

Geography, Climate, and Habitat Shape the Microbiome of the Endangered Rock Gnome Lichen (Cetradonia linearis)
 and
and
Abstract
:1. Introduction
2. Methods
2.1. Sampling, DNA Extractions, and Amplicon Sequencing
2.2. Environmental Data
2.3. Microbiome Analyses in QIIME2
2.4. Core Microbiome Characterization
3. Results
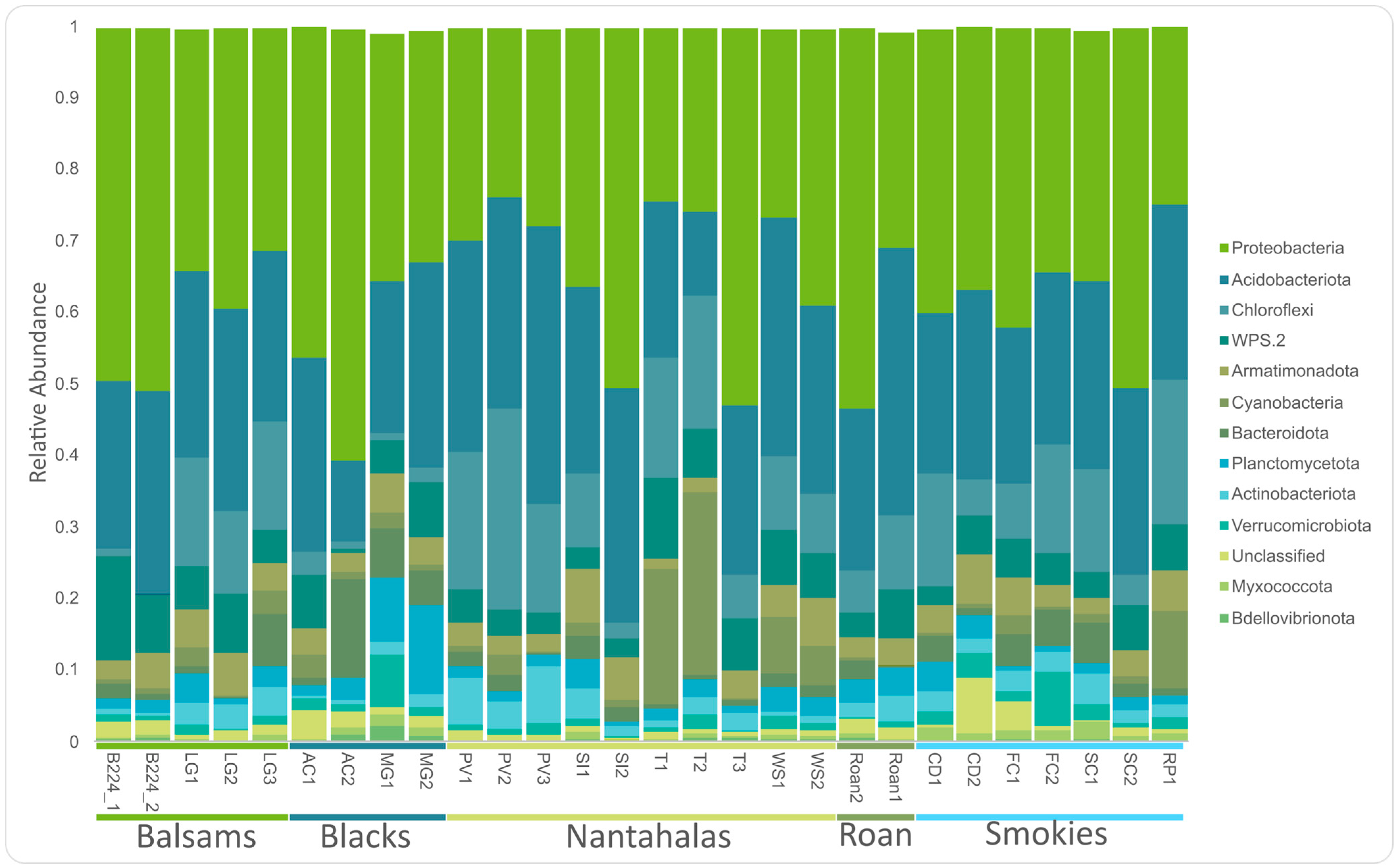
3.1. Core Microbiome
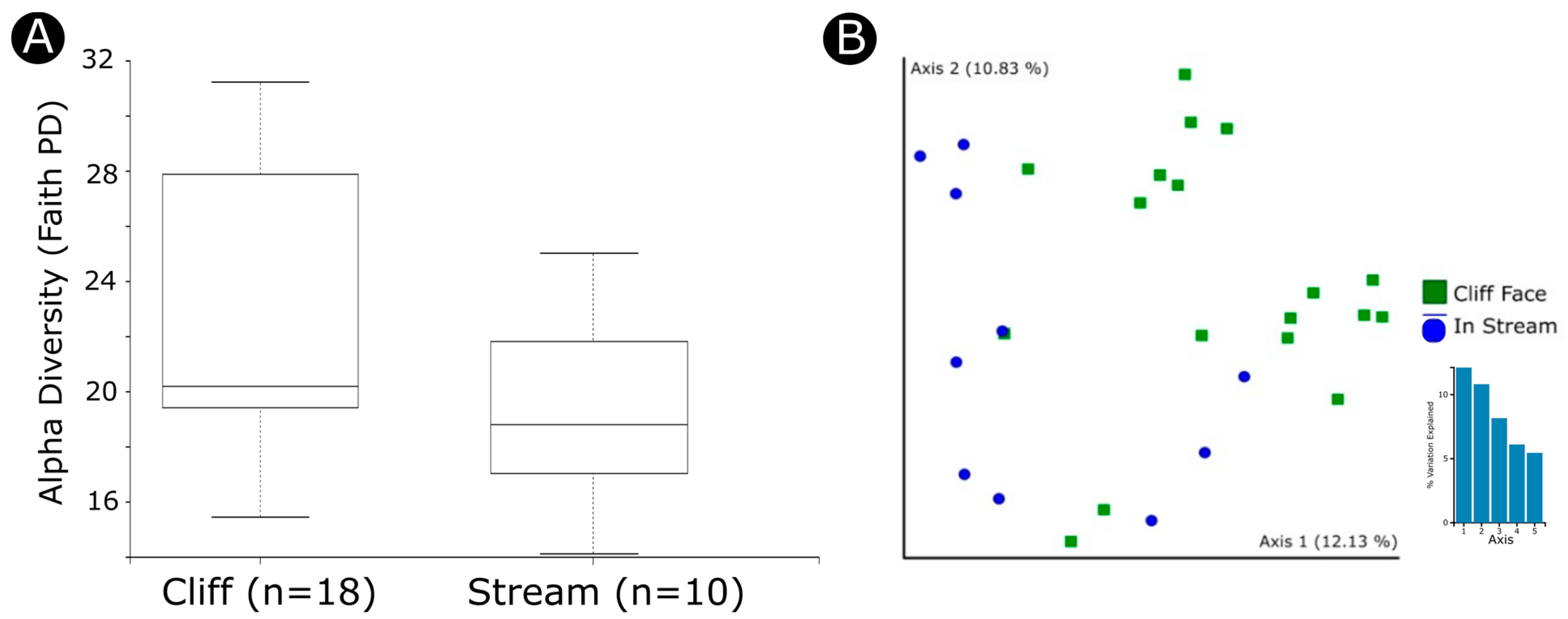
3.2. Factors Influencing Diversity and Community Composition
4. Discussion
4.1. Dominant Groups in the Microbiome of Cetradonia linearis Are Consistent with Previous Reports in the Family Cladoniaceae
4.2. Both Mycobiont and Bacterial Communities Are Geographically Structured
4.3. Environmental Factors Significantly Impact the Bacterial Microbiome
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Redford, K.H.; Segre, J.A.; Salafsky, N.; del Rio, C.M.; McAloose, D. Conservation and the Microbiome. Conserv. Biol. 2012, 26, 195–197. [Google Scholar] [CrossRef]
- Carthey, A.J.R.; Blumstein, D.T.; Gallagher, R.V.; Tetu, S.G.; Gillings, M.R. Conserving the holobiont. Funct. Ecol. 2019, 34, 764–776. [Google Scholar] [CrossRef]
- Loiola, M.; Silva, A.E.T.; Krull, M.; Barbosa, F.A.; Galvão, E.H.; Patire, V.F.; Cruz, I.C.S.; Barros, F.; Hatje, V.; Meirelles, P.M. Mangrove microbial community recovery and their role in early stages of forest recolonization within shrimp ponds. Sci. Total Environ. 2023, 855, 158863. [Google Scholar] [CrossRef]
- Zheng, F.; Mou, X.; Zhang, J.; Zhang, T.; Xia, L.; Yin, S.; Wu, L.; Leng, X.; An, S.; Zhao, D. Gradual Enhancement of the Assemblage Stability of the Reed Rhizosphere Microbiome with Recovery Time. Microorganisms 2022, 10, 937. [Google Scholar] [CrossRef] [PubMed]
- Dove, N.C.; Klingeman, D.M.; Carrell, A.A.; Cregger, M.A.; Schadt, C.W. Fire alters plant microbiome assembly patterns: Integrating the plant and soil microbial response to disturbance. New Phytol. 2021, 230, 2433–2446. [Google Scholar] [CrossRef] [PubMed]
- Coban, O.; De Deyn, G.B.; Van Der Ploeg, M. Soil microbiota as game-changers in restoration of degraded lands. Science 2022, 375, abe0725. [Google Scholar] [CrossRef]
- Liddicoat, C.; Siegfried, K.L.; Bissett, A.; Borrett, R.J.; Ducki, L.C.; Peddle, S.D.; Bullock, P.; Dobrowolski, M.P.; Grigg, A.; Tibbett, M.; et al. Next generation restoration metrics: Using soil eDNA bacterial community data to measure trajectories towards rehabilitation targets. J. Environ. Manag. 2022, 310, 114748. [Google Scholar] [CrossRef]
- Perry, E.K.; Digby, A.; Taylor, M.W. The Low-Diversity Fecal Microbiota of the Critically Endangered Kākāpō is Robust to Anthropogenic Dietary and Geographic Influences. Front. Microbiol. 2017, 8, 2033. [Google Scholar] [CrossRef]
- Waite, D.W.; Deines, P.; Taylor, M.W. Gut Microbiome of the Critically Endangered New Zealand Parrot, the Kakapo (Strigops habroptilus). PLoS ONE 2012, 7, e35803. [Google Scholar] [CrossRef]
- West, A.G.; Digby, A.; Lear, G.; Kākāpō Recovery Team; Kākāpō Aspergillosis Research Consortium; Taylor, M.W. Influence of management practice on the microbiota of a critically endangered species: A longitudinal study of kākāpō chick feces and associated nest litter. Anim. Microbiome 2022, 4, 55. [Google Scholar] [CrossRef]
- Jiménez, R.R.; Carfagno, A.; Linhoff, L.; Gratwicke, B.; Woodhams, D.C.; Chafran, L.S.; Bletz, M.C.; Carly, B.B.; Muletz-Wolz, R. Inhibitory Bacterial Diversity and Mucosome Function Differentiate Susceptibility of Appalachian Salamanders to Chytrid Fungal Infection. Appl. Environ. Microbiol. 2022, 88, e01818-21. [Google Scholar] [CrossRef]
- Walke, J.B.; Belden, L.K. Harnessing the Microbiome to Prevent Fungal Infections: Lessons from Amphibians. PLoS Pathog. 2016, 12, e1005796. [Google Scholar] [CrossRef] [PubMed]
- Goulet, T.L. Most corals may not change their symbionts. Mar. Ecol. Prog. Ser. 2006, 321, 1–7. [Google Scholar] [CrossRef]
- Buerger, P.; Alvarez-Roa, C.; Coppin, C.W.; Pearce, S.L.; Chakravarti, L.J.; Oakeshott, J.G.; Edwards, O.R.; Van Oppen, M.J.H. Heat-evolved microalgal symbionts increase coral bleaching tolerance. Sci. Adv. 2020, 6, eaba2498. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.D.; Reiter, N.; Peakall, R. Orchid conservation: From theory to practice. Ann. Bot. 2020, 126, 345–362. [Google Scholar] [CrossRef]
- Huang, Y.-Y.; Carballo-Bolaños, R.; Kuo, C.-Y.; Keshavmurthy, S. Leptoria phrygia in Southern Taiwan shuffles and switches symbionts to resist thermal-induced bleaching. Sci. Rep. 2020, 10, 7808. [Google Scholar] [CrossRef]
- Tian, F.; Liao, X.-F.; Wang, L.-H.; Bai, X.-X.; Yang, Y.-B.; Luo, Z.-Q.; Yan, F.-X. Isolation and identification of beneficial orchid mycorrhizal fungi in Paphiopedilum barbigerum (Orchidaceae). Plant Signal. Behav. 2021, 17, 2005882. [Google Scholar] [CrossRef]
- Masako, F.; Chihiro, M.; Tatsuki, Y.; Shintaro, K.; Kenji, S.; Takahiro, Y.; Masahide, Y.; Hironori, K. Relative effectiveness of Tulasnella fungal strains in orchid mycorrhizal symbioses between germination and subsequent seedling growth. Symbiosis 2020, 81, 53–63. [Google Scholar]
- Altinkaynak, H.; Ozkoc, I. Isolation and molecular characterization of plant growth promoting bacteria from the rhizosphere of orchids in Turkey. Rhizosphere 2020, 16, 100280. [Google Scholar] [CrossRef]
- Herrera, H.; Sanhueza, T.; Novotná, A.; Charles, T.C.; Arriagada, C. Isolation and Identification of Endophytic Bacteria from Mycorrhizal Tissues of Terrestrial Orchids from Southern Chile. Diversity 2020, 12, 55. [Google Scholar] [CrossRef]
- Kaur, J.; Sharma, J. Orchid Root Associated Bacteria: Linchpins or Accessories? Front. Plant Sci. 2021, 12, 661966. [Google Scholar] [CrossRef]
- Hawksworth, D.L.; Grube, M. Reflections on lichens as ecosystems. New Phytol. 2023, 241, 972–973. [Google Scholar] [CrossRef]
- Sanders, W.B. The disadvantages of current proposals to redefine lichens. New Phytol. 2023, 241, 969–971. [Google Scholar] [CrossRef]
- Jüriado, I.; Kaasalainen, U.; Jylhä, M.; Rikkinen, J. Relationships between mycobiont identity, photobiont specificity and ecological preferences in the lichen genus Peltigera (Ascomycota) in Estonia (northeastern Europe). Fungal Ecol. 2019, 39, 45–54. [Google Scholar] [CrossRef]
- Leavitt, S.D.; Kraichak, E.; Vondrak, J.; Nelsen, M.P.; Sohrabi, M.; Perez-Ortega, S.; Clair, L.L.S.; Lumbsch, H.T. Cryptic diversity and symbiont interactions in rock-posy lichens. Mol. Phylogenet. Evol. 2016, 99, 261–274. [Google Scholar] [CrossRef]
- Steinová, J.; Škaloud, P.; Yahr, R.; Bestová, H.; Muggia, L. Reproductive and dispersal strategies shape the diversity of mycobiont-photobiont association in Cladonia lichens. Mol. Phylogenetics Evol. 2019, 134, 226–237. [Google Scholar] [CrossRef]
- Rolshausen, G.; Grande, F.D.; Sadowska-Deś, A.D.; Otte, J.; Schmitt, I. Quantifying the climatic niche of symbiont partners in a lichen symbiosis indicates mutualist-mediated niche expansions. Ecography 2018, 41, 1380–1392. [Google Scholar] [CrossRef]
- Cernava, T.; Aschenbrenner, I.A.; Soh, J.; Sensen, C.W.; Grube, M.; Berg, G. Plasticity of a holobiont: Desiccation induces fasting-like metabolism within the lichen microbiota. ISME J. 2019, 13, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Brime, S.; Muggia, L.; Maier, S.; Grube, M.; Wedin, M. Bacterial communities in an optional lichen symbiosis are determined by substrate, not algal photobionts. FEMS Microbiol. Ecol. 2019, 95, fiz012. [Google Scholar] [CrossRef]
- Aschenbrenner, I.; Cernava, T.; Berg, G.; Grube, M. Understanding Microbial Multi-Species Symbiosis. Front. Microbiol. 2016, 7, 180. [Google Scholar] [CrossRef]
- Cardinale, M.; Steinová, J.; Rabensteiner, J.; Berg, G.; Grube, M. Age, sun and substrate: Triggers of bacterial communities in lichens. Environ. Microbiol. Rep. 2011, 4, 23–28. [Google Scholar] [CrossRef]
- Bates, S.T.; Cropsey, G.W.G.; Caporaso, J.G.; Knight, R.; Fierer, N. Bacterial Communities Associated with the Lichen Symbiosis. Appl. Environ. Microbiol. 2011, 77, 1309–1314. [Google Scholar] [CrossRef]
- Hodkinson, B.; Lutzoni, F. A Microbiotic Survey of Lichen-Associated Bacteria Reveals a New Lineage from the Rhizobiales. Symbiosis 2009, 49, 163–180. [Google Scholar] [CrossRef]
- Cardinale, M.; Puglia, A.M.; Grube, M. Molecular analysis of lichen-associated bacterial communities. FEMS Microbiol. Ecol. 2006, 57, 484–495. [Google Scholar] [CrossRef]
- Grube, M.; Berg, G. Microbial consortia of bacteria and fungi with focus on the lichen symbiosis. Fungal Biol. Rev. 2009, 23, 72–85. [Google Scholar] [CrossRef]
- Aschenbrenner, I.; Cardinale, M.; Berg, G.; Grube, M. Microbial cargo: Do bacteria on symbiotic propagules reinforce the microbiome of lichens? Environ. Microbiol. 2014, 16, 3743–3752. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Garcia, M.; Villarreal, J.C.A. Bacterial community of reindeer lichens differs between northern and southern lichen woodlands. Can. J. Forest Res. 2022, 52, 662–673. [Google Scholar] [CrossRef]
- Vijayakumar, V.R.; Saravanan, K.; Somasundaram, M.; Jayaraj, R.; Annamalai, P.; Nooruddin, T.; Dharumadurai, D. Metagenomic analysis of lichen-associated bacterial community profiling in Roccella montagnei. Arch. Microbiol. 2022, 204, 54. [Google Scholar] [CrossRef]
- Sierra, M.A.; Danko, D.C.; Sandoval, T.A.; Pishchany, G.; Moncada, B.; Kolter, R.; Mason, C.E.; Zambrano, M.M. The Microbiomes of Seven Lichen Genera Reveal Host Specificity, a Reduced Core Community and Potential as Source of Antimicrobials. Front. Microbiol. 2020, 11, 398. [Google Scholar] [CrossRef]
- West, N.J.; Parrot, D.; Fayet, C.; Grube, M.; Tomasi, S.; Suzuki, M.T. Marine cyanolichens from different littoral zones are associated with distinct bacterial communities. PeerJ 2018, 17, e5208. [Google Scholar] [CrossRef]
- Hodkinson, B.; Gottel, N.; Schadt, C.; Lutzoni, F. Photoautotrophic symbiont and geography are major factors affecting highly structured and diverse bacterial communities in the lichen microbiome. Environ. Microbiol. 2012, 14, 147–161. [Google Scholar] [CrossRef]
- U.S. Fish and Wildlife Service. Rock Gnome Lichen (Gymnoderma lineare) 5-Year Review: Summary and Evaluation. 2020. Available online: https://ecos.fws.gov/docs/five_year_review/doc6561.pdf (accessed on 9 March 2023).
- Allen, J.L.; Lendemer, J.C.; McMullin, T. Cetradonia linearis; The IUCN Red List of Threatened Species: Cambridge, UK, 2015. [Google Scholar] [CrossRef]
- Zhou, Q.-M.; Wei, J.-C.; Ahti, T.; Stenroos, S.; Högnabba, F. The systematic position of Gymnoderma and Cetradonia based on SSU rDNA sequences. J. Hattori Bot. Lab. 2006, 100, 871–880. [Google Scholar]
- Wei, J.; Ahti, T. Cetradonia, a new genus in the new family Cetradoniaceae (Lecanorales, Ascomycota). Lichenologist 2002, 34, 19–31. [Google Scholar] [CrossRef]
- Allen, J.L.; McKenzie, S.K.; Sleith, R.S.; Alter, S.E. First genome-wide analysis of the endangered, endemic lichen Cetradonia linearis reveals isolation by distance and strong population structure. Am. J. Bot. 2018, 105, 1556–1567. [Google Scholar] [CrossRef] [PubMed]
- Alonso-García, M.; Villarreal, A.J.C.; McFarland, K.; Goffinet, B. Population genomics and phylogeography of a clonal bryophyte with spatially separated sexes and extreme sex ratios. Front. Plant Sci. 2020, 11, 495. [Google Scholar] [CrossRef]
- Crespi, E.J.; Rissler, L.J.; Browne, R.A. Testing Pleistocene refugia theory: Phylogeographical analysis of Desmognathus wrighti, a high-elevation salamander in the southern Appalachians. Mol. Ecol. 2003, 12, 969–984. [Google Scholar] [CrossRef]
- Keith, R.; Hedin, M. Extreme mitochondrial population subdivision in southern Appalachian paleoendemic spiders (Araneae: Hypochilidae: Hypochilus), with implications for species delimitation. J. Arachnol. 2012, 40, 167–181. [Google Scholar] [CrossRef]
- Hijmans, R.J.; Van Etten, J.; Mattiuzzi, M.; Cheng, J.; Sumner, M.; Greenberg, J.A. Raster: Geographic Data Analysis and Modeling; R Package Version 2.9-23; R Core Team R Foundation for Statistical Computing: Viena, Austria, 2019. [Google Scholar]
- Hijmans, R.J.; Cameron, S.E.; Parra, J.L.; Jones, P.G.; Jarvis, A. Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 2005, 25, 1965–1978. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Niu, B.; Chen, Q.; Hu, Y.; Yang, Y.; Song, L.; Wang, J.; Zhang, G. Temperature and Precipitation Drive Elevational Patterns of Microbial Beta Diversity in Alpine Grasslands. Microb. Ecol. 2022, 84, 1141–1153. [Google Scholar] [CrossRef]
- Nottingham, A.T.; Fierer, N.; Turner, B.L.; Whitaker, J.; Ostle, N.J.; McNamara, N.P.; Bardgett, R.D.; Leff, J.W.; Salinas, N.; Silman, M.R.; et al. Microbes follow Humboldt: Temperature drives plant and soil microbial diversity patterns from the Amazon to the Andes. Ecology 2018, 99, 2455–2466. [Google Scholar] [CrossRef]
- Truu, M.; Ostonen, I.; Preem, J.-K.; Lõhmus, K.; Nõlvak, H.; Ligi, T.; Rosenvald, K.; Parts, K.; Kupper, P.; Truu, J. Elevated Air Humidity Changes Soil Bacterial Community Structure in the Silver Birch Stand. Front. Microbiol. 2017, 8, 557. [Google Scholar] [CrossRef]
- Li, H.; Yang, Q.; Li, J.; Gao, H.; Li, P.; Zhou, H. The impact of temperature on microbial diversity and AOA activity in the Tengchong Geothermal Field, China. Sci. Rep. 2015, 5, 17056. [Google Scholar] [CrossRef]
- Root, H.T.; McCune, B.; Jovan, S. Lichen communities and species indicate climate thresholds in southeast and south-central Alaska, USA. Bryologist 2014, 117, 241–252. [Google Scholar] [CrossRef]
- Pearson, L.C. Influence of Temperature and Humidity on Distribution of Lichens in a Minnesota Bog. Ecology 1969, 50, 740–746. [Google Scholar] [CrossRef]
- Vančurová, L.; Malíček, J.; Steinová, J.; Škaloud, P. Choosing the Right Life Partner: Ecological Drivers of Lichen Symbiosis. Front. Microbiol. 2021, 12, 769304. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson AJ, A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Shetty, S.; Lahti, L. Microbiomeutilities: Microbiomeutilities: Utilities for Microbiome Analytics; R Package Version 1.00.17; R Core Team R Foundation for Statistical Computing: Viena, Austria, 2022. [Google Scholar]
- Inkscape Project. Inkscape. 2020. Available online: https://inkscape.org (accessed on 9 March 2023).
- Zinger, L.; Shahnavaz, B.; Baptist, F.; Geremia, R.A.; Choler, P. Microbial diversity in alpine tundra soils correlates with snow cover dynamics. ISME J. 2009, 3, 850–859. [Google Scholar] [CrossRef]
- Schiff, S.L.; Tsuji, J.M.; Wu, L.; Venkiteswaran, J.J.; Molot, L.A.; Elgood, R.J.; Paterson, M.J.; Neufeld, J.D. Millions of Boreal Shield Lakes can be used to Probe Archaean Ocean Biogeochemistry. Sci. Rep. 2017, 7, 46708. [Google Scholar] [CrossRef]
- Yergeau, E.; Newsham, K.K.; Pearce, D.A.; Kowalchuk, G.A. Patterns of bacterial diversity across a range of Antarctic terrestrial habitats. Environ. Microbiol. 2007, 9, 2670–2682. [Google Scholar] [CrossRef]
- Sigurbjörnsdóttir, M.A.; Andrésson, Ó.S.; Vilhelmsson, O. Analysis of the Peltigera Membranacea Metagenome Indicates That Lichen-Associated Bacteria Are Involved in Phosphate Solubilization. Microbiology 2015, 161, 989–996. [Google Scholar] [CrossRef]
- Navarro-Noya, Y.E.; Jiménez-Aguilar, A.; Valenzuela-Encinas, C.; Alcántara-Hernández, R.J.; Ruíz-Valdiviezo, V.M.; Ponce-Mendoza, A.; Luna-Guido, M.; Marsch, R.; Dendooven, L. Bacterial Communities in Soil under Moss and Lichen-Moss Crusts. Geomicrobiol. J. 2013, 31, 152–160. [Google Scholar] [CrossRef]
- Hoang, K.L.; Choi, H.; Gerardo, N.M.; Morran, L.T. Coevolution’s conflicting role in the establishment of beneficial associations. Evolution 2022, 76, 1073–1081. [Google Scholar] [CrossRef]
- IUCN. The IUCN Red List of Threatened Species. Version 2022-2. 2022. Available online: https://www.iucnredlist.org (accessed on 9 March 2023).
- Noh, H.-J.; Lee, Y.M.; Park, C.H.; Lee, H.K.; Cho, J.-C.; Hong, S.G. Microbiome in Cladonia squamosa Is Vertically Stratified According to Microclimatic Conditions. Front. Microbiol. 2020, 11, 268. [Google Scholar] [CrossRef]
- Park, C.H.; Kim, K.M.; Kim, O.-S.; Jeong, G.; Hong, S.G. Bacterial communities in Antarctic lichens. Antarct. Sci. 2016, 28, 455–461. [Google Scholar] [CrossRef]
- Cardinale, M.; de Castro, J.V.; Müller, H.; Berg, G.; Grube, M. In situ analysis of the bacterial community associated with the reindeer lichen Cladonia arbuscula reveals predominance of Alphaproteobacteria. FEMS Microbiol. Ecol. 2008, 66, 63–71. [Google Scholar] [CrossRef]
- Yan, D.; Hu, D.; Li, K.; Li, B.; Zeng, X.; Chen, J.; Li, Y.; Wronski, T. Effects of Chronic Stress on the Fecal Microbiome of Malayan Pangolins (Manis javanica) Rescued from the Illegal Wildlife Trade. Curr. Microbiol. 2021, 78, 1017–1025. [Google Scholar] [CrossRef]
- Amato, K.R.; Kuthyar, S.; Ekanayake-Weber, M.; Salmi, R.; Snyder-Mackler, N.; Wijayathunga, L.; Vandercone, R.; Lu, A. Gut microbiome, diet, and conservation of endangered langurs in Sri Lanka. Biotropica 2020, 52, 981–990. [Google Scholar] [CrossRef]
- Mezzasoma, A.; Coleine, C.; Sannino, C.; Selbmann, L. Endolithic Bacterial Diversity in Lichen-Dominated Communities Is Shaped by Sun Exposure in McMurdo Dry Valleys, Antarctica. Microb. Ecol. 2022, 83, 328–339. [Google Scholar] [CrossRef]
- Shigyo, N.; Umeki, K.; Hirao, T. Seasonal Dynamics of Soil Fungal and Bacterial Communities in Cool-Temperate Montane Forests. Front. Microbiol. 2019, 10, 1944. [Google Scholar] [CrossRef]
- Du, C.; Xu, C.-Y.; Jian, J.-S.; He, W.-X.; Hou, L.; Geng, Z.-C. Seasonal dynamics of bacterial communities in a Betula albosinensis forest. Eur. J. Soil Sci. 2018, 69, 666–674. [Google Scholar] [CrossRef]
- Rolshausen, G.; Grande, F.D.; Otte, J.; Schmitt, I. Lichen holobionts show compositional structure along elevation. Mol. Ecol. 2022, 32, 6619–6630. [Google Scholar] [CrossRef]
- Yu, X.; Yu, K.; Liao, Z.; Chen, B.; Deng, C.; Yu, J.; Yao, Q.; Qin, Z.; Liang, J. Seasonal fluctuations in symbiotic bacteria and their role in environmental adaptation of the scleractinian coral Acropora pruinosa in high-latitude coral reef area of the South China Sea. Sci. Total Environ. 2021, 792, 148438. [Google Scholar] [CrossRef]
- Bulan, E.D.; Wilantho, A.; Krainara, P.; Viyakarn, V.; Chavanich, S.; Somboonna, N. Spatial and Seasonal Variability of Reef Bacterial Communities in the Upper Gulf of Thailand. Front. Mar. Sci. 2018, 5, 441. [Google Scholar] [CrossRef]
- Sharp, K.H.; Pratte, Z.A.; Kerwin, A.H.; Rotjan, R.D.; Stewart, F.J. Season, but not symbiont state, drives microbiome structure in the temperate coral Astrangia poculata. Microbiome 2017, 5, 120. [Google Scholar] [CrossRef]
- Goreau, T.J.F.; Hayes, R.L. Global warming triggers coral reef bleaching tipping point. Ambio 2021, 50, 1137–1140. [Google Scholar] [CrossRef]
- Ziegler, M.; Seneca, F.O.; Yum, L.K.; Palumbi, S.R.; Voolstra, C.R. Bacterial community dynamics are linked to patterns of coral heat tolerance. Nat. Commun. 2017, 8, 14213. [Google Scholar] [CrossRef]
- Kuek, F.W.I.; Lim, L.-F.; Ngu, L.-H.; Mujahid, A.; Lim, P.-T.; Leaw, C.-P.; Müller, M. The potential roles of bacterial communities in coral defense: A case study at Talang-talang reef. Ocean Sci. J. 2015, 50, 269–282. [Google Scholar] [CrossRef]
- Cernava, T.; Erlacher, A.; Aschenbrenner, I.A.; Krug, L.; Lassek, C.; Riedel, K.; Grube, M.; Berg, G. Deciphering Functional Diversification Within the Lichen Microbiota by Meta-omics. Microbiome 2017, 5, 82. [Google Scholar] [CrossRef]
- Cernava, T.; Müller, H.; Aschenbrenner, I.A.; Grube, M.; Berg, G. Analyzing the Antagonistic Potential of the Lichen Microbiome Against Pathogens by Bridging Metagenomic with Culture Studies. Front. Microbiol. 2015, 6, 620. [Google Scholar] [CrossRef]
- Cernava, T.; Vasfiu, Q.; Erlacher, A.; Francesconi, K.; Grube, M.; Berg, G. Adaptions of Lichen Microbiota Functioning Under Persistent Exposure to Arsenic Contamination. Front. Microbiol. 2018, 9, 2959. [Google Scholar] [CrossRef]
- Erlacher, A.; Cernava, T.; Cardinale, M.; Soh, J.; Sensen, C.W.; Grube, M.; Berg, G. Rhizobiales as functional and endosymbiotic members in the lichen symbiosis of Lobaria pulmonaria L. Front. Microbiol. 2015, 6, 53. [Google Scholar] [CrossRef]
- Grube, M.; Cernava, T.; Jung, S.; Fuchs, S.; Aschenbrenner, I.; Lassek, C.; Wegner, U.; Becher, D.; Riedel, K.; Sensen, C.W.; et al. Exploring functional contexts of symbiotic sustain within lichen-associated bacteria by comparative omics. ISME J. 2015, 9, 412–424. [Google Scholar] [CrossRef]
- Bijlsma, R.; Loeschcke, V. Genetic erosion impedes adaptive responses to stressful environments. Evol. Appl. 2012, 5, 117–129. [Google Scholar] [CrossRef]
- Henry, L.P.; Bruijning, M.; Forsberg, S.K.G.; Ayroles, J.F. The microbiome extends host evolutionary potential. Nat. Commun. 2021, 12, 5141. [Google Scholar] [CrossRef]
- Carthey, A.J.R.; Gillings, M.R.; Blumstein, D.T. The Extended Genotype: Microbially Mediated Olfactory Communication. Trends Ecol. Evol. 2018, 33, 885–894. [Google Scholar] [CrossRef]
- Zilber-Rosenberg, I.; Rosenberg, E. Role of microorganisms in the evolution of animals and plants: The hologenome theory of evolution. FEMS Microbiol. Rev. 2008, 32, 723–735. [Google Scholar] [CrossRef]
- Coelho, L.; Afonso, M.; Jesus, F.; Campos, I.; Abrantes, N.; Gonçalves, F.J.M.; Serpa, D.; Marques, S.M. Effects of Eucalypt ashes from moderate and high severity wildfires on the skin microbiome of the Iberian frog (Rana iberica). Environ. Pollut. 2022, 313, 120065. [Google Scholar] [CrossRef]
- MacKnight, N.J.; Cobleigh, K.; Lasseigne, D.; Chaves-Fonnegra, A.; Gutting, A.; Dimos, B.; Antoine, J.; Fuess, L.; Ricci, C.; Butler, C.; et al. Microbial dysbiosis reflects disease resistance in diverse coral species. Commun. Biol. 2021, 4, 679. [Google Scholar] [CrossRef]
- Maes, P.W.; Rodrigues, P.A.P.; Oliver, R.; Mott, B.M.; Anderson, K.E. Diet-related gut bacterial dysbiosis correlates with impaired development, increased mortality and Nosema disease in the honeybee (Apis mellifera). Mol. Ecol. 2016, 25, 5439–5450. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mountain Range | Sample Name | 50 (%) | 50 Quartile | 75 (%) | 75 Quartile | 90 (%) | 90 Quartile |
|---|---|---|---|---|---|---|---|
| Roan Highlands | ROAN.2 | 0.446 | 4 | 0.327 | 4 | 0.186 | 4 |
| ROAN.1 | 0.369 | 3 | 0.251 | 3 | 0.152 | 3 | |
| Balsams | B224.1 | 0.483 | 4 | 0.353 | 4 | 0.261 | 4 |
| LG.2 | 0.453 | 4 | 0.276 | 4 | 0.200 | 4 | |
| B224.2 | 0.448 | 4 | 0.3435 | 4 | 0.301 | 4 | |
| LG.3 | 0.345 | 3 | 0.239 | 3 | 0.115 | 3 | |
| LG.1 | 0.287 | 2 | 0.184 | 2 | 0.078 | 1 | |
| Blacks | MG.1 | 0.336 | 3 | 0.241 | 3 | 0.117 | 3 |
| AC.1 | 0.327 | 2 | 0.271 | 4 | 0.172 | 4 | |
| MG.2 | 0.304 | 2 | 0.246 | 3 | 0.111 | 2 | |
| AC.2 | 0.158 | 1 | 0.086 | 1 | 0.041 | 1 | |
| Nantahalas | SI.2 | 0.519 | 4 | 0.406 | 4 | 0.247 | 4 |
| PV.1 | 0.371 | 4 | 0.159 | 1 | 0.098 | 2 | |
| WS.2 | 0.351 | 3 | 0.238 | 3 | 0.127 | 3 | |
| T3 | 0.338 | 3 | 0.219 | 2 | 0.125 | 3 | |
| WS.1 | 0.326 | 2 | 0.2242 | 2 | 0.099 | 2 | |
| SI.1 | 0.319 | 2 | 0.233 | 3 | 0.106 | 2 | |
| PV.2 | 0.316 | 2 | 0.138 | 1 | 0.0873 | 2 | |
| PV.3 | 0.247 | 1 | 0.123 | 1 | 0.0715 | 1 | |
| T1 | 0.220 | 1 | 0.188 | 2 | 0.106 | 2 | |
| T2 | 0.148 | 1 | 0.105 | 1 | 0.0465 | 1 | |
| Smokies | SC2 | 0.532 | 4 | 0.353 | 4 | 0.227 | 4 |
| FC1 | 0.356 | 3 | 0.214 | 2 | 0.111 | 2 | |
| RP2 | 0.25 | 2 | 0.160 | 2 | 0.120 | 3 | |
| CD.2 | 0.247 | 1 | 0.175 | 2 | 0.079 | 1 | |
| SC.1 | 0.203 | 1 | 0.119 | 1 | 0.066 | 1 | |
| FC2 | 0.159 | 1 | 0.113 | 1 | 0.060 | 1 |
| Category | Beta Diversity Metric | Pseudo F | p-Value |
|---|---|---|---|
| Habitat (stream vs. cliff) | Bray-Curtis | 2.038 | 0.001 |
| Jaccard | 1.534 | 0.002 | |
| Unweighted unifrac | 1.659 | 0.006 | |
| Weighted unifrac | 1.189 | 0.267 | |
| Location | Bray-Curtis | 2.015 | 0.001 |
| Jaccard | 1.581 | 0.001 | |
| Unweighted unifrac | 1.560 | 0.001 | |
| Weighted unifrac | 2.102 | 0.001 | |
| Mountain Range | Bray-Curtis | 1.431 | 0.004 |
| Jaccard | 1.288 | 0.001 | |
| Unweighted unifrac | 1.245 | 0.006 | |
| Weighted unifrac | 1.726 | 0.023 |
| Category | Alpha Diversity Metric | H | p-Value |
|---|---|---|---|
| Habitat (stream vs. cliff) | Shannon | 1.014 | 0.313996 |
| Evenness | 0.230 | 0.63161 | |
| Faith | 3.864 | 0.049322 | |
| Location | Shannon | 11.874 | 0.45582 |
| Evenness | 15.803 | 0.20043 | |
| Faith | 20.766 | 0.053914 | |
| Mountain Range | Shannon | 2.993 | 0.558939 |
| Evenness | 0.670 | 0.951372 | |
| Faith | 6.416 | 0.17016 |
| Variable | Beta Diversity Metric | Spearman Rho | p-Value |
|---|---|---|---|
| Bio8 | Bray-Curtis | 0.023 | 0.751 |
| Mean temperature of the wettest quarter | Jaccard | 0.030 | 0.667 |
| Unweighted unifrac | 0.079 | 0.269 | |
| Weighted unifrac | −0.062 | 0.395 | |
| Bio10 | Bray-Curtis | 0.214 | 0.001 |
| Mean temperature of the warmest quarter | Jaccard | 0.205 | 0.004 |
| Unweighted unifrac | 0.138 | 0.022 | |
| Weighted unifrac | 0.267 | 0.001 | |
| Bio12 | Bray-Curtis | −0.043 | 0.463 |
| Annual precipitation | Jaccard | −0.0291 | 0.637 |
| Unweighted unifrac | −0.088 | 0.115 | |
| Weighted unifrac | −0.113 | 0.064 |
| Variable | Alpha Diversity Metric | Spearman Rho | p-Value |
|---|---|---|---|
| Bio8 | Shannon | −0.049 | 0.8061 |
| Mean temperature of the wettest quarter | Evenness | −0.046 | 0.8158 |
| Faith | −0.153 | 0.4357 | |
| Bio10 | Shannon | −0.083 | 0.6766 |
| Mean temperature of the warmest quarter | Evenness | 0.233 | 0.2326 |
| Faith | −0.369 | 0.0536 | |
| Bio12 | Shannon | 0.070 | 0.07213 |
| Annual precipitation | Evenness | −0.152 | 0.4382 |
| Faith | 0.330 | 0.0862 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paulsen, J.; Allen, J.L.; Morris, N.; Dorey, J.; Walke, J.B.; Alter, S.E. Geography, Climate, and Habitat Shape the Microbiome of the Endangered Rock Gnome Lichen (Cetradonia linearis). Diversity 2024, 16, 178. https://doi.org/10.3390/d16030178
Paulsen J, Allen JL, Morris N, Dorey J, Walke JB, Alter SE. Geography, Climate, and Habitat Shape the Microbiome of the Endangered Rock Gnome Lichen (Cetradonia linearis). Diversity. 2024; 16(3):178. https://doi.org/10.3390/d16030178
Chicago/Turabian StylePaulsen, Julianna, Jessica L. Allen, Nathan Morris, Jenna Dorey, Jenifer B. Walke, and S. Elizabeth Alter. 2024. "Geography, Climate, and Habitat Shape the Microbiome of the Endangered Rock Gnome Lichen (Cetradonia linearis)" Diversity 16, no. 3: 178. https://doi.org/10.3390/d16030178