
A Study of the Adsorption Properties of Individual Atoms on the Graphene Surface: Density Functional Theory Calculations Assisted by Machine Learning Techniques
 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Crystal Structure and Calculations Method
2.2. Machine Learning Databases and Models
3. Results and Discussion
3.1. Correlation Analysis and Selection of Eigenvalues
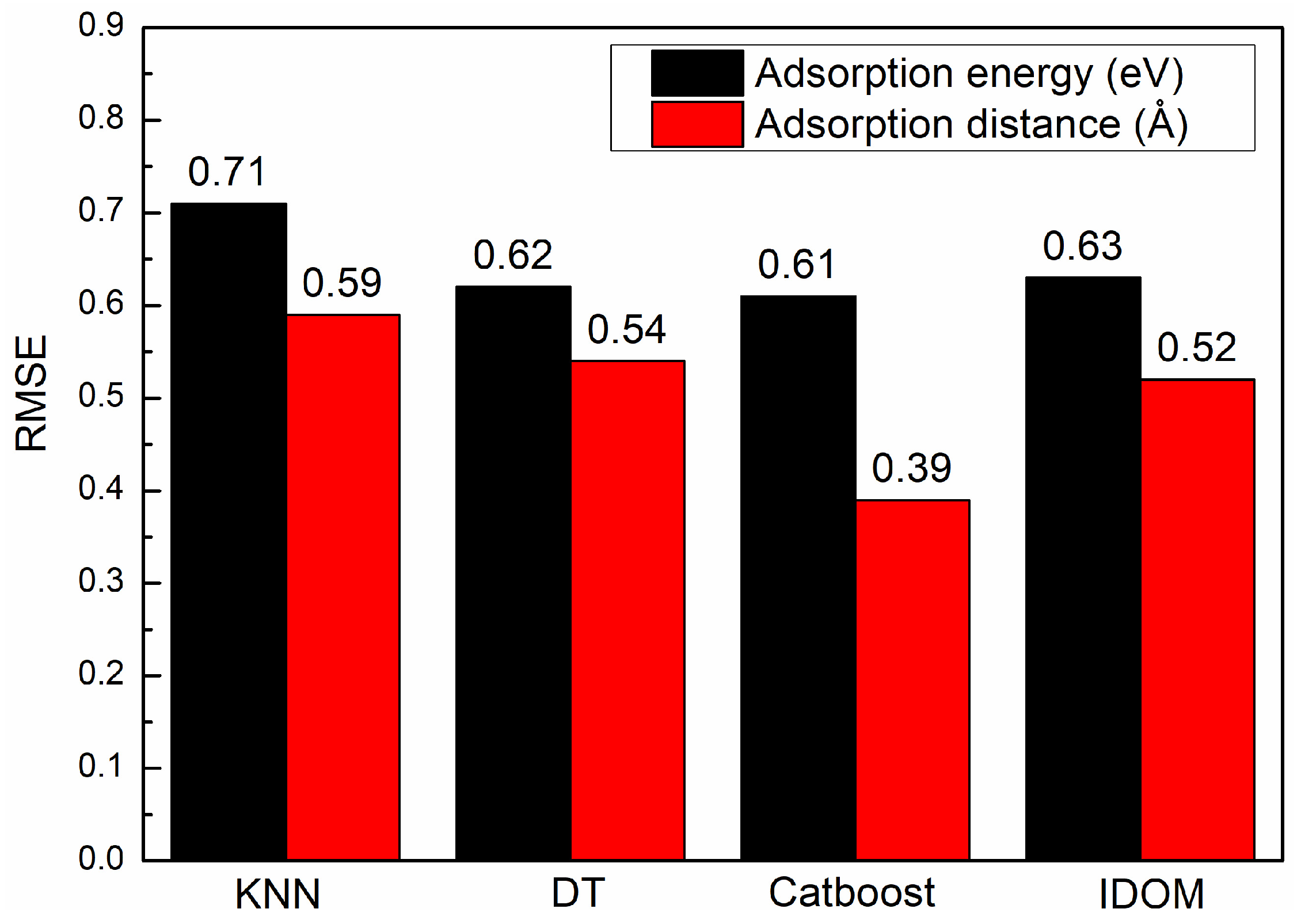
3.2. Machine Learning Model Building and Selection
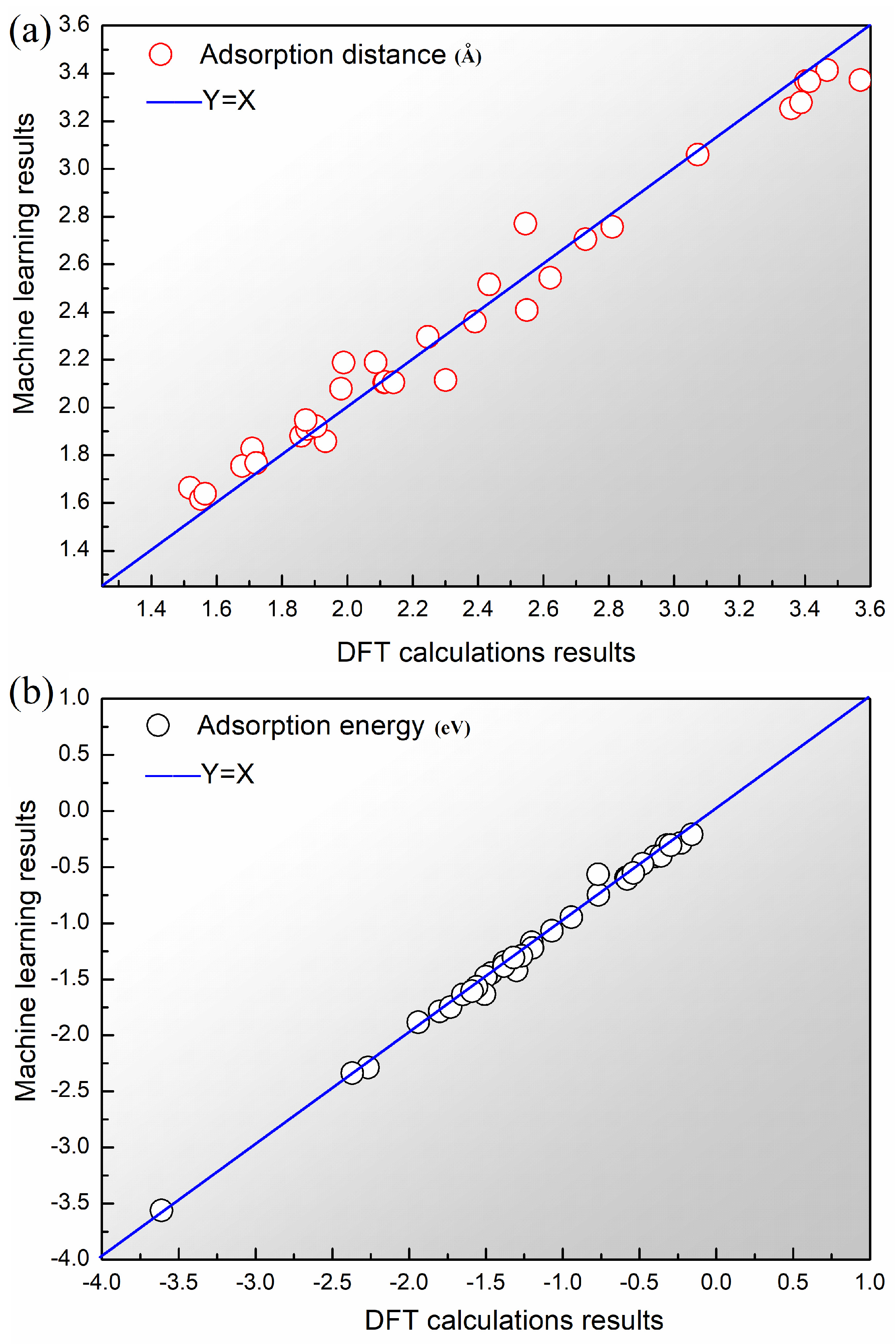
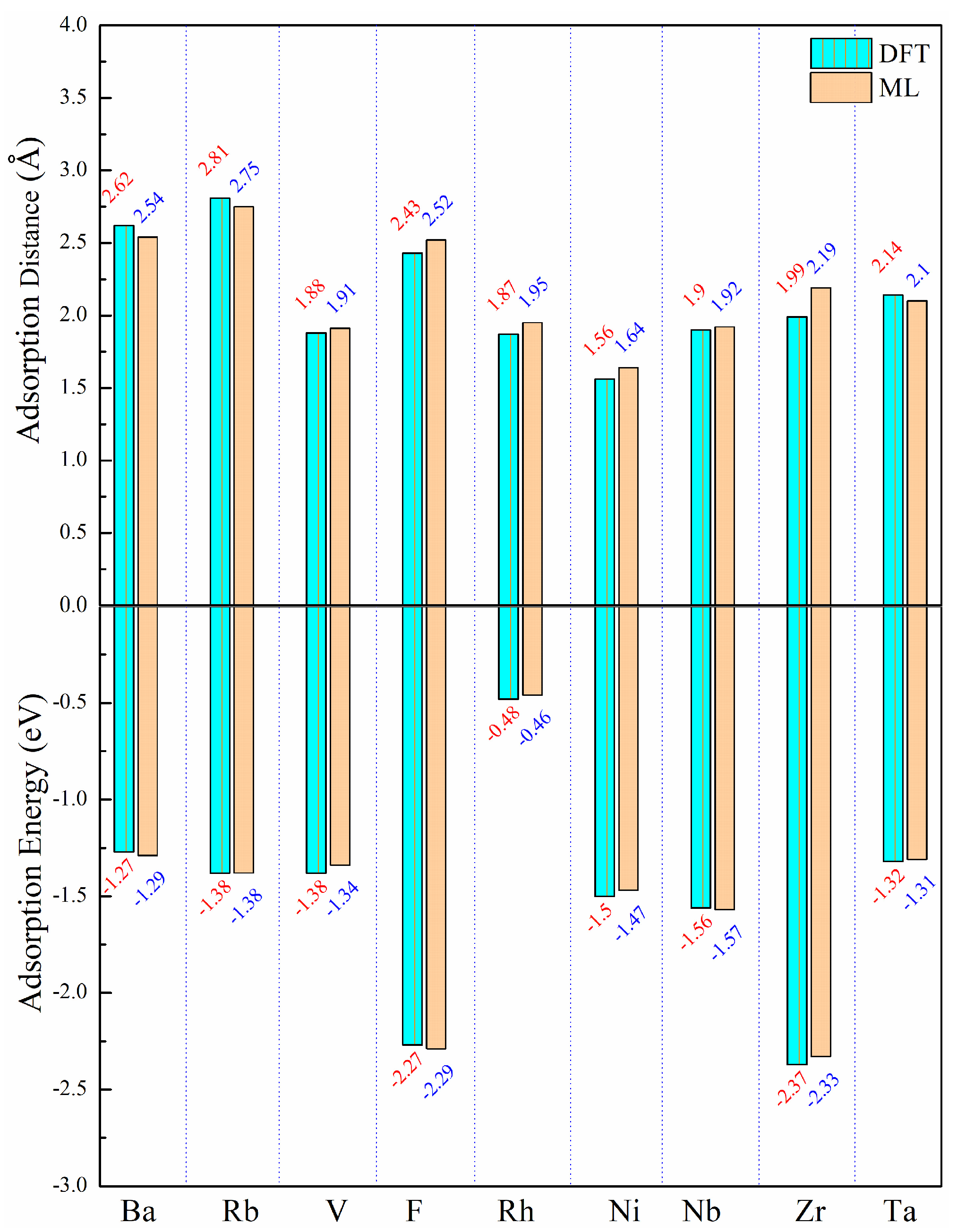
3.3. Prediction and Verification of Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, M.; Lim, M.K.; Qu, Y.C.; Ni, D.; Xiao, Z. Supply chain risk management with machine learning technology: A literature review and future research directions. Comput. Ind. Eng. 2023, 175, 108859. [Google Scholar] [CrossRef]
- Wang, X.Z.; Liu, A.; Kara, S. Machine learning for engineering design toward smart customization: A systematic review. J. Manuf. Syst. 2022, 65, 391–405. [Google Scholar] [CrossRef]
- Melnikov, A.A.; Nautrup, H.P.; Krenn, M.; Dunjko, V.; Tiersch, M.; Zeilinger, A.; Briegel, H.J. Active learning machine learns to create new quantum experiments. Proc. Natl. Acad. Sci. USA 2018, 115, 1221–1226. [Google Scholar] [CrossRef]
- Manley, K.; Egoh, B.N. Mapping and modeling the impact of climate change on recreational ecosystem services using machine learning and big data. Environ. Res. Lett. 2022, 17, 054025. [Google Scholar] [CrossRef]
- Ward, L.; Agrawal, A.; Choudhary, A.; Wolverton, C. A general-purpose machine learning framework for predicting properties of inorganic materials. npj Comput. Mater. 2016, 2, 16028. [Google Scholar] [CrossRef]
- Stein, H.S.; Guevarra, D.; Newhouse, P.F.; Soedarmadji, E.; Gregoire, J.M. Machine learning of optical properties of materials-predicting spectra from images and images from spectra. Chem. Sci. 2019, 10, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Z.; Zhang, H.W.; Li, S.R.; Zhu, W.D.; Ke, Y.L. Machine learning and materials informatics approaches for predicting transverse mechanical properties of unidirectional CFRP composites with microvoids. Mater. Des. 2022, 224, 111340. [Google Scholar] [CrossRef]
- Schutt, K.T.; Glawe, H.; Brockherde, F.; Sanna, A.; Muller, K.R.; Gross, E.K.U. How to represent crystal structures for machine learning: Towards fast prediction of electronic properties. Phys. Rev. B 2014, 89, 163–168. [Google Scholar] [CrossRef]
- Pasti, I.A.; Jovanovic, A.; Dobrota, A.S.; Mentus, S.V.; Johansson, B.; Skorodumova, N.V. Atomic adsorption on pristine graphene along the Periodic Table of Elements—From PBE to non-local functionals. Appl. Surf. Sci. 2018, 436, 433–440. [Google Scholar] [CrossRef]
- Pasti, I.A.; Jovanovic, A.; Dobrota, A.S.; Mentus, S.V.; Johansson, B.; Skorodumova, N.V. Atomic adsorption on graphene with a single vacancy: Systematic DFT study through the periodic table of elements. Phys. Chem. Chem. Phys. 2018, 20, 858–865. [Google Scholar] [CrossRef]
- Mashhadzadeh, A.H.; Ghanbari, M.; Koochaki, A.; Seyyedbarzegar, S.; Ahangari, M.G. Experiment and theory for acetylene adsorption in transformer oil. J. Mol. Struct. 2021, 1230, 129860. [Google Scholar] [CrossRef]
- Balachandran, P.V.; Xue, D.Z.; Theiler, J.; Hogden, J.; Lookman, T. Adaptive Strategies for Materials Design using Uncertainties. Sci. Rep. 2016, 6, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanaka, Y. Material synthesis and design from first principle calculations and machine learning. Comput. Mater. Sci. 2016, 112, 364–367. [Google Scholar] [CrossRef]
- Rupp, M.; Tkatchenko, A.; Muller, K.R.; Lilienfeld, O.A. Fast and Accurate Modeling of Molecular Atomization Energies with Machine Learning. Phys. Rev. Lett. 2012, 108, 058301. [Google Scholar] [CrossRef] [PubMed]
- Scholkopf, B.; Smola, A.J. Learning with Kernels: Support Vector Machines Regularization, Optimization Andbeyond; MIT Press: Cambridge, MA, USA, 2003. [Google Scholar]
- Isayev, O.; Oses, C.; Toher, C.; Gossett, E.; Curtarolo, S.; Tropsha, A. Universal fragment descriptors for predicting properties of inorganic crystals. Nat. Commun. 2017, 8, 15679. [Google Scholar] [CrossRef]
- Lee, C.G.; Wei, X.D.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2009, 6, 11–19. [Google Scholar]
- Xu, Y.; Sheng, K.; Li, C.; Shi, G. Self-assembled graphene hydrogel via a one-step hydrothermal process. ACS Nano 2010, 4, 4324–4330. [Google Scholar] [CrossRef]
- Tang, H.Y.; Menabde, S.G.; Anwar, T.; Kim, J.; Jang, M.S.; Tagliabue, G. Photo-modulated optical and electrical properties of graphene. Nanophotonics 2022, 11, 917–940. [Google Scholar] [CrossRef]
- Li, W.; Cheng, G.J.; Liang, Y.R.; Tian, B.Y.; Liang, X.L.; Peng, L.M.; Walker, A.R.H.; Gundlach, D.J.; Nguyen, N.V. Broadband optical properties of graphene by spectroscopic ellipsometry. Carbon 2016, 99, 348–353. [Google Scholar] [CrossRef]
- Sun, Y.W.; Papageorgiou, D.G.; Humphreys, C.J.; Dunstan, D.J.; Puech, P.; Proctor, J.E.; Bousige, C.; Machon, D.; San-Miguel, A. Mechanical properties of graphene. Appl. Phys. Rev. 2021, 8, 021310. [Google Scholar] [CrossRef]
- Papageorgiou, D.G.; Kinloch, I.A.; Young, R.J. Mechanical properties of graphene and graphene-based nanocomposites. Prog. Mater. Sci. 2017, 90, 75–127. [Google Scholar] [CrossRef]
- Wang, D.Z.; Xiao, Z.B. Revealing the Al/L12-Al3Zr inter-facial properties: Insights from first-principles calculations. Vacuum 2022, 195, 110620. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Xu, L.L.; Zheng, H.F.; Xu, B.; Liu, G.Y.; Zhang, S.L.; Zheng, H.B. Suppressing Nonradiative Recombination by Electron-Donating Substituents in 2D Conjugated Triphenylamine Polymers toward Efficient Perovskite Optoelectronics. Nano Lett. 2023, 23, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1998, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.O.; Gunnarsson, O. The density functional formalism, its applications and prospects. Rev. Mod. Phys. 1989, 61, 689–746. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Huang, J.T.; Liu, Y.; Chen, J.Y.; Lai, Z.H.; Hu, J.; Zhou, F.; Li, M.W.; Zhu, J.C. Adsorption behaviour of Al atoms on the surface of perfect and defective graphene: A first principle study. Mol. Phys. 2022, 120, e2121231. [Google Scholar] [CrossRef]
- Mu, Y.S.; Liu, X.D.; Wang, L.D. A Pearson’s correlation coefficient based decision tree and its parallel implementation. Inf. Sci. 2018, 435, 40–58. [Google Scholar] [CrossRef]
- Tang, M.C.; Zhang, D.Z.; Wang, D.Y.; Deng, J.; Kong, D.T.; Zhang, H. Performance prediction of 2D vertically stacked MoS2-WS2 heterostructures base on first-principles theory and Pearson correlation coefficient. Appl. Surf. Sci. 2022, 596, 153498. [Google Scholar] [CrossRef]
- Chen, W.C.; Schmidt, J.N.; Yan, D.; Vohra, Y.K.; Chen, C.C. Machine learning and evolutionary prediction of superhard B-C-N compounds. npj Comput. Mater. 2021, 7, 114. [Google Scholar] [CrossRef]
- Qiao, L.; Liu, Y.; Zhu, J.C. Application of generalized regression neural network optimized by fruit fly optimization algorithm for fracture toughness in a pearlitic steel. Eng. Fract. Mech. 2020, 235, 107105. [Google Scholar] [CrossRef]
- Sutojo, T.; Rustad, S.; Akrom, M.; Syukur, A.; Shidik, G.F.; Dipojono, H.K. A machine learning approach for corrosion small datasets. npj Mater. Degrad. 2023, 7, 18. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MSE | RMSE | MAPE | ||
|---|---|---|---|---|
| Adsorption distance | 0.0096 | 0.9752 | 0.0982 | 3.6181 |
| Adsorption energy | 0.0028 | 0.9946 | 0.0525 | 4.5995 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Chen, M.; Xue, J.; Li, M.; Cheng, Y.; Lai, Z.; Hu, J.; Zhou, F.; Qu, N.; Liu, Y.; et al. A Study of the Adsorption Properties of Individual Atoms on the Graphene Surface: Density Functional Theory Calculations Assisted by Machine Learning Techniques. Materials 2024, 17, 1428. https://doi.org/10.3390/ma17061428
Huang J, Chen M, Xue J, Li M, Cheng Y, Lai Z, Hu J, Zhou F, Qu N, Liu Y, et al. A Study of the Adsorption Properties of Individual Atoms on the Graphene Surface: Density Functional Theory Calculations Assisted by Machine Learning Techniques. Materials. 2024; 17(6):1428. https://doi.org/10.3390/ma17061428
Chicago/Turabian StyleHuang, Jingtao, Mo Chen, Jingteng Xue, Mingwei Li, Yuan Cheng, Zhonghong Lai, Jin Hu, Fei Zhou, Nan Qu, Yong Liu, and et al. 2024. "A Study of the Adsorption Properties of Individual Atoms on the Graphene Surface: Density Functional Theory Calculations Assisted by Machine Learning Techniques" Materials 17, no. 6: 1428. https://doi.org/10.3390/ma17061428