Neuroprotective Actions of Dietary Choline

Department of Pathology and Laboratory Medicine, Boston University School of Medicine, 72 East Concord Street, Boston, MA 02118, USA
*
Author to whom correspondence should be addressed.
Nutrients 2017, 9(8), 815; https://doi.org/10.3390/nu9080815
Submission received: 20 June 2017
/
Revised: 21 July 2017
/
Accepted: 25 July 2017
/
Published: 28 July 2017
(This article belongs to the Special Issue Dietary Choline)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Choline is an essential nutrient for humans. It is a precursor of membrane phospholipids (e.g., phosphatidylcholine (PC)), the neurotransmitter acetylcholine, and via betaine, the methyl group donor S-adenosylmethionine. High choline intake during gestation and early postnatal development in rat and mouse models improves cognitive function in adulthood, prevents age-related memory decline, and protects the brain from the neuropathological changes associated with Alzheimer’s disease (AD), and neurological damage associated with epilepsy, fetal alcohol syndrome, and inherited conditions such as Down and Rett syndromes. These effects of choline are correlated with modifications in histone and DNA methylation in brain, and with alterations in the expression of genes that encode proteins important for learning and memory processing, suggesting a possible epigenomic mechanism of action. Dietary choline intake in the adult may also influence cognitive function via an effect on PC containing eicosapentaenoic and docosahexaenoic acids; polyunsaturated species of PC whose levels are reduced in brains from AD patients, and is associated with higher memory performance, and resistance to cognitive decline.
Keywords:
Alzheimer’s disease; autism; brain; choline; epilepsy; DNA methylation; memory; nutrition; pregnancy1. Choline: An Essential Nutrient for Humans
Choline is an essential nutrient; that is, together with essential amino acids, fatty acids, vitamins and minerals, it must be obtained from the diet to maintain health [1]. Because choline is present in many foods, a variety of diets can satisfy the need for this nutrient. For the purposes of estimating choline intake by people, researchers use a database available at the US Department of Agriculture web site that lists dietary compounds that contain the choline moiety (i.e., free choline, glycerophosphocholine, phosphocholine, phosphatidylcholine, sphingomyelin and betaine—a metabolite of choline) [2]. Because all of those compounds provide bioavailable choline, studies on choline intake generally refer to the total dietary choline. Adequate Intake (AI) recommendations, issued by the Food and Nutrition Board of the Institute of Medicine of the National Academy of Sciences, call for an average daily intake of 7.5 mg of choline per kg of body weight, but are higher during pregnancy and lactation [1], given the needs of the fetus and nursing baby [3,4,5]. Reported pathological effects of low choline consumption include liver dysfunction in adult men [6], muscle damage [7], and apoptotic death of lymphocytes [8]. Moreover, higher consumption of choline or betaine (an oxidized choline metabolite and a methyl group donor) in adults is associated with reduced risk of some cancers [9,10,11,12,13] (reviewed in [14]).
Choline has been shown to exert neuroprotective effects in both animal and human studies. High choline intake during the perinatal period is neuroprotective in a variety of animal models of neuronal dysfunction, including that resulting from aging [15,16,17], seizures [18,19,20,21], prenatal alcohol exposure [22,23,24,25,26,27] and the genetic disorders Rett syndrome [28,29,30,31] and Down syndrome [32,33,34,35,36,37,38]. Dietary supplementation with choline-containing compounds, alone or in combination with other methyl donors such as folate and vitamin B12, was also found to ameliorate neurological impairments in animal models of stroke [39], and ischemia [40]. Moreover, high choline consumption during pregnancy correlated with reduced risk of neural tube defects in humans [41,42]. This association was less robust in more recent studies—possibly as a result of the addition of choline to vitamin supplements routinely taken by pregnant women, and variations in total daily caloric intake and reported ranges of choline consumption, in the different studies [43,44]. Dietary intake of choline in adult human subjects has been shown to correlate with cognitive function [45], and treatment with certain choline-containing compounds showed a limited tendency to reduce cognitive impairment in human vascular dementias in small clinical trials [46].
In this review, we summarize the results of studies in animal models and humans on the effects of perinatal choline nutrition on brain development, on cognitive function in the adult, and on the progression of several neurodegenerative diseases. We also discuss emerging evidence for neuroprotective effects of dietary choline consumption during adulthood. While we focus on choline, it is important to recognize that other nutrients such as folate, vitamin B12 and vitamin B6, influence the metabolism of choline, and their availability may modify the requirements for choline.
2. Choline Nutrition during Development and Cognitive Function
2.1. Protection against Age-Related Memory Decline and Advancement of Hippocampal Development
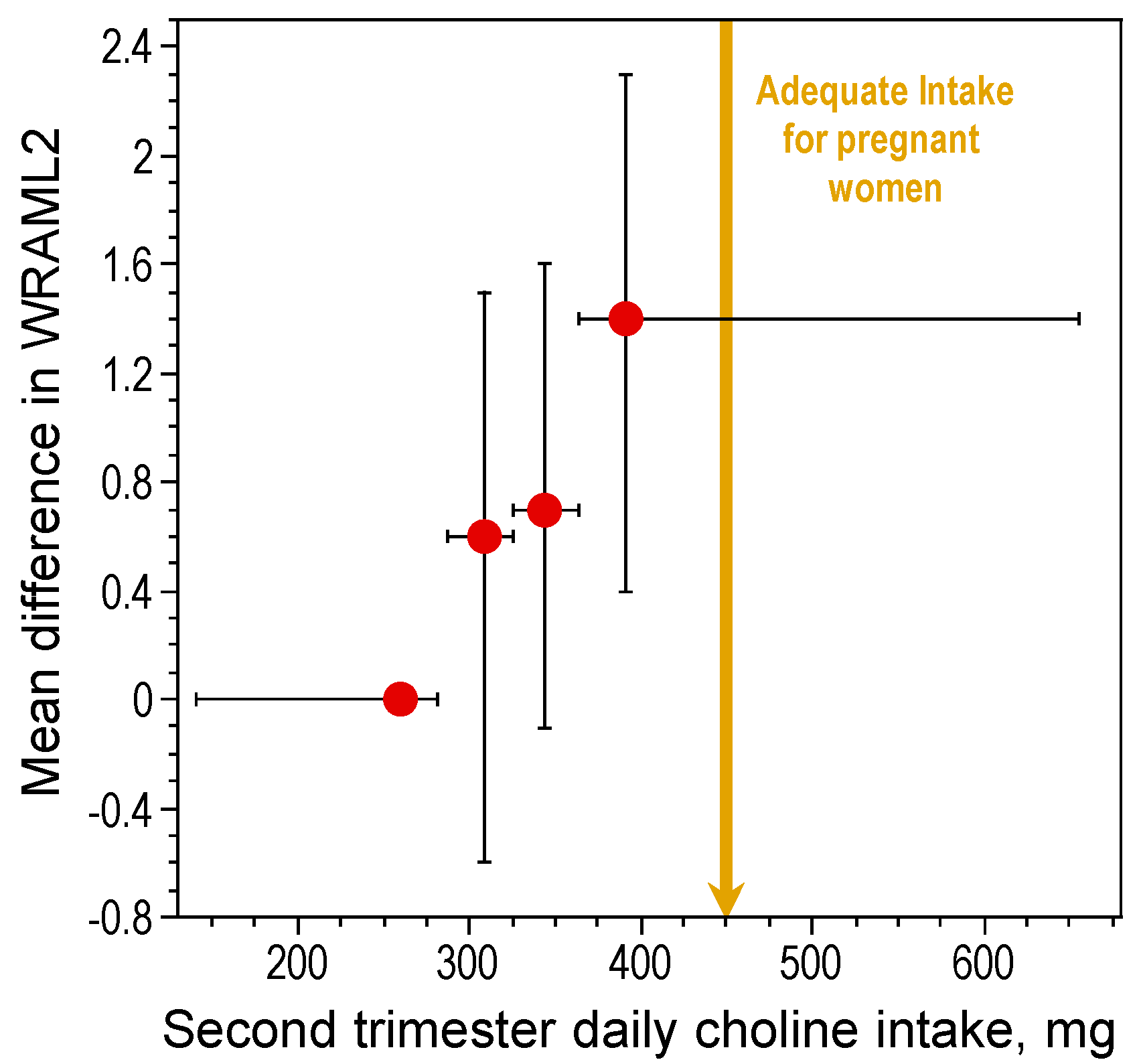
In rats, high maternal choline consumption at specific times during pregnancy (Embryonic Days 11–17) enhances cognitive ability of the offspring throughout life [15,17,47,48,49,50,51,52,53]. One important developmental benchmark, the ability to navigate using relational cues in a Morris water maze, was found to occur three days earlier in prenatally choline-supplemented rats [51]. The acquisition of this ability is thought to signal the onset of hippocampal function [54]. Thus, prenatal choline supplementation causes an approximately three-day advancement in hippocampal development. While it is not possible to conduct a similar, well controlled study in humans, data obtained by project Viva in Massachusetts show that maternal choline intake within the AI range during pregnancy was associated with better memory function in children at seven years of age as compared to children of mothers whose consumption was approximately 50% of the AI levels [55] (Figure 1). Thus, in humans, as in animal models, higher gestational consumption of choline has long-term positive effects on cognition during childhood. However, it is not yet known if these effects persist into adulthood and old age in humans. Using a rat model, Meck et al. [17] altered choline availability to rats during seven timeframes spanning Embryonic Day (E) 6 through Postnatal Day (P) 75 and examined spatial memory ability in the perinatally-treated adults. Two sensitive periods were identified, E12–17 and P16–30, during which choline supplementation facilitated spatial memory. Moreover, choline supplementation during E12–17 only, prevented the memory decline normally observed in aged (26 months old) rats [17].
2.2. Cellular, Transcriptomic, Proteomic and Epigenetic Correlates of the Neuroprotective Action of Choline
During fetal development, maternal choline deficiency inhibits hippocampal precursor cell proliferation and stimulates apoptosis in the hippocampus [56], whereas gestational choline supplementation stimulates hippocampal cell division [57]. In the adult, neurogenesis in the dentate gyrus of the hippocampus, a process that occurs throughout life [58,59,60], is enhanced by prenatal choline supplementation, and impaired by prenatal choline deficiency [20,61,62]. This effect on adult neurogenesis was associated with increased hippocampal concentrations of the trophic factors nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), insulin-like growth factor 2 (IGF2), and vascular endothelial growth factor (VEGF) [20,61,62,63,64,65], increased size of basal forebrain cholinergic neurons [66]—cells required for normal cognitive function [67,68]—and elevated acetylcholine synthesis and release from these neurons [17,65,69]. Prenatal choline supplementation increased the phosphorylation of mitogen-activated protein kinase (MAPK) and 3′,5′-cyclic adenosine monophosphate (cAMP) response element binding protein (CREB) in response to activation of glutamate receptors in hippocampus, [51]. The MAPK signaling cascade, and its downstream targets including CREB, are central players in the processes underlying synaptic plasticity, and learning and memory [70]. The induction in hippocampus of long term potentiation, an electrophysiological correlate of synaptic plasticity [71], was similarly sensitive to prenatal choline availability [49,72]. Gene expression analysis identified 530 hippocampal and 815 cerebral cortical mRNA species whose levels were affected by prenatal choline status [64]. The protein products of a subset of these genes, including calcium/calmodulin-dependent kinase 1 and IGF2, participate in signaling pathways involved in memory processes and thus may contribute to the observed choline-induced changes in cognitive performance (reviewed in [73]).
Advances in the field of epigenetics have provided a framework to explain how heritable changes in gene expression patterns may be propagated during development and throughout adulthood without altering the primary DNA sequence. The methylation of CpG sequences within the regulatory elements of genes changes their expression via an interaction with a complex network of proteins, including transcription factors [74]. Following DNA replication, the unmethylated daughter strand in hemimethylated DNA is symmetrically methylated by the enzyme DNA methyltransferase 1 (DNMT1) [75], thus recapitulating the parent methylation pattern in the daughter cells. The process of DNA methylation is dynamic [76] and responsive to changes in the environment, including alterations in nutrient availability. In particular, DNA methylation is modulated by the availability of nutrients that serve as methyl group donors and cofactors, such as choline, betaine, methionine, folic acid and vitamin B12, via their influence on tissue levels of S-adenosylmethionine (SAM) (the methyl group donor for most enzymatic methylation reactions) (see [77] for a review). Kovacheva et al. [78] evaluated DNA methylation parameters in liver and cerebral cortex on E17 in rats exposed in utero to varying amounts of choline (via the diet supplied to pregnant dams) beginning on E11. The investigators focused on the differentially methylated region 2 (DMR2) of the Igf2 gene, which undergoes a dramatic change in methylation status during development [79]. DMR2 methylation was increased in choline-deficient embryos, as compared to the control and choline-supplemented rats, and was accompanied by a compensatory increase in the expression of the maintenance DNA methylase Dnmt1. Because DNA methylation is highly dynamic in adult brain and may modulate the expression of genes involved in the regulation of synaptic plasticity [80,81,82,83,84] and learning and memory [85,86,87,88,89,90,91,92,93,94,95,96], it is possible that dietary availability of methyl donors such as choline in the adult will also prove to be an important determinant of cognitive function.
2.3. Neuroprotective Actions of Perinatal Choline in Animal Models of Disease
2.3.1. Epilepsy
Status epilepticus, a period of prolonged seizures, is a neurological condition that produces multiple degenerative and regenerative changes in the hippocampus [97]. These changes are accompanied by cognitive deficits in hippocampal-dependent tasks [97]. Several studies tested the effects of high choline intake in rat models of chemically-evoked epilepsy. In pilocarpine- [18] and kainic acid- [19,21] induced status epilepticus, prenatal choline supplementation attenuated the deficits in visual-spatial memory. In addition, these protective actions of choline were accompanied by a decrease in seizure-induced hippocampal neurodegeneration, and a reduction in compensatory proliferation of cells within the dentate gyrus [98]. Moreover, choline supplementation prevented hippocampal loss of GAD65 mRNA [98], which encodes a form of the γ-aminobutyric acid (GABA)-synthesizing enzyme, glutamic acid decarboxylase, that is localized to the nerve terminals. As in the other rodent models [20,29,30,61,65,98,99], prior to seizures choline supplementation also increased the levels of several trophic factors in the hippocampus: specifically, NGF, BDNF and IGF1, indicating that this nutritional treatment may establish a neuroprotective hippocampal milieu that attenuates the neuropathological response to and/or helps facilitate recovery from seizures to protect cognitive function.
2.3.2. Down Syndrome
Down syndrome, a common form of mental retardation, affects approximately 1 in 700 births in the United States [100]. The disorder is caused by trisomy of the whole or a part of chromosome 21, and the resulting increase in expression of the genes encoded on the extra chromosome [101]. Strupp and colleagues [32,33,34,35,36,37,38] used a mouse model of Down syndrome (Ts65Dn mice) to test the hypothesis that choline supplementation from conception to weaning could prevent some of the neurological and cognitive deficits observed in these mice. The genome of Ts65Dn mice was engineered to carry a third copy of the distal region of mouse chromosome 16, which contains approximately 94 genes orthologous to the Down syndrome critical region of the human chromosome 21 [102]. The adult offspring of choline-supplemented Ts65Dn dams performed significantly better than control Ts65Dn mice in several visual attention tasks [32,35,36]. In some of these tasks, the choline-supplemented Ts65Dn mice did not differ from the wild type controls [32]. The improvement in spatial cognition correlated with normalization of hippocampal neurogenesis in choline-supplemented Ts65Dn mice [35]. It is interesting to note that a number of genes on chromosome 21 are involved in epigenetic mechanisms, including those encoding the DNA methyltransferase-like protein DNMT3L [101], which is required for the establishment of maternal genomic imprints [103], and also stimulates de novo methylation by DNMT3A [104]. In frontal cortex neurons purified from Down syndrome patients and controls, 272 genes showed differential methylation of CpG islands; the majority of which were hypermethylated in Down syndrome relative to controls [105,106]. An earlier report demonstrated that global DNA methylation, measured at E17, was reduced in frontal cortex of perinatally choline-supplemented rats relative to control and choline deficient embryos, and this was reflected in a significant reduction in mRNA levels of the Dnmt3l and Dnmt3a [78] These observations raise the possibility that choline supplementation in Down syndrome models may improve cognitive function in part by opposing the hypermethylation that otherwise occurs in neurons of these animals.
2.3.3. Rett Syndrome
Rett syndrome is a genetic neurological disorder of childhood that represents a common (approximately 1 in 10,000 births) form of mental retardation and almost exclusively affects girls [31]. It is usually caused by a mutation in the X chromosome-linked methyl-CpG-binding protein 2 (Mecp2) gene encoding a protein with possible roles in gene transcription, chromatin organization, alternative splicing, and miRNA processing [107]. Several mouse models with inactivating mutations of Mecp2 have been developed. These mice resemble the human disease in that they are apparently normal at birth, but develop severe neurological symptoms within weeks. In a series of studies, Berger-Sweeney and colleagues tested the effects of perinatal choline supplementation in two mouse models of Rett syndrome [28,29,30,108,109]. Rett syndrome model mice were supplemented with choline or saccharine (controls) via the mothers’ milk from birth to weaning. Choline supplementation improved motor coordination and locomotor activity of Mecp2-null males, and grip strength in females, but did not alter cognitive deficits in either sex [28]. These improvements in locomotor function were accompanied by increases in total brain volume in females, and in cerebellar volume in males [108]. Postnatal choline supplementation increased striatal NGF expression in both wild type and Mecp2 null mice [29] and increased the brain levels of N-acetyl aspartate, a marker of neuronal integrity, in nuclear magnetic resonance spectroscopy assays [109]. These results are consistent with an effect of choline on enhanced neuronal proliferation and survival in mutant mice. In mice with a different Mecp2 mutation, early postnatal choline treatment prevented deficits in locomotor activity [30], and restored the activity of the acetylcholine-synthesizing enzyme, choline acetyltransferase (CHAT), in the striatum. In both mouse models, choline supplementation increased mRNA expression of NGF and BDNF in the cerebral cortex and hippocampus [30,31]. Taken together, these data suggest that nutritional supplementation with choline may improve neuronal function in Rett syndrome patients and thus constitutes a potential therapy for this disease.
2.3.4. Schizophrenia
To evaluate the possibility that perinatal choline treatment could be useful in preventing certain psychiatric disorders, Stevens et al. [110] studied the effects of choline supplementation in the DBA/2 mouse strain, which is extensively used as a model of schizophrenia [111]—a disease with 60–80% heritability [112,113]. These mice resemble human schizophrenia patients in that they show reduced levels of hippocampal α7 nicotinic receptors, and deficits in sensory inhibition in response to repeated auditory stimuli [114]. DBA/2 dams were placed on control or choline-supplemented diets from mating until weaning. The offspring of dams maintained on the control diet displayed the characteristic abnormality in sensory processing that is also present in patients with schizophrenia, whereas prenatally choline-supplemented mice exhibited a normal sensory processing phenotype [110], suggesting that this nutritional treatment may reduce the risk of schizophrenia. This effect of choline was not seen in DBA/2 mice with one or zero copies of the α7 nicotinic receptor [114], suggesting that perinatal choline exerts its effect on sensory processing through an action on this receptor.
2.3.5. Alzheimer’s Disease
Mellott et al. [62] used a mouse model of AD to examine the effects of choline supplementation from conception to weaning on AD pathology. This APPswe/PS1dE9 (APP.PS1; MGI ID: 3524957) AD model mouse strain was engineered to express murine App with the human Aβ amino acid sequence harboring mutations that cause a familial form of AD (the Swedish mutation APPK595N/M596L; APPswe) together with a mutated form of PSEN1 (PS1 with exon 9 deleted; PS1dE9) [115]. Though no model of AD fully recapitulates the human disease [116], APP.PS1 mice are characterized by: (1) high production of amyloid Aβ peptides in brain and accumulation of amyloid plaques by 4–6 months of age [117]; (2) cognitive impairments [118,119,120,121,122]; (3) cholinergic defects [123,124,125,126,127,128]; and (4) evidence of abnormal methylation of several genes [129]. Perinatal choline supplementation significantly reduced the average number of Aβ42 plaques in both 9- and 12-month old APP.PS1 female and male mice. While the number of Aβ42 plaques increased with age in the control APP.PS1 mice, the plaque number was more stable in choline-supplemented mice, suggesting that Aβ42 synthesis, clearance, and/or aggregation may be altered in these mice to prevent additional plaque formation. A decline in cholinergic function and diminished expression of the cholinergic marker, CHAT, is apparent in aged humans and animals [130,131,132], in patients with Alzheimer’s disease (AD) [133,134,135,136], and in animal models of AD [118,123,124,125,126,127,128,135,137]. Thus, it has been postulated that abnormal cholinergic neurotransmission, due to dysfunction and/or degeneration of the septo-hippocampal cholinergic system, contributes to the memory deficits seen in advanced age and in AD [131,135,136]. At 9- and 12-months, CHAT protein levels were significantly decreased in the APP.PS1 female and male mice from the control group. Perinatal choline supplementation prevented this decrease, suggesting that choline supplementation may rescue cholinergic function in AD mice. APP.PS1 mice were also characterized by hippocampal gliosis [128,137,138,139]. This gliosis was nearly eliminated by perinatal choline supplementation. Given that activation of glial cells in AD and in AD mouse models may be initiated by Aβ peptides [140], it is possible that reduced gliosis in perinatally choline-supplemented APP.PS1 mice is secondary to the amelioration of the amyloidosis seen in these animals although it is also possible that high choline intake during development may have long-term anti-inflammatory actions in brain. While this study used an AD model caused by genes mutated in the familial human disease, the vast majority of AD cases are sporadic, with no known causes, and even though AD prevalence is alarming, affecting over 30% of individuals over 85 years of age [141], the disease does not appear to be an inevitable result of aging. Some of the factors that prevent or forestall AD may be genetic; e.g., non-carriers of the APOE ε4 allele [142,143,144,145] or individuals who inherit the rare APP A673T allele [146] may be somewhat protected. The study by Mellott et al. [62] suggests that vulnerability to AD may be modified by early-life nutrition.
2.3.6. Autism Spectrum Disorder
Autism spectrum disorder (ASD) is a heterogeneous group of developmental conditions characterized by deficient social interactions, deficits in verbal and nonverbal communication, stereotyped, repetitive patterns of behaviors and interests, and cognitive inflexibility in childhood, frequently continuing into adolescence and adulthood [147,148,149,150,151]. ASD has high heritability and genetic studies identified multiple loci that may increase the risk of ASD [152]. However, ASD is not a simple genetic disorder and environmental factors such as maternal nutrition during the periconceptual period, and throughout pregnancy and nursing, may contribute to its etiology [153,154]. While autism may be uniquely human, animal models have been developed to mimic some of the components of the disorder. The inbred BTBR T+Itpr3tf/J mice display autism-like behavioral phenotypes, with deficits in reciprocal social interactions [155,156], impaired communication, and repetitive behaviors, as compared with the high sociability and low self-grooming of the reference strain C57BL/6J (B6) [157,158,159,160]. Langley et al. [161] demonstrated that maternal dietary choline supplementation in BTBR mice during pregnancy and lactation alleviated anxiety-like behaviors and improved deficits in social behavior in adolescent and adult progeny. Interestingly, the behavioral phenotype-related genetic quantitative trait loci in BTBR mice contain genes associated with cholinergic neurotransmission (Chrna3, Chrna5, and Chrnb4), choline (Pcyt1b, Chpt1, Pld1, and Ppap2c) and folate metabolism (Mthfd1, Mthfs, and Slc19a1), as well as DNA methylation (Dnmt3l and Mecp2). Note that the expression of Dnmt3l was modulated by choline availability in another study [78], and that mutations in MECP2 cause Rett syndrome (see above). Overall, the results suggested that high choline intake during early development can prevent or reduce deficits in social behavior and anxiety in an autistic mouse model.
2.4. Neuroprotective Effects of Choline Intake in Adults: Implications for Cognitive Aging and Alzheimer’s Disease
Despite choline’s recently acquired status as an essential nutrient [1], and its presence in many foods [2], at least 75% of American adults consume less than the recommended Adequate Intake levels (see above) [9,45,73,162,163]). A recent National Health and Nutrition Examination Survey (NHANES) found that only 4% of men and 2% of women over the age of 71 years meet the AI value [164]. This was also found to be the case in a report from the Framingham Heart Study (FHS) Offspring Cohort (FHS Generation 2), which measured cognitive performance and dietary choline intake in 1391 dementia-free subjects (age 61 ± 9 SD). Approximately 75% of the subjects in this study consumed less than the recommended AI levels of choline [45], as estimated using a Food Frequency Questionnaire in which participants reported on which foods they had eaten for the prior 12-month period.
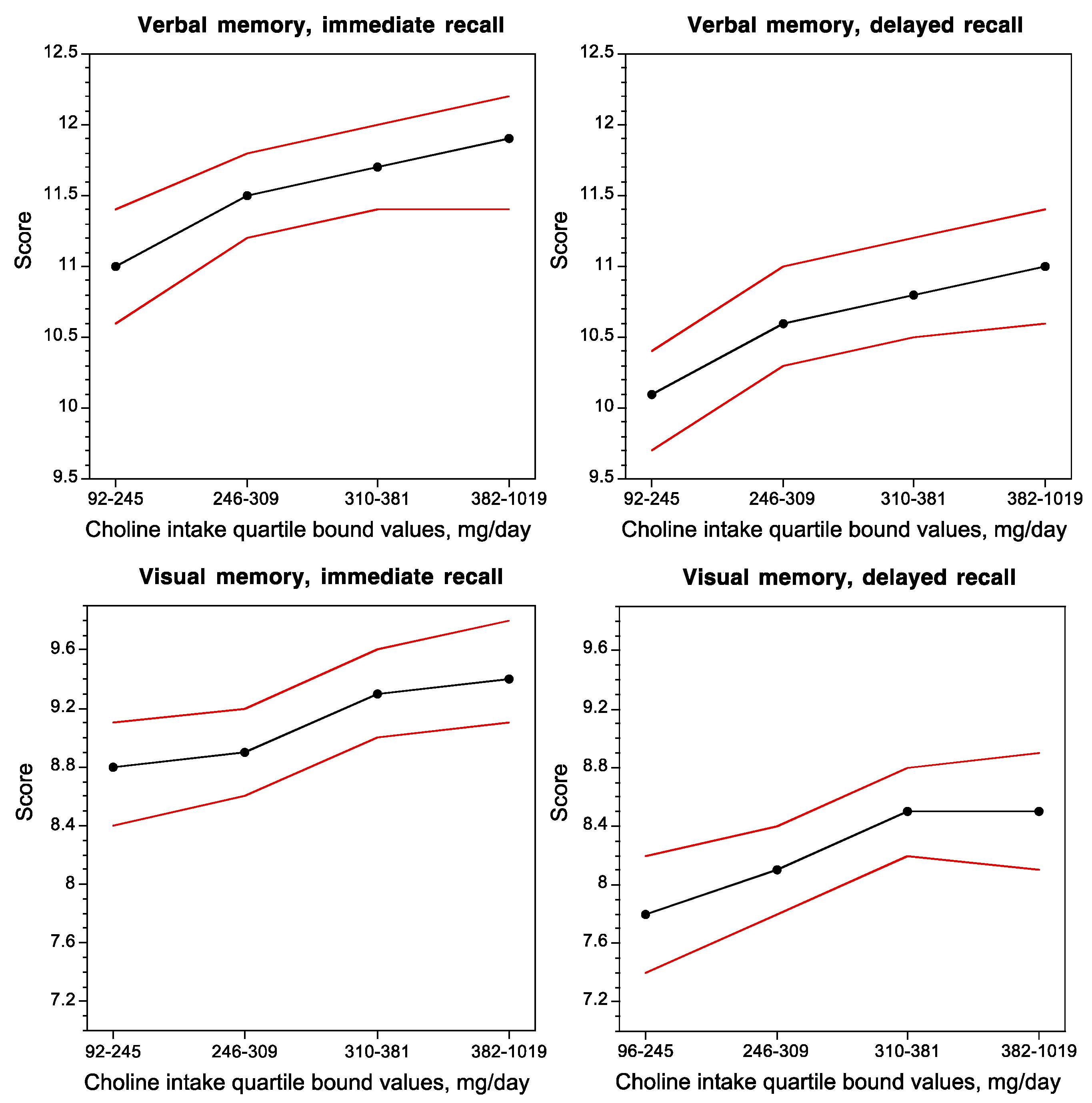
Concurrent choline intake was positively correlated with the performance of subjects on verbal and visual memory tasks (Figure 2) [45], and inversely correlated with white-matter hyperintensity volume, a brain magnetic resonance imaging measurement that is associated with impaired cognitive function and Alzheimer’s disease [45].
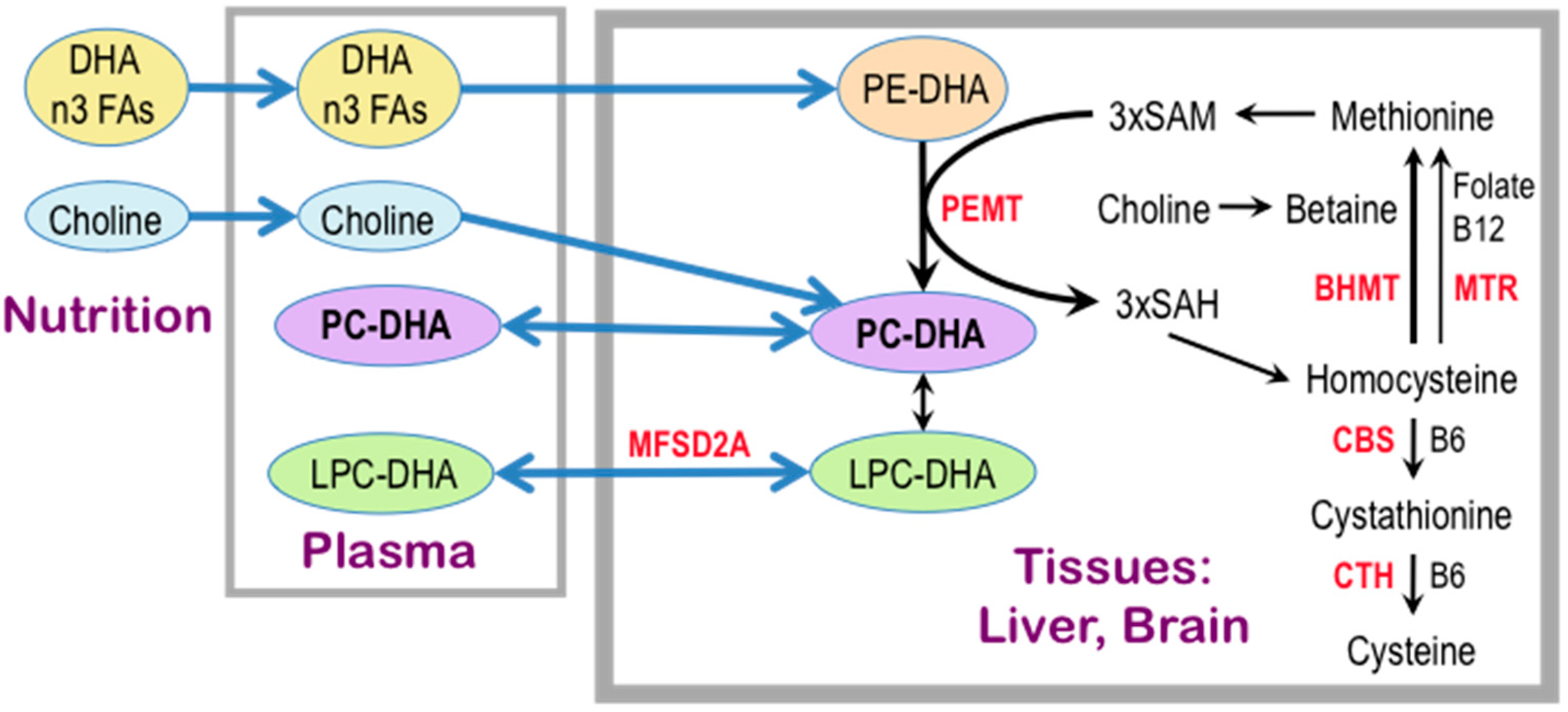
One possible explanation for the effect of concurrent choline intake on cognition in adults lies in its function as a precursor of the phospholipid phosphatidylcholine (PC), a major constituent of all biological membranes, including those in neurons and glial cells. Evidence that phospholipid metabolism is abnormal in AD originated with postmortem brain sample studies dating to the 1980s and 1990s [165,166,167,168] which showed reduced levels of PC and phosphatidylethanolamine (PE) and increased levels of their metabolites, glycerophosphocholine and glycerophosphoethanolamine, respectively, in the cerebral cortex of AD patients as compared to age-matched controls and to patients with Down syndrome, Parkinson’s disease and Huntington’s disease [167,168]. These data, and additional evidence that brain regions generally free of plaques and tangles, such as the caudate and the cerebellum, are similarly affected [167], led to the hypothesis that the phospholipid defect is specific to AD and widespread within the brain. A large body of evidence, reported since that time, is consistent with this concept (e.g., [169,170,171,172,173,174,175,176]). Moreover, there is a dramatic and specific reduction in molecular species of PC containing docosahexaenoic acid [DHA, 22:6n-3] (PC-DHA) levels in the temporal cortex gray matter of AD patients as compared to controls [177]. The advent of metabolomic techniques that permit untargeted large-scale analyses of hundreds of compounds combined with the intense need to discover plasma biomarkers for AD resulted in the surprising finding that lipid abnormalities in AD are not confined to the brain but are also evident in the peripheral blood plasma [176,178,179,180,181,182,183,184,185], suggesting that a blood lipidomic biomarker can be developed for prodromal AD [176,178,179,180,181,182,183,186,187], and importantly, that a lipid defect might be a driver of this disease. In particular, the plasma concentrations of PC species with 5 and 6 double bonds are consistently reported as reduced in prodromal and/or frank AD [178,181,182,183,186,188]. Data obtained by studies of erythrocyte membrane lipids further support these notions. Because erythrocytes live for an average of 120 days, their lipid profiles reflect whole body lipid metabolism over several months [189,190] as opposed to the snapshot view obtained from a plasma assay. In a study by Selley [191], erythrocytes from AD patients had reductions in concentrations of PC of 23% and of PC-DHA of 60% as compared to controls. Moreover, lower levels of erythrocyte phospholipid n-3 fatty acid (eicosapentaenoic [EPA, 20:5n-3] and DHA) content in a cohort of elderly subjects were associated with faster cognitive decline than in those with high n-3 FA erythrocyte content [192]. Similarly, in an elderly cohort, subjects within the lowest quartile of erythrocyte DHA concentrations had poorer memory test performance than individuals in the higher quartiles [193]. A positive correlation was observed between DHA intake and plasma PC-DHA concentrations in an FHS Generation 1 study, and was associated with a 50% reduction of dementia risk in this cohort [178]. Interestingly, in an intervention study of young women consuming 200 mg/day of DHA, increasing choline intake from 480 mg/day to 930 mg/day for six weeks increased PC-DHA concentrations in plasma and erythrocytes by 50% and 20%, respectively [194]. Conversely, plasma PC-DHA concentrations dropped in subjects placed on a choline-deficient diet [195]. Together these studies show that circulating PC-DHA levels are subject to regulation by the dietary supply of DHA and choline. Moreover, EPA and DHA are transported across the blood-brain-barrier from plasma to brain in the form of the lysophosphatidylcholines (LPC), LPC-EPA, and LPC-DHA, in a process that is catalyzed by a specific transporter, major facilitator superfamily domain containing 2a (MFSD2A) [196,197,198,199] (Figure 3). Thus, plasma levels of these LPC species may govern the rate of the supply of these essential fatty acids to brain, and there are indications of reduced plasma levels of LPC associated with AD [183,186]. Intervention studies using polyunsaturated fatty acids or DHA to treat dementia have been reviewed recently [200], and efficacy has been observed in some of those studies. Not surprisingly, early intervention works better, and the response to treatment may be attenuated in subjects with the APOE4 genotype [201,202]—the major genetic risk factor for AD [203]. Taken together, the evidence suggests that an adequate dietary supply of choline and DHA is necessary to maintain plasma and brain PC-DHA levels, and may possibly help to slow the onset of cognitive decline with aging, and potentially to delay or ameliorate the pathogenetic process in AD.
3. Choline Availability, Phosphatidylethanolamine N-methyltransferase, and Cognitive Ability
Most of the polyunsaturated PCs in mammals, including PC-DHA, are synthesized by phosphatidylethanolamine N-methyltransferase (PEMT) [195,204,205,206,207], which methylates phosphatidylethanolamine (PE) to generate PC using SAM as the methyl donor (Figure 3). The main site of the PEMT-catalyzed synthesis of PC-DHA is liver [195,204,205,206], which secretes it into the circulation as a component of very low density lipoproteins. PEMT activity in brain is relatively low [208], and the polyunsaturated species of PC (including PC-EPA and PC-DHA) [207] that are synthesized there are adequate for local needs only. PEMT activity in brain is higher during the perinatal period and declines in adulthood [209]. The PEMT reaction consumes three molecules of SAM for every PC molecule produced, and generates three molecules of S-adenosylhomocysteine (SAH). SAH is an inhibitor of PEMT (reviewed in [210]) and so, for PEMT to be active, SAH must be rapidly hydrolyzed to homocysteine and then remethylated to generate methionine and subsequently, SAM. Alternatively, homocysteine may be catabolized to cysteine via pyridoxal phosphate (vitamin B6)-dependent transsulfuration [211]. Some of the SAH-derived hepatic homocysteine enters the circulation. This results in a quantitatively significant link between hepatic PEMT activity and plasma levels of homocysteine [212]. High blood levels of homocysteine are associated with high plasma levels of SAH [213,214], whereas low plasma SAM/SAH ratio strongly correlates with low levels of plasma PC-DHA [195], and an increased risk of AD [215]. In 2002, a prospective evaluation of dementia-free FHS Generation 1 subjects showed that elevations in plasma homocysteine concentrations over eight years prior to the onset of clinical dementia predicted the subsequent development of AD [215]. A meta-analysis confirmed these results [216]. In a study of 29 AD patients and 26 controls, the high plasma SAH and homocysteine concentrations that characterized the AD patients, were associated with reduced levels of erythrocyte PC-DHA [191]. There are two enzymatic pathways that cause the remethylation of homocysteine to methionine: one is catalyzed by the vitamin B12-requiring 5-methyltetrahydrofolate-homocysteine S-methyltransferase (MTR), in which methyltetrahydrofolate is used as a methyl donor, and the other is catalyzed by betaine:homocysteine S-methyltransferase (BHMT) (reviewed in [73]). Betaine can be derived from the diet [2] and it is also produced in the liver by choline dehydrogenase [217]. Thus, in this way, choline is not the direct precursor of the PC headgroup, but rather, once oxidized to form betaine, donates a methyl group to homocysteine to form methionine, which is then converted to SAM (Figure 3). This SAM can be used by PEMT to generate the DHA molecular species of PC. In sum, dietary choline and its metabolite betaine, as well as folate and vitamins B6 and B12, via regulation of homocysteine and methionine levels, play central roles in the maintenance of adequate levels of PC-DHA in liver, plasma, and brain, and this in turn may moderate the risk of AD in human populations.
The importance of PEMT in cognitive function is further underscored by evidence that polymorphisms in PEMT affecting activity may be related to AD risk. The SNP rs7946 G523A causing the amino acid substitution V175M is associated with increased risk of AD in a study of a Chinese cohort [218]. The association was particularly significant in women and individuals who are not carriers of the APOE4 gene. In vitro, the PEMT175M enzyme, encoded by the A allele associated with AD, is 35% less active than the PEMT175V isoform [219], suggesting that carriers of the A allele may have a defect in PC-DHA production. Given that AD is more common in women than in men (based on FHS data, it was estimated that after the age of 65, one in five women and one in ten men are destined to develop AD [220]), the female-specific association between the rs7946 PEMT SNP and AD is particularly intriguing because, depending on another PEMT SNP (rs12325817 present in an intron), PEMT expression can be modulated by estrogen [195,219,221,222,223,224]. In individuals with the rs12325817 G allele, hepatic PEMT expression is induced by estrogen whereas in persons with the C allele this response is absent [223]. It has been proposed that during pregnancy and lactation the estrogen-induced PEMT generates PC and choline necessary for the normal development of the fetus and baby [222]. Indeed, young women on choline-deficient diets are resistant to liver and muscle dysfunction because of the high activity of their estrogen-regulated PEMT, which produces choline de novo [222]. In contrast, these symptoms are typically seen in men and postmenopausal women on such diets [6,225]. These observations reinforce earlier studies in animals that showed that levels of PEMT in the brain are regulated by the dietary supply of choline in a sexually dimorphic fashion, i.e., choline deficiency increases PEMT activity in brains of female, but not male, rats [226]. Thus, the production of PC-DHA can be modulated by: (1) diet; (2) intrinsic PEMT activity determined by its amino acid sequence encoded by specific PEMT alleles; and (3) sexually dimorphic regulation of the amount of PEMT enzyme expressed in the liver (and possibly in brain [226]) due to PEMT allele-specific responsiveness to estrogen. These data point to strong associations between dietary intake of DHA and choline, brain PEMT activity, sex, and phenotypes associated with cognitive impairment and AD.
Although the studies summarized in the preceding paragraphs underscore the potential benefits of adequate dietary choline intake, less is known about the potential harms of excess choline supplementation [227]. The addition of methylation pathway components such as folate, choline, and B vitamins to certain food products as well as dietary supplements has coincided with an increased incidence of diseases related to altered DNA methylation including cancer, autism spectrum disorders, and some neurological disorders (reviewed in [228]). A prospective study of male health professionals reported an increased risk of lethal prostate cancer in subjects in the highest quintile of choline consumption, even after adjusting for the presence of other nutrients that could increase cancer risk [229]. In contrast, several studies reported that high choline intake was associated with reduced incidence of breast [9], colorectal [230] and liver [13] cancer. It is clear that more studies in humans will be necessary to refine our understanding of what constitutes optimal choline intake at various stages of life, and how this may be affected by polymorphisms in the genes responsible for choline metabolism.
4. Conclusions
High choline intake during gestation and the early postnatal period has been shown to enhance cognitive performance in childhood, adulthood and into old age in multiple animal models and in some human studies. Moreover, choline is neuroprotective in a variety of experimental models of neuronal damage. Choline intake in adulthood may also be critical for normal cognitive function in people. The maternal choline supply during pregnancy modifies fetal DNA [78,231] and histone methylation [232], implicating an epigenomic mechanism in these long-term effects. While these epigenomic mechanisms may also operate in the adult, the effects of dietary choline both during development and in the adult brain may also be mediated, at least in part, by an influence on the peripheral and central metabolism of polyunsaturated species of PC. Taken together, the available evidence strongly supports the notion that adequate choline intake during pregnancy, and throughout life, is an important determinant of brain development, cognitive performance in the adult, and resistance to cognitive decline associated with aging and neurodegenerative disease.
Acknowledgments
Some of the studies reviewed here were supported by N.I.H. grants AG009525 and CA120488 to J.K.B. and a grant from the Simons Foundation to T.J.M.
Author Contributions
J.K.B., B.E.S. and T.J.M. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Food and Nutrition Board. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Panthotenic Acid, Biotin, and Cholin; National Academy Press: Washington, DC, USA, 1998. [Google Scholar]
- Patterson, K.Y.; Bhagwat, A.S.; Williams, J.R.; Howe, J.C.; Holden, J.M.; Zeisel, S.H.; Da Costa, C.A.; Mar, H. USDA Database for the Choline Content of Common Foods. Release Two. Available online: http://www.ars.usda.gov/Services/docs.htm?docid=6232 (accessed on 26 August 2017).
- Garner, S.C.; Mar, M.H.; Zeisel, S.H. Choline distribution and metabolism in pregnant rats and fetuses are influenced by the choline content of the maternal diet. J. Nutr. 1995, 125, 2851–2858. [Google Scholar] [PubMed]
- Holmes-McNary, M.Q.; Cheng, W.L.; Mar, M.H.; Fussell, S.; Zeisel, S.H. Choline and choline esters in human and rat milk and in infant formulas. Am. J. Clin. Nutr. 1996, 64, 572–576. [Google Scholar] [PubMed]
- Zeisel, S.H.; Char, D.; Sheard, N.F. Choline, phosphatidylcholine and sphingomyelin in human and bovine milk and infant formulas. J. Nutr. 1986, 116, 50–58. [Google Scholar] [PubMed]
- Zeisel, S.H.; Da Costa, K.-A.; Franklin, P.D.; Alexander, E.A.; Lamont, J.T.; Sheard, N.F.; Beiser, A. Choline, an essential nutrient for humans. FASEB J. 1991, 5, 2093–2098. [Google Scholar] [CrossRef]
- Da Costa, K.A.; Badea, M.; Fischer, L.M.; Zeisel, S.H. Elevated serum creatine phosphokinase in choline-deficient humans: Mechanistic studies in C2C12 mouse myoblasts. Am. J. Clin. Nutr. 2004, 80, 163–170. [Google Scholar] [PubMed]
- Da Costa, K.A.; Niculescu, M.D.; Craciunescu, C.N.; Fischer, L.M.; Zeisel, S.H. Choline deficiency increases lymphocyte apoptosis and DNA damage in humans. Am. J. Clin. Nutr. 2006, 84, 88–94. [Google Scholar] [PubMed]
- Xu, X.; Gammon, M.D.; Zeisel, S.H.; Lee, Y.L.; Wetmur, J.G.; Teitelbaum, S.L.; Bradshaw, P.T.; Neugut, A.I.; Santella, R.M.; Chen, J. Choline metabolism and risk of breast cancer in a population-based study. FASEB J. 2008, 22, 2045–2052. [Google Scholar] [CrossRef] [PubMed]
- Ibiebele, T.I.; Hughes, M.C.; Pandeya, N.; Zhao, Z.; Montgomery, G.; Hayward, N.; Green, A.C.; Whiteman, D.C.; Webb, P.M. High intake of folate from food sources is associated with reduced risk of esophageal cancer in an Australian population. J. Nutr. 2011, 141, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.; Rahbar, M.H.; Hallman, D.M.; Hernandez, L.M.; Spitz, M.R.; Forman, M.R.; Gorlova, O.Y. Associations between dietary intake of choline and betaine and lung cancer risk. PLoS ONE 2013, 8, e54561. [Google Scholar]
- Zhang, C.X.; Pan, M.X.; Li, B.; Wang, L.; Mo, X.F.; Chen, Y.M.; Lin, F.Y.; Ho, S.C. Choline and betaine intake is inversely associated with breast cancer risk: A two-stage case-control study in China. Cancer Sci. 2013, 104, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.F.; Chen, X.L.; Zhou, Z.G.; Zhang, Y.J.; Lan, Q.Y.; Liao, G.C.; Chen, Y.M.; Zhu, H.L. Higher dietary intakes of choline and betaine are associated with a lower risk of primary liver cancer: A case-control study. Sci. Rep. 2017, 7, 679. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S. Choline, Other Methyl-Donors and Epigenetics. Nutrients 2017, 9, 445. [Google Scholar] [CrossRef] [PubMed]
- Meck, W.H.; Williams, C.L. Simultaneous temporal processing is sensitive to prenatal choline availability in mature and aged rats. Neuroreport 1997, 8, 3045–3051. [Google Scholar] [CrossRef] [PubMed]
- Meck, W.H.; Williams, C.L. Metabolic imprinting of choline by its availability during gestation: Implications for memory and attentional processing across the lifespan. Neurosci. Biobehav. Rev. 2003, 27, 385–399. [Google Scholar] [CrossRef]
- Meck, W.H.; Williams, C.L.; Cermak, J.M.; Blusztajn, J.K. Developmental periods of choline sensitivity provide an ontogenetic mechanism for regulating memory capacity and age-related dementia. Front. Integr. Neurosci. 2007, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Z.; Cermak, J.M.; Tandon, P.; Sarkisian, M.R.; Stafstrom, C.F.; Neill, J.C.; Blusztajn, J.K.; Holmes, G.L. Protective effects of prenatal choline supplementation on seizure-induced memory impairment. J. Neurosci. 2000, 20, RC109. [Google Scholar] [PubMed]
- Holmes, G.L.; Yang, Y.; Liu, Z.; Cermak, J.M.; Sarkisian, M.R.; Stafstrom, C.E.; Neill, J.C.; Blusztajn, J.K. Seizure-induced memory impairment is reduced by choline supplementation before or after status epilepticus. Epilepsy Res. 2002, 48, 3–13. [Google Scholar] [CrossRef]
- Glenn, M.J.; Kirby, E.D.; Gibson, E.M.; Wong-Goodrich, S.J.; Mellott, T.J.; Blusztajn, J.K.; Williams, C.L. Age-related declines in exploratory behavior and markers of hippocampal plasticity are attenuated by prenatal choline supplementation in rats. Brain Res. 2008, 1237, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Wong-Goodrich, S.J.; Mellott, T.J.; Liu, B.; Blusztajn, J.K.; Williams, C.L. Water maze experience and prenatal choline supplementation differentially promote long-term hippocampal recovery from seizures in adulthood. Hippocampus 2011, 21, 584–608. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; La Fiette, M.H.; Quinn, V.R.; Riley, E.P. Neonatal choline supplementation ameliorates the effects of prenatal alcohol exposure on a discrimination learning task in rats. Neurotoxicol. Teratol. 2000, 22, 703–711. [Google Scholar] [CrossRef]
- Thomas, J.D.; Biane, J.S.; O’Bryan, K.A.; O’Neill, T.M.; Dominguez, H.D. Choline supplementation following third-trimester-equivalent alcohol exposure attenuates behavioral alterations in rats. Behav. Neurosci. 2007, 121, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.H.; Williams, J.K.; Thomas, J.D. Choline supplementation attenuates learning deficits associated with neonatal alcohol exposure in the rat: Effects of varying the timing of choline administration. Brain Res. 2008, 1237, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Tran, T.D. Choline supplementation mitigates trace, but not delay, eyeblink conditioning deficits in rats exposed to alcohol during development. Hippocampus 2011, 22, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Otero, N.K.; Thomas, J.D.; Saski, C.A.; Xia, X.; Kelly, S.J. Choline Supplementation and DNA Methylation in the Hippocampus and Prefrontal Cortex of Rats Exposed to Alcohol during Development. Alcohol. Clin. Exp. Res. 2012, 36, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.D.; Thomas, J.D. Adolescent Choline Supplementation Attenuates Working Memory Deficits in Rats Exposed to Alcohol During the Third Trimester Equivalent. Alcohol. Clin. Exp. Res. 2016, 40, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Nag, N.; Berger-Sweeney, J.E. Postnatal dietary choline supplementation alters behavior in a mouse model of Rett syndrome. Neurobiol. Dis. 2007, 26, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Nag, N.; Mellott, T.J.; Berger-Sweeney, J.E. Effects of postnatal dietary choline supplementation on motor regional brain volume and growth factor expression in a mouse model of Rett syndrome. Brain Res. 2008, 1237, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Ricceri, L.; De Filippis, B.; Fuso, A.; Laviola, G. Cholinergic hypofunction in MeCP2–308 mice: Beneficial neurobehavioural effects of neonatal choline supplementation. Behav. Brain Res. 2011, 221, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Ricceri, L.; De Filippis, B.; Laviola, G. Rett syndrome treatment in mouse models: Searching for effective targets and strategies. Neuropharmacology 2013, 68, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.; Chen, M.; Gandhy, S.U.; Strawderman, M.; Levitsky, D.A.; Maclean, K.N.; Strupp, B.J. Perinatal choline supplementation improves cognitive functioning and emotion regulation in the Ts65Dn mouse model of Down syndrome. Behav. Neurosci. 2010, 124, 346–361. [Google Scholar] [CrossRef] [PubMed]
- Kelley, C.M.; Powers, B.E.; Velazquez, R.; Ash, J.A.; Ginsberg, S.D.; Strupp, B.J.; Mufson, E.J. Maternal choline supplementation differentially alters the basal forebrain cholinergic system of young-adult Ts65Dn and disomic mice. J. Comp. Neurol. 2014, 522, 1390–1410. [Google Scholar] [CrossRef] [PubMed]
- Strupp, B.J.; Powers, B.E.; Velazquez, R.; Ash, J.A.; Kelley, C.M.; Alldred, M.J.; Strawderman, M.; Caudill, M.A.; Mufson, E.J.; Ginsberg, S.D. Maternal Choline Supplementation: A Potential Prenatal Treatment for Down Syndrome and Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, R.; Ash, J.A.; Powers, B.E.; Kelley, C.M.; Strawderman, M.; Luscher, Z.I.; Ginsberg, S.D.; Mufson, E.J.; Strupp, B.J. Maternal choline supplementation improves spatial learning and adult hippocampal neurogenesis in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 2013, 58, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Powers, B.E.; Kelley, C.M.; Velazquez, R.; Ash, J.A.; Strawderman, M.S.; Alldred, M.J.; Ginsberg, S.D.; Mufson, E.J.; Strupp, B.J. Maternal choline supplementation in a mouse model of Down syndrome: Effects on attention and nucleus basalis/substantia innominata neuron morphology in adult offspring. Neuroscience 2017, 340, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Ash, J.A.; Velazquez, R.; Kelley, C.M.; Powers, B.E.; Ginsberg, S.D.; Mufson, E.J.; Strupp, B.J. Maternal choline supplementation improves spatial mapping and increases basal forebrain cholinergic neuron number and size in aged Ts65Dn mice. Neurobiol. Dis. 2014, 70, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Kelley, C.M.; Ash, J.A.; Powers, B.E.; Velazquez, R.; Alldred, M.J.; Ikonomovic, M.D.; Ginsberg, S.D.; Strupp, B.J.; Mufson, E.J. Effects of Maternal Choline Supplementation on the Septohippocampal Cholinergic System in the Ts65Dn Mouse Model of Down Syndrome. Curr. Alzheimer Res. 2016, 13, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Jadavji, N.M.; Emmerson, J.T.; MacFarlane, A.J.; Willmore, W.G.; Smith, P.D. B-vitamin and choline supplementation increases neuroplasticity and recovery after stroke. Neurobiol. Dis. 2017, 103, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Borges, A.A.; El-Batah, P.N.; Yamashita, L.F.; Santana Ados, S.; Lopes, A.C.; Freymuller-Haapalainen, E.; Coimbra, C.G.; Sinigaglia-Coimbra, R. Neuroprotective effect of oral choline administration after global brain ischemia in rats. Nutr. Neurosci. 2015, 18, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Shaw, G.M.; Carmichael, S.L.; Yang, W.; Selvin, S.; Schaffer, D.M. Periconceptional dietary intake of choline and betaine and neural tube defects in offspring. Am. J. Epidemiol. 2004, 160, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, S.L.; Witte, J.S.; Shaw, G.M. Nutrient pathways and neural tube defects: A semi-Bayesian hierarchical analysis. Epidemiology 2009, 20, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, S.L.; Yang, W.; Shaw, G.M. Periconceptional nutrient intakes and risks of neural tube defects in California. Birth Defects Res. 2010, 88, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Lavery, A.M.; Brender, J.D.; Zhao, H.; Sweeney, A.; Felkner, M.; Suarez, L.; Canfield, M.A. Dietary intake of choline and neural tube defects in Mexican Americans. Birth Defects Res. 2014, 100, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Poly, C.; Massaro, J.M.; Seshadri, S.; Wolf, P.A.; Cho, E.; Krall, E.; Jacques, P.F.; Au, R. The relation of dietary choline to cognitive performance and white-matter hyperintensity in the Framingham Offspring Cohort. Am. J. Clin. Nutr. 2011, 94, 1584–1591. [Google Scholar] [CrossRef] [PubMed]
- Parnetti, L.; Mignini, F.; Tomassoni, D.; Traini, E.; Amenta, F. Cholinergic precursors in the treatment of cognitive impairment of vascular origin: Ineffective approaches or need for re-evaluation? J. Neurol. Sci. 2007, 257, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Meck, W.H.; Smith, R.A.; Williams, C.L. Pre- and postnatal choline supplementation produces long-term facilitation of spatial memory. Dev. Psychobiol. 1998, 21, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Schenk, F.; Brandner, C. Indirect effect of peri- and postnatal choline treatment on place-learning abilities in rat. Psychobiology 1995, 23, 302–313. [Google Scholar]
- Jones, J.P.; Meck, W.H.; Williams, C.L.; Wilson, W.A.; Swartzwelder, H.S. Choline availability to the developing rat fetus alters adult hippocampal long-term potentiation. Dev. Brain Res. 1999, 118, 159–167. [Google Scholar] [CrossRef]
- Tees, R.C.; Mohammadi, E. The effects of neonatal choline dietary supplementation on adult spatial and configural learning and memory in rats. Dev. Psychobiol. 1999, 35, 226–240. [Google Scholar] [CrossRef]
- Mellott, T.J.; Williams, C.L.; Meck, W.H.; Blusztajn, J.K. Prenatal choline supplementation advances hippocampal development and enhances MAPK and CREB activation. FASEB J. 2004, 18, 545–547. [Google Scholar] [CrossRef] [PubMed]
- Buhusi, C.V.; Lamoureux, J.A.; Meck, W.H. Prenatal choline supplementation increases sensitivity to contextual processing of temporal information. Brain Res. 2008, 1237, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.K.; MacDonald, C.J.; Williams, C.L.; Meck, W.H. Prenatal choline supplementation alters the timing, emotion, and memory performance (TEMP) of adult male and female rats as indexed by differential reinforcement of low-rate schedule behavior. Learn. Mem. 2008, 15, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Castro, C.A.; Rudy, J.W. Early-life malnutrition selectively retards the development of distal- but not proximal-cue navigation. Dev. Psychobiol. 1987, 20, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Boeke, C.E.; Gillman, M.W.; Hughes, M.D.; Rifas-Shiman, S.L.; Villamor, E.; Oken, E. Choline intake during pregnancy and child cognition at age 7 years. Am. J. Epidemiol. 2013, 177, 1338–1347. [Google Scholar] [CrossRef] [PubMed]
- Albright, C.D.; Friedrich, C.B.; Brown, E.C.; Mar, M.H.; Zeisel, S.H. Maternal dietary choline availability alters mitosis, apoptosis and the localization of TOAD-64 protein in the developing fetal rat septum. Dev. Psychobiol. 1999, 115, 123–129. [Google Scholar] [CrossRef]
- Craciunescu, C.N.; Albright, C.D.; Mar, M.H.; Song, J.; Zeisel, S.H. Choline availability during embryonic development alters progenitor cell mitosis in developing mouse hippocampus. J. Nutr. 2003, 133, 3614–3618. [Google Scholar] [PubMed]
- Clelland, C.D.; Choi, M.; Romberg, C.; Clemenson, G.D., Jr.; Fragniere, A.; Tyers, P.; Jessberger, S.; Saksida, L.M.; Barker, R.A.; Gage, F.H.; et al. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science 2009, 325, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Saitoh, Y.; Takashima, N.; Murayama, A.; Niibori, Y.; Ageta, H.; Sekiguchi, M.; Sugiyama, H.; Inokuchi, K. Adult neurogenesis modulates the hippocampus-dependent period of associative fear memory. Cell 2009, 139, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, J.T.; Schafer, S.T.; Gage, F.H. Adult Neurogenesis in the Hippocampus: From Stem Cells to Behavior. Cell 2016, 167, 897–914. [Google Scholar] [CrossRef] [PubMed]
- Glenn, M.J.; Gibson, E.M.; Kirby, E.D.; Mellott, T.J.; Blusztajn, J.K.; Williams, C.L. Prenatal choline availability modulates hippocampal neurogenesis and neurogenic responses to enriching experiences in adult female rats. Eur. J. Neurosci. 2007, 25, 2473–2482. [Google Scholar] [CrossRef] [PubMed]
- Mellott, T.J.; Huleatt, O.M.; Shade, B.N.; Pender, S.M.; Liu, Y.B.; Slack, B.E.; Blusztajn, J.K. Perinatal choline supplementation reduces amyloidosis and increases choline acetyltransferae expression in the hippocampus of the APPswePS1dE9 Alzheimer’s disease model mice. PLoS ONE 2017, 12, e0170450. [Google Scholar]
- Sandstrom, N.J.; Loy, R.; Williams, C.L. Prenatal choline supplementation increases NGF levels in the hippocampus and frontal cortex of young and adult rats. Brain Res. 2002, 947, 9–16. [Google Scholar] [CrossRef]
- Mellott, T.J.; Follettie, M.T.; Diesl, V.; Hill, A.A.; Lopez-Coviella, I.; Blusztajn, J.K. Prenatal choline availability modulates hippocampal and cerebral cortical gene expression. FASEB J. 2007, 21, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Napoli, I.; Blusztajn, J.K.; Mellott, T.J. Prenatal choline supplementation in rats increases the expression of IGF2 and its receptor IGF2R and enhances IGF2-induced acetylcholine release in hippocampus and frontal cortex. Brain Res. 2008, 1237, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.L.; Meck, W.H.; Heyer, D.; Loy, R. Hypertrophy of basal forebrain neurons and enhanced visuospatial memory in perinatally choline-supplemented rats. Brain Res. 1998, 794, 225–238. [Google Scholar] [CrossRef]
- Fibiger, H.C. Cholinergic mechanisms in learning, memory and dementia: A review of recent evidence. Trends Neurosci. 1991, 14, 220–223. [Google Scholar] [CrossRef]
- Sarter, M.; Parikh, V. Choline transporters, cholinergic transmission and cognition. Nat. Rev. Neurosci. 2005, 6, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Cermak, J.M.; Holler, T.; Jackson, D.A.; Blusztajn, J.K. Prenatal availability of choline modifies development of the hippocampal cholinergic system. FASEB J. 1998, 12, 349–357. [Google Scholar] [PubMed]
- Sweatt, J.D. The neuronal MAP kinase cascade: A biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 2001, 76, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, R.A. A Brief History of Long-Term Potentiation. Neuron 2017, 93, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Pyapali, G.K.; Turner, D.A.; Williams, C.L.; Meck, W.H.; Swartzwelder, H.S. Prenatal dietary choline supplementation decreases the threshold for induction of long-term potentiation in young adult rats. J. Neurophysiol. 1998, 79, 1790–1796. [Google Scholar] [PubMed]
- Blusztajn, J.K.; Mellott, T.J. Neuroprotective actions of perinatal choline nutrition. Clin. Chem. Lab. Med. 2013, 51, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. 2009, 10, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef] [PubMed]
- Blusztajn, J.K.; Mellott, T.J. Choline nutrition programs brain development via DNA and histone methylation. Cent. Nerv. Syst. Agents Med. Chem. 2012, 12, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Kovacheva, V.P.; Mellott, T.J.; Davison, J.M.; Wagner, N.; Lopez-Coviella, I.; Schnitzler, A.C.; Blusztajn, J.K. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J. Biol. Chem. 2007, 282, 31777–31788. [Google Scholar] [CrossRef] [PubMed]
- Lopes, S.; Lewis, A.; Hajkova, P.; Dean, W.; Oswald, J.; Forne, T.; Murrell, A.; Constancia, M.; Bartolomei, M.; Walter, J.; et al. Epigenetic modifications in an imprinting cluster are controlled by a hierarchy of DMRs suggesting long-range chromatin interactions. Hum. Mol. Genet. 2003, 12, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.G.; Chang, Q.; Lin, Y.X.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.P.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent Bdnf gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Levenson, J.M.; Roth, T.L.; Lubin, F.D.; Miller, C.A.; Huang, I.C.; Desai, P.; Malone, L.M.; Sweatt, J.D. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 2006, 281, 15763–15773. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.D.; Kavalali, E.T.; Monteggia, L.M. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J. Neurosci. 2008, 28, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Yossifoff, M.; Kisliouk, T.; Meiri, N. Dynamic changes in DNA methylation during thermal control establishment affect CREB binding to the brain-derived neurotrophic factor promoter. Eur. J. Neurosci. 2008, 28, 2267–2277. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Sweatt, J.D. Covalent modification of DNA regulates memory formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Lubin, F.D.; Roth, T.L.; Sweatt, J.D. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 2008, 28, 10576–10586. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Webb, W.M.; Sanchez, R.G.; Perez, G.; Butler, A.A.; Hauser, R.M.; Rich, M.C.; O’Bierne, A.L.; Jarome, T.J.; Lubin, F.D. Dynamic association of epigenetic H3K4me3 and DNA 5hmC marks in the dorsal hippocampus and anterior cingulate cortex following reactivation of a fear memory. Neurobiol. Learn. Mem. 2017, 142, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, Q.; Duan, X.; York, P.; Chen, G.D.; Yin, P.; Zhu, H.; Xu, M.; Chen, P.; Wu, Q.; et al. The 5-Hydroxymethylcytosine (5hmC) Reader UHRF2 Is Required for Normal Levels of 5hmC in Mouse Adult Brain and Spatial Learning and Memory. J. Biol. Chem. 2017, 292, 4533–4543. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.J.; Rahn, E.J.; Paulukaitis, B.S.; Savell, K.E.; Kordasiewicz, H.B.; Wang, J.; Lewis, J.W.; Posey, J.; Strange, S.K.; Guzman-Karlsson, M.C.; et al. Tcf4 Regulates Synaptic Plasticity, DNA Methylation, and Memory Function. Cell Rep. 2016, 16, 2666–2685. [Google Scholar] [CrossRef] [PubMed]
- Penner, M.R.; Parrish, R.R.; Hoang, L.T.; Roth, T.L.; Lubin, F.D.; Barnes, C.A. Age-related changes in Egr1 transcription and DNA methylation within the hippocampus. Hippocampus 2016, 26, 1008–1020. [Google Scholar] [CrossRef] [PubMed]
- Halder, R.; Hennion, M.; Vidal, R.O.; Shomroni, O.; Rahman, R.U.; Rajput, A.; Centeno, T.P.; van Bebber, F.; Capece, V.; Garcia Vizcaino, J.C.; et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat. Neurosci. 2016, 19, 102–110. [Google Scholar] [PubMed]
- Roth, E.D.; Roth, T.L.; Money, K.M.; SenGupta, S.; Eason, D.E.; Sweatt, J.D. DNA methylation regulates neurophysiological spatial representation in memory formation. Neuroepigenetics 2015, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Adachi, M.; Na, E.S.; Monteggia, L.M. Selective role for DNMT3a in learning and memory. Neurobiol. Learn. Mem. 2014, 115, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, A.; Dawlaty, M.M.; Seo, J.; Cheng, A.W.; Meng, J.; Le, T.; Faull, K.F.; Jaenisch, R.; Tsai, L.H. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 2013, 79, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Kaas, G.A.; Zhong, C.; Eason, D.E.; Ross, D.L.; Vachhani, R.V.; Ming, G.L.; King, J.R.; Song, H.; Sweatt, J.D. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 2013, 79, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Acharya, M.M.; Hattiangady, B.; Shetty, A.K. Progress in neuroprotective strategies for preventing epilepsy. Prog. Neurobiol. 2008, 84, 363–404. [Google Scholar] [CrossRef] [PubMed]
- Wong-Goodrich, S.J.; Mellott, T.J.; Glenn, M.J.; Blusztajn, J.K.; Williams, C.L. Prenatal choline supplementation attenuates neuropathological response to status epilepticus in the adult rat hippocampus. Neurobiol. Dis. 2008, 30, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Wong-Goodrich, S.J.; Glenn, M.J.; Mellott, T.J.; Blusztajn, J.K.; Meck, W.H.; Williams, C.L. Spatial memory and hippocampal plasticity are differentially sensitive to the availability of choline in adulthood as a function of choline supply in utero. Brain Res. 2008, 1237, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.E.; Mai, C.T.; Canfield, M.A.; Rickard, R.; Wang, Y.; Meyer, R.E.; Anderson, P.; Mason, C.A.; Collins, J.S.; Kirby, R.S.; et al. Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004–2006. Birth Defects Res. 2010, 88, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Karmiloff-Smith, A.; Al-Janabi, T.; D’Souza, H.; Groet, J.; Massand, E.; Mok, K.; Startin, C.; Fisher, E.; Hardy, J.; Nizetic, D.; et al. The importance of understanding individual differences in Down syndrome. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.H.; Irving, N.G.; Moran, T.H.; Wohn, A.; Kitt, C.; Sisodia, S.S.; Schmidt, C.; Bronson, R.T.; Davisson, M.T. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 1995, 11, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Bourc'his, D.; Xu, G.L.; Lin, C.S.; Bollman, B.; Bestor, T.H. Dnmt3L and the establishment of maternal genomic imprints. Science 2001, 294, 2536–2539. [Google Scholar] [CrossRef] [PubMed]
- Chedin, F.; Lieber, M.R.; Hsieh, C.L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc. Natl. Acad. Sci. USA 2002, 99, 16916–16921. [Google Scholar] [CrossRef] [PubMed]
- Mendioroz, M.; Do, C.; Jiang, X.; Liu, C.; Darbary, H.K.; Lang, C.F.; Lin, J.; Thomas, A.; Abu-Amero, S.; Stanier, P.; et al. Trans effects of chromosome aneuploidies on DNA methylation patterns in human Down syndrome and mouse models. Genome Biol. 2015, 16, 263. [Google Scholar] [CrossRef] [PubMed]
- Do, C.; Xing, Z.; Yu, Y.E.; Tycko, B. Trans-acting epigenetic effects of chromosomal aneuploidies: Lessons from Down syndrome and mouse models. Epigenomics 2017, 9, 189–207. [Google Scholar] [CrossRef] [PubMed]
- Fasolino, M.; Zhou, Z. The Crucial Role of DNA Methylation and MeCP2 in Neuronal Function. Genes 2017, 8, 141. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.C.; Agarwal, S.; Wang, K.; Berger-Sweeney, J.; Kolodny, N.H. Longitudinal brain MRI study in a mouse model of Rett Syndrome and the effects of choline. Neurobiol. Dis. 2008, 31, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.C.; Kolodny, N.H.; Nag, N.; Berger-Sweeney, J.E. Neurochemical changes in a mouse model of Rett syndrome: Changes over time and in response to perinatal choline nutritional supplementation. J. Neurochem. 2009, 108, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K.E.; Adams, C.E.; Yonchek, J.; Hickel, C.; Danielson, J.; Kisley, M.A. Permanent improvement in deficient sensory inhibition in DBA/2 mice with increased perinatal choline. Psychopharmacology 2008, 198, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Singer, P.; Feldon, J.; Yee, B.K. Are DBA/2 mice associated with schizophrenia-like endophenotypes? A behavioural contrast with C57BL/6 mice. Psychopharmacology 2009, 206, 677–698. [Google Scholar] [PubMed]
- Greenwood, T.A.; Lazzeroni, L.C.; Calkins, M.E.; Freedman, R.; Green, M.F.; Gur, R.E.; Gur, R.C.; Light, G.A.; Nuechterlein, K.H.; Olincy, A.; et al. Genetic assessment of additional endophenotypes from the Consortium on the Genetics of Schizophrenia Family Study. Schizophr. Res. 2016, 170, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Kumar, A.; Agarwal, S.; Phadke, S.R.; Jaiswal, Y. Genetic insight of schizophrenia: Past and future perspectives. Gene 2014, 535, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K.E.; Choo, K.S.; Stitzel, J.A.; Marks, M.J.; Adams, C.E. Long-term improvements in sensory inhibition with gestational choline supplementation linked to alpha7 nicotinic receptors through studies in Chrna7 null mutation mice. Brain Res. 2014, 1552, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Slunt, H.H.; Ratovitski, T.; Jenkins, N.A.; Copeland, N.G.; Borchelt, D.R. Co-expression of multiple transgenes in mouse CNS: A comparison of strategies. Biomol. Eng. 2001, 17, 157–165. [Google Scholar] [CrossRef]
- Götz, J.; Ittner, L.M. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat. Rev. 2008, 9, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: Evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Savonenko, A.; Xu, G.M.; Melnikova, T.; Morton, J.L.; Gonzales, V.; Wong, M.P.; Price, D.L.; Tang, F.; Markowska, A.L.; Borchelt, D.R. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: Relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol. Dis. 2005, 18, 602–617. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Widi, G.A.; Gimbel, D.A.; Harel, N.Y.; Lee, D.H.; Strittmatter, S.M. Subcutaneous Nogo receptor removes brain amyloid-beta and improves spatial memory in Alzheimer’s transgenic mice. J. Neurosci. 2006, 26, 13279–13286. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, T.P.; Brown, R.E. Visuo-spatial learning and memory deficits on the Barnes maze in the 16-month-old APPswe/PS1dE9 mouse model of Alzheimer’s disease. Behav. Brain Res. 2009, 201, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Gimbel, D.A.; Nygaard, H.B.; Coffey, E.E.; Gunther, E.C.; Lauren, J.; Gimbel, Z.A.; Strittmatter, S.M. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 2010, 30, 6367–6374. [Google Scholar] [CrossRef] [PubMed]
- Kemppainen, S.; Rantamaki, T.; Jeronimo-Santos, A.; Lavasseur, G.; Autio, H.; Karpova, N.; Karkkainen, E.; Staven, S.; Miranda, H.V.; Outeiro, T.F.; et al. Impaired TrkB receptor signaling contributes to memory impairment in APP/PS1 mice. Neurobiol. Aging 2011, 33, 1122.e23–1122.e39. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.E.; Dar, S.; Ikonomovic, M.D.; Dekosky, S.T.; Mufson, E.J. Cholinergic forebrain degeneration in the APPswe/PS1DeltaE9 transgenic mouse. Neurobiol. Dis. 2007, 28, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Machova, E.; Rudajev, V.; Smyckova, H.; Koivisto, H.; Tanila, H.; Dolezal, V. Functional cholinergic damage develops with amyloid accumulation in young adult APPswe/PS1dE9 transgenic mice. Neurobiol. Dis. 2010, 38, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Payette, D.J.; Xie, J.; Guo, Q. Reduction in CHT1-mediated choline uptake in primary neurons from presenilin-1 M146V mutant knock-in mice. Brain Res. 2007, 1135, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Niidome, T.; Hongo, H.; Akaike, A.; Kihara, T.; Sugimoto, H. Impaired muscarinic regulation of excitatory synaptic transmission in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Eur. J. Pharmacol. 2008, 583, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Nikolajsen, G.N.; Jensen, M.S.; West, M.J. Cholinergic axon length reduced by 300 meters in the brain of an Alzheimer mouse model. Neurobiol. Aging 2011, 32, 1927–1931. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.M.; Norman, T.A.; Haydar, T.F.; Slack, B.E.; Leeman, S.E.; Blusztajn, J.K.; Mellott, T.J. BMP9 ameliorates amyloidosis and the cholinergic defect in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, 19567–19572. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mut, J.V.; Aso, E.; Panayotis, N.; Lott, I.; Dierssen, M.; Rabano, A.; Urdinguio, R.G.; Fernandez, A.F.; Astudillo, A.; Martin-Subero, J.I.; et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain 2013, 136, 3018–3027. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Reisberg, B.; Zaudig, M.; Petersen, R.C.; Ritchie, K.; Broich, K.; Belleville, S.; Brodaty, H.; Bennett, D.; Chertkow, H.; et al. Mild cognitive impairment. Lancet 2006, 367, 1262–1270. [Google Scholar] [CrossRef]
- Haense, C.; Kalbe, E.; Herholz, K.; Hohmann, C.; Neumaier, B.; Krais, R.; Heiss, W.D. Cholinergic system function and cognition in mild cognitive impairment. Neurobiol. Aging 2012, 33, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Sarter, M.; Bruno, J.P. Developmental origins of the age-related decline in cortical cholinergic function and associated cognitive abilities. Neurobiol. Aging 2004, 25, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Bowen, D.M.; Allen, S.J.; Benton, J.S.; Goodhardt, M.J.; Haan, E.A.; Palmer, A.M.; Sims, N.R.; Smith, C.C. T.; Spillane, J.A.; Esiri, M.M.; et al. Biochemical assessment of serotonergic and cholinergic dysfunction and cerebral atrophy in Alzheimer’s disease. J. Neurochem. 1983, 41, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Mufson, E.J.; Counts, S.E.; Perez, S.E.; Ginsberg, S.D. Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev. Neurother. 2008, 8, 1703–1718. [Google Scholar] [PubMed]
- Grothe, M.; Heinsen, H.; Teipel, S.J. Atrophy of the cholinergic Basal forebrain over the adult age range and in early stages of Alzheimer’s disease. Biol. Psychiatry 2012, 71, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Mellott, T.J.; Pender, S.M.; Burke, R.M.; Langley, E.A.; Blusztajn, J.K. IGF2 Ameliorates Amyloidosis, Increases Cholinergic Marker Expression and Raises BMP9 and Neurotrophin Levels in the Hippocampus of the APPswePS1dE9 Alzheimer’s Disease Model Mice. PLoS ONE 2014, 9, e94287. [Google Scholar]
- Kamphuis, W.; Mamber, C.; Moeton, M.; Kooijman, L.; Sluijs, J.A.; Jansen, A.H.; Verveer, M.; de Groot, L.R.; Smith, V.D.; Rangarajan, S.; et al. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS ONE 2012, 7, e42823. [Google Scholar]
- Kamphuis, W.; Orre, M.; Kooijman, L.; Dahmen, M.; Hol, E.M. Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer’s disease mouse model. Glia 2012, 60, 615–629. [Google Scholar] [PubMed]
- Osborn, L.M.; Kamphuis, W.; Wadman, W.J.; Hol, E.M. Astrogliosis: An integral player in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2016, 144, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Hebert, L.E.; Scherr, P.A.; Bienias, J.L.; Bennett, D.A.; Evans, D.A. Alzheimer disease in the US population -Prevalence estimates using the 2000 census. Arch. Neurol. 2003, 60, 1119–1122. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to b-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; Van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease—A meta-analysis. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; De Deyn, P.; Berr, C.; et al. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol. Psychiatry 2011, 16, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Losh, M.; Piven, J. Social-cognition and the broad autism phenotype: Identifying genetically meaningful phenotypes. J. Child Psychol. Psychiatry Allied Discip. 2007, 48, 105–112. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic Criteria from DSM-IV-TR; American Psychiatric Association: Washington, DC, USA, 2000. [Google Scholar]
- Lord, C.; Leventhal, B.L.; Cook, E.H., Jr. Quantifying the phenotype in autism spectrum disorders. Am. J. Med. Genet. 2001, 105, 36–38. [Google Scholar] [CrossRef]
- Lord, C.; Risi, S.; DiLavore, P.S.; Shulman, C.; Thurm, A.; Pickles, A. Autism from 2 to 9 years of age. Arch. Gen. Psychiatry 2006, 63, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Volkmar, F.R.; Lord, C.; Bailey, A.; Schultz, R.T.; Klin, A. Autism and pervasive developmental disorders. J. Child Psychol. Psychiatry Allied Discip. 2004, 45, 135–170. [Google Scholar] [CrossRef]
- De la Torre-Ubieta, L.; Won, H.; Stein, J.L.; Geschwind, D.H. Advancing the understanding of autism disease mechanisms through genetics. Nat. Med. 2016, 22, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Suren, P.; Roth, C.; Bresnahan, M.; Haugen, M.; Hornig, M.; Hirtz, D.; Lie, K.K.; Lipkin, W.I.; Magnus, P.; Reichborn-Kjennerud, T.; et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. JAMA 2013, 309, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.J.; Hansen, R.L.; Hartiala, J.; Allayee, H.; Schmidt, L.C.; Tancredi, D.J.; Tassone, F.; Hertz-Picciotto, I. Prenatal vitamins, one-carbon metabolism gene variants, and risk for autism. Epidemiology 2011, 22, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Pobbe, R.L.; Defensor, E.B.; Pearson, B.L.; Bolivar, V.J.; Blanchard, D.C.; Blanchard, R.J. General and social anxiety in the BTBR T + tf/J mouse strain. Behav. Brain Res. 2011, 216, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Pobbe, R.L.; Pearson, B.L.; Defensor, E.B.; Bolivar, V.J.; Blanchard, D.C.; Blanchard, R.J. Expression of social behaviors of C57BL/6J versus BTBR inbred mouse strains in the visible burrow system. Behav. Brain Res. 2010, 214, 443–449. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, H.G.; Kusek, G.K.; Yang, M.; Phoenix, J.L.; Bolivar, V.J.; Crawley, J.N. Autism-like behavioral phenotypes in BTBR T + tf/J mice. Genes Brain Behav. 2008, 7, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.L.; Tolu, S.S.; Barkan, C.L.; Crawley, J.N. Repetitive self-grooming behavior in the BTBR mouse model of autism is blocked by the mGluR5 antagonist MPEP. Neuropsychopharmacology 2010, 35, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Wohr, M.; Roullet, F.I.; Crawley, J.N. Reduced scent marking and ultrasonic vocalizations in the BTBR T + tf/J mouse model of autism. Genes Brain Behav. 2011, 10, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Scattoni, M.L.; Zhodzishsky, V.; Chen, T.; Caldwell, H.; Young, W.S.; McFarlane, H.G.; Crawley, J.N. Social approach behaviors are similar on conventional versus reverse lighting cycles, and in replications across cohorts, in BTBR T + tf/J, C57BL/6J, and vasopressin receptor 1B mutant mice. Front. Behav. Neurosci. 2007, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Langley, E.A.; Krykbaeva, M.; Blusztajn, J.K.; Mellott, T.J. High maternal choline consumption during pregnancy and nursing alleviates deficits in social interaction and improves anxiety-like behaviors in the BTBR T + Itpr3tf/J mouse model of autism. Behav. Brain Res. 2015, 278, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Jensen, H.H.; Batres-Marquez, S.P.; Carriquiry, A.; Schalinske, K.L. Choline in the diets of the US population: NHANES, 2003–2004. FASEB J. 2007, 21, LB46. [Google Scholar]
- Cho, E.; Holmes, M.D.; Hankinson, S.E.; Willett, W.C. Choline and betaine intake and risk of breast cancer among post-menopausal women. Br. J. Cancer 2010, 102, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.C.; Fulgoni, V.L., 3rd. Assessment of Total Choline Intakes in the United States. J. Am. Coll. Nutr. 2016, 35, 108–112. [Google Scholar] [PubMed]
- Barany, M.; Chang, Y.C.; Arus, C.; Rustan, T.; Frey, W.H. Increased glycerol-3-phosphorylcholine in post-mortem Alzheimer’s brain. Lancet 1985, 1, 517. [Google Scholar] [CrossRef]
- Pettegrew, J.W.; Panchalingam, K.; Moossy, J.; Martinez, J.; Rao, G.; Boller, F. Correlation of phosphorus-31 magnetic resonance spectroscopy and morphologic findings in Alzheimer’s disease. Arch. Neurol. 1988, 45, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Blusztajn, J.K.; Lopez Gonzalez-Coviella, I.; Logue, M.; Growdon, J.H.; Wurtman, R.J. Levels of phospholipid catabolic intermediates, glycerophosphocholine and glycerophosphoethanolamine, are elevated in brains of Alzheimer’s disease but not of Down’s syndrome patients. Brain Res. 1990, 536, 240–244. [Google Scholar] [CrossRef]
- Nitsch, R.M.; Blusztajn, J.K.; Pittas, A.G.; Slack, B.E.; Growdon, J.H.; Wurtman, R.J. Evidence for a membrane defect in Alzheimer disease brain. Proc. Natl. Acad. Sci. USA 1992, 89, 1671–1675. [Google Scholar] [CrossRef] [PubMed]
- Wells, K.; Farooqui, A.A.; Liss, L.; Horrocks, L.A. Neural membrane phospholipids in Alzheimer disease. Neurochem. Res. 1995, 20, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, L.; Rafique, S.; Xuereb, J.H.; Rapoport, S.I.; Gershfeld, N.L. Disease and anatomic specificity of ethanolamine plasmalogen deficiency in Alzheimer’s disease brain. Brain Res. 1995, 698, 223–226. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Rapoport, S.I.; Horrocks, L.A. Membrane phospholipid alterations in Alzheimer’s disease: Deficiency of ethanolamine plasmalogens. Neurochem. Res. 1997, 22, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.Z.; Wang, Y.A.; Cairns, N.J.; Lantos, P.L.; Dallner, G.; Sindelar, P.J. Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with Alzheimer disease. J. Neuropathol. Exp. Neurol. 1999, 58, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Han, X.L.; Holtzman, D.M.; McKeel, D.W., Jr. Plasmalogen deficiency in early Alzheimer’s disease subjects and in animal models: Molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 2001, 77, 1168–1180. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Fabelo, N.; Santpere, G.; Puig, B.; Marin, R.; Ferrer, I.; Diaz, M. Lipid alterations in lipid rafts from Alzheimer’s disease human brain cortex. J. Alzheimers Dis. 2010, 19, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Ma, K.; Gao, F.; Kim, H.W.; Rapoport, S.I.; Rao, J.S. Disturbed choline plasmalogen and phospholipid fatty acid concentrations in Alzheimer’s disease prefrontal cortex. J. Alzheimers Dis. 2011, 24, 507–517. [Google Scholar] [PubMed]
- Cunnane, S.C.; Schneider, J.A.; Tangney, C.; Tremblay-Mercier, J.; Fortier, M.; Bennett, D.A.; Morris, M.C. Plasma and brain fatty acid profiles in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 2012, 29, 691–697. [Google Scholar] [PubMed]
- Yuki, D.; Sugiura, Y.; Zaima, N.; Akatsu, H.; Takei, S.; Yao, I.; Maesako, M.; Kinoshita, A.; Yamamoto, T.; Kon, R.; et al. DHA-PC and PSD-95 decrease after loss of synaptophysin and before neuronal loss in patients with Alzheimer’s disease. Sci. Rep. 2014, 4, 7130. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.J.; Bongard, V.; Beiser, A.S.; Lamon-Fava, S.; Robins, S.J.; Au, R.; Tucker, K.L.; Kyle, D.J.; Wilson, P.W.; Wolf, P.A. Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and Alzheimer disease: The Framingham Heart Study. Arch. Neurol. 2006, 63, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Rozen, S.; Boyle, S.H.; Hellegers, C.; Cheng, H.; Burke, J.R.; Welsh-Bohmer, K.A.; Doraiswamy, P.M.; Kaddurah-Daouk, R. Metabolomics in early Alzheimer’s disease: Identification of altered plasma sphingolipidome using shotgun lipidomics. PLoS ONE 2011, 6, e21643. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Dutta, T.; Persson, X.M.; Mielke, M.M.; Petersen, R.C. Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer’s disease using metabolomics. PLoS ONE 2013, 8, e63644. [Google Scholar] [CrossRef] [PubMed]
- Whiley, L.; Sen, A.; Heaton, J.; Proitsi, P.; Garcia-Gomez, D.; Leung, R.; Smith, N.; Thambisetty, M.; Kloszewska, I.; Mecocci, P.; et al. Evidence of altered phosphatidylcholine metabolism in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; Macarthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Dominguez, R.; Garcia-Barrera, T.; Gomez-Ariza, J.L. Combination of metabolomic and phospholipid-profiling approaches for the study of Alzheimer’s disease. J. Proteomics 2014, 104, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Klavins, K.; Koal, T.; Dallmann, G.; Marksteiner, J.; Kemmler, G.; Humpel, C. The ratio of phosphatidylcholines to lysophosphatidylcholines in plasma differentiates healthy controls from patients with Alzheimer’s disease and mild cognitive impairment. Alzheimers Dement. 2015, 1, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Olazaran, J.; Gil-de-Gomez, L.; Rodriguez-Martin, A.; Valenti-Soler, M.; Frades-Payo, B.; Marin-Munoz, J.; Antunez, C.; Frank-Garcia, A.; Acedo-Jimenez, C.; et al. A blood-based, 7-metabolite signature for the early diagnosis of Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 1157–1173. [Google Scholar] [PubMed]
- Fiandaca, M.S.; Zhong, X.; Cheema, A.K.; Orquiza, M.H.; Chidambaram, S.; Tan, M.T.; Gresenz, C.R.; FitzGerald, K.T.; Nalls, M.A.; Singleton, A.B.; et al. Plasma 24-metabolite Panel Predicts Preclinical Transition to Clinical Stages of Alzheimer’s Disease. Front. Neurol. 2015, 6, 237. [Google Scholar] [CrossRef] [PubMed]
- Proitsi, P.; Kim, M.; Whiley, L.; Simmons, A.; Sattlecker, M.; Velayudhan, L.; Lupton, M.K.; Soininen, H.; Kloszewska, I.; Mecocci, P.; et al. Association of blood lipids with Alzheimer’s disease: A comprehensive lipidomics analysis. Alzheimers Dement. 2016, 13, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Nevado-Holgado, A.; Whiley, L.; Snowden, S.G.; Soininen, H.; Kloszewska, I.; Mecocci, P.; Tsolaki, M.; Vellas, B.; Thambisetty, M.; et al. Association between Plasma Ceramides and Phosphatidylcholines and Hippocampal Brain Volume in Late Onset Alzheimer’s Disease. J. Alzheimers Dis. 2016. [Google Scholar] [CrossRef] [PubMed]
- Katan, M.B.; Deslypere, J.P.; van Birgelen, A.P.; Penders, M.; Zegwaard, M. Kinetics of the incorporation of dietary fatty acids into serum cholesteryl esters, erythrocyte membranes, and adipose tissue: An 18-month controlled study. J. Lipid Res. 1997, 38, 2012–2022. [Google Scholar] [PubMed]
- Harris, W.S.; Thomas, R.M. Biological variability of blood omega-3 biomarkers. Clin. Biochem. 2010, 43, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Selley, M.L. A metabolic link between S-adenosylhomocysteine and polyunsaturated fatty acid metabolism in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1834–1839. [Google Scholar] [CrossRef] [PubMed]
- Heude, B.; Ducimetiere, P.; Berr, C. Cognitive decline and fatty acid composition of erythrocyte membranes—The EVA Study. Am. J. Clin. Nutr. 2003, 77, 803–808. [Google Scholar] [PubMed]
- Tan, Z.S.; Harris, W.S.; Beiser, A.S.; Au, R.; Himali, J.J.; Debette, S.; Pikula, A.; Decarli, C.; Wolf, P.A.; Vasan, R.S.; et al. Red blood cell omega-3 fatty acid levels and markers of accelerated brain aging. Neurology 2012, 78, 658–664. [Google Scholar] [CrossRef] [PubMed]
- West, A.A.; Yan, J.; Jiang, X.; Perry, C.A.; Innis, S.M.; Caudill, M.A. Choline intake influences phosphatidylcholine DHA enrichment in nonpregnant women but not in pregnant women in the third trimester. Am. J. Clin. Nutr. 2013, 97, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, K.A.; Sanders, L.M.; Fischer, L.M.; Zeisel, S.H. Docosahexaenoic acid in plasma phosphatidylcholine may be a potential marker for in vivo phosphatidylethanolamine N-methyltransferase activity in humans. Am. J. Clin. Nutr. 2011, 93, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.L.; Silver, D.L. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 2014, 509, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Alakbarzade, V.; Hameed, A.; Quek, D.Q.; Chioza, B.A.; Baple, E.L.; Cazenave-Gassiot, A.; Nguyen, L.N.; Wenk, M.R.; Ahmad, A.Q.; Sreekantan-Nair, A.; et al. A partially inactivating mutation in the sodium-dependent lysophosphatidylcholine transporter MFSD2A causes a non-lethal microcephaly syndrome. Nat. Genet. 2015, 47, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Guemez-Gamboa, A.; Nguyen, L.N.; Yang, H.; Zaki, M.S.; Kara, M.; Ben-Omran, T.; Akizu, N.; Rosti, R.O.; Rosti, B.; Scott, E.; et al. Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat. Genet. 2015, 47, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Betsholtz, C. Lipid transport and human brain development. Nat. Genet. 2015, 47, 699–701. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H. Effects of N-3 Polyunsaturated Fatty Acids on Dementia. J. Clin. Med. Res. 2017, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.F.; Raman, R.; Thomas, R.G.; Yurko-Mauro, K.; Nelson, E.B.; Van Dyck, C.; Galvin, J.E.; Emond, J.; Jack, C.R., Jr.; Weiner, M.; et al. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: A randomized trial. JAMA 2010, 304, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Frautschy, S.A.; Cole, G.M. What was lost in translation in the DHA trial is whom you should intend to treat. Alzheimers Res. Ther. 2011, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.M.; Zhu, X.; Zeisel, S.H. Phosphatidylethanolamine-N-methyltransferase activity and dietary choline regulate liver-plasma lipid flux and essential fatty acid metabolism in mice. J. Nutr. 2003, 133, 3386–3391. [Google Scholar] [PubMed]
- DeLong, C.J.; Shen, Y.J.; Thomas, M.J.; Cui, Z. Molecular distinction of phosphatidylcholine synthesis between the CDP-choline pathway and phosphatidylethanolamine methylation pathway. J. Biol. Chem. 1999, 274, 29683–29688. [Google Scholar] [CrossRef] [PubMed]
- Pynn, C.J.; Henderson, N.G.; Clark, H.; Koster, G.; Bernhard, W.; Postle, A.D. The specificity and rate of human and mouse liver and plasma phosphatidylcholine synthesis analysed in vivo. J. Lipid Res. 2011, 52, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Blusztajn, J.K.; Holbrook, P.G.; Lakher, M.; Liscovitch, M.; Maire, J.-C.; Mauron, C.; Richardson, U.I.; Tacconi, M.T.; Wurtman, R.J. Relationships between acetylcholine release and membrane phosphatidylcholine turnover in brain and in cultured cholinergic neurons. In Phospholipids in the Nervous System: Biochemical and Molecular Pharmacology; Horrocks, L.A., Freysz, L., Toffano, G., Eds.; Liviana Press: Padua, Italy; Springer: Berlin, Germany, 1986; pp. 283–290. [Google Scholar]
- Blusztajn, J.K.; Zeisel, S.H.; Wurtman, R.J. Synthesis of lecithin (phosphatidylcholine) from phosphatidylethanolamine in bovine brain. Brain Res. 1979, 179, 319–327. [Google Scholar] [CrossRef]
- Blusztajn, J.K.; Zeisel, S.H.; Wurtman, R.J. Developmental changes in the activity of phosphatidylethanolamine N-methyltransferases in rat brain. Biochem. J. 1985, 232, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Vance, D.E. Phospholipid methylation in mammals: From biochemistry to physiological function. Biochim. Biophys. Acta 2014, 1838, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Ueland, P.M.; Ulvik, A.; Rios-Avila, L.; Midttun, O.; Gregory, J.F. Direct and Functional Biomarkers of Vitamin B6 Status. Annu. Rev. Nutr. 2015, 35, 33–70. [Google Scholar] [CrossRef] [PubMed]
- Mudd, S.H.; Brosnan, J.T.; Brosnan, M.E.; Jacobs, R.L.; Stabler, S.P.; Allen, R.H.; Vance, D.E.; Wagner, C. Methyl balance and transmethylation fluxes in humans. Am. J. Clin. Nutr. 2007, 85, 19–25. [Google Scholar] [PubMed]
- Chen, N.C.; Yang, F.; Capecci, L.M.; Gu, Z.; Schafer, A.I.; Durante, W.; Yang, X.F.; Wang, H. Regulation of homocysteine metabolism and methylation in human and mouse tissues. FASEB J. 2010, 24, 2804–2817. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Hine, R.J.; James, S.J. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J. Biol. Chem. 2000, 275, 29318–29323. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.F.; Wolf, P.A. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Wald, D.S.; Kasturiratne, A.; Simmonds, M. Serum homocysteine and dementia: Meta-analysis of eight cohort studies including 8669 participants. Alzheimers Dement. 2011, 7, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.R.; Craciunescu, C.N.; Guo, Z.; Thresher, R.J.; Blusztajn, J.K.; Zeisel, S.H. Deletion of murine choline dehydrogenase results in diminished sperm motility. FASEB J. 2010, 24, 2752–2761. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.H.; Zhao, H.L.; Zhang, Z.X.; Zhang, J.W. PEMT G523A (V175M) is associated with sporadic Alzheimer’s disease in a Chinese population. J. Mol. Neurosci. 2012, 46, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; da Costa, K.A.; Fischer, L.M.; Kohlmeier, M.; Kwock, L.; Wang, S.; Zeisel, S.H. Polymorphism of the PEMT gene and susceptibility to nonalcoholic fatty liver disease (NAFLD). FASEB J. 2005, 19, 1266–1271. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, G.; Wolf, P.A.; Beiser, A.S.; Au, R.; Seshadri, S. Risk estimations, risk factors, and genetic variants associated with Alzheimer’s disease in selected publications from the Framingham Heart Study. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S439–S445. [Google Scholar]
- Resseguie, M.; Song, J.; Niculescu, M.D.; da Costa, K.A.; Randall, T.A.; Zeisel, S.H. Phosphatidylethanolamine N-methyltransferase (PEMT) gene expression is induced by estrogen in human and mouse primary hepatocytes. FASEB J. 2007, 21, 2622–2632. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.M.; da Costa, K.A.; Galanko, J.; Sha, W.; Stephenson, B.; Vick, J.; Zeisel, S.H. Choline intake and genetic polymorphisms influence choline metabolite concentrations in human breast milk and plasma. Am. J. Clin. Nutr. 2010, 92, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Resseguie, M.E.; da Costa, K.A.; Galanko, J.A.; Patel, M.; Davis, I.J.; Zeisel, S.H. Aberrant estrogen regulation of PEMT results in choline deficiency-associated liver dysfunction. J. Biol. Chem. 2011, 286, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, K.A.; Corbin, K.D.; Niculescu, M.D.; Galanko, J.A.; Zeisel, S.H. Identification of new genetic polymorphisms that alter the dietary requirement for choline and vary in their distribution across ethnic and racial groups. FASEB J. 2014, 28, 2970–2978. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.M.; da Costa, K.A.; Kwock, L.; Stewart, P.W.; Lu, T.S.; Stabler, S.P.; Allen, R.H.; Zeisel, S.H. Sex and menopausal status influence human dietary requirements for the nutrient choline. Am. J. Clin. Nutr. 2007, 85, 1275–1285. [Google Scholar] [PubMed]
- Johnson, P.I.; Blusztajn, J.K. Sexually dimorphic activation of liver and brain phosphatidylethanolamine N-methyltransferase by dietary choline deficiency. Neurochem. Res. 1998, 23, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H. Is maternal diet supplementation beneficial? Optimal development of baby depends on mother’s diet. Am. J. Clin. Nutr. 2008, 89, 685S–687S. [Google Scholar] [PubMed]
- Shorter, K.R.; Felder, M.R.; Vrana, P.B. Consequences of dietary methyl donor supplements: Is more always better? Prog. Biophys. Mol. Biol. 2015, 118, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Richman, E.L.; Kenfield, S.A.; Stampfer, M.J.; Giovannucci, E.L.; Zeisel, S.H.; Willett, W.C.; Chan, J.M. Choline intake and risk of lethal prostate cancer: Incidence and survival. Am. J. Clin. Nutr. 2012, 96, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Willett, W.C.; Colditz, G.A.; Fuchs, C.S.; Wu, K.; Chan, A.T.; Zeisel, S.H.; Giovannucci, E.L. Dietary choline and betaine and the risk of distal colorectal adenoma in women. J. Natl. Cancer Inst. 2007, 99, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Kovacheva, V.P.; Davison, J.M.; Mellott, T.J.; Rogers, A.E.; Yang, S.; O’Brien, M.J.; Blusztajn, J.K. Raising gestational choline intake alters gene expression in DMBA-evoked mammary tumors and prolongs survival. FASEB J. 2008, 23, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Davison, J.M.; Mellott, T.J.; Kovacheva, V.P.; Blusztajn, J.K. Gestational choline supply regulates methylation of histone H3, expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a) and DNA methylation of their genes in rat fetal liver and brain. J. Biol. Chem. 2009, 284, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Relationship between intake of choline during the second trimester of pregnancy and offspring visual memory assessed by Wide Range Assessment of Memory and Learning, Second Edition (WRAML2). The top quartile choline intake was associated with a child WRAML2 score 1.4 points higher than the bottom quartile (P-trend = 0.003). Figure created from data in [55].
Figure 1.
Relationship between intake of choline during the second trimester of pregnancy and offspring visual memory assessed by Wide Range Assessment of Memory and Learning, Second Edition (WRAML2). The top quartile choline intake was associated with a child WRAML2 score 1.4 points higher than the bottom quartile (P-trend = 0.003). Figure created from data in [55].
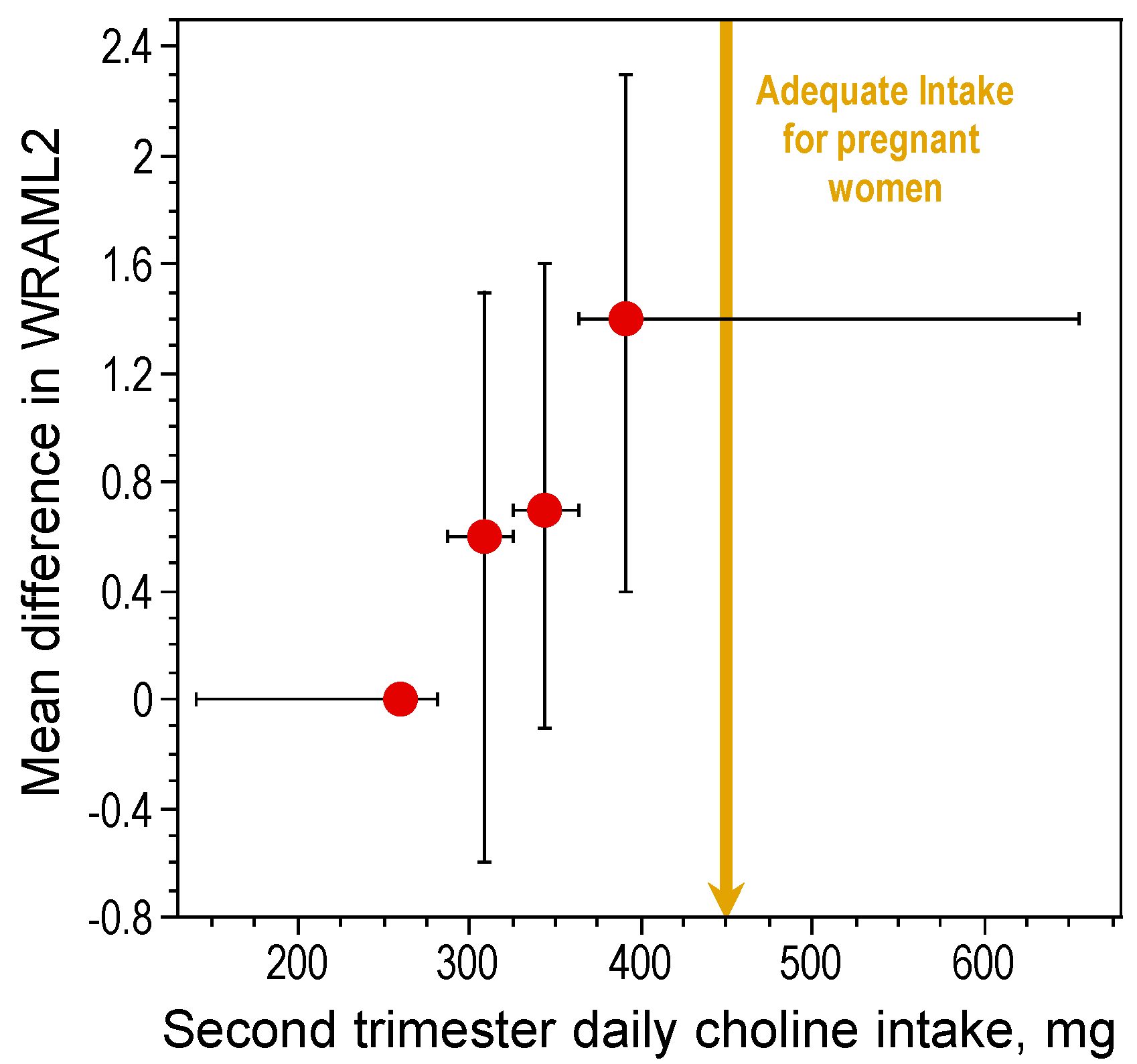
Figure 2.
Dose–response relationship between the average daily choline intake and verbal and visual memory performance in non-demented adults. Red lines indicate 95% confidence interval. Figure created from data in [45].
Figure 2.
Dose–response relationship between the average daily choline intake and verbal and visual memory performance in non-demented adults. Red lines indicate 95% confidence interval. Figure created from data in [45].
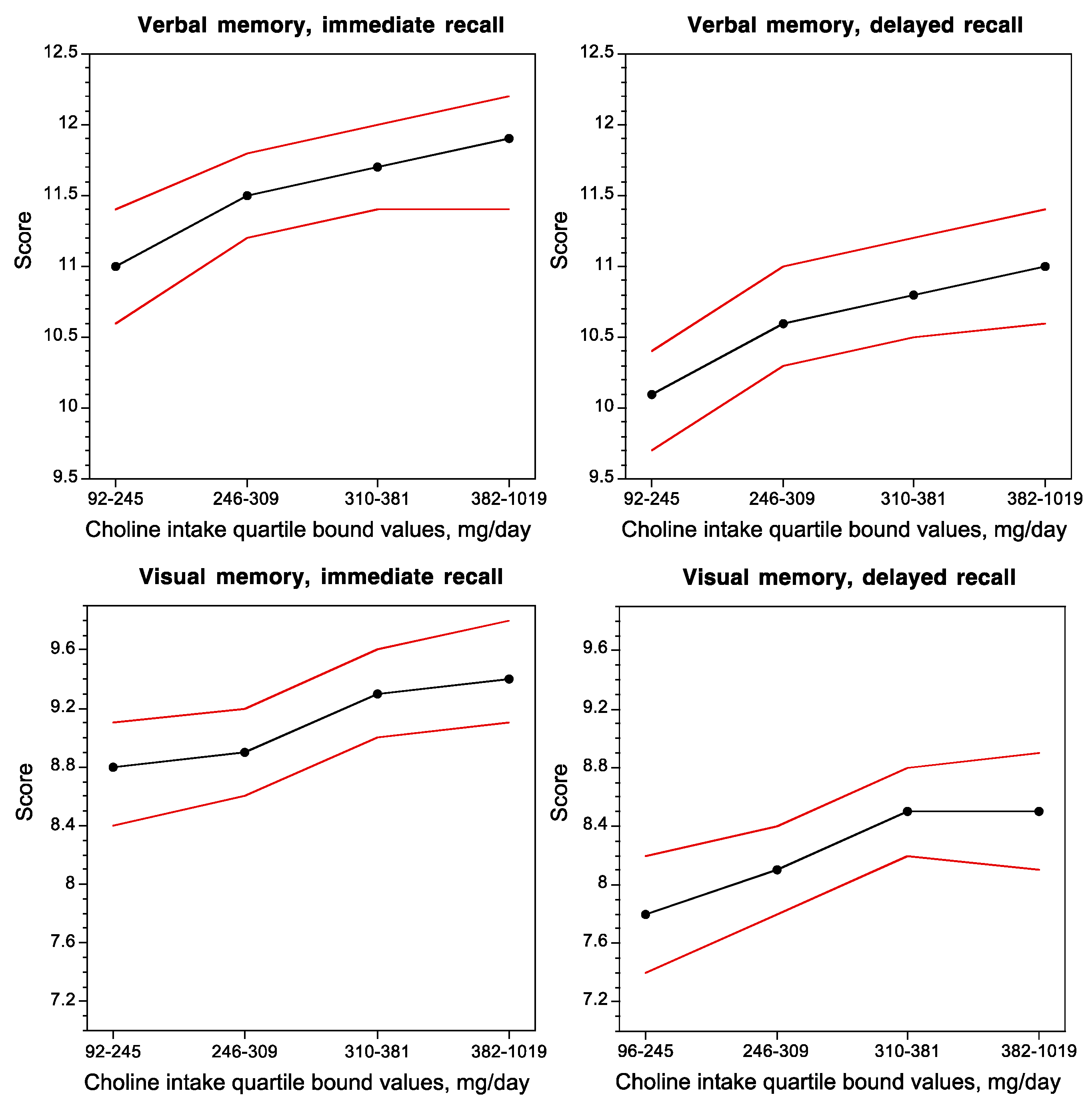
Figure 3.
PC-DHA levels depend on nutrition, liver and brain synthesis by PEMT and transport of LPC-DHA across the blood-brain barrier catalyzed by MFSD2A. Plasma PC levels and PC FA molecular species (e.g., PC-DHA) depend on hepatic PC metabolism, the dietary supply of choline and n-3 FA, including DHA, and PC and LPC flux between plasma and tissues (e.g., brain). Hepatic PEMT activity is high and the liver produces large amounts of PC-DHA by methylating PE for the needs of the entire organism. This PC-DHA is secreted by the liver into the circulation as a component of lipoproteins. Brain PEMT activity is relatively low and neuronal and glial PEMT produces PC-DHA to sustain local membranes. PC synthesis by PEMT requires three molecules of SAM that is synthesized from the amino acid, methionine. In addition to PC, PEMT generates SAH that is hydrolyzed to homocysteine. The latter can be converted back to methionine by two pathways: one catalyzed by betaine:homocysteine S-methyltransferase (BHMT) and another catalyzed by vitamin B12-requiring 5-methyltetrahydrofolate-homocysteine S-methyltransferase (MTR), in which methyltetrahydrofolate is used as a methyl donor. Betaine is the product of enzymatic oxidation of choline. Thus, dietary choline is used to produce PC directly and to convert homocysteine to methionine and subsequently to SAM. In this fashion the methyl groups derived from dietary choline can be utilized by PEMT to generate PC-DHA. Recent evidence indicates that the main source of brain DHA is LPC-DHA—the circulating deacylated metabolite of PC-DHA. The transport of LPC-DHA from plasma to the brain is catalyzed by MFSD2A. The blue arrows indicate flux and the black arrows represent enzymatic pathways. Protein names are in red. Dietary DHA is primarily stored in PE in tissues and only a small fraction is incorporated directly into PC. Homocysteine can also be converted to cysteine via cystathionine in a transsulfuration pathway that includes two vitamin B6-requiring enzymes, CBS and CTH. Abbreviations: BHMT, betaine:homocysteine S-methyltransferase; CBS, cystathionine-beta-synthase; CTH, cystathionine gamma-lyase; DHA, docosahexaenoic acid; FA, fatty acid; LPC, lysophosphatidylcholine; MFSD2A, major facilitator superfamily domain containing 2a; MTR, 5-methyltetrahydrofolate-homocysteine S-methyltransferase; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PEMT, phosphatidylethanolamine N-methyltransferase; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine.
Figure 3.
PC-DHA levels depend on nutrition, liver and brain synthesis by PEMT and transport of LPC-DHA across the blood-brain barrier catalyzed by MFSD2A. Plasma PC levels and PC FA molecular species (e.g., PC-DHA) depend on hepatic PC metabolism, the dietary supply of choline and n-3 FA, including DHA, and PC and LPC flux between plasma and tissues (e.g., brain). Hepatic PEMT activity is high and the liver produces large amounts of PC-DHA by methylating PE for the needs of the entire organism. This PC-DHA is secreted by the liver into the circulation as a component of lipoproteins. Brain PEMT activity is relatively low and neuronal and glial PEMT produces PC-DHA to sustain local membranes. PC synthesis by PEMT requires three molecules of SAM that is synthesized from the amino acid, methionine. In addition to PC, PEMT generates SAH that is hydrolyzed to homocysteine. The latter can be converted back to methionine by two pathways: one catalyzed by betaine:homocysteine S-methyltransferase (BHMT) and another catalyzed by vitamin B12-requiring 5-methyltetrahydrofolate-homocysteine S-methyltransferase (MTR), in which methyltetrahydrofolate is used as a methyl donor. Betaine is the product of enzymatic oxidation of choline. Thus, dietary choline is used to produce PC directly and to convert homocysteine to methionine and subsequently to SAM. In this fashion the methyl groups derived from dietary choline can be utilized by PEMT to generate PC-DHA. Recent evidence indicates that the main source of brain DHA is LPC-DHA—the circulating deacylated metabolite of PC-DHA. The transport of LPC-DHA from plasma to the brain is catalyzed by MFSD2A. The blue arrows indicate flux and the black arrows represent enzymatic pathways. Protein names are in red. Dietary DHA is primarily stored in PE in tissues and only a small fraction is incorporated directly into PC. Homocysteine can also be converted to cysteine via cystathionine in a transsulfuration pathway that includes two vitamin B6-requiring enzymes, CBS and CTH. Abbreviations: BHMT, betaine:homocysteine S-methyltransferase; CBS, cystathionine-beta-synthase; CTH, cystathionine gamma-lyase; DHA, docosahexaenoic acid; FA, fatty acid; LPC, lysophosphatidylcholine; MFSD2A, major facilitator superfamily domain containing 2a; MTR, 5-methyltetrahydrofolate-homocysteine S-methyltransferase; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PEMT, phosphatidylethanolamine N-methyltransferase; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine.
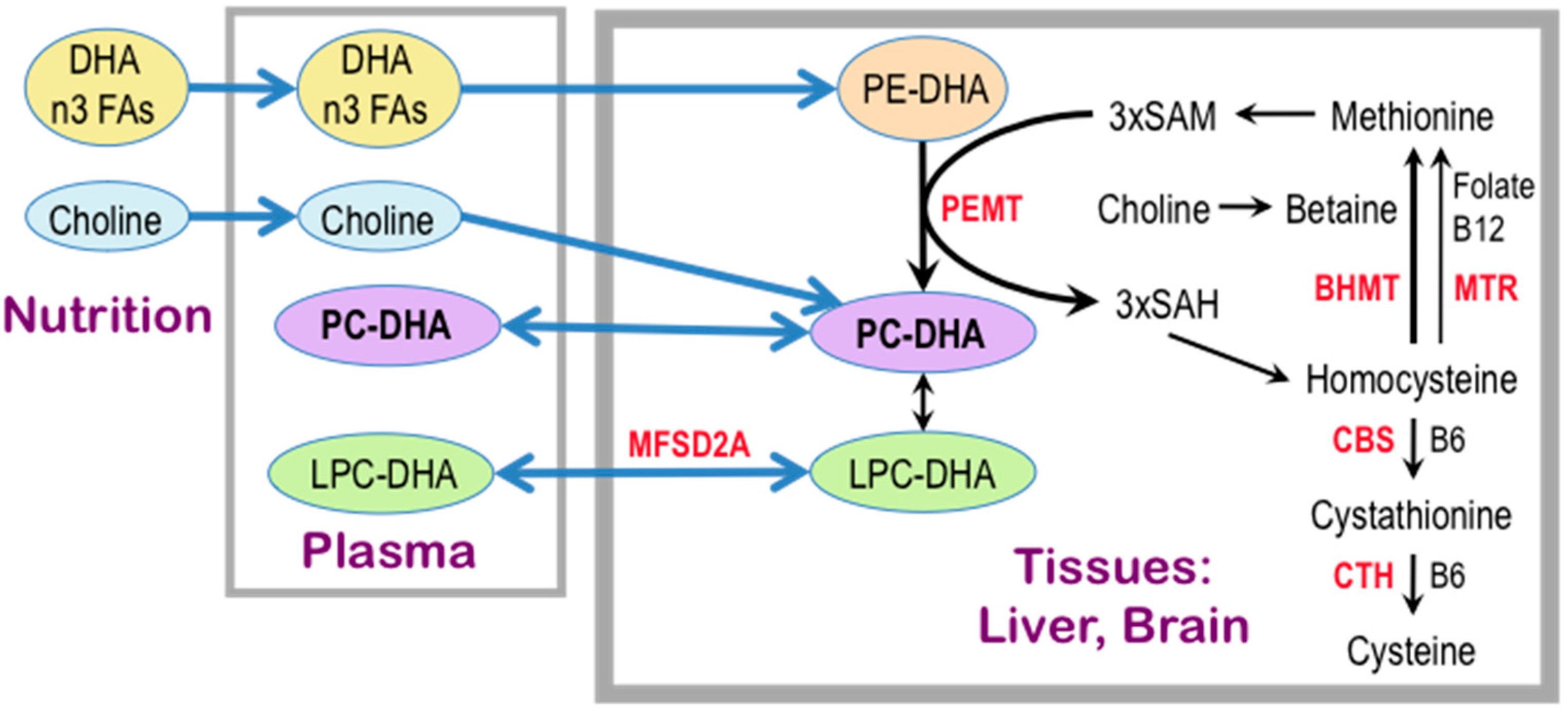
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Blusztajn, J.K.; Slack, B.E.; Mellott, T.J. Neuroprotective Actions of Dietary Choline. Nutrients 2017, 9, 815. https://doi.org/10.3390/nu9080815
AMA Style
Blusztajn JK, Slack BE, Mellott TJ. Neuroprotective Actions of Dietary Choline. Nutrients. 2017; 9(8):815. https://doi.org/10.3390/nu9080815
Chicago/Turabian StyleBlusztajn, Jan Krzysztof, Barbara E. Slack, and Tiffany J. Mellott. 2017. "Neuroprotective Actions of Dietary Choline" Nutrients 9, no. 8: 815. https://doi.org/10.3390/nu9080815
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.