
A Novel Long Noncoding RNA in Osteocytes Regulates Bone Formation through the Wnt/β-Catenin Signaling Pathway
 ,
,
Abstract
:1. Introduction
2. Results
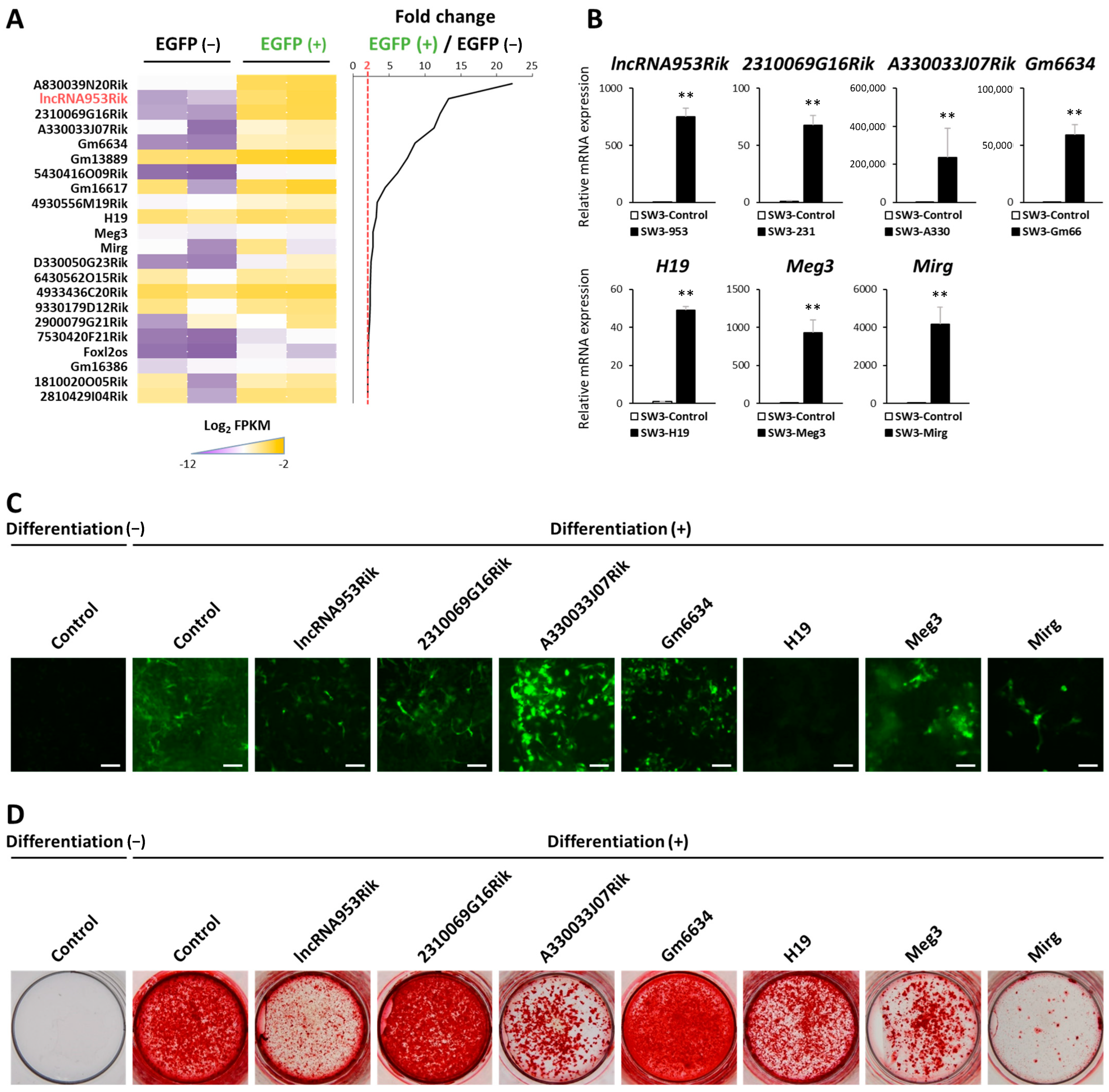
2.1. Identification and Characterization of lncRNAs in Osteocytes
2.2. lncRNA953Rik Inhibits Osteogenesis
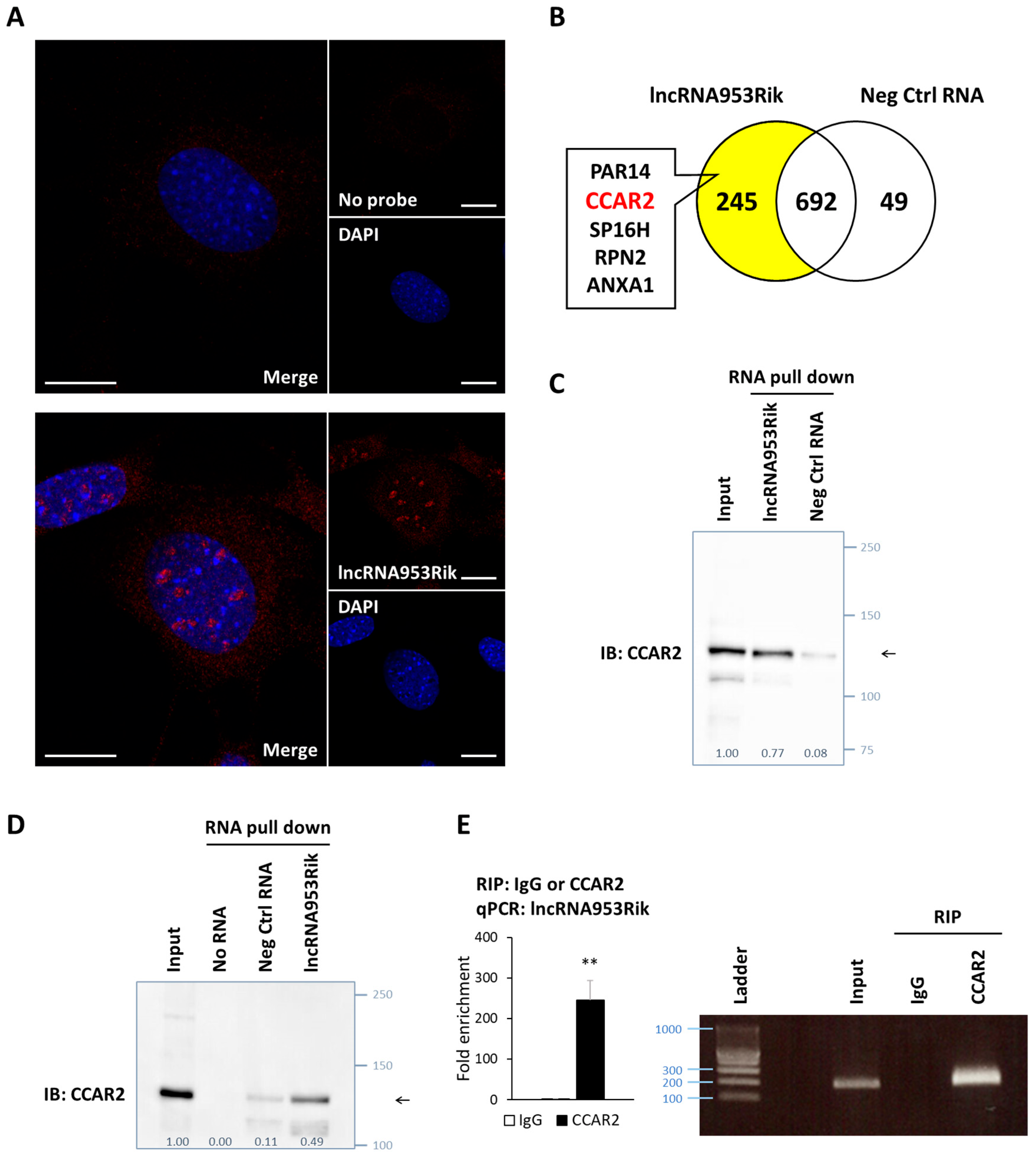
2.3. lncRNA953Rik Binds to CCAR2
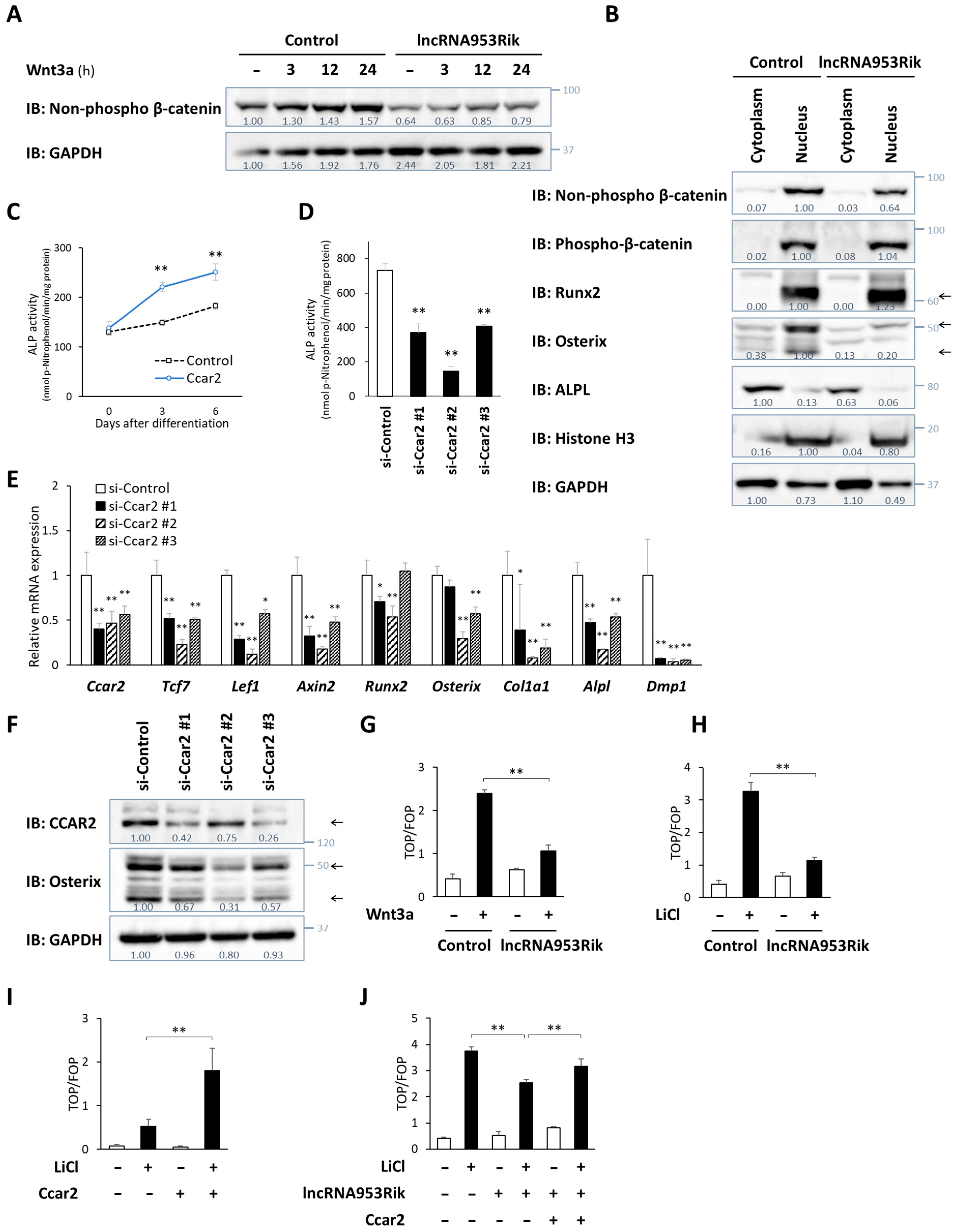
2.4. lncRNA953Rik Suppresses Wnt/β-Catenin Signaling
2.5. lncRNA953Rik Suppresses Osterix Transcription by Sequestering CCAR2 from HDAC1
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. Alkaline Phosphatase (ALP) Assay
4.3. ALP Staining
4.4. Alizarin Red S Staining
4.5. RNA Extraction, Reverse Transcription and qPCR
4.6. Immunoblotting
4.7. Cell Fractionation
4.8. Antisense Oligonucleotide-Mediated Knockdown
4.9. siRNA-Mediated Knockdown
4.10. Mice
4.11. Isolation of Osteocytes
4.12. RNA-seq
4.13. FISH
4.14. RNA Pull-Down
4.15. Silver Staining
4.16. Mass Spectrometry
4.17. RNA Immunoprecipitation (RIP)
4.18. Luciferase Assay
4.19. Immunoprecipitation
4.20. Chromatin Immunoprecipitation (ChIP)
4.21. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef] [PubMed]
- The ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef]
- Zhang, P.; Wu, S.; He, Y.; Li, X.; Zhu, Y.; Lin, X.; Chen, L.; Zhao, Y.; Niu, L.; Zhang, S.; et al. LncRNA-Mediated Adipogenesis in Different Adipocytes. Int. J. Mol. Sci. 2022, 23, 7488. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lyu, Y.; Xiang, R.; Yang, J. Long Noncoding RNAs in the Pathogenesis of Insulin Resistance. Int. J. Mol. Sci. 2022, 23, 16054. [Google Scholar] [CrossRef]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef]
- Lee, N.K.; Sowa, H.; Hinoi, E.; Ferron, M.; Ahn, J.D.; Confavreux, C.; Dacquin, R.; Mee, P.J.; McKee, M.D.; Jung, D.Y.; et al. Endocrine regulation of energy metabolism by the skeleton. Cell 2007, 130, 456–469. [Google Scholar] [CrossRef]
- Oury, F.; Sumara, G.; Sumara, O.; Ferron, M.; Chang, H.; Smith, C.E.; Hermo, L.; Suarez, S.; Roth, B.L.; Ducy, P.; et al. Endocrine regulation of male fertility by the skeleton. Cell 2011, 144, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Ensrud, K.E.; Thompson, D.E.; Cauley, J.A.; Nevitt, M.C.; Kado, D.M.; Hochberg, M.C.; Santora, A.C., 2nd; Black, D.M. Prevalent Vertebral Deformities Predict Mortality and Hospitalization in Older Women with Low Bone Mass. J. Am. Geriatr. Soc. 2000, 48, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.D.; Center, J.R.; Eisman, J.A.; Nguyen, T.V. Bone loss, weight loss, and weight fluctuation predict mortality risk in elderly men and women. J. Bone Miner. Res. 2007, 22, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yoshida, H. Low bone mineral density at femoral neck is a predictor of increased mortality in elderly Japanese women. Osteoporos. Int. 2010, 21, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Huang, X.; Jin, F.; Wang, H.; Hao, Y.; Tang, T.; Dai, K. Bone mineral density and all-cause, cardiovascular and stroke mortality: A meta-analysis of prospective cohort studies. Int. J. Cardiol. 2013, 166, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef]
- Kennedy, O.D.; Herman, B.C.; Laudier, D.M.; Majeska, R.J.; Sun, H.B.; Schaffler, M.B. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone 2012, 50, 1115–1122. [Google Scholar] [CrossRef]
- Brunkow, M.E.; Gardner, J.C.; Van Ness, J.; Paeper, B.W.; Kovacevich, B.R.; Proll, S.; Skonier, J.E.; Zhao, L.; Sabo, P.; Fu, Y.-H.; et al. Bone Dysplasia Sclerosteosis Results from Loss of the SOST Gene Product, a Novel Cystine Knot–Containing Protein. Am. J. Hum. Genet. 2001, 68, 577–589. [Google Scholar] [CrossRef]
- Balemans, W.; Patel, N.; Ebeling, M.; Van Hul, E.; Wuyts, W.; Lacza, C.; Dioszegi, M.; Dikkers, F.G.; Hildering, P.; Willems, P.J.; et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 2002, 39, 91–97. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [PubMed]
- Ominsky, M.S.; Niu, Q.-T.; Li, C.; Li, X.; Ke, H.Z. Tissue-Level Mechanisms Responsible for the Increase in Bone Formation and Bone Volume by Sclerostin Antibody. J. Bone Miner. Res. 2014, 29, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, S.; Ishii, K.; Amizuka, N.; Li, M.; Kobayashi, T.; Kohno, K.; Ito, M.; Takeshita, S.; Ikeda, K. Targeted Ablation of Osteocytes Induces Osteoporosis with Defective Mechanotransduction. Cell Metab. 2007, 5, 464–475. [Google Scholar] [CrossRef]
- Xiao, Z.; Dallas, M.; Qiu, N.; Nicolella, D.; Cao, L.; Johnson, M.; Bonewald, L.; Quarles, L.D. Conditional deletion of Pkd1 in osteocytes disrupts skeletal mechanosensing in mice. FASEB J. 2011, 25, 2418–2432. [Google Scholar] [CrossRef] [PubMed]
- Qiu, N.; Xiao, Z.; Cao, L.; Buechel, M.M.; David, V.; Roan, E.; Quarles, L.D. Disruption of Kif3a in osteoblasts results in defective bone formation and osteopenia. J. Cell Sci. 2012, 125, 1945–1957. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, S.; Tang, C.; Chen, W. Upregulation of long non-coding RNA HIF 1α-anti-sense 1 induced by transforming growth factor-β-mediated targeting of sirtuin 1 promotes osteoblastic differentiation of human bone marrow stromal cells. Mol. Med. Rep. 2015, 12, 7233–7238. [Google Scholar] [CrossRef]
- Liang, W.-C.; Fu, W.-M.; Wang, Y.-B.; Sun, Y.-X.; Xu, L.-L.; Wong, C.-W.; Chan, K.-M.; Li, G.; Waye, M.M.-Y.; Zhang, J.-F. H19 activates Wnt signaling and promotes osteoblast differentiation by functioning as a competing endogenous RNA. Sci. Rep. 2016, 6, 20121. [Google Scholar] [CrossRef]
- Jin, C.; Jia, L.; Huang, Y.; Zheng, Y.; Du, N.; Liu, Y.; Zhou, Y. Inhibition of lncRNA MIR31HG Promotes Osteogenic Differentiation of Human Adipose-Derived Stem Cells. Stem Cells 2016, 34, 2707–2720. [Google Scholar] [CrossRef]
- Yu, E.J.; Kim, S.-H.; Kim, H.J.; Heo, K.; Ou, C.-Y.; Stallcup, M.R.; Kim, J.H. Positive regulation of β-catenin-PROX1 signaling axis by DBC1 in colon cancer progression. Oncogene 2016, 35, 3410–3418. [Google Scholar] [CrossRef]
- Glass, D.A., 2nd; Karsenty, G. Molecular bases of the regulation of bone remodeling by the canonical Wnt signaling pathway. Curr. Top Dev. Biol. 2006, 73, 43–84. [Google Scholar] [CrossRef]
- Bonewald, L.F.; Johnson, M.L. Osteocytes, mechanosensing and Wnt signaling. Bone 2008, 42, 606–615. [Google Scholar] [CrossRef]
- Duan, P.; Bonewald, L. The role of the wnt/β-catenin signaling pathway in formation and maintenance of bone and teeth. Int. J. Biochem. Cell Biol. 2016, 77, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Moon, R.T.; Bowerman, B.; Boutros, M.; Perrimon, N. The Promise and Perils of Wnt Signaling Through β-Catenin. Science 2002, 296, 1644–1646. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Visweswaran, M.; Pohl, S.; Arfuso, F.; Newsholme, P.; Dilley, R.; Pervaiz, S.; Dharmarajan, A. Multi-lineage differentiation of mesenchymal stem cells—To Wnt, or not Wnt. Int. J. Biochem. Cell Biol. 2015, 68, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Clément-Lacroix, P.; Ai, M.; Morvan, F.; Roman-Roman, S.; Vayssière, B.; Belleville, C.; Estrera, K.; Warman, M.L.; Baron, R.; Rawadi, G. Lrp5-independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17406–17411. [Google Scholar] [CrossRef] [PubMed]
- Galli, C.; Piemontese, M.; Lumetti, S.; Manfredi, E.; Macaluso, G.M.; Passeri, G. GSK3b-inhibitor lithium chloride enhances activation of Wnt canonical signaling and osteoblast differentiation on hydrophilic titanium surfaces. Clin. Oral Implant. Res. 2012, 24, 921–927. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Escande, C.; Nin, V.; Chini, E.N. HDAC3 Is Negatively Regulated by the Nuclear Protein DBC1. J. Biol. Chem. 2010, 285, 40830–40837. [Google Scholar] [CrossRef]
- Choi, H.; Kim, T.-H.; Yang, S.; Lee, J.-C.; You, H.-K.; Cho, E.-S. A Reciprocal Interaction between β-Catenin and Osterix in Cementogenesis. Sci. Rep. 2017, 7, 8160. [Google Scholar] [CrossRef]
- Neer, R.M.; Arnaud, C.D.; Zanchetta, J.R.; Prince, R.; Gaich, G.A.; Reginster, J.-Y.; Hodsman, A.B.; Eriksen, E.F.; Ish-Shalom, S.; Genant, H.K.; et al. Effect of Parathyroid Hormone (1-34) on Fractures and Bone Mineral Density in Postmenopausal Women with Osteoporosis. N. Engl. J. Med. 2001, 344, 1434–1441. [Google Scholar] [CrossRef]
- Body, J.-J.; Gaich, G.A.; Scheele, W.H.; Kulkarni, P.M.; Miller, P.D.; Peretz, A.; Dore, R.K.; Correa-Rotter, R.; Papaioannou, A.; Cumming, D.C.; et al. A Randomized Double-Blind Trial to Compare the Efficacy of Teriparatide [Recombinant Human Parathyroid Hormone (1–34)] with Alendronate in Postmenopausal Women with Osteoporosis. J. Clin. Endocrinol. Metab. 2002, 87, 4528–4535. [Google Scholar] [CrossRef]
- Nakamura, T.; Sugimoto, T.; Nakano, T.; Kishimoto, H.; Ito, M.; Fukunaga, M.; Hagino, H.; Sone, T.; Yoshikawa, H.; Nishizawa, Y.; et al. Randomized Teriparatide [Human Parathyroid Hormone (PTH) 1–34] Once-Weekly Efficacy Research (TOWER) Trial for Examining the Reduction in New Vertebral Fractures in Subjects with Primary Osteoporosis and High Fracture Risk. J. Clin. Endocrinol. Metab. 2012, 97, 3097–3106. [Google Scholar] [CrossRef] [PubMed]
- Kawane, T.; Komori, H.; Liu, W.; Moriishi, T.; Miyazaki, T.; Mori, M.; Matsuo, Y.; Takada, Y.; Izumi, S.; Jiang, Q.; et al. Dlx5 and Mef2 Regulate a Novel Runx2 Enhancer for Osteoblast-Specific Expression. J. Bone Miner. Res. 2014, 29, 1960–1969. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Kida, K.; Yamaguchi, A.; Hata, K.; Ichida, F.; Meguro, H.; Aburatani, H.; Nishimura, R.; Yoneda, T. BMP2 Regulates Osterix through Msx2 and Runx2 during Osteoblast Differentiation. J. Biol. Chem. 2008, 283, 29119–29125. [Google Scholar] [CrossRef] [PubMed]
- Felber, K.; Elks, P.M.; Lecca, M.; Roehl, H.H. Expression of osterix Is Regulated by FGF and Wnt/β-Catenin Signalling during Osteoblast Differentiation. PLoS ONE 2015, 10, e0144982. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Xuan, M.; Zhang, M.; Yao, Q.; Zhang, K.; Zhang, X.; Guo, J.; Song, L. Postnatal deletion of β-catenin in osterix-expressing cells is necessary for bone growth and intermittent PTH-induced bone gain. J. Bone Miner. Metab. 2018, 36, 560–572. [Google Scholar] [CrossRef]
- Liu, Q.; Li, M.; Wang, S.; Xiao, Z.; Xiong, Y.; Wang, G. Recent Advances of Osterix Transcription Factor in Osteoblast Differentiation and Bone Formation. Front. Cell Dev. Biol. 2020, 8, 601224. [Google Scholar] [CrossRef]
- Matsumoto, A.; Pasut, A.; Matsumoto, M.; Yamashita, R.; Fung, J.; Monteleone, E.; Saghatelian, A.; Nakayama, K.I.; Clohessy, J.G.; Pandolfi, P.P. mTORC1 and muscle regeneration are regulated by the LINC00961-encoded SPAR polypeptide. Nature 2017, 541, 228–232. [Google Scholar] [CrossRef]
- Woo, S.M.; Rosser, J.; Dusevich, V.; Kalajzic, I.; Bonewald, L.F. Cell line IDG-SW3 replicates osteoblast-to-late-osteocyte differentiation in vitro and accelerates bone formation in vivo. J. Bone Miner. Res. 2011, 26, 2634–2646. [Google Scholar] [CrossRef]
- Fukuda, T.; Ochi, H.; Sunamura, S.; Haiden, A.; Bando, W.; Inose, H.; Okawa, A.; Asou, Y.; Takeda, S. MicroRNA-145 regulates osteoblastic differentiation by targeting the transcription factor Cbfb. FEBS Lett. 2015, 589, 3302–3308. [Google Scholar] [CrossRef]
- Hirakawa, H.; Gatanaga, H.; Ochi, H.; Fukuda, T.; Sunamura, S.; Oka, S.; Takeda, S.; Sato, S. Antiretroviral Therapy Containing HIV Protease Inhibitors Enhances Fracture Risk by Impairing Osteoblast Differentiation and Bone Quality. J. Infect. Dis. 2017, 215, 1893–1897. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, S.; Niwa, H.; Tashiro, F.; Sano, S.; Kondoh, G.; Takeda, J.; Tabayashi, K.; Miyazaki, J.-I. A novel reporter mouse strain that expresses enhanced green fluorescent protein upon Cre-mediated recombination. FEBS Lett. 2000, 470, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xie, Y.; Zhang, S.; Dusevich, V.; Bonewald, L.; Feng, J. DMP1-targeted Cre Expression in Odontoblasts and Osteocytes. J. Dent. Res. 2007, 86, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward/Reverse | Sequence |
|---|---|---|
| 2310069G16Rik | Forward | 5′-AATAATCACGTGGTGCGGCAG-3′ |
| Reverse | 5′-CCGCCGCACGTGTTCCGAAGCCC-3′ | |
| lncRNA953Rik | Forward | 5′-CATGCGAGGGACTGCTGAT-3′ |
| Reverse | 5′-GGTGCTGAGAAGGCAAAGAT-3′ | |
| A330033J07Rik | Forward | 5′-GTTGAGCGCGATAATGCAAA-3′ |
| Reverse | 5′-CTCCATAAGCTGTGCGTTGA-3′ | |
| Alpl | Forward | 5′-TTCCTGGGAGATGGTATG-3′ |
| Reverse | 5′-TTATATGTCTTGGAGAGGGC-3′ | |
| Axin2 | Forward | 5′-TGTGAGATCCACGGAAACAG-3′ |
| Reverse | 5′-TGGCTGGTGCAAAGACATAG-3′ | |
| Ccar2 | Forward | 5′-GAGGATCAACCCACTTCC-3′ |
| Reverse | 5′-AAGACTCGCTGCTTTTCC-3′ | |
| Col1a1 | Forward | 5′-AGATGTAGGAGTCGAGGGAC-3′ |
| Reverse | 5′-CATAGCCATAGGACATCTGG-3′ | |
| Dkk1 | Forward | 5′-TTGACAACTACCAGCCCTAC-3′ |
| Reverse | 5′-GAAAATGGCTGTGGTCAG-3′ | |
| Dmp1 | Forward | 5′-AAGAGAGGACGGGTGATTTG-3′ |
| Reverse | 5′-TCCGTGTGGTCACTATTTGC-3′ | |
| Gapdh | Forward | 5′-ACCCAGAAGACTGTGGATGG-3′ |
| Reverse | 5′-ACCCAGAAGACTGTGGATGG-3′ | |
| Gm6634 | Forward | 5′-CTTGGTTAAAGTGGATTACATTGAT-3′ |
| Reverse | 5′-TGCTGAAGATGTCACCACTG-3′ | |
| H19 | Forward | 5′-GCCTTCTTGAACACCATGG-3′ |
| Reverse | 5′-CAGACATGAGCTGGGTAGCA-3′ | |
| Krm1 | Forward | 5′-ACCGAGTGCAATAGTGTC-3′ |
| Reverse | 5′-GAGTCCCTGATATCAAACAG-3′ | |
| Krm2 | Forward | 5′-GCGCATAACTTCTGTAGG-3′ |
| Reverse | 5′-CTTTCAGAGCCACAGAAG-3′ | |
| Lef1 | Forward | 5′-GAAGGAAAGCATCCAGAC-3′ |
| Reverse | 5′-GGCACTTTATTTGATGTCC-3′ | |
| Meg3 | Forward | 5′-CCTGGATTAGGCCAAAGCC-3′ |
| Reverse | 5′-AGTCTTGGGTCCAGCATGTC-3′ | |
| Mirg | Forward | 5′-CCTCTGCTGGACAGCTTCAG-3′ |
| Reverse | 5′-CATAGGCAGGGTTCCTTGAA-3′ | |
| Osteocalcin | Forward | 5′-TCTGACAAAGCCTTCATGTCCA-3′ |
| Reverse | 5′-CGGTCTTCAAGCCATACTGGTC-3′ | |
| Osterix | Forward | 5′-GTCCTCTCTGCTTGAGGA-3′ |
| Reverse | 5′-AGGAGAGAGGAGTCCATTG-3′ | |
| Runx2 | Forward | 5′-GCCGGGAATGATGAGAACTA-3′ |
| Reverse | 5′-ATGCGCCCTAAATCACTGAG-3′ | |
| Sfrp1 | Forward | 5′-TGCTCAAATGTGACAAGTTC-3′ |
| Reverse | 5′-CAGCTTCAAGGGTTTCTTC-3′ | |
| Sfrp4 | Forward | 5′-TATGATGGTGCAAGAAAG-3′ |
| Reverse | 5′-TAGGTGACAAAGACTTGAAG-3′ | |
| Tcf7 | Forward | 5′-AGCTTTCTCCACTCTACG-3′ |
| Reverse | 5′-GAGGTCAGAGAATAAAATCC-3′ | |
| Wif1 | Forward | 5′-TGGTCTGTGTGTCACTCC-3′ |
| Reverse | 5′-GCATTTACCTCCATTTCG-3′ |
| Antisense Oligonucleotide | Sequence |
|---|---|
| Anti-lncRNA953Rik #1 | 5′-CTTGCGTCTTAAATTC-3′ |
| Anti-lncRNA953Rik #2 | 5′-TCATGCGTTAACTTGC-3′ |
| Anti-lncRNA953Rik #3 | 5′-ACAGGTCATTAAGGAC-3′ |
| Negative control | 5′-AACACGTCTATACGC-3′ |
| siRNA | Forward/Reverse | Sequence |
|---|---|---|
| si-Ccar2 #1 | Forward | 5′-AGAGGCUACUGAACAGGCUCCUGAU-3′ |
| Reverse | 5′-AUCAGGAGCCUGUUCAGUAGCCUCU-3′ | |
| si-Ccar2 #2 | Forward | 5′-CCUCAUCAAUGUGGGAAGCCUGUUA-3′ |
| Reverse | 5′-UAACAGGCUUCCCACAUUGAUGAGG-3′ | |
| si-Ccar2 #3 | Forward | 5′-GCGGGAUGAUGGAGAGGACGAAUUU-3′ |
| Reverse | 5′-AAAUUCGUCCUCUCCAUCAUCCCGC-3′ |
| Gene | Forward/Reverse | Sequence |
|---|---|---|
| lncRNA953Rik | Forward | 5′-GATCACTAATACGACTCACTATAGGGAATCTCAAGGAAGCTCTTT-3′ |
| Reverse | 5′-TTCAATTACCATGGAGGTTTA-3′ | |
| Negative control | Forward | 5′-GATCACTAATACGACTCACTATAGGGAACTACGGCTACACTAGAA-3′ |
| Reverse | 5′-TTACTTTTAAAGTTCTGCTATGT-3′ |
| Location | Forward/Reverse | Sequence |
|---|---|---|
| Osterix −222 bases | Forward | 5′-CTCATTGGATCCGGAGTCTTCT-3′ |
| Reverse | 5′-TGTCTGTAGGGATCCACCCTCTA-3′ | |
| Osterix −2022 bases | Forward | 5′-TAGAGCATTCCCTTTGGG-3′ |
| Reverse | 5′-AGCAGGACTGAGGGACAG-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arai, M.; Ochi, H.; Sunamura, S.; Ito, N.; Nangaku, M.; Takeda, S.; Sato, S. A Novel Long Noncoding RNA in Osteocytes Regulates Bone Formation through the Wnt/β-Catenin Signaling Pathway. Int. J. Mol. Sci. 2023, 24, 13633. https://doi.org/10.3390/ijms241713633
Arai M, Ochi H, Sunamura S, Ito N, Nangaku M, Takeda S, Sato S. A Novel Long Noncoding RNA in Osteocytes Regulates Bone Formation through the Wnt/β-Catenin Signaling Pathway. International Journal of Molecular Sciences. 2023; 24(17):13633. https://doi.org/10.3390/ijms241713633
Chicago/Turabian StyleArai, Makoto, Hiroki Ochi, Satoko Sunamura, Nobuaki Ito, Masaomi Nangaku, Shu Takeda, and Shingo Sato. 2023. "A Novel Long Noncoding RNA in Osteocytes Regulates Bone Formation through the Wnt/β-Catenin Signaling Pathway" International Journal of Molecular Sciences 24, no. 17: 13633. https://doi.org/10.3390/ijms241713633