Kaddour Chabane, Sandrine Hayette, Claire Bardel, Gilles Salles, Pierre Sujobert, and Sarah Huet. 2018. "What Does This Mutation Mean? The Tools and Pitfalls of Variant Interpretation in Lymphoid Malignancies" International Journal of Molecular Sciences 19, no. 4: 1251. https://doi.org/10.3390/ijms19041251