






Comprehensive Overview of Homogeneous Gold-Catalyzed Transformations of π-Systems for Application Scientists

Laboratory of Medicinal Chemistry, Section of Pharmaceutical Chemistry, Faculty of Pharmacy, National and Kapodistrian University of Athens, Panepistimiopolis Zografou, 15771 Athens, Greece
*
Author to whom correspondence should be addressed.
Catalysts 2023, 13(6), 921; https://doi.org/10.3390/catal13060921
Submission received: 20 April 2023
/
Revised: 15 May 2023
/
Accepted: 16 May 2023
/
Published: 23 May 2023
(This article belongs to the Special Issue Exclusive Papers of the Editorial Board Members and Topical Advisory Panel Members of Catalysts in Section "Catalysis in Organic and Polymer Chemistry")
Abstract
:We present an overview of fundamental catalytic reactions of nucleophiles with π-systems in relation to gold chemistry. We present examples of reactions with gold-activated π-systems, alkynyl or allenyl moieties, and the regulation of their reactivity due to the presence of an electron-donating or -withdrawing group. The reactions describe furnished hard-to-reach heterocyclic building blocks for medicinal chemistry purposes. Important gold(I) or gold(III) complexes that are used as catalysts are presented. We examine the activation of such π-systems using gold(I) or gold(III) catalysts and the corresponding divergent catalytic transformations. We provide examples of divergent catalysis using gold(I) catalyst and other metal catalysts (Pt, Ag, Pd, Rh, Sc, Cu) or by changing the ligands in gold(I) catalyst complexes. We also discuss the role of the solvent, counterions and additives in gold(I)-catalyzed reactions. We mention, in a few cases, characteristic experimental or computational studies of these gold-catalyzed reactions of nucleophiles with π-systems.
1. Introduction
Homogenous gold catalysis blossoms year after year, according to the increasing number of publications [1,2,3,4,5], because of its beneficial traits such as the mild reaction conditions and the easiness of use. Thus, it is a powerful tool in the hands of synthetic chemists who work on organic and bioorganic synthesis or material science.
Gold has remarkable properties that set it apart from other metals. The bulk metal is of course best known for its inert character and resistance to oxidation and chemical attack and has therefore long been regarded as unpromising for catalytic applications. Being the most electronegative of metallic elements (2.54 on the Pauling scale), almost identical to carbon, gold forms highly covalent, hydrolytically stable, Au–C bonds. This is also the reason why the exploration of its organometallic chemistry has so long lagged behind work on other noble metals [6]. This situation has now drastically changed.
The position of gold in the periodic table is unique and gold exists as a catalyst both in gold(I) and gold(III) forms. Although many transition metals are commonly used as catalysts, gold reveals divergent chemical properties resulting from the differences between its electronic structure and the electronic structure of the other metals.
In the gold element, due to the relativistic effect, 1s orbital contracts, and so do all s and p atomic orbitals, reducing atomic radius and increasing ionization energies [7]. However, for most of the elements, the contraction of the atomic radius is not as significant as it is for the elements with filled 4f and 5d orbitals. Thus, for the elements such as platinum, gold and mercury with the electron structure [Xe]4f145d96s1, [Xe]4f145d106s1 and [Xe]4f145d106s2, respectively, relativistic effects have a high impact on atomic radius due to the contraction of 6s atomic orbital [8]. The contraction of the s and p atomic orbitals implies better shielding for the electrons of the d and f orbitals. Therefore, the nuclear attraction on the electrons of d and f orbitals is decreased. Consequently, the d and f orbitals expand while the s and p orbitals contract. In the case of gold with the electron structure [Xe]4f145d106s1, 6s orbital contraction and 5d orbital expansion render gold a stronger Lewis acid with higher electronegativity compared with copper or silver.
Gold(I) catalysts are abundant either as inorganic gold (AuCl) or in a complex with organic ligands. The ligand of the gold complex controls its Lewis acidity. The contraction of the 6s orbital strengthens the Au–ligand bond [9], and a comparative study between gold(I) and silver(I) with phosphine ligand revealed that the covalent character of the bond is stronger in the Au(I) complex [10,11]. Consequently, according to the HSAB concept (“hard and soft (Lewis) acids and bases”) [12,13], Au(I) catalysts are soft acids due to their extended radius and diffused charge and form bonds with a more covalent character. Thus, these catalysts prefer reacting with soft bases such as π-systems, e.g., alkynes, alkenes, allenes and “soft” atoms such as p and s. While gold(I) species [LAu]+ are known as carbophilic electrophiles and also preferentially bind to “soft” bases, gold(III) is a “hard” Lewis acid. This is reflected in the stability of their OH and F compounds; whereas the first isolable gold(I) hydroxide and fluoride complexes LAuX (X = OH, F; L = N-heterocyclic carbene NHC) [14] were only reported since 2005, examples of structurally characterized hydroxo [15] and fluoro [16,17] complexes of gold(III) have been known for several decades.
In the oxidation state +1, with a filled d shell, gold behaves rather like a main group element and forms linear, two-coordinate complexes that show a marked reluctance to interact with donor ligands perpendicular to the molecular axis. Gold in the oxidation state III, on the other hand, displays all the characteristics of a transition metal, adopts almost exclusively the square-planar coordination geometry that is so familiar from other heavy metal cations with d8 electronic structure and is distinctly different in terms of structure and reactivity from gold(I) compounds. Moreover, the intensive relativistic effects on gold(I), which are diminished on gold(III), contract the bonds’ length of the gold(I) complexes.
In the first part of this review, we provided cases for the gold-catalyzed reactions of nucleophiles with π-systems. In the second part, we described the divergent catalysis for gold(I) and gold(III) complexes (Figure 1). In the third part, we presented the divergent gold (I) catalysis over other metal catalysts and the effect of ligands and counterions in gold(I) complexes and of solvent in gold(I) catalysis. In the reaction schemes described in this review, we gave the relevant citation where appropriate as well as the corresponding authors in parenthesis.
Many detailed as well as comprehensive reviews are available in the literature from experts in the field of gold-catalyzed-reactions in solution [12,18,19,20,21,22,23,24,25,26,27,28] and those that include involving π-systems [4,27,29,30], as well as their mechanisms and intermediates studied experimentally or often using Density Functional Theory (DFT) calculations [26,27,31,32,33,34,35,36,37,38]. We reviewed the gold-catalyzed reactions of nitrogen, oxygen and carbon nucleophiles with π-systems having alkynyl or allenyl moieties with the electron-donating group, e.g., ynamides, ynols, allenamides and allenyl ethers. In addition, we described reactions with electron-withdrawing group π-systems, e.g., alkynyl carbonyl and allenyl carbonyl derivatives. We presented the major categories of gold(I)- and gold(III)-π-system complexes in catalysis, e.g., gold(I)– and gold(III)–carbene complexes and gold(I)– and gold(III)–π-alkene and –π-alkyne complexes. We introduced divergent catalysis in reactions with π-systems using gold(I) versus gold (III) complexes or gold(I) versus other metals or by changing the ligands in gold(I) complexes. We described the effect of counterions, solvents or additives on the catalytic cycle of reactions with π-systems. We directed this review both for organic or medicinal or theoretical chemists that wish to enter the amazing field of homogeneous gold catalysis with π-systems as reactants. The synthetic protocols described will inspire medicinal chemists since they can lead to hard-to-reach heterocycles that are parts of drugs and natural products.
2. Gold-Catalyzed Reactions of Nucleophiles with π-Systems
2.1. General Description of Reactivity
A major part of homogenous gold catalysis is related to activated alkynes, allenes and alkenes. Gold complexes with the carbon π-system (double or triple C–C bond) are exposed to the attack of nucleophilic moieties according to Scheme 1.
In the cases that an asymmetric and polarized substituted π-system is involved in a gold-catalyzed reaction, the transformation reveals regio-, stereo- and chemoselectivity. Regio-, stereo- and chemoselectivity are related to the nature of the substituents and orientation of the carbon π-system. Substituents can be divided into electron-donating groups (EDGs) and electron-withdrawing groups (EWGs). Below, we described the functionalization of π-systems with an EDG, such as ynamides, ynols, allenamides and allenyl ethers and the functionalization of π-systems with an EWG such as alkynyl carbonyl and allenyl carbonyl derivatives.
2.2. Gold-Catalyzed Functionalization of Activated π-Systems with an Electron-Donating Group
2.2.1. Functionalization of Ynamides
General Reactivity Profile
Ynamides 2a are classified as particularly activated alkynes bearing an electron-donating group (Scheme 2).
The activation process of the ynamides follows a first step of activation by gold that forms a keteniminium intermediate 2b. The keteniminium intermediate is a polarized and electrophilic species. In the second step, variable nucleophiles can approach and attack the α-carbon regioselectively to form compound 2c. Some examples of the reaction of ynamides with N-, O- and C-based nucleophiles were commented on in the next paragraphs.
Functionalization with Nitrogen Nucleophiles
The reaction between oxazole 3b and ynamide 3a is shown in Scheme 3A, and the mechanism of the reaction is described in Scheme 3B [39].
It was suggested that the first step of the mechanism of the reaction is the activation of ynamide 3a by the gold(I) catalyst. Accordingly, isoxazole’s 3b nitrogen attacks the α-carbon of the activated complex 3e, forming 3f, and then the α-imino gold carbene intermediates 3g after the breaking of the N–O+ bond in 3f. A nucleophilic attack at the carbocation carbon of the α-imino gold carbene 3g from the activated C–C bond in 3g can lead to the cyclization of the α-imino gold intermediate 3g to form 3h, which, after deauration, leads to 2-amino pyrroles 3c/3d.
Similar reactions have been also carried out [28], for example, between anthranils 4b and ynamides 4a to afford indoles 4c (Scheme 4) [40] using catalyst IPrAuCl (4d)/AgNTf2 (IPr is the N-heterocyclic carbene (NHC) ancillary ligand 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene and NTf is the trifluoromethanesulfonyl group).
Other examples are the Au(I)-catalyzed reactions between sufoximines 5b with ynamides 5a to afford 5c using Ph3PAuCl/AgOTf (Scheme 5) [41] and imines 6b with 1-alkynyltriazenes 6a to form 1,3-diaminopyrazoles 6c with catalyst JohnPhosAuCl (6d)/AgNTf2, see Scheme 6 (JohnPhos = 2-(di-tert-butylphosphino)biphenyl) [42].
We investigated [43] the reaction of anthranils 7b with ynamides 7a as well as apolar alkynes (terminal or central) using DFT calculations. When the alkyne was terminal, the reaction afforded selectively the 2-substituted 7-acyl-indole 7c compared with the 3-substituted 7-acyl-indole indole 7n (Scheme 7).
The DFT calculations showed that the observed regioselectivity seemed to be connected to the irreversible formation of the key α-imino gold carbene intermediate (7e or 7j) common to both reaction profiles in Scheme 7, respectively, through the initial regioselective nucleophilic attack of the N atom of anthranil 7b onto the ynamide 7a fragment.
In another paper, we also compared [44] the reaction between anthranil, 1,2,4-oxadiazole, or 4,5-dihydro-1,2,4-oxadiazole, and the ynamide, PhC≡C-N(Ts)Me, to afford proceeding via the formation of the aforementioned α-imino gold carbene intermediate, which, after intramolecular capture, regioselectively produced 2-amino-3-phenyl-7-acyl indoles, N-acyl-5-aminoimidazoles, or N-alkyl-4-aminoimidazoles, respectively. In all cases, the regioselectivity of the substituents at 2, 3 in the 7-acyl-indole ring and 4, 5 in the substituted imidazole ring is decided at the first transition state, involving the attack of nitrogen on the C1 or C2 carbon of the activated ynamide. A subsequent and steep energy drop furnishes the key α-imino gold carbene. These features are more pronounced for anthranil and 4,5-dihydro-1,2,4-oxadiazole reactions.
Functionalization with Oxygen Nucleophiles
Activated ynamides 8a can react with O-based nucleophiles 8b (Scheme 8), such as pyridine N-oxides (Scheme 9) and sulfoxides, forming an intermediate α-oxo gold carbene intermediate 8d after the cleavage of the bond between the oxygen and leaving group X in 8c (Scheme 8) [4].
Scheme 9 shows the synthesis of α,β-unsaturated carbonyl derivatives 9c from the reaction of pyridine N-oxide 9b and ynamides 9a catalyzed by different forms of Au(III).
Ynamides 10a react with alcohols and ethers either intermolecularly or intramolecularly forming fused imidazole heterocycles, i.e., diaminofurans. It is noteworthy that the reaction of Scheme 10 follows the opposite regioselectivity. That the formation of the six-member ring in 10b, versus the five-member ring, is favored is possibly due to the easier approach of the OH group in 10a to the β-carbon of alkyne giving a 6-endo-dig attack [46].
2,5-Diaminofurans 11b were formed by the reaction of water with diynamides 11a catalyzed by Ph3PAuNTf2 [47] (Scheme 11).
In the synthesis of dihydrobenzoxepines 12c by the formal [4+3] cycloaddition between ynamides 12a and epoxides 12b catalyzed by IPrAuNTf2, retention of stereochemistry was observed (Scheme 12) [48]. Moreover, the reaction proceeded under a wide variety of substituents not only on ynamide 12a but also on epoxide 12b. The proposed mechanism of the reaction is that an initial nucleophilic attack of the epoxide 12b onto the activated ynamide leads to oxonium intermediate 12d. Possibly, the orientation of the attack is very strict due to the conformation constraint that the aryl group induces the oxiranyl ring orientation. In addition, a synchronous breaking of the bond C–O in the oxirane 12d and the formation of the bond C–C between the aryl group and oxirane can contribute to the conservation of the stereochemical conformation [48].
Functionalization with Carbon Nucleophiles
In gold(I)-catalyzed reactions, an activated ynamide can participate in an intramolecular or intermolecular cycloisomerization through a formal cycloaddition.
An example of gold(I)-catalyzed cycloisomerization is the reaction of the ynamide in Scheme 13. Using either (ArO)3P)AuCl/AgOTf or [(JohnPhos)Au(NCMe)]SbF6 as a gold(I) catalyst, the activated ynamide 13a is attacked at the α-carbon by the nucleophilic C2 indole carbon forming a six-member ring condensed with indole regioselectively. At the second step, a 5-exo-dig or 6-endo-dig cyclization leads to the formation of the two types of fused indole N-heterocycles 13b or 13c. The role of the R3 alkyl group is critical. A sizeable R3 group favors a 5-exo-dig attack while a small one urges the 6-endo-dig path [49].
A variation of the previous reaction is the synthesis of sulfone-containing pyrrolo[2,1-a]isoquinolines 13d from diynamides 13a using IPrAuNTf2 [51]. The nucleophilic attack to the activated ynamide group is succeeded by benzene in Scheme 14 but can also be carried out by heterocyclic arenes, e.g., an indole ring. An electron-donating group on the phenyl ring in Scheme 14 or otherwise a heterocyclic arene increases the yield of the reaction. It was suggested that at the first step of the reaction, the Au(I)-activated ynamide is attacked at the α-carbon by the benzene ring forming a C–C bond in 13b. In the second step, the activated second alkyne moiety reacts intramolecularly through a 5-endo-dig cyclization with the nucleophilic nitrogen. It is noteworthy that the path to the formation of pyrrolo[2,1-a]isoquinolines 13d proceeds through a two-step [1,2]sulfonyl shift.
Intramolecular Hydride Shift of Keteniminium Intermediates
Functionalization of ynamides by an electrophilic gold(I) complex may be used for the formation of polycyclic compounds via an allene intermediate or can lead to an allenamide in the case that cyclization is not feasible.
In the case where allenamide 15d is the final product, the presence of a benzyloxy group in ynamide 15a is necessary to act as a hydride donor. Thus, the Au(I)-activation of ynamide 15a with catalyst [(JohnPhos)Au(NCMe)]SbF6 [52] forming keteniminium 15b is shown in Scheme 15. Then, a transformation can proceed through a [1,5]-hydride shift from the benzyloxy group to the keteniminium 15b. The unstable intermediate 15c intervenes between allenamide 15d and keteniminium 15b. The concerted elimination of a molecule of benzaldehyde and catalyst from 15c leads to the allenamide product 15d [52].
2.2.2. Functionalization of Ynol Derivatives
General Reactivity Profile
Although ynols are reactive as regards the nucleophilic attack at the α-carbon (Scheme 16), similarly to ynamides, there are fewer methods in the literature compared to ynamides.
Often, most of the examples include ynols derivatives, such as alkynylsilylynol ethers or alkynylaryl ethers. Functionalization of ynols can be succeeded with C-, N- and O-based nucleophiles (Scheme 16).
Functionalization with O-Based Nucleophiles
Phenol derivatives 17c can be formed by the gold(I)-catalyzed reaction of terminal arylalkynyl ethers or ynols 17a with 2,5-disubstituted furans 17b (Scheme 17) catalyzed by IPrAuCl/NaBArF (BArF is a non-coordinating anion, i.e., the tetrakis[3,5-bis(trifluoromethyl)phenyl]borate). This reaction is sensitive to steric and electronic effects associated with the ynol adduct. In the case of aliphatic ynol ethers, the reaction proceeds in low yield.
The synthesis of the benzofuranoquinoline 18c was carried out by the reaction of arylethynyl ether 18a, an ynol with an electron-donating group, and an anthranil derivative 18b using JohnPhosAuCl/AgSbF6 as a catalyst (Scheme 18) [54]. It was suggested that the reaction proceeds via an α-amino gold carbene intermediate 18d, which is then subjected to an intramolecular nucleophilic attack from aryloxy group to carbene. Next, an intramolecular condensation leads to the benzofuranoquinoline 18c.
2.2.3. Functionalization of Allenamides and Allenyl Ethers
General Reactivity Profile
Synthetic methods using allenyl derivatives are less abundant than methods using yne derivatives because of the unstable nature of the former and the difficulties of manipulation. However, allenyl derivatives offer interesting chemical characteristics when they are used as substrates for gold(I)-catalyzed reactions. There are two available positions on allenes, a central and a terminal one (α and γ), for the formation of a new C–C, C–N or C–O bond. A general reactivity profile for gold(I)-catalyzed reactions of allenes 19a with nucleophiles is based on the presence of an N-, O-, or S-electron-donating group X in allene 19a that transforms allene 19a to an aurated alkenyl-intermediate 19b. The latter is sensitive to both an intermolecular and an intramolecular nucleophilic attack on positions α or β to afford 19c or 19d, respectively (Scheme 19) [55].
Functionalization with C-Based Nucleophiles
The benzyloxy group can be part of an allene following an Au(I)-catalyzed intramolecular cyclization. In Scheme 20, the activation of allene 20a by AuCl led to the cyclopropyl intermediate 20b. Next, the C–C bond rearrangement and deauration afforded the cyclohexadiene derivative 20c, which is easily aromatized to the benzyloxybenezene derivatives 20d by means of the leaving group R2COO− [56].
Another example of an Au(I)-catalyzed functionalization with a C-based nucleophile is based on the formal [4+2] cycloaddition between an allene 21b and 2-alkenylindoles 21a, leading to tetrahydrocarbazole derivatives 21d (Scheme 21) [56]. The suggested mechanism starts with the nucleophilic attack by the electron-rich C-3 carbon of the indole ring at the terminal position γ-carbon of allene 21b activated by Au(I) using the JohnPhosAuNTf2 catalyst. A second step is a nucleophilic attack between the alkenyl side chain group and the β-carbon of the allene in 21c which leads to the cyclization product 21d.
The experimental conditions and Au(I) catalyst are critical for determining which product of the reaction is formed. For instance, the usage of AuCl3 at a low temperature (−70 to −50 °C) with a sub-stoichiometric amount of allenamide produced indole 21g as the main product. On the contrary, a higher temperature (−20 °C) and JohnPhosAuNTf2 urged the reaction to the formation of the isomer 21f. Excess of allenamide led to the disubstituted product 21f due to one more nucleophilic addition at the γ-position of another gold(I)-activated allenamide 21e [57].
Scheme 21.
Alkenylindoles 21a as gold-catalyzed reaction partners in [4+2] cycloadditions with allenamide 21b to afford indoles 21f with JohnPhosAuNTf2 or 21g with AuCl3 (Vicente 2013) [57].
Scheme 21.
Alkenylindoles 21a as gold-catalyzed reaction partners in [4+2] cycloadditions with allenamide 21b to afford indoles 21f with JohnPhosAuNTf2 or 21g with AuCl3 (Vicente 2013) [57].

Functionalization with O- and N-Based Nucleophiles
The most common Au(I)-catalyzed functionalizations with O- and N-nucleophiles are the intramolecular ones, e.g., the formation of the dihydrofuran 22c or pyrrole 23c starting from allenes 22a [58] or 23a [59] with a vicinal hydroxyl or amino group, respectively. The first steps of both reactions are probably similar.
Thus, in the first case (Scheme 22), a nucleophilic attack of the hydroxyl group to the α-carbon of the activated allene with Ph3PAuCl/AgBF4 catalyst can form intermediate 22b. After protodeauration, the dihydrofuran 22c was formed in good yields and within a short reaction time (e.g., 5 min).
The second Ph3PAuNTf2-catalyzed reaction (Scheme 23) followed a differentiated path after protodeauration.
Thus, after the nucleophilic attack of the amino group to the α-carbon of allene 23a and protodeauration, concerted steps of the elimination of the phosphate group and aromatization led to the formation of pyrrole derivatives 23c.
Examples of intermolecular N-, O- and S-functionalization reactions by N-, O- and S-based nucleophiles are not very common. For example, a reaction can proceed with the regioselective Au(I)-catalyzed addition of pyrazole, triazole or benzotriazole 24b to allene 24a under heating and microwave irradiation using Ph3PAuNTf2 as a catalyst to afford 24c [60]. The presence of co-catalyst PtCl2 and the excess of the N-based nucleophile leads to double hydroamination products 24d, as is described in Scheme 24 [60].
2.3. Gold-Catalyzed Functionalization of Activated π-Systems with a Withdrawing Group
2.3.1. Activated π-Systems
Compared to the π-systems with EDGs discussed in Section 2.2, π-systems connected with EWGs, e.g., alkynes, allenes and alkenes are shown in Scheme 25.
Alkynes with EWG are a carbonyl group directly connected to α-carbon; the alkyne derivative can be a ketone, a carboxylic acid, an ester or an amide. Alternatively, if the EWG is an imine group instead of a carbonyl, then the substrate can be an imine, a hydrazone or an oxime. The EWG can be also a halogen, a boronate, a sulfoxide-sulfone or a fluorine group. These π-systems connected with EWGs show different chemistry. These polarized π-systems activated by electrophilic gold(I) catalysts can present regioselectivity in the reaction with nucleophiles. Thus, alkynes, allenes and alkenes bearing EWGs are rendered susceptible to nucleophilic attack at the β-position of the π-system [4].
2.3.2. Alkynyl Carbonyl Derivatives
General Reactivity Profile
Alkynyl carbonyl derivatives 26a are susceptible to a gold-catalyzed nucleophilic attack at the β-position of the π-system. If carbonyl’s X = EWG, see Scheme 26, the alkyne β-carbon becomes electron-poor, and, after Au-activation, intermediated 26b is formed, and a nucleophilic attack can easily be carried out to form a large variety of products 26c. C-, N- and O-based nucleophiles can be applied to regioselective reactions with alkynyl carbonyl derivatives 26a (Scheme 26).
Functionalization with C-Based Nucleophiles
Alkene or alkyne groups are used as C-based nucleophiles to electron-deficient activated alkynes. There are several examples of intramolecular or intermolecular cyclizations, cycloisomerizations and cycloadditions.
The Au(I)-catalyzed cyclization of 1,6-enyne 27a that bears an ester or amide EWG group tethered to the alkyne is shown in Scheme 27.
First, the Au(I)-activated alkyne using Ph3PAuSbF6 as a catalyst was subjected to a nucleophilic attack by the adjacent alkene to the electron-deficient β-carbon. After a 6-endo-dig cyclization and formation of the intermediate 27b, a 1,2-carbon shift led to the cyclobutane condensed bicyclic compound 27d.
On the other hand, an intermolecular nucleophilic attack to the Au(I)-activated electron-deficient alkyne, using JohnPhosAuCl/AgSbF6 as a catalyst, between a propiolic acid 28b and alkene 28a led to the unsaturated δ-lactones 28d (Scheme 28) [62]. This is an [4+2] annulation reaction that proceeded with the step of a nucleophilic attack to the β-carbon of activated alkyne forming a cyclopropyl gold carbene 28c. Then, the nucleophilic carbonyl oxygen of intermediate 28c attacked the cyclopropyl ring, which opens, forming the unsaturated δ-lactone 28d.
Gold(Ι)-catalyzed functionalization of an alkynyl–carbonyl compound can be succeeded via the formation of enols or enol derivatives, with the enol group acting as EWG being the tautomeric form of a ketone group. The enol or enol derivative can be generated in situ unless it exists in a native form when it is more stable than the ketone tautomer. One method to synthesize an enol or enol ester is the attack of the oxygen nucleophile to the gold(I)-activated terminal alkyne. Scheme 29 shows the synthesis of α-pyrones 29e from the reaction between alkynyl-carboxylic acids 29a and alkynes 29b, using the catalyst Ph3PAuCl/AgOTf, as an example of the functionalization of an alkene bearing an enol group as EWG, see 29c [63]. The gold(I)-activated alkyne accepts the nucleophilic attack of the carboxylic group of the propiolic acid forming enol 29c at the C2 carbon. The 6-endo-dig cyclization (Scheme 29), i.e., the intramolecular nucleophilic attack of the enol carbon to the Au(I)-activated β-carbon of the alkyne moiety and protodeauration of the intermediate 29d can lead to α-pyrone 29e [63].
A variation of the previous method is based on the replacement of enol ester moiety in 29c with allenol ester moiety in 30b [63,64] (Scheme 30). The first step of the mechanism of this reaction is a gold(I)-catalyzed [3,3]-rearrangement to form the allenyl ester 30b from 30a using the catalyst Ph3PAuCl/AgSbF6. In the next step, the gold(I) catalyst activates the triple bond of the alkyne, while the β-carbon of the allenyl group carries out a nucleophilic attack to the β-carbon of alkyne moiety (6-endo-dig cyclization). After the formation of the cyclic product 30c, the reaction may follow different paths depending on the conditions. The presence of alcohol in the reaction mixture urges the reaction to the formation of esters 30d. Instead of esterification, arylation takes place forming compound 30e if instead of the nucleophilic alcohol an aromatic compound is present. If an additional nucleophile is absent, the alkenyl-α-pyrone 30f is formed due to the redistribution of the π-electrons and deauration of intermediate 30c.
Functionalization with O-Based Nucleophiles
Many reactions have been performed using O-based nucleophiles, such as alcohols, ketones and aldehydes for the functionalization of alkynyl carbonyl derivatives. The chemical transformation can be hydrofunctionalizations, cyclizations, isomerizations or oxidations and can be encountered in a reactions cascade. A typical example of intramolecular cyclization is the hydroalkoxylation of alkynones or alkynyl carboxylic acid derivatives for the formation of oxygen-containing heterocycles that include the reaction of an O-based nucleophile with the gold(I)-activated alkyne moiety. As is shown in Scheme 31, the formation of oxazepinone 31b proceeds via the activation of alkynyl carboxamide 31a by the gold(I) catalyst [66]. This is followed by the nucleophilic attack of the hydroxyl group to the β-carbon of the alkynyl bond, i.e., a 7-endo-dig cyclization that forms oxazepinone 31b. The combination of the gold(I) catalyst Ph3PAuCl with AgOTf provided excellent yields. However, Ph3PAuCl alone can also catalyze the reaction [66].
The Au(III)-catalyzed synthesis of 5-, 6- and 7-member ring cyclic acetals 32e and exocyclic enol ethers 32b–32d is similar to the above method [67] (Scheme 32). Despite the E-isomer that is favored, a mixture of E- and Z-isomers is obtained. According to the proposed path, after the nucleophilic attack of the hydroxyl group on the Au(I)-activated alkyne in 32a, the Z-enol ether 32b is formed. Isomerization of the Z-enol ether 32b to the intermediate oxocarbenium 32c is the path that leads to the formation of E-isomer 32d, while isomers 32b and 32c are in equilibrium. Both of the structures 32b and 32c provide the substrate for the nucleophilic attack of the alcohol R6OH resulting in the acetal 32e [67,68].
When the alcohol nucleophile is absent, the cyclic acetals bearing enol ester groups 33b can be obtained from alkynyl esters 33a (Scheme 33).
Some O-based nucleophiles, e.g., nitro or sulfoxide groups, pyridine N-oxide or quinoline N-oxide (see −O-LG+ in Scheme 34), can oxidize the alkynyl carbonyl substrate 34a to the reactive intermediate 34b through an attack from the O-nucleophilic atom at the β-carbon of the gold-activated alkyne moiety of the alkynyl carbonyl derivative 34a. Intermediate 34b can react according to two alternative paths. The first one is the cleavage of the bond O–LG forming an α,α′-dioxo gold carbene 34c. The alternative path is that of a synchronous nucleophilic attack at the α-carbon of the alkyne with O–LG bond cleavage, forming the 1,3-diketo compound 34d. A very interesting point of the reaction is that the oxidation state of the catalyst favors one path or the other. That is, the first path is favored by electron-rich gold(I) catalysts, while the second path is feasible with gold(III) catalysts.
Application of the above mechanism is the intramolecular C-H functionalization of aryl groups. Thus, the N-alkynoyl anilines 35a are transformed to 3-acyl 2-oxyindoles 35d under the action of 2-bromopyridine N-oxide (Scheme 35) [69]. The suggested path consists of an oxidation step to an α,α′-dioxo gold carbene 35b, followed by an electrophilic aromatic substitution to the adjacent aromatic ring of aniline resulting in 35c. Finally, protodeauration and aromatization of the condensed rings lead to the 3-acyl 2-oxyindole 35d [69]. The method is tolerant to a variety of substituted anilines and several alkynamide moieties.
Functionalization with N-Based Nucleophiles
Many synthetic methods have been developed using a variety of N-based nucleophiles for the functionalization of the alkynyl carbonyl derivatives. These methods can be applied for synthesizing compounds with industrial interest or natural products.
An example of the Au(I)-intermolecular functionalization of alkynyl esters or alkynones 36b by N-based nucleophiles, generated by an aldehyde 36a and an aliphatic amine (e.g., methylamine in Scheme 36), is the synthesis of 1,4-dihydropyridines 36f. The proposed path includes a 1,4-addition of methylamine to an alkynyl ester (or alkynone) 36b forming an enamine 36c. Enamine 36c can react with one more molecule of alkynyl ester (or alkynone) 36b with the aid of a Ph3PAuCl/AgOTf catalyst to afford dienamine 36d. Compound 36d can react with aldehyde 36a to afford 36e, which, after redistribution of π-electrons, can furnish dihydropyridine 36f (Scheme 36).
The pros of the method are the plurality of the substrates that can be used. Aldehyde 36a can bear an alkyl, alkenyl or aryl group. Additionally, the reaction is feasible either with alkynones or alkynyl esters.
Anilines provide another example of an N-based nucleophile that can functionalize both alkynones or alkynyl esters or alkynyl amides in Au(I)-catalyzed reactions [68]. The method allows the use of a large variety of anilines including α-keto anilines 37a (see Scheme 37) that provide a carbonyl group instead of the aldehyde molecule in Scheme 36. In the case of a Ph3PAuCl/AgOTf-catalyzed reaction of α-keto anilines 37a with alkynones 37b, the 3-acyl-kinolines 37c are produced in good yield [68].
2.3.3. Allenyl Carbonyl Derivatives
General Reactivity Profile
An example of the reactivity profile of allenyl carbonyl derivatives is shown in Scheme 38. Thus, allenyl carbonyl derivatives are very useful for the formation of furans 38d and butenolides 38e [55]. The gold-catalyzed cycloisomerization of allenones 38a (R1 = alkyl group) affords 38b, which, after deauration, produces furans 38d, while the gold-catalyzed cycloisomerization of allenoates 38a (R1 = alkoxy group) forms butenolides 38e after fragmentation and deauration (Scheme 38) [55].
Functionalization with O-Based Nucleophiles
The gold(III)-catalyzed furan synthesis from allenones 39a starts with the activation of the allenyl group by AuCl3 (Scheme 39). The carbonyl oxygen of allenone 39a acts as a nucleophile undergoing a 5-endo-trig cyclization to form an oxonium intermediate 39b. An aromatization step and then a protodeauration step furnished 2-alkyl furans 39c. However, the main disadvantage of the method is that the reaction can step further with a 1,4-addition step from furan 39c as a nucleophile resulting in an α,β-unsaturated ketone 39d [71]. The mechanism of the reaction was studied with the use of DFT calculations not only for a variety of allyl-ketones [72,73] but also using a gold(I) catalyst [74].
More sophisticated methods have been developed that avoid by-products. A method for synthesis of the multi-functionalized 3-alkynylfurans 40e from allenones 40a and the hypervalent iodine (1-[(triisopropylsilyl) ethynyl]-1,2-benziodoxol-3(1H)-one) (TIPS-EBX) reagent 40b acting as an oxidizing reagent is based on a possible Au(I)/Au(III) redox cycle (Scheme 40) [75,76].
According to DFT calculations [76], at the first step of the proposed mechanism, allenone 40a undergoes a nAu(I)-cyclization using AuCl and 2-pyridine carboxylic acid [76] forming the intermediate 40c. Accordingly, gold(I) is oxidized to a gold(III) state by TIPS-EBX forming the intermediate 40d, in which an alkynyl-TIPS moiety is connected to gold(III). Finally, a reductive elimination step transfers the alkynyl-TIPS moiety on the furan ring forming the 3-alkynylfuran 40e (Scheme 40) [76]. The synthesis of γ-butyrolactones derivatives can be carried out by the Au(I)-catalyzed cyclization of allenoates 41a, which proceeds to the intermediate 41b and then to the gold(I)-butenolides 41c (Scheme 41).
For the transformation shown in Scheme 41 various transition metals/electrophilic reagents can be used, e.g., the mixture of gold(I) phosphine complex with AgOTf in stoichiometric amounts. The R3PAuCl/AgOTf catalyst renders the reaction in Scheme 41 feasible at room temperature. DFT calculations supported the proposed mechanism [78].
Gold(I)-butenolides 42a are valuable substrates because the C–AuPPh3 bond in the gold(I) complex can be substituted with various groups leading to many different products (Scheme 42). Not only iodination and protodeauration can be applied but also dimerization [79,80] via a redox process using SelectFluor (1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate)) a reagent that is used as a fluorine donor.
Moreover, transmetallation of gold (I)-butenolides 43a can be carried out either with palladium(II) complexes, e.g., dppfPdCl2 (dppf=1,1′-bis(diphenyl-phosphino)ferrocene). The palladium(II) complex intermediates, e.g., 43b, affected cross-coupling reactions introducing a variety of aryl, alkenyl, allyl and benzyl groups to the position of C-Au(I), either via an intramolecular or an intermolecular reaction leading to 43c (Scheme 43).
Functionalization with Other Nucleophiles
Compared to the typical cyclization of allenyl butenoates 41a to lactone derivatives, e.g., 42b–e and 43c, allenoate esters with a second functional group that can be nucleophilic, e.g., 44a, can react with an alternative gold(I)-catalyzed intramolecular nucleophilic addition (Scheme 44).
Nucleophilic addition can proceed to the α- or β-position of the allene moiety depending on the nature of the nucleophilic group and the size of the formed ring. As shown in Scheme 44, the reaction can furnish dihydrofurans 44c, pyrroles 44d or tetrahydropyridines 44e using Ph3PAuCl/AgOTf, Ph3PAuNTf2 or Cy3PAuNTf2, respectively. To facilitate the intramolecular nucleophilic attack in the allenoate ester molecule 44a, methyl or ethyl esters are commonly used [84,85,86,87,88].
ΕΒΧ hypervalent iodine alkyne transfer reagents are a category of alkynes with unique features due to the highly polarized carbon-halogen bond which implies stronger regioselectivity compared with other alkyne substrates in the gold(I)-catalyzed reactions of haloalkynes [89,90]. Additionally, redox reactions and [1,2]–X rearrangements are very rare on all other types of alkynes. These haloalkynes give inter- and intramolecular functionalization with C, N- and O-based nucleophiles, as has been thoroughly studied. Special emphasis must be given to the case of alkynyl iodoniums that operate as excellent alkynyl transfer agents. Scheme 45 shows the alkynyl iodonium complex reagent TIPS-EBX and its relevance to the functionalization via gold(I)-catalyzed C-H reactions [91,92,93,94,95,96,97,98].
3. Gold(I)– and Gold(III)–π-System Complexes in Catalysis
3.1. Gold–Carbon Bond in Gold(I)– and Gold(III)–Carbene Complexes
Carbene transfer reactions are often used with gold catalysis [28,32,34,39,99,100,101]. Regarding this, we provided a comparison of the stability and the traits of carbenes formed by gold(I) and gold(III). The bonding in these AuL+ species resulted from (a) the lone pair of the carbon ligand forming a strong σ bond with an orbital of appropriate symmetry of gold to form a carbocation-like structure 46b and (b) the π-backbonding of the metal d-orbital from gold to an empty p-orbital of carbon to form a singlet carbene like structure 46a (Scheme 46).
Thus, the stabilization of the carbene-like structure 46a is related to the π-backdonation from gold(I) [5,27,37,102]. Consequently, in a gold carbene intermediate, the degree of σ- and π-bonding determines the gold-coordinated carbocation and gold-stabilized singlet carbene character depending on the substituents in the carbene (Scheme 46). Thus, the ancillary ligand (L) has a critical role in the stability of the carbene regulating the reactivity profile of the catalyst via the control of the π-backbonding. Strongly σ-donating and weakly π-acidic ligands, such as N-heterocyclic carbene (NHC) ligands [100,103] or cyclic(alkyl)(amino)carbenes (CAAC) [104], with an enhanced σ-donor character of gold-carbon bond, are expected to improve the carbene-like reactivity. The bulky N-heterocyclic carbene (NHC) ancillary ligand 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr) [105,106] is the most important NHC ligand in the field of homogeneous catalysis. The replacement of one of the electronegative amino substituents of NHCs by a strong σ-donor alkyl group results in CAAC ligands that are even more electron-rich [104]. In contrast, phosphines that are weak σ-donating and π-acidic ligands enhance the carbocation-like character of the intermediates [5].
Experiments were performed to investigate the change of the HOMO character in the carbene that directs an anti-bonding σ interaction of the fully occupied sp2 orbitals in the singlet carbene with an empty gold(I) 5d orbitals to the LUMO character in the carbene that directs the interaction of the empty p orbital of the carbene with a doubly occupied gold(I) 5d orbital. Such experimental evidence on the nature of the gold(I)-carbon bond, i.e., a strong Au–C σ|bond vs. a significant Au=C backbonding, was provided by Straub [107], in which the gold(I) carbene complex 47b, as shown in Scheme 47, was synthesized. The strong Au–C σ|bond and the significant Au=C backbonding are consistent with a major impact of relativistic effects on gold’s valence shell, that is, higher energy for gold’s 5d orbitals and lower energy for gold’s 6s orbitals. This gold(I)–carbene complex 47b, with the IPr as NHC ligand, was the first metal complex without heteroatom donor substituents linked to the metal and with high carbenoid (Au=C) character in contrast to other complexes with a predominant ammonium ylide or oxonium ylide ligand character. According to DFT calculations on the gold(III)–carbene complex in Scheme 47, the Au–C bond has a single bond character and the carbene carbon has a nucleophilic character. That is, it can react with electrophiles such as CS2 and PhNCS [108,109].
3.2. Gold(I)– and Gold(III)–π-Alkene and –π-Alkyne Complexes
Gold(I) or gold(III) can activate π-bonds. The ability of gold(I) to activate alkenes was demonstrated with the isolation of gold(I)–alkene complexes. The IPrAu(I) pre-catalyst forms complexes with alkenes such as norbornadiene and styrene, which have been isolated and characterized [110,111]. Afterwards, many alkyl phosphine–gold(I)–π-alkene complexes were synthesized and composed by various alkenes (Scheme 49) [110,111,112,113] shortly after the first gold(III)–π-alkene complexes were synthesized (Scheme 49) [114,115,116]. However, contrary to gold(I) complexes, the gold(III)–π-alkene complexes were not as stable and abundant. For some of them, the efforts to be crystallized were successful. For example, for the Tilset complex [114] (Scheme 49), both X-rays and calculations revealed a weak metal d→π*(C=C) backbonding but were significant enough to stabilize the Au(III) bis(alkene) complex. More recently, the isolation and characterization of the Bourissou complex [116] (Scheme 49) confirmed previous mechanistic proposals for its existence in various works where the complex was formed [5,114,115,116,117].
Similarly, to alkenes, alkynes also form with gold(I) π-complexes. Not only stable NHC–gold(I) complexes with alkynes but also phosphine-type ligands with gold(I) have been formed. Some of the stable π-alkyne–gold(I) complexes’ structures are depicted in Scheme 50 [118,119,120,121].
Comparison of the activation of alkynes by gold(I) and gold(III) led to the conclusion that π-backdonation is very weak in gold(III) complexes. Since the stability of the gold(III)–π-alkyne complex depends on the π-donation from the triple bond, due to the reduced backbonding capacity of gold(III), one of the alkyne-C atoms is charged positively so that it is rendered more susceptible to a nucleophilic attack. The resulting polarization of the C≡C bond is much larger for gold(III)– than for gold(I)–alkyne complexes [122,123]. Thus, the attempts for the isolation of similar to gold(III)–π-alkyne complexes (Scheme 50) were unsuccessful since gold(III) can’t form a stable complex and is reduced to gold(I) or gold(0) spontaneously [124]. The synthesis of gold(III) alkyne complexes 51b succeeded with the use of C^N and C^C donor ligands as depicted in Scheme 51 [122,125].
3.3. Divergent Catalysis for Gold(I) and Gold (III) Complexes in Reactions with π-Systems
There are many uncertainties regarding the structure and the oxidation state of actual catalytic species in the catalysis of reactions with π-systems using gold(III) [2,124]. For example, experimental data have shown the reduction of gold(III) to gold(I) during the catalytic cycle due to the instability of gold(III) complexes as mentioned before [124].
However, gold(I) compared to gold(III) remains more suspicious for catalytic activity species [124]. In the case of gold(I), this is due to other species that are present in the reaction mixture with the catalyst, such as counterions, which can interfere with the reaction mechanism [126], or cofactors that can provide in situ the gold(I) catalyst [127]. Additionally, gold(I) shows a tendency to form multi-metallic aggregates, even with other gold(I) ions, a phenomenon known as aurophilicity [2].
Many experimental data on catalyzed reactions by both oxidation states of gold exist, where in many cases the same reactant can lead to different products depending on the gold oxidation state. The divergent catalysis between gold(I) and gold(III) and the mechanistic explanation is a topic of interest.
Cycloisomerization of indole-tethered alkynes 52a is an example of a gold-catalyzed regiodivergent reaction [128]. The transformation proceeds at the first step with the triple bond activation either by a gold(I) or a gold(III) catalyst. However, gold(I) JohnPhosAu(MeCN)SbF6 catalyst favors the 6-exo-dig cyclization, while gold(III) AuCl3 catalyst favors the 7-endo-dig cyclization via a nucleophilic attack by indole C3 in both cases (Scheme 52). The spiranic intermediates 52b/52d undergo a 1,2-alkyl shift followed by a re-aromatization and protodeauration step, forming the azepino[4,5-b]indole 52c derivatives and the eight-member ring derivative 52e, respectively [128] (Scheme 52).
Cycloisomerization of allenic hydroxylamines 53a is another divergent catalytic reaction, in which the choice of catalyst gold(I) or gold(III) urges the reaction to the formation of a five-member or six-member ring, respectively [129]. Phosphine ligand-gold(I) catalysts promote the 5-endo-dig cyclization to form dihydroisoxazoles 53b, while the 6-endo-trig cyclization is favored by using the AuCl3 catalyst to furnish 3,6-dihydro-1,2-oxazine 53c as the major product (Scheme 53) [129]. We investigated [130] these alternative pathways of divergent catalysis using DFT calculations.
Gold-catalyzed cycloisomerization of 1,6-diyne esters 54a can lead either to 1H-cyclopenta[b]naphthalene 54b after treatment with IPrAu(PhCN)SbF6 catalyst or to cyclopentenyl diketone 54c after treatment with PicAuCl2 catalyst [131], possibly due to the different Lewis acidity between gold(I) and gold(III) catalysts (Scheme 54).
According to the proposed mechanism, and supported by DFT calculations [132], the gold-activated alkyne 54a undergoes a 1,3-acyloxy shift to 54d, which furnishes the intermediate 54e after a 5-exo-dig cyclization. From that point, divergent catalysis begins. Gold(I) catalyst urges the intermediate to an intramolecular Friedel–Crafts cyclization [132] to furnish 1H-cyclopenta[b]naphthalene 54b [131,132], while the gold(III) catalyst induces a 1,5-acyloxy shift [132], which produced the cis-cyclopenten-2-yl δ-diketone 54c without by-products [131,132]. In Scheme 55, both gold(I) and gold(III) catalysts bear NHC ligands and the same counterions. Starting from ketene acetal 55a and cinnamaldehyde 55b, the NHC gold(I) catalyst IPrAuCl 4d, which is activated by AgOTf, promotes the formation of Mukaiyama aldol product 55c by a 1,2-addition, while gold(III) complex 55e leads to the products of 1,4-addition 55d, in a Mukaiyama–Michael reaction [133].
4. Divergent Gold(I) Catalysis
4.1. Differences between the Electronic Structure of Gold and Other Metals
There has been a spectacular rise in the application of soluble gold(I) catalysts in synthesis. Their development has been the subject of numerous reviews [18,19,20,21,22,23,24,25,26,27,134]. While gold(I) prefers reacting with alkynes and alkenes, the formation of an Au(I)-alkyne complex is favored over an Au(I)-alkene complex, and, consequently, a variety of alkyne-catalyzed versus alkene-catalyzed reactions exists. That trait of Au(I) is referred to in the bibliography as “alkynophylicity”. A possible explanation of the “alkynophylicity” of Au(I) is that due to the lower energy of LUMO of Au(I)-alkyne complex compared to the energy of LUMO of Au(I)-alkene the addition of a nucleophile to the activated complex is easier in the first case [7].
Of course, in the field of gold(I) catalysis, the type of ligand is a factor that affects the selectivity of the catalyst not only due to the drift of Lewis acidity of the catalyst but also to its geometry. In the next chapter, we will examine the impact of ligands on gold(I) catalysis.
The reactivity of metal complexes is directed by the trend in metal–ligand bond energies. For gold, depending on the nature of the O-ligand, the sequence is Au-H > Au-O > Au-C or Au-H > Au-C > Au-O, whereas for other metals, including its neighbor in the periodic table, platinum(II), the trend Pt-O > Pt-H > Pt-C is observed [135]. Oxygen and fluoride ligands tend to act as good leaving groups and are utilized with good effect in ligand substitution and catalytic reactions. The bond dissociation energies of gold(I) compounds tend to be larger than those of gold(III) but follow the same trend.
Due to the relativistic effect that is intense in gold(I), unique catalytic properties are present in comparison with other transition metal catalysis. Thus, reactions of divergent gold(I) catalysis [18], using the same starting material but a different gold(I) catalyst, can afford different products, since each catalyst can activate selectively different functional groups leading the system to discrete pathways. However, the prediction of divergent catalysis, due to different metal catalysts, is still not feasible and remains empirical [136,137,138].
4.2. Gold(I) versus Other Metal Catalysts
4.2.1. Au versus Pt
An example of divergent catalysis between gold and platinum is the reaction between isoxazoles 56a and ynamides 56b. This reaction leads either to 2-aminopyrroles 56g using the gold(I) catalyst ((2,4-tBu2C6H3O)3PAuNTf2)) [139] or to 1,3-oxazepines 56i′ with platinum(II) catalyst PtCl2 [140]. With both catalysts, after the activation of ynamide 56b by the catalyst, follows the nucleophilic attack of isoxazole 56a and N–O bond cleavage, leading to the α-imino metal carbene intermediate 56d or 56d′, respectively (Scheme 56). Then, divergent steps are observed through a nucleophilic attack at the metal-coordinated carbocation of 56d or 56d′ favoring a 1,5-cyclization forming 2-aminopyrrole 56g for the gold catalyst or 1,7-cyclization forming 1,3-oxazepine 56i′ (Scheme 56). We have confirmed these mechanistic steps for the Au(I)-catalyzed reaction between 1,2,4-oxadiazoles and ynamides [44].
It was proposed that the observed regioselectivity of the reaction is controlled by steric factors during the nucleophilic attack at the metal-coordinated carbocation of intermediate 56d/56d′. The linear structure of the gold(I) catalyst renders the intermediate 56d less sterically hindered and the nucleophilic attack by the carbon–carbon double bond is favored. On the contrary Pt(II)-coordinated carbocation in intermediate 56d′ is more sterically hindered due to the bigger size of platinum making only the nucleophilic attack from the less sterically exposed oxygen feasible. Although the platinum(II) catalyst is harder for Lewis acid than gold(I), the steric factor plays a dominant role here.
4.2.2. Au versus Ag
The cycloisomerization reaction of indolyl ynones 57a catalyzed by a gold(I) or silver(I) catalyst is another example [141]. Gold (I) catalysis (Ph3PAuNTf2) furnishes the carbazole 57e, while silver catalysis (AgOTf) furnishes the spirocyclic product 57c′ (Scheme 57).
According to the proposed mechanism, both catalysts, Ph3PAuNTf2 or AgOTf, activate the alkyne group urging the system to a 5-endo-dig cyclization to form a spirocyclic intermediate 57b or 57b′. In the case of silver(Ι) catalysis, the path ends with the desilveration and formation of the spyrocyclic product 57c′. However, in the case of gold(I) catalysis, the activation of the alkyne follows a cascade of ring expansion, aromatization and deauration that leads to the formation of carbazole 57e (Scheme 57).
4.2.3. Au versus Pd
The Au(I) (MeDalPhosAuCl)-catalyzed reaction of N-phenylpyrrole 58a with iodobenzene 58b led, selectively, to the C3-substituted pyrrole 58c [142]. The Pd(II) (PdCl2)-catalyzed version of the same reaction [143] furnished, selectively, the C2-substituted pyrrole 58d (Scheme 58).
Noteworthy is the role of the MeDalPhos ligand in a gold complex that enables the gold catalyst to oscillate during the catalytic cycle between oxidative states I (59a) and III (59c) [142] (Scheme 59), adding to the reaction traits of high reactivity and selectivity using variant aryl halides 59b as substrates [144,145,146].
4.2.4. Au versus Rh
The alkynylation of isoquinolones 60a catalyzed by gold(I) or rhodium(II) [147] leads to C4 or C8 C-H alkyne insertion, respectively (Scheme 60) [147]. In the reaction between isoquinolone 60a and 1-[(triisopropylsilyl)ethynyl]-1,2-benziodoxol-3(1H)-one (TIPS-EBX) 60b, the latter undergoes a selectively carbophilic activation of the alkyne group when gold(I) catalyst is present. Then, a C-H insertion at C4 is favored, and the α,β-elimination of gold is the last step in furnishing product 60e.
On the contrary, the rhodium(II) catalyst promotes C-H activation of the C8 position of isoquinolones 60a. The activated complex undergoes the insertion of TIPS-EBX to form the intermediate 60d. Product 60e′ is obtained after the reductive elimination of rhodium. DFT calculations studies on the mechanism and regioselectivity of the reaction, as well as on the role of TIPS-EBX, have been carried out [148].
4.2.5. Au versus Sc
The reaction between 1-(1-alkynyl)-cyclo-propyl-ketones 61a and nitrones 61b is either [4+3] cycloaddition furnishing products 61e or [3+3] cycloaddition furnishing products 61e′, depending on whether the applied catalyst is Sc(III) or Au(I), respectively (Scheme 61) [149].
The Sc(OTf)3/Phen catalyst activates the carbonyl group of ketone 61a chemoselectively, facilitating the nucleophilic attack of nitrone 61b onto a cyclopropyl ring. Then, the intermediate 61d′ undergoes a formal [3+3] cycloaddition to afford a tetrahydro-1,2-oxazine derivative 61e′.
On the contrary, the Ph3PAuOTf catalyst activates the alkyne group in ketone 61a selectively, altering the mechanism of the reaction to an f 5-endo-dig nucleophilic attack of carbonyl oxygen on the activated alkyne group. This follows a nucleophilic attack of nitrone 61b on the cyclopropyl ring, and, finally, the formal [4+3] cycloaddition affords a 5,7-fused bicyclic furo[3,4-d][1,2]-oxazepine 61e (Scheme 61).
It is noteworthy that according to mechanistic studies, both pathways, either [3+3] or [3+4] cycloaddition steps, proceed via a chair-like transition state, see structures 61d or 61d′, respectively (Scheme 61) [149]. Studies of the mechanism in the gold(I)-catalyzed reaction using experimental kinetics shed light on the stereoselectivity [150], while DFT calculations supported a mechanism via the formation of an oxonium ion [151].
4.2.6. Au versus Cu
The reaction of O-propargylic oximes 62b with maleimides 62a in the presence of gold(I) (Ph3PAuNTf2) [152] and copper(II) [CuCl(cod)]2 [153] catalysts is shown in Scheme 62.
The reaction begins with the activation of the alkyne group of O-propargylic oxime 62b, which follows the nucleophilic attack of nitrogen to the activated alkyne group to produce via a 5-endo-dig cyclization the common intermediate 62c/62c′ similarly by both catalysts. The intermediate 62c/62c′ follows a different path regarding the applied catalyst. Copper(II) catalyst lowers the activation energy for the cleavage of the C–O bond to form N-allenyl-nitrone 62d′. N-allenyl-nitrone 62d′ reacts with maleimide 62a through a [3+2] cycloaddition forming N-allenylisoxazolidone 62e′, which undergoes a 1,3-oxygen shift to the oxazepine derivative 62f′ [152].
On the contrary, the gold(I) catalyst maintains the energy barrier for the bond C–O cleavage high. The reaction prefers an intermolecular methylene transfer to form diene 62d, which reacts with maleimide 62a through to form isoxazole 62e (Scheme 62).
4.3. The Effect of the Ligand in Gold(I) Catalyst Complex in Divergent Catalytic Paths
Gold(I)-catalyzed reactions present sensitivity to various factors that can affect the formation of the product. Factors such as counterions and ligands in the gold(I) complex can affect the reaction mechanism and, consequently, the type and yield of the products.
A reaction that showed the key role of the ligand in the gold(I) catalyst complex is the hydration-oxacyclization of skipped diynones 63a (Scheme 63) [154]. Depending on the reaction conditions and the ligand in the gold(I) complex, a five- or six-member heterocyclic ring can be formed. These two divergent pathways of the reaction are depicted in Scheme 63. According to the first pathway, the Ph3PAuCl/AgSbF6 catalyst in dioxane/water at 100 °C leads to the formation of 3(2H)-furanones 63b as a major product. Ceteris paribus, changing the ligand of the gold(I) catalyst from Ph3PAuCl/AgSbF6 to IPrAuNTf2 urges the reaction to the formation of the 4-pyrones 63c as a major product. It is proposed that the observed regioselectivity resulted from a combination of both ligand and counter-anion types.
The suggested mechanism consists of two steps [154]: the first step is the hydration of one of two alkyne groups in diynone 63a, and the second step is the intramolecular endo-oxacyclization followed by protodeauration. The regioselectivity of the reaction is controlled by the hydration step of diyne in diynone 63a, see Scheme 63. Thus, using the IPrAuNTf2 catalyst after a Michael addition of water at the end carbon of the activated alkyne group due to the steric crowding induced by the IPr group, a 6-endo-dig cyclization leads to the formation of 4-pyrones 63c. On the contrary, using the Ph3PAuCl/AgSbF6 catalyst, a Michael addition of water at the other carbon of the activated alkyne group, which is in α-position to carbonyl, urges the transformation to the 3(2H)-furanone 63b.
Ligands in gold(I) complexes may regulate the ability of the catalyst to activate π-systems. An example that reveals the key role of the electron density of the ligand in a gold(I) complex is provided by the cyclization reaction of N-(3-iodoprop-2-ynyl)-N-tosylanilines 64a (Scheme 64) [155].
Applying (2,4-tBu2C6H3O)3PAuCl in the cyclization reaction of N-(3-iodoprop-2-ynyl)-N-tosylanilines 64a affords the expected product of hydroarylation, i.e., the 4-iodide quinoline 64b. Substitution of (2,4-tBu2C6H3O)3P with the electron-rich substituent IPr changes the reaction path, leading to the formation of 3-iodide quinoline 64c. That is, an isomerization step of 1,2-iodide shift interferes before the cyclization step. Indeed, the IPrAuNTf2-catalyzed reaction proceeds via the formation of a gold(I)-vinylidine intermediate 64d. Moreover, the role of the R substituent in the N-phenyl group affects the electron density of the ligand. Electron-rich R substituents favor the formation of 4-iodide-derivatives 64b, whereas electron-withdrawing R substituents favor the path through the 1,2-iodide shift [155].
The cycloisomerization of alkynones 65a (Scheme 65) is a case of a chemoselective Au(I)-catalyzed reaction, in which the difference in the electronic properties of the ligand in the gold(I) catalyst complex affects the electrophilicity of gold(I), resulting in divergent paths [156].
In general, electron-rich ligands, such as IPr, favor the transformation of alkynones 65a to benzofurans 65c, and electron-deficient ligands, such as Ph3P, favor the transformation of alkynones to 6,7-dihydrobenzofunan-4(5H)-ones 65b. Thus, the IPrAu+ catalyst results in benzofurans 65c formation, whereas the use of the Ph3PAu+ catalyst leads to the formation of 6,7-dihydrobenzofunan-4(5H)-ones 65b.
For the first step of the divergent reaction, it is proposed that the IPr ligand as an electron-rich group suppresses the electrophilicity of the gold center and transforms it into a soft Lewis acid. Thus, only the alkyne moiety is activated by the gold(I) catalyst and becomes susceptible to the nucleophilic attack by the adjacent carbonyl oxygen. After deprotonation and protodeauration, the intermediate 65j is obtained. A methanol molecule contributes to the epoxide ring opening, forming the intermediate 65k that undergoes aromatization in the presence of HCl to benzofuran 65c. On the other hand, the Ph3P group is not as electron-rich a ligand as IPr, and the Ph3PAu+ catalyst is a harder Lewis acid than IPrAu+. Consequently, the gold(I) catalyst activates both alkyne and epoxy moiety. The nucleophilic attack of methanol to both carbons of the epoxide ring leads to two isomers 65e/65f, which, after a 5-endo-dig cyclization reaction, undergo elimination of water or methanol and protodeauration to furnish the 6,7-dihydrobenzofunan-4(5H)-ones 65b.
Another case of regulation of the activity of gold(I) catalyst according to the electron density of the ligand is the divergent cycloisomerization of ortho-(propargyloxy)aryl methylenecyclopropanes 66a [157]. According to Scheme 66, ortho-(propargyloxy)aryl methylenecyclopropanes 66a can be transformed to methylenecyclopropane 2H-chromene 66c derivatives using the catalyst (4-CF3C6H4)3PAuSbF6 or to cyclobutene-substituted 2H-chromenes 66b using the catalyst IPrAuSbF6.
Although at first glance it seems to be a dual path reaction, in fact, it is a one-way reaction with an additional step of expansion of the cyclopropane ring to cyclobutene. First, the gold(I) catalyst activates the alkyne group that accepts the intramolecular nucleophilic attack of the tethered aryl group. Methylenecyclopropane-2H-chromene 66c is formed after protodeauration of the intermediate 66e. After the formation of the 2H-chromene 66c derivative, the regulatory role of the ligand of the gold(I) complex begins. If the ligand is electron-deficient, such as (4-CF3C6H4)3P, the reaction stops at this step. However, in the case that the ligand is electron-rich, such as IPr, the electron-rich complex IPrAuSbF6 effectively activates the double bond of methylenecyclopropane 66c. Due to the activation of the double bond, a carbocation 66f is formed that leads to tandem alkyl swift and 1,2-H swift furnishing and, finally, a cyclobutene-substituted 2H-chromene 66b [157]. The suggested reaction mechanism was confirmed experimentally. The methylenecyclopropane-2H-chromene 66c transformed to cyclobutene-substituted 2H-chromene 66b under the action of the IPrAuSbF6 catalyst [157] (Scheme 66). The activation of alkyne moiety via π-complex formation is usually observed. However, an alternative way of dual σ,π-activation has been reported and is supported by experimental data derived from mechanistic studies by NMR [157]. The gold(I)-catalyzed heterocyclization of 1-(orthoethynylaryl) urea 67a is a case in which the ligand the in gold(I) complex also determined the activation mode of the alkynyl moiety (Scheme 67) [158]. The use of the IPrAu+ catalyst leads to the transformation of 1-(orthoethynylaryl) urea 67a to quinazolin-2-one 67b. Steric crowding is the dominant factor for which the IPr ligand induces a π-mode activation of the alkyne group. Thus, steric hindrance between the bulky ligand of the gold(I) complex and substrate activates the alkyne group unsymmetrically, rendering benzylic carbon more electrophilic and consequently more susceptible to the nucleophilic attack of N-3 via a 6-exo-dig cyclization furnishing quinazolin-2-one 67b [158], see Scheme 67. The alternative path of this reaction is the transformation of 1-(orthoethynylaryl)urea 67a to N-substituted indoles 67c [158]. The reaction proceeds via a dual σ,π-activation of a triple bond. Due to the less sizeable ligand (tBu)3P, the activation of alkyne moiety is symmetrical, and the acidic proton is substituted by a molecule of the catalyst, see Scheme 67. Finally, the dual-activated intermediate furnishes via 5-endo-dig cyclization indole 67c.
The regulating role of the ligand is presented in the [5+2] and [5+1] annulation reactions between 1,2-benzoisoxazoles 68b and ynamides 68a (Scheme 68).
The [5+2] annulation reaction is favored when the IPrAuCl/AgNTf2 catalyst is used. On the contrary, JohnPhosAuCl/AgNTf2 turns the balance to the [5+1] annulation. Thus, the reaction between 1,2-benzoisoxazoles 68b and ynamides 68a is rendered chemodivergent due to the ligands of the gold(I) [159]. The reaction initiates with a nucleophilic attack of the N of 1,2-benzoisoxazole onto the activated alkyne moiety of ynamide 68a. Intermediate 68e may follow the path to 68c or 68d, depending on the applied catalyst. The IPr ligand in the ligand IPrAuCl is more electron-rich than the phoshine ligand in the complex JohnPhosAuCl. The former catalyst favors the path to 68c due to its better ability to stabilize the gold(I)-carbene intermediate 68f. The alternative path of the reaction starts from the less sterically hindered conformation 68e′ and includes a 1,2-alkene swift and N–O bond cleavage, followed by a 1,2-sulfonamide shift, and after deauration, the product 68d is formed [159].
The gold(I)-catalyzed divergent cycloaddition products of allene-dienes 69a also reveals that the electronic properties of the gold(I) complex catalyst are regulated by its ligand, according to findings from the DFT calculations. Thus, [4+3] cycloaddition is the result of the action of JohnPhosAuCl/AgSbF6 catalyst on allene-dienes 69a. Allene’s activation by gold(I) promotes the formation of cycloheptene gold(I)-carbene intermediate 69e, following a 1,2-hydrid shift that furnishes cycloheptadiene 69c (Scheme 69).
Changing the electron properties of ligands in the gold(I) complex of the catalyst using (2,4-tBu2C6H3O)3PAuCl/AgSbF6 renders the Au(I) complex electron-deficient, and [4+2] cycloaddition is favored [161]. Despite the initial assumption that, due to the lack of the backbonding ability of the gold(I) center, the formation of carbocation 69d is favored directly from the activated allene-diene 69a, the DFT calculations did not suggest the formation of 69d. According to the DFT calculations, the potential energy surface of the reaction to 69c is featured by low energy barriers for both of the catalysts. Then, a question arises of why different catalysts alter the products of the reaction. The solution of the riddle is the ability of the electron-deficient catalyst to convert intermediate 69e to intermediate 69d via a 1,2-alkyl shift (ring contraction) while JohnPhosAuCl favors the 1,2-hydrid shift to form 69c [160].
One year later, the regulating role of ligands was studied in a similar work for the gold(I)-catalyzed cycloisomerization reaction of allene-ene 70a [160] (Scheme 70). A formal [3+2] cycloisomerization is dominant when the L2 ligand is present. On the contrary, a formal [2+2] cycloisomerization is favored by the L1 ligand. After the first common step of cyclization, the intermediate 70e is formed. Then, the two divergent cycloisomerization paths are observed. The first one is the 5-endo-trig cyclization to 70g intermediate, which is converted to 70h due to 1,2-Me-shift and, finally, to 70c/70d after deauration. Alternatively, cyclization of 68e to 68f intermediate and deauration leads to 70b.
The key role of the ligands L1 and L2 in the mechanism of the reaction is possibly related to the π-acceptor ability of NHC (N-heterocyclic carbene) [162]. Although both ligands (L1 and L2) are featured by a similar σ-donor ability, L1 has a higher π-acceptor ability due to its lower LUMO energy. Consequently, Au(I)–L2 complex is electron-rich compared with the electron-deficient Au(I)–L1 system. Thus, L2 favors [3+2] cycloaddition and L1 [2+2], see Scheme 70 [162].
A divergent reaction, in which not only the ligands of the gold(I) catalyst, but also a combination of factors such as solvent, temperature and counterion affect the balance of products, is presented in Scheme 71.
The 3-propargylindoles 71a can be converted according to Nazarov or iso-Nazarov cyclization [163,164]. A 3-propargylindole 71a, after the first common step of 1,2-indole shift to form 71d, is converted into the gold(I)-carbene intermediate 71e. The transformation can be carried out either using Ph3PAuNTf2 in dichloromethane at rt or with (2,4-tBu2C6H3O)3PAuCl/AgOTf in toluene at 0 °C. The second step of the transformation is differentiated according to factors of the reaction, such as the nature of ligand in gold(I) complex, solvent, counterion and temperature. Using Ph3PAuNTf2/DCM/rt turns the balance to the iso-Nazarov reaction and 3-(inden-2-yl)indoles 71c formation. In contrast, the combination (2,4-tBu2C6H3O)3PAuCl/AgOTf/toluene/0 °C favors a Nazarov cyclization that furnishes 3-(inden-2-yl)indoles 71b [163,164]. The mechanistic aspects of this reaction were investigated experimentally and with DFT calculations [164,165,166].
4.4. The Role of the Solvent and Counterions in Gold(I)-Catalyzed Reactions
4.4.1. General Description
Gold(I) catalysts are commercially available in inactive forms that are afterwards activated in situ. The inactive forms are less stable but can be stabilized by the appropriate counterions. Nowadays, a large variety of counterions are available, for example, halogen anions (Cl−, Br−, I−), oxygen-based ions (OTs−, OMs−), nitrogen-based ions (NTf2−), carbon-based ions (CN−), boron-based ions (BF4−) and fluorinated ions (SbF6−, PF6−) [126].
During a catalytic cycle, the gold(I) complex dissociates to the gold(I) cation and counterion, while the gold(I) cation associates with the substrate. This dissociation equilibrium is described with the following equation:
LAu+X− + S ⇄ LAu+S− + X−
Consequently, the affinity of gold(I) for the counterion/substrate and the polarity of the solvent are of critical importance [167]. Solvents with a low dielectric constant (benzene, toluene, dichloromethane or dichloroethane) urge the system of catalyst/counterion to exist as a contact ion pair. Then, the counterion remains close to the reaction center and possibly participates in the mechanism of the reaction. On the other hand, polar solvents with a high dielectric constant (alcohols, nitromethane or acetonitrile) dissociate the system of catalyst/counterion, solvate the ions and keep the counterion away from the catalytic center. As a result, the contribution of the counterion to the mechanism or the reaction is minor [168].
It is almost clear that in the case of polar solvents, the role of counterion is out of interest. However, in the case of non-polar solvents used in a gold(I)-catalyzed reaction of a nucleophile with the triple bond of an alkyne, the counterion can affect the following four steps: (1) catalyst activation, (2) alkyne activation by gold(I), (3) nucleophilic attack to the activated triple bond and (4) protodeauration (Scheme 72).
During the catalytic cycle (Scheme 72a), the counterion is displaced from a region close to gold(I) (inner sphere ion pair ISIP) to a region far away from gold(I) (outer sphere ion pair OSIP). The sooner the counterion passes from ISIP to OSIP, the higher the catalyst activity is. The energy barrier between the two states is related to the affinity of the counterion with the gold(I) cation. Counterions, such as Cl−, that bind strongly with gold(I), reduce catalytic activity, impeding the formation of the gold(I)/alkyne complex. Thus, the counterion affinity is inversely proportional to the catalyst activity [169].
The model becomes more complicated if an active proton is involved. The kind and the strength of the interaction between a counterion and an active proton, e.g., from -OH, or -NH2 groups of the nucleophile, may change the reaction rate. If the counterion could act as an acceptor to a hydrogen bond, then the nucleophilicity of the attacking nucleophile increases. The interacting counterion with the active proton should orientate properly (Scheme 72b) at the OSIP to allow the nucleophilic attack.
At the last step of a catalytic cycle, i.e., the protodeauration, gold(I) must dissociate from the substrate to be regenerated; a proton must replace it. The counterion can become involved in breaking an X–H bond and transferring the proton. However, sometimes counterions play an undefined role in the stability of gold(I) catalysts. To predict the chemical traits of counterions, the dissociation energy of counterions from gold(I) catalysts have been calculated as gold(I) affinity index values (Table 1). Moreover, a model of a hydrogen bond formation of a counterion with phenol has been developed and also provides a hydrogen bond basicity index. Using these two indexes, it is possible to predict how the counterion could affect a gold(I)-catalyzed reaction.
For example, in the case of a reaction without active hydrogen (Scheme 73), a counterion with a low gold affinity index can be related to a high reactivity catalyst. That is, the low affinity of counterion leads to the easier formation of a gold(I)–substrate complex and, consequently, to faster kinetics of the Au(I)-catalyzed reaction [170].
For the investigation of the counterion effect, on the other hand, the presence of an active proton makes the mechanistic profile more complicated, as described in Scheme 74.
Then, high gold(I) affinity index values are accompanied by a low gold(I) catalytic activity; however, high gold(I) affinity index values correspond to an ability for a strong hydrogen bond of the counterion. However, the ability to have a strong hydrogen bond is necessary for counterions that participate in a reaction with active protons. The balance between these two contrasting features is not so clear. A good yield is received as a result of a good balance between basicity and the gold coordination ability of the counterion (Table 2).
The exact mechanism of the counterions’ participation in asymmetric synthesis is not well defined, and it was suggested that a chiral counterion, e.g., the (R)-3,3′-bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate ((R)-TRIP) 75 (Scheme 75) in the enantioselective functionalization of allenes (Scheme 76) can induce an asymmetric synthesis through a dinuclear gold(I) complex. Chiral counterions can help with the correct orientation of the substrate to induce enantioselectivity. Moreover, chiral ligands combined with chiral counterions may provide higher enantioselectivity results [172].
4.4.2. Examples of Divergent Gold(I)-Catalyzed Paths
Counterions can differentiate the mechanism of the Au(I)-catalyzed reaction. Thus, a divergent cycloisomerization of homopropargylic ketones due to the counterions SbF6− or OTf− has been reported (Scheme 77) [173]. It is noteworthy that the solvent also may play a crucial role in the mechanism of this reaction affecting the catalytic activity of the gold(I) complex.
In the case of TfO− counterion, the reaction starts with a first step of alkyne activation by the gold(I) catalyst, followed by a 5-endo-dig cyclization. A carbonyl oxygen nucleophilic attack to the activated triple bond forms the intermediate 77i. Next, the 1,2-hydride shift leads to product 77c. The 1,2-hydride shift consists of two steps: a deprotonation and a protodeauration step in which both are catalyzed by TfO− [173].
The second path of the reaction where the SbF6− counterion is present is dominated by a 1,2-Si shift. Similarly, the reaction starts with the activation of a triple bond by the gold(I) complex. However, an isomerization step to form propargyl-allenyl intermediate 77e is preceded by a cyclization step. Possibly, due to SbF6− counterion and water molecules, this isomerization can take place. Next, a cyclization step leads to the intermediate 77f and then to 77g. Finally, the 1,2-Si shift and deauration lead to the formation of the 3-silyl-substituted furan 77b. The 1,2-Si shift was suggested by DFT calculations as the more kinetically favorable step [173].
Another example of the critical role of counterions on the mechanism of the Au(I)-catalyzed reaction is the reaction of C3-alkyl indoles with allenamides [174,175]. According to Scheme 78, 2,3-disubstituted indoles 78a react with allenamides 78b to form either the C3-alkylation product 78c or the N-alkylation product 78d. The reaction proceeds with the catalyst (2,4-tBu2C6H3O)3PAuCl and either AgOTf or AgTFA as ion chloride scavengers. The N-alkylation product 78d is favored using TfO− counterion, while the C3-alkylation product 78c and dearomatization of indole is favored by TFA− counterion.
The TFA− counterion has a much stronger hydrogen bonding ability than the TfO− counterion. Consequently, these two counterions present different coordinating tendencies, thereby changing the mechanism of the reaction. The intermediate 78e represents a structure in which the counterion reveals its regulating role. The TFA− counterion forms a strong hydrogen bond with N-H and, by weakening the nucleophilicity of N the reaction, proceeds with the nucleophilic attack to the allene 78b, with C3 carbon of indole forming the product 78c. In contrast, TfO−, with a low ability for hydrogen bonding, facilitates the nucleophilic attack to activate allenamide 78b by the N of the intermediate 78e to form the N-alkylation product 78d [174,175].
4.5. The Role of Additives in Gold(I)-Catalyzed Reactions
4.5.1. General Issues
Additives are compounds that accompany catalysts and increase their activity. The role of the additives is not clear enough. It has been proposed that an additive can act as:
(a) a hydrogen bond acceptor; (b) a gold(I) catalyst activator; (c) an acidic co-catalyst [126] and (d) a hydrogen bond acceptor.
4.5.2. Additives as Hydrogen Bond Acceptors
4.5.3. Additives as Gold(I) Catalyst Activators
In many cases, an inactive gold(I) catalyst is used in its chloride form. Silver salts work as chloride scavengers, releasing gold(I) cationic catalysts according to the following reaction:
LAuCl + AgOTf → LAuOTf + AgCl ↓
Excess of silver salt increases the efficiency of catalysis [177].
Another category of catalyst activators is metal triflates, such as Yb(OTf)3. Yb(OTf)3 is a Lewis acid that complexes with the chloride anion of the catalyst, e.g., (PMe3)AuCl, and releases the active gold(I) form. It is remarkable that the catalyst is more efficient when it is activated by the additive than in its active form (PMe3)AuOTf [178], as has been demonstrated in the conversion of epoxy alkynes 81a to compounds 81b (Scheme 81).
4.5.4. Additives as Acidic Co-Catalysts
Acidic co-catalysts could have a synergistic effect in gold(I)-catalyzed reactions, assisting gold(I) at a not defined, still, mechanism. An acidic co-catalyst, e.g., M=Ga in Scheme 82, can act as a Lewis acid or a Brönsted–Lowry acid, see structures 82a or 82b, respectively, to increase cationic gold(I) acidity. Indeed, DFT calculations showed that the energy difference between HOMO and LUMO is reduced due to the synergy of gallium with the gold(I) catalyst [179].
Another way that an acidic co-catalyst can act as a Lewis acid is revealed through gallium salts that have a synergistic effect on the gold(I)-catalyzed reaction of diketone 83a with alkyne 83b, which is activated by a gold(I) catalyst to form the α,α′-vinyl diketone 83c. A suggested explanation is that gallium cations stabilize the enolic form of diketone 83d via chelation and increase the nucleophilicity of diketone (Scheme 83).
4.5.5. Additives as Hydrogen Bond Donors
An acid can facilitate a gold(I)-catalyzed reaction through the hydrogen bonding donor effect to the protodeauration step of the gold(I)-catalyzed reaction (Scheme 84) [77].
As the acid gets stronger, the conversion gets faster. The relation between the strength of the acid and the reaction rate is shown in Table 3.
5. Conclusions
Gold(I) catalysts, in contrast to gold(III) or other metal catalysts, are “soft acids” that have a high affinity for π-systems; we reviewed the gold-catalyzed reactions of nitrogen, oxygen and carbon nucleophiles with π-systems having alkynyl or allenyl moieties with EDG or EWG. The presence of EDG or EWG on π-systems is the main factor for the orientation of the nucleophilic attack on the carbons of π-systems. We described reactions of ynamides, ynols, allenamides and allenyl ethers or alkynyl carbonyl and allenyl carbonyl derivatives, etc. Factors such as counterions, solvents or additives interfere with the catalytic cycle, changing the mechanism and pathway of these reactions.
We presented the gold(I) and gold(III) catalyst complexes and examples of divergent catalysis with π-systems for gold(I) versus gold(III) catalysts. Additionally, we showed that divergent catalysis can be achieved by regulating the acidity of the gold(I) catalyst complexes by changing its ligands and by using other metals, e.g., Pt(II), Ag(I), Pd(II), Rh(II), Sc(III) and Cu(II). The use of gold as a catalyst is desirable when it has a similar activity as a more expensive catalyst or when it shows a higher activity or a higher selectivity than less expensive catalysts. Indeed, gold often reacts much faster than other transition metals that, in principle, can catalyze the same reaction. In addition, in many cases of gold, a completely new chemical transformation is possible using the gold catalyst.
This work provides an introductory but comprehensive review for organic or medicinal or theoretical organic chemists for transformations of π-systems in relation to gold chemistry. The reactions correspond to powerful and efficient methods for the rapid assembly of valuable natural or non-natural products. The described reactions provide building blocks of drugs and natural products, e.g., indoles, pyrroles, isoxazoles, carbazoles, 4-pyrones, 3(2H)-furanones, unsaturated δ-lactones, α-pyrones, oxazepinones, diamino furans, aminopyrroles, oxazepines, dihydrobenzoxepines,, dihydroisoxazoles, dihydrofurans, dihydropyridines, tetrahydropyridines, dihydro-1,2-oxazines, tetrahydro-1,2-oxazines, pyrrolo[2,1-a]isoquinolines and 1,3-oxazepine furo[3,4-d][1,2]-oxazepine.
Despite these achievements, there is still room for further exploration: (a) Experimental or theoretical works that seek to describe the mechanism of catalyzed reactions and may pave the way for new types of catalysts. (b) Catalytic asymmetric versions have been rarely demonstrated and are always challenging in gold catalysis.
Author Contributions
I.S. and A.K. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not appliable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mato, M.; Franchino, A.; García-Morales, C.; Echavarren, A.M. Gold-Catalyzed Synthesis of Small Rings. Chem. Rev. 2021, 121, 8613–8684. [Google Scholar] [CrossRef] [PubMed]
- Rocchigiani, L.; Bochmann, M. Recent Advances in Gold(III) Chemistry: Structure, Bonding, Reactivity, and Role in Homogeneous Catalysis. Chem. Rev. 2021, 121, 8364–8451. [Google Scholar] [CrossRef]
- Reyes, R.L.; Iwai, T.; Sawamura, M. Construction of Medium-Sized Rings by Gold Catalysis. Chem. Rev. 2021, 121, 8926–8947. [Google Scholar] [CrossRef]
- Campeau, D.; Leo, D.F.; Mansour, A.; Muratov, K.; Gagosz, F. Gold-Catalyzed Reactions of Specially Activated Alkynes, Allenes, and Alkenes. Chem. Rev. 2021, 121, 8756–8867. [Google Scholar] [CrossRef] [PubMed]
- Herrera, R.P.; Concepción Gimeno, M. Main Avenues in Gold Coordination Chemistry. Chem. Rev. 2021, 121, 8311–8363. [Google Scholar] [CrossRef] [PubMed]
- Raubenheimer, H.G.; Schmidbaur, H. The Late Start and Amazing Upswing in Gold Chemistry. J. Chem. Educ. 2014, 91, 2024–2036. [Google Scholar] [CrossRef]
- Gorin, D.; Toste, F. Relativistic Effects in Homogeneous Gold Catalysis. Nature 2007, 446, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Desclaux, J.P. Relativistic Dirac–Fock Expectation Values for Atoms with Z = 1 to Z = 120. At. Data Nucl. Data Tables 1973, 12, 311–406. [Google Scholar] [CrossRef]
- Desclaux, J.P.; Pyykkö, P. Dirac–Fock One-Center Calculations—Molecules CuH, AgH and AuH Including P-Type Symmetry Functions. Chem. Phys. Lett. 1976, 39, 300–303. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Hermann, H.L.; Schmidbaur, H. Stability of the Gold(I)–Phosphine Bond. A Comparison with Other Group 11 Elements. Inorg. Chem. 2003, 42, 1334–1342. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Boyd, P.D.W.; Burrell, A.K.; Robinson, W.T.; Taylor, M.J. Relativistic Effects in Gold Chemistry. 3. Gold(I) Complexes. Inorg. Chem. 1990, 29, 3593–3607. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and Soft Acids and Bases, HSAB, Part 1: Fundamental Principles. J. Chem. Educ. 1968, 45, 581. [Google Scholar] [CrossRef]
- Nelson, D.J.; Nolan, S.P. Hydroxide Complexes of the Late Transition Metals: Organometallic Chemistry and Catalysis. Coord. Chem. Rev. 2017, 353, 278–294. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.G.; Schelbach, R.; Schwarzmann, E. Hydroxy Complexes of Gold 2. Calcium Aurates [1]. Z. Naturforsch. 1987, 42, 522–524. [Google Scholar] [CrossRef]
- Einstein, F.W.B.; Rao, P.R.; Trotter, J.; Bartlett, N. The Crystal Structure of Gold Trifluoride. J. Chem. Soc. A Inorg. Phys. Theor. 1967, 478–482. [Google Scholar] [CrossRef]
- Réffy, B.; Kolonits, M.; Schulz, A.; Klapötke, T.M.; Hargittai, M. Intriguing Gold TrifluorideMolecular Structure of Monomers and Dimers: An Electron Diffraction and Quantum Chemical Study. J. Am. Chem. Soc. 2000, 122, 3127–3134. [Google Scholar] [CrossRef]
- Chintawar, C.C.; Yadav, A.K.; Kumar, A.; Sancheti, S.P.; Patil, N.T. Divergent Gold Catalysis: Unlocking Molecular Diversity through Catalyst Control. Chem. Rev. 2021, 121, 8478–8558. [Google Scholar] [CrossRef]
- Wang, T.; Hashmi, A.S.K. 1,2-Migrations onto Gold Carbene Centers. Chem. Rev. 2021, 121, 8948–8978. [Google Scholar] [CrossRef]
- Wang, Y.M.; Lackner, A.D.; Toste, F.D. Development of Catalysts and Ligands for Enantioselective Gold Catalysis. Acc. Chem. Res. 2014, 47, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Zi, W.; Toste, D.F. Recent Advances in Enantioselective Gold Catalysis. Chem. Soc. Rev. 2016, 45, 4567–4589. [Google Scholar] [CrossRef]
- Hashmi, A.S.K. Dual Gold Catalysis. Acc. Chem. Res. 2014, 47, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K. Gold-Catalyzed Organic Reactions. In Inventing Reactions. Topics in Organometallic Chemistry; Gooßen, L., Ed.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 44. [Google Scholar]
- Hashmi, A.S.K. Homogeneous Catalysis by Gold. Gold Bull. 2004, 37, 51–65. [Google Scholar]
- Zheng, Z.; Ma, X.; Cheng, X.; Zhao, K.; Gutman, K.; Li, T.; Zhang, L. Homogeneous Gold-Catalyzed Oxidation Reactions. Chem. Rev. 2021, 121, 8979–9038. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, C.M.; Sekine, K.; Koshikawa, T.; Tanaka, K.; Hashmi, A.S.K. Homogeneous and Heterogeneous Gold Catalysis for Materials Science. Chem. Rev. 2021, 121, 9113–9163. [Google Scholar] [CrossRef] [PubMed]
- Faza, O.N.; López, C.S. Computational Approaches to Homogeneous Gold Catalysis. In Homogeneous Gold Catalysis. Topics in Current Chemistry; Springer: Cham, Switzerland, 2014; Volume 357, pp. 213–285. [Google Scholar]
- Ye, L.-W.; Zhu, X.-Q.; Sahani, R.L.; Xu, Y.; Qian, P.-C.; Liu, R.-S. Nitrene Transfer and Carbene Transfer in Gold Catalysis. Chem. Rev. 2021, 121, 9039–9112. [Google Scholar] [CrossRef]
- Dorel, R.; Echavarren, A.M. Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev. 2015, 115, 9028–9072. [Google Scholar] [CrossRef] [Green Version]
- Debrouwer, W.; Heugebaert, T.S.A.; Roman, B.I.; Stevens, C.V. Homogeneous Gold-Catalyzed Cyclization Reactions of Alkynes with N- and S-Nucleophiles. Adv. Synth. Catal. 2015, 357, 2975–3006. [Google Scholar] [CrossRef]
- Jiménez-Núñez, E.; Echavarren, A.M. Gold-Catalyzed Cycloisomerizations of Enynes: A Mechanistic Perspective. Chem. Rev. 2008, 108, 3326–3350. [Google Scholar] [CrossRef]
- Harris, R.J.; Widenhoefer, R.A. Gold Carbenes, Gold-Stabilized Carbocations, and Cationic Intermediates Relevant to Gold-Catalysed Enyne Cycloaddition. Chem. Soc. Rev. 2016, 45, 4533–4551. [Google Scholar] [CrossRef]
- Lein, M.; Rudolph, M.; Hashmi, S.K.; Schwerdtfeger, P. Homogeneous Gold Catalysis: Mechanism and Relativistic Effects of the Addition of Water to Propyne. Organometallics 2010, 29, 2206–2210. [Google Scholar] [CrossRef] [Green Version]
- Dorel, R.; Echavarren, A.M. Gold-Catalyzed Reactions via Cyclopropyl Gold Carbene-like Intermediates. J. Org. Chem. 2015, 80, 7321–7332. [Google Scholar] [CrossRef]
- López-Carrillo, V.; Huguet, N.; Mosquera, Á.; Echavarren, A.M. Nature of the Intermediates in Gold(I)-Catalyzed Cyclizations of 1,5-Enynes. Chem. –A Eur. J. 2011, 17, 10972–10978. [Google Scholar] [CrossRef] [PubMed]
- Seidel, G.; Fürstner, A. Structure of a Reactive Gold Carbenoid. Angew. Chem. Int. Ed. 2014, 53, 4807–4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benitez, D.; Shapiro, N.D.; Tkatchouk, E.; Wang, Y.; Goddard, W.A.; Toste, F.D. A Bonding Model for Gold(I) Carbene Complexes. Nat. Chem. 2009, 1, 482–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephen, A.; Hashmi, K. Homogeneous Gold Catalysis beyond Assumptions and Proposals-Characterized Intermediates. Angew. Chem. Int. Ed. 2010, 49, 5232–5241. [Google Scholar]
- Zhou, A.-H.; He, Q.; Shu, C.; Yu, Y.-F.; Liu, S.; Zhao, T.; Zhang, W.; Lu, X.; Ye, L.-W. Atom-Economic Generation of Gold Carbenes: Gold-Catalyzed Formal [3 + 2] Cycloaddition Between Ynamides and Isoxazoles. Chem. Sci. 2015, 6, 1265–1271. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Huang, L.; Xie, J.; Rudolph, M.; Rominger, F.; Hashmi, A.S.K. Gold-Catalyzed C-H Annulation of Anthranils with Alkynes: A Facile, Flexible, and Atom-Economical Synthesis of Unprotected 7-Acylindoles. Angew. Chem. Int. Ed. 2016, 55, 794–797. [Google Scholar] [CrossRef]
- Jeanbourquin, L.N.; Scopelliti, R.; Fadaei-Tirani, F.; Severin, K. Gold-Catalyzed Synthesis of 1,3-Diaminopyrazoles from 1-Alkynyltriazenes and Imines. Helv. Chim. Acta 2017, 100, e1700186. [Google Scholar] [CrossRef]
- Chen, Z.; Huang, J.; Wang, Z. Transition-Metal-Catalyzed Hydrosulfoximination and Oxidation Reaction for the Synthesis of Sulfoximine Derivatives. J. Org. Chem. 2016, 81, 9308–9314. [Google Scholar] [CrossRef]
- Stylianakis, I.; Litinas, I.; Nieto Faza, O.; Kolocouris, A.; Silva López, C. On the Mechanism of the Au(I)-Mediated Addition of Alkynes to Anthranils to Furnish 7-Acylindoles. J. Phys. Org. Chem. 2022, 35, e4333. [Google Scholar] [CrossRef]
- Stylianakis, I.; Litinas, I.; Kolocouris, A.; Silva, C. Formation and Intramolecular Capture of -Imino Gold Carbenoids in the Au(I)-Catalyzed [3 + 2] Reaction of Anthranils, 1,2,4-Oxadiazoles, and 4,5-Dihydro-1,2,4-Oxadiazoles with Ynamides. Catalysts 2022, 12, 915. [Google Scholar] [CrossRef]
- Davies, P.W.; Cremonesi, A.; Martin, N. Site-Specific Introduction of Gold-Carbenoids by Intermolecular Oxidation of Ynamides or Ynol Ethers. Chem. Commun. 2011, 47, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Laroche, C.; Kerwin, S.M. Efficient, Regioselective Access to Bicyclic Imidazo[1,2-x]-Heterocycles via Gold- and Base-Promoted Cyclization of 1-Alkynylimidazoles. J. Org. Chem. 2009, 74, 9229–9232. [Google Scholar] [CrossRef]
- Kramer, S.; Madsen, J.L.H.; Rottländer, M.; Skrydstrup, T. Access to 2,5-Diamidopyrroles and 2,5-Diamidofurans by Au(I)-Catalyzed Double Hydroamination or Hydration of 1,3-Diynes. Org. Lett. 2010, 12, 2758–2761. [Google Scholar] [CrossRef]
- Karad, S.N.; Bhunia, S.; Liu, R.-S. Retention of Stereochemistry in Gold-Catalyzed Formal [4 + 3] Cycloaddition of Epoxides with Arenynamides. Angew. Chem. Int. Ed. 2012, 51, 8722–8726. [Google Scholar] [CrossRef]
- Baldwin, J.E. Rules for Ring Closure. J. Chem. Soc. Chem. Commun. 1976, 18, 734–736. [Google Scholar] [CrossRef]
- Liu, C.; Sun, Z.; Xie, F.; Liang, G.; Yang, L.; Li, Y.; Cheng, M.; Lin, B.; Liu, Y. Gold(I)-Catalyzed Pathway-Switchable Tandem Cycloisomerizations to Indolizino[8,7-b] Indole and Indolo[2,3-a] Quinolizine Derivatives. Chem. Commun. 2019, 55, 14418–14421. [Google Scholar] [CrossRef]
- Liu, J.; Chakraborty, P.; Zhang, H.; Zhong, L.; Wang, Z.-X.; Huang, X. Gold-Catalyzed Atom-Economic Synthesis of Sulfone-Containing Pyrrolo[2,1-a]Isoquinolines from Diynamides: Evidence for Consecutive Sulfonyl Migration. ACS Catal. 2019, 9, 2610–2617. [Google Scholar] [CrossRef]
- Zhao, Q.; Gagosz, F. Synthesis of Allenamides and Structurally Related Compounds by a Gold-Catalyzed Hydride Shift Process. Adv. Synth. Catal. 2017, 359, 3108–3113. [Google Scholar] [CrossRef]
- Zeiler, A.; Ziegler, M.J.; Rudolph, M.; Rominger, F.; Hashmi, A.S.K. Scope and Limitations of the Intermolecular Furan-Yne Cyclization. Adv. Synth. Catal. 2015, 357, 1507–1514. [Google Scholar] [CrossRef]
- Patil, M.D.; Liu, R.S. Direct Access to Benzofuro[2,3-b]Quinoline and 6H-Chromeno[3,4-b]Quinoline Cores Through Gold-Catalyzed Annulation of Anthranils with Arenoxyethynes and Aryl Propargyl Ethers. Org. Biomol. Chem. 2019, 17, 4452–4455. [Google Scholar] [CrossRef] [PubMed]
- Krause, N.; Winter, C. Gold-Catalyzed Nucleophilic Cyclization of Functionalized Allenes: A Powerful Access to Carboand Heterocycles. Chem. Rev. 2011, 111, 1994–2009. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, L. AuCl-Catalyzed Synthesis of Benzyl-Protected Substituted Phenols: A Formal [3+3] Approach. Org. Lett. 2007, 9, 4627–4630. [Google Scholar] [CrossRef] [PubMed]
- Pirovano, V.; Decataldo, L.; Rossi, E.; Vicente, R. Gold-Catalyzed Synthesis of Tetrahydrocarbazole Derivatives through an Intermolecular Cycloaddition of Vinyl Indoles and N-Allenamides. Chem. Commun. 2013, 49, 3594–3596. [Google Scholar] [CrossRef] [Green Version]
- Hyland, C.J.T.; Hegedus, L.S. Gold-Catalyzed and NIodosuccinimide-Mediated Cyclization of γ-Substituted Allenamides. J. Org. Chem. 2006, 71, 8658–8660. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, A.; Iino, A.; Ishikawa, S.; Aoki, T.; Terada, M. Efficient Synthesis of Polysubstituted Pyrroles Based on [3 + 2] Cycloaddition Strategy Utilizing [1,2]-Phospha-Brook Rearrangement under Brønsted Base Catalysis. Chem. –A Eur. J. 2018, 24, 15246–15253. [Google Scholar] [CrossRef]
- Alonso, J.M.; Muñoz, M.P. Platinum and Gold Catalysis: à la Carte Hydroamination of Terminal Activated Allenes with Azoles. Org. Lett. 2019, 21, 7639–7644. [Google Scholar] [CrossRef]
- Lee, Y.T.; Kang, Y.K.; Chung, Y.K. Au(I)-Catalyzed Cycloisomerization Reaction of Amide- or Ester-Tethered 1,6-Enynes to Bicyclo[3.2.0]Hept-6-En-2-Ones. J. Org. Chem. 2009, 74, 7922–7934. [Google Scholar] [CrossRef]
- Yeom, H.-S.; Koo, J.; Park, H.-S.; Wang, Y.; Liang, Y.; Yu, Z.-X.; Shin, S. Gold-Catalyzed Intermolecular Reactions of Propiolic Acids with Alkenes: [4 + 2] Annulation and Enyne Cross Metathesis. J. Am. Chem. Soc. 2012, 134, 208–211. [Google Scholar] [CrossRef]
- Luo, T.; Dai, M.; Zheng, S.-L.; Schreiber, S.L. Syntheses of α-Pyrones Using Gold-Catalyzed Coupling Reactions. Org. Lett. 2011, 13, 2834–2836. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Schreiber, S.L. Gold(I)-Catalyzed Coupling Reactions for the Synthesis of Diverse Small Molecules Using the Build/Couple/Pair Strategy. J. Am. Chem. Soc. 2009, 131, 5667–5674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, T.; Schreiber, S.L. Complex α-Pyrones Synthesized by a Gold-Catalyzed Coupling Reaction. Angew. Chem. Int. Ed. 2007, 46, 8250–8253. [Google Scholar] [CrossRef]
- Peshkov, A.A.; Nechaev, A.A.; Pereshivko, O.P.; Goeman, J.L.; Van der Eycken, J.; Peshkov, V.A.; Van der Eycken, E.V. Gold and Silver-Catalyzed 7-Endo-Dig Cyclizations for the Synthesis of Oxazepines. Chem. –A Eur. J. 2015, 19, 4190–4197. [Google Scholar]
- Diéguez-Vázquez, A.; Tzschucke, C.C.; Crecente-Campo, J.; McGrath, S.; Ley, S.V. AuCl3-Catalyzed Hydroalkoxylation of Conjugated Alkynoates: Synthesis of Five- and Six-Membered Cyclic Acetals. Eur. J. Org. Chem. 2009, 2009, 1698–1706. [Google Scholar] [CrossRef]
- Cai, S.; Zeng, J.; Bai, Y.; Liu, X.-W. Access to Quinolines through Gold-Catalyzed Intermolecular Cycloaddition of 2-Aminoaryl Carbonyls and Internal Alkynes. J. Org. Chem. 2012, 77, 801–807. [Google Scholar] [CrossRef]
- Qian, D.; Zhang, J. Catalytic Oxidation/C−H Functionalization of N-Arylpropiolamides by Means of Gold Carbenoids: Concise Route to 3-Acyloxindoles. Chem. Commun. 2012, 48, 7082–7084. [Google Scholar] [CrossRef]
- Wang, S.; Chen, H.; Zhao, H.; Cao, H.; Li, Y.; Liu, Q. Gold-Catalyzed Multicomponent Reaction: Facile Strategy for the Synthesis of N-Substituted 1,4-Dihydropyridines by Using Activated Alkynes, Aldehydes, and Methanamine. Eur. J. Org. Chem. 2013, 2013, 7300–7304. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Schwarz, L.; Choi, J.-H.; Frost, T.M. A New Gold-Catalyzed C−C Bond Formation. Angew. Chem. Int. Ed. 2000, 39, 2285–2288. [Google Scholar] [CrossRef]
- Fang, R.; Yang, L.; Wang, Y. A DFT Study on the Mechanism of Gold(III)-Catalyzed Synthesis of Highly Substituted Furansvia [3,3]-Sigmatropic Rearrangements and/or [1,2]-Acyloxy Migration Based on Propargyl Ketones. Org. Biomol. Chem. 2011, 9, 2760–2770. [Google Scholar] [CrossRef]
- Yang, L.; Fang, R.; Wang, Y. On the Mechanism of AuCl3-Catalyzed Synthesis of Highly Substituted Furans from 2-(1-Alkynyl)-2-Alken-1-Ones with Nucleophiles: A DFT Study. Comput. Theor. Chem. 2011, 965, 180–185. [Google Scholar] [CrossRef]
- Zhang, J.; Shen, W.; Li, L.; Li, M. Gold(I)-Catalyzed Cycloaddition of 1-(1-Alkynyl)Cyclopropyl Ketones with Nucleophiles to Yield Substituted Furans: A DFT Study. Organometallics 2009, 28, 3129–3139. [Google Scholar] [CrossRef]
- Li, Y.; Brand, J.P.; Waser, J. Gold-Catalyzed Regioselective Synthesis of 2- and 3-Alkynyl Furans. Angew. Chem. Int. Ed. 2013, 52, 6743–6747. [Google Scholar] [CrossRef] [Green Version]
- Ghari, H.; Li, Y.; Roohzadeh, R.; Caramenti, P.; Waser, J.; Ariafard, A. Gold-Catalyzed Domino Cyclization-Alkynylation Reactions with EBX Reagents: New Insights into the Reaction Mechanism. Dalton Trans. 2017, 46, 12257–12262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.-P.; Xu, B.; Mashuta, M.S.; Hammond, G.B. Synthesis and Structural Characterization of Stable Organogold(I) Compounds. Evidence for the Mechanism of Gold-Catalyzed Cyclizations. J. Am. Chem. Soc. 2008, 130, 17642–17643. [Google Scholar] [CrossRef]
- Liu, L.-P.; Hammond, G.B. Reactions of Cationic Gold(I) with Allenoates: Synthesis of Stable Organogold(I) Complexes and Mechanistic Investigations on Gold-Catalyzed Cyclizations. Chem. –Asian J. 2009, 4, 1230–1236. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Ramamurthi, T.D.; Todd, M.H.; Tsang, A.S.K.; Graf, K. Gold-Catalysis: Reactions of Organogold Compounds with Electrophiles. Aust. J. Chem. 2010, 63, 1619–1626. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Molinari, L. Effective Transmetalation from Gold to Iron or Ruthenium. Organometallics 2011, 30, 3457–3460. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Lothschutz, C.; Dopp, R.; Rudolph, M.; Ramamurthi, T.D.; Rominger, F. Gold and Palladium Combined for Cross-Coupling. Angew. Chem. Int. Ed. 2009, 48, 8243–8246. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Dopp, R.; Lothschutz, C.; Rudolph, M.; Riedel, D.; Rominger, F. Scope and Limitations of Palladium-Catalyzed Cross-Coupling Reactions with Organogold Compounds. Adv. Synth. Catal. 2010, 352, 1307–1314. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Lothschutz, C.; Döpp, R.; Ackermann, M.; De Buck Becker, J.; Rudolph, M.; Scholz, C.; Rominger, F. On Homogeneous Gold/Palladium Catalytic Systems. Adv. Synth. Catal. 2012, 354, 133–147. [Google Scholar] [CrossRef]
- Tap, A.; Blond, A.; Wakchaure, V.N.; List, B. Chiral Allenes via Alkynylogous Mukaiyama Aldol Reaction. Angew. Chem. Int. Ed. 2016, 55, 8962–8965. [Google Scholar] [CrossRef] [PubMed]
- Selig, P.; Turockin, A.; Raven, W. Guanidine-Catalyzed γ-Selective Morita−Baylis−Hillman Reactions on α,γ-Dialkyl-Allenoates: Access to Densely Substituted Heterocycles. Synlett 2013, 24, 2535–2539. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Han, J.; Hammond, G.B.; Xu, B. Revisiting the Influence of Silver in Cationic Gold Catalysis: A Practical Guide. Org. Lett. 2015, 17, 4534–4537. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, A.; Ozawa, R.; Terada, M. Synthesis of Trisubstituted Allenamides Utilizing 1,2-Rearrangement of Dialkoxyphosphoryl Moiety under Brønsted Base Catalysis. Chem. Lett. 2019, 48, 1164–1167. [Google Scholar] [CrossRef]
- Liu, K.; Zhu, C.; Min, J.; Peng, S.; Xu, G.; Sun, J. Stereodivergent Synthesis of N-Heterocycles by Catalyst-Controlled, Activity-Directed Tandem Annulation of Diazo Compounds with Amino Alkynes. Angew. Chem. Int. Ed. 2015, 54, 12962–12967. [Google Scholar] [CrossRef]
- Wodrich, M.D.; Caramenti, P.; Waser, J. Alkynylation of Thiols with Ethynylbenziodoxolone (EBX) Reagents: α- Or β-π-Addition? Org. Lett. 2016, 18, 60–63. [Google Scholar] [CrossRef]
- Frei, R.; Waser, J. A Highly Chemoselective and Practical Alkynylation of Thiols. J. Am. Chem. Soc. 2013, 135, 9620–9623. [Google Scholar] [CrossRef] [Green Version]
- Brand, J.P.; Waser, J. Direct Alkynylation of Thiophenes: Cooperative Activation of TIPS-EBX with Gold and Bronsted Acids. Angew. Chem. Int. Ed. 2010, 49, 7304–7307. [Google Scholar] [CrossRef] [Green Version]
- Brand, J.P.; Chevalley, C.; Scopelliti, R.; Waser, J. Ethynyl Benziodoxolones for the Direct Alkynylation of Heterocycles: Structural Requirement, Improved Procedure for Pyrroles, and Insights into the Mechanism. Chem. –A Eur. J. 2012, 18, 5655–5666. [Google Scholar] [CrossRef] [Green Version]
- Brand, J.P.; Waser, J. Para-Selective Gold-Catalyzed Direct Alkynylation of Anilines. Org. Lett. 2012, 14, 744–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Waser, J. Zinc-Gold Cooperative Catalysis for the Direct Alkynylation of Benzofurans. Beilstein J. Org. Chem. 2013, 9, 1763–1767. [Google Scholar] [CrossRef]
- Li, Y.; Xie, F.; Li, X. Formal Gold- and Rhodium-Catalyzed Regiodivergent C-H Alkynylation of 2-Pyridones. J. Org. Chem. 2016, 81, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Shinde, P.S.; Patil, N.T. Gold-Catalyzed Dehydrazinative C(Sp)-S Coupling Reactions of Arylsulfonyl Hydrazides with Ethynylbenziodoxolones for Accessing Alkynyl Sulfones. Eur. J. Org. Chem. 2017, 2017, 3512–3515. [Google Scholar] [CrossRef]
- Banerjee, S.; Patil, N.T. Exploiting the Dual Role of Ethynylbenziodoxolones in Gold-Catalyzed C(Sp)-C(Sp) Cross-Coupling Reactions. Chem. Commun. 2017, 53, 7937–7940. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Zhang, Y.; Xia, L. Gold(I)- and Rhodium(III)-Catalyzed Formal Regiodivergent C-H Alkynylation of 1-Arylpyrazolones. Org. Biomol. Chem. 2018, 16, 2860–2864. [Google Scholar] [CrossRef] [PubMed]
- Marion, N.; Nolan, S.P. N-Heterocyclic Carbenes in Gold Catalysis. Chem. Soc. Rev. 2008, 37, 1776. [Google Scholar] [CrossRef]
- Herrmann, W.A. N-Heterocyclic Carbenes: A New Concept in Organometallic Catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Ung, G.; Soleilhavoup, M.; Bertrand, G. Gold(III)- versus Gold(I)-Induced Cyclization: Synthesis of Six-Membered Mesoionic Carbene and Acyclic (Aryl)(Heteroaryl) Carbene Complexes. Angew. Chem. Int. Ed. 2013, 52, 758–761. [Google Scholar] [CrossRef] [Green Version]
- Echavarren, A. Carbene or Cation? Nat. Chem. 2009, 1, 431–433. [Google Scholar] [CrossRef]
- Littke, A.F.; Fu, G.C. Palladium-Catalyzed Coupling Reactions of Aryl Chlorides. Angew. Chem. Int. Ed. 2002, 41, 4176–4211. [Google Scholar] [CrossRef]
- Lavallo, V.; Canac, Y.; Präsang, C.; Donnadieu, B.; Bertrand, G. Stable Cyclic (Alkyl)(Amino)Carbenes as Rigid or Flexible, Bulky, Electron-Rich Ligands for Transition-Metal Catalysts: A Quaternary Carbon Atom Makes the Difference. Angew. Chem. Int. Ed. 2005, 44, 5705–5709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Stevens, E.D.; Nolan, S.P.; Petersen, J.L. Olefin Metathesis-Active Ruthenium Complexes Bearing a Nucleophilic Carbene Ligand. J. Am. Chem. Soc. 1999, 121, 2674–2678. [Google Scholar] [CrossRef]
- Arduengo, A.J. Looking for Stable Carbenes: The Difficulty in Starting Anew. Acc. Chem. Res. 1999, 32, 913–921. [Google Scholar] [CrossRef]
- Hussong, M.W.; Hoffmeister, W.T.; Rominger, F.; Straub, B.F. Copper and Silver Carbene Complexes without Heteroatom-Stabilization: Structure, Spectroscopy, and Relativistic Effects. Angew. Chem. Int. Ed. 2015, 54, 10331–10335. [Google Scholar] [CrossRef] [PubMed]
- Cantat, T.; Ricard, L.; Le Floch, P.; Mézailles, N.N.M. Phosphorus-Stabilized Geminal Dianions. Organometallics 2006, 25, 4965–4976. [Google Scholar] [CrossRef]
- Pujol, A.; Lafage, M.; Rekhroukh, F.; Saffon-Merceron, N.; Amgoune, A.; Bourissou, D.; Nebra, N.; Fustier-Boutignon, M.; Mézailles, N. A Nucleophilic Gold(III) Carbene Complex. Angew. Chem. Int. Ed. 2017, 56, 12264. [Google Scholar] [CrossRef]
- de Frémont, P.; Marion, N.; Nolan, S.P. Cationic NHC–Gold(I) Complexes: Synthesis, Isolation, and Catalytic Activity. J. Organomet. Chem. 2009, 694, 551–560. [Google Scholar] [CrossRef]
- Zuccaccia, D.; Belpassi, L.; Tarantelli, F.; Macchioni, A. Ion Pairing in Cationic Olefin−Gold(I) Complexes. J. Am. Chem. Soc. 2009, 131, 3170–3171. [Google Scholar] [CrossRef]
- Hooper, T.N.; Green, M.; McGrady, J.E.; Patel, J.R.; Russell, C.A. Synthesis and Structural Characterisation of Stable Cationic Gold(I) Alkene Complexes. Chem. Commun. 2009, 26, 3877–3879. [Google Scholar] [CrossRef]
- Brown, T.J.; Dickens, N.J.; Widenhoefer, R.A. Syntheses and X-ray Crystal Structures of Cationic, Two-Coordinate Gold(I) π-Alkene Complexes That Contain a Sterically Hindered o-Biphenylphosphine Ligand. Chem. Commun. 2009, 42, 6451–6453. [Google Scholar] [CrossRef] [PubMed]
- Langseth, E.; Scheuermann, M.L.; Balcells, D.; Kaminsky, W.; Goldberg, K.I.; Eisenstein, O.; Heyn, R.H.; Tilset, M. Generation and Structural Characterization of a Gold(III) Alkene Complex. Angew. Chem. Int. Ed. 2013, 125, 1704–1707. [Google Scholar] [CrossRef]
- Savjani, N.; Roşca, D.A.; Schormann, M.; Bochmann, M. Gold(III) Olefin Complexes. Angew. Chem. Int. Ed. 2013, 52, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Rekhroukh, F.; Estevez, L.; Bijani, C.; Miqueu, K.; Amgoune, A.; Bourissou, D. Coordination–Insertion of Norbornene at Gold: A Mechanistic Study. Organometallics 2016, 35, 995–1001. [Google Scholar] [CrossRef]
- Balcells, D.; Eisenstein, O.; Tilset, M.; Nova, A. Coordination and Insertion of Alkenes and Alkynes in AuIII Complexes: Nature of the Intermediates from a Computational Perspective. Dalton Trans. 2016, 45, 5504–5513. [Google Scholar] [CrossRef] [Green Version]
- Akana, J.A.; Bhattacharyya, K.X.; Müller, P.; Sadighi, J.P. Reversible C−F Bond Formation and the Au-Catalyzed Hydrofluorination of Alkynes. J. Am. Chem. Soc. 2007, 129, 7736–7737. [Google Scholar] [CrossRef]
- Lavallo, V.; Frey, G.D.; Donnadieu, B.; Soleilhavoup, M.; Bertrand, G. Homogeneous Catalytic Hydroamination of Alkynes and Allenes with Ammonia. Angew. Chem. Int. Ed. 2008, 47, 5224–5228. [Google Scholar] [CrossRef] [Green Version]
- Brown, T.J.; Widenhoefer, R.A. Synthesis and Equilibrium Binding Studies of Cationic, Two-Coordinate Gold(I) π-Alkyne Complexes. J. Organomet. Chem. 2011, 696, 1216–1220. [Google Scholar] [CrossRef] [Green Version]
- Hooper, T.N.; Green, M.; Russell, C.A. Cationic Au(I) Alkyne Complexes: Synthesis, Structure and Reactivity. Chem. Commun. 2010, 46, 2313–2315. [Google Scholar] [CrossRef]
- Rocchigiani, L.; Fernandez-Cestau, J.; Agonigi, G.; Chambrier, I.; Budzelaar, P.H.; Bochmann, M. Gold(III) Alkyne Complexes: Bonding and Reaction Pathways. Angew. Chem. Int. Ed. 2017, 56, 13861–13865. [Google Scholar] [CrossRef] [Green Version]
- Gregori, L.; Sorbelli, D.; Belpassi, L.; Tarantelli, F.; Belanzoni, P. Alkyne Activation with Gold(III) Complexes: A Quantitative Assessment of the Ligand Effect by Charge-Displacement Analysis. Inorg. Chem. 2019, 58, 3115–3129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, G.; Zhan, L.; Yang, X.; Wang, J.; Wei, Y.; Xu, S.; Shi, M.; Zhang, J. Gold(I) or Gold(III) as Real Intermediate Species in Gold-Catalyzed Cycloaddition Reactions of Enynal/Enynone? ACS Catal. 2020, 10, 6682–6690. [Google Scholar] [CrossRef]
- Chambrier, I.; Rocchigiani, L.; Hughes, D.L.; Budzelaar, P.M.H.; Bochmann, M. Thermally Stable Gold(III) Alkene and Alkyne Complexes: Synthesis, Structures, and Assessment of the Trans-Influence on Gold–Ligand Bond Enthalpies. Chem. –A Eur. J. 2018, 24, 11467–11474. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Li, T.; Mudshinge, S.R.; Xu, B.; Hammond, G.B. Optimization of Catalysts and Conditions in Gold(I) Catalysis—Counterion and Additive Effects. Chem. Rev. 2021, 121, 8452–8477. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.T.; Boeck, F.; Hintermann, L. Hidden Brønsted Acid Catalysis: Pathways of Accidental or Deliberate Generation of Triflic Acid from Metal Triflates. J. Org. Chem. 2011, 76, 9353–9361. [Google Scholar] [CrossRef]
- Ferrer, C.; Echavarren, A.M. Gold-Catalyzed Intramolecular Reaction of Indoles with Alkynes: Facile Formation of Eight-Membered Rings and an Unexpected Allenylation. Angew. Chem. Int. Ed. 2006, 45, 1105–1109. [Google Scholar] [CrossRef]
- Winter, C.; Krause, N. Structural Diversity through Gold Catalysis: Stereoselective Synthesis of N-Hydroxypyrrolines, Dihydroisoxazoles, and Dihydro-1,2-Oxazines. Angew. Chem. Int. Ed. 2009, 48, 6339–6342. [Google Scholar] [CrossRef]
- Kiriakidi, S.; Nieto Faza, O.; Kolocouris, A.; López, C.S. Governing Effects in the Mechanism of the Gold-Catalyzed Cycloisomerization of Allenic Hydroxylamine Derivatives. Org. Biomol. Chem. 2017, 15, 5920–5926. [Google Scholar] [CrossRef]
- Li, D.; Rao, W.; Tay, G.L.; Ayers, B.J.; Chan, P.W.H. Gold-Catalyzed Cycloisomerization of 1,6-Diyne Esters to 1H-Cyclopenta[b]Naphthalenes, Cis-Cyclopenten-2-Yl δ-Diketones, and Bicyclo[3.2.0]Hepta-1,5-Dienes. J. Org. Chem. 2014, 79, 11301–11315. [Google Scholar] [CrossRef]
- Li, Y.; Tu, P.-C.; Zhou, L.; Kirillov, A.M.; Fang, R.; Yang, L. How Does the Catalyst Affect the Reaction Pathway? DFT Analysis of the Mechanism and Selectivity in the 1,6-Diyne Ester Cycloisomerization. Organometallics 2018, 37, 261–270. [Google Scholar] [CrossRef]
- Wu, C.Y.; Horibe, T.; Jacobsen, C.B.; Toste, F.D. Stable Gold(III) Catalysts by Oxidative Addition of a Carbon–Carbon Bond. Nature 2015, 517, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrach, G.; Valicsek, Z.; Horváth, O. Water-Soluble Silver(II) and Gold(III) Porphyrins: The Effect of Structural Distortion on the Photophysical and Photochemical Behavior. Inorg. Chem. Commun. 2011, 14, 1756–1761. [Google Scholar] [CrossRef]
- Roşca, D.-A.; Wright, J.A.; Bochmann, M. An Element through the Looking Glass: Exploring the Au–C, Au–H and Au–O Energy Landscape. Dalton Trans. 2015, 44, 20785–20807. [Google Scholar] [CrossRef] [Green Version]
- Trost, B.M. Selectivity: A Key to Synthetic Efficiency. Science 1983, 219, 245–250. [Google Scholar] [CrossRef]
- Kumar, R.R.; Kagan, H.B. Regioselective Reactions on a Chiral Substrate Controlled by the Configuration of a Chiral Catalyst. Adv. Synth. Catal. Synth. Catal. 2010, 352, 231–242. [Google Scholar] [CrossRef]
- Mahatthananchai, J.; Dumas, A.M.; Bode, J.W. Catalytic Selective Synthesis. Angew. Chem. Int. Ed. 2012, 51, 10954–10990. [Google Scholar] [CrossRef]
- Xiao, X.-Y.; Zhou, A.-H.; Shu, C.; Pan, F.; Li, T.; Ye, L.-W. Atom-Economic Synthesis of Fully Substituted 2-Aminopyrroles via Gold-Catalyzed Formal [3 + 2] Cycloaddition between Ynamides and Isoxazoles. Chem. –Asian J. 2015, 10, 1854–1858. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.-B.; Xiao, X.-Y.; Sun, Q.; Zhou, B.; Zhu, X.-Q.; Yan, J.-Z.; Lu, X.; Ye, L.-W. Highly Site Selective Formal [5+2] and [4+2] Annulations of Isoxazoles with Heterosubstituted Alkynes by Platinum Catalysis: Rapid Access to Functionalized 1,3-Oxazepines and 2,5-Dihydropyridines. Angew. Chem. Int. Ed. 2017, 56, 605–609. [Google Scholar] [CrossRef]
- Liddon, J.T.; James, M.J.; Clarke, A.K.; O’brien, P.; Taylor, R.J.; Unsworth, W.P. Catalyst-Driven Scaffold Diversity: Selective Synthesis of Spirocycles, Carbazoles and Quinolines from Indolyl Ynones. Chem. –A Eur. J. 2016, 22, 8777–8780. [Google Scholar] [CrossRef] [Green Version]
- Zeineddine, A.; Estévez, L.; Mallet-Ladeira, S.; Miqueu, K.; Amgoune, A.; Bourissou, D. Rational Development of Catalytic Au(I)/Au(III) Arylation Involving Mild Oxidative Addition of Aryl Halides. Nat. Commun. 2017, 8, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Amaike, K.; Maceiczyk, R.M.; Itami, K.; Yamaguchi, J. β-Selective CSH Arylation of Pyrroles Leading to Concise Syntheses of Lamellarins C and I. J. Am. Chem. Soc. 2014, 136, 13226–13232. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Zeineddine, A.; Sosa Carrizo, E.D.; Miqueu, K.; Saffon-Merceron, N.; Amgoune, A.; Bourissou, D. Catalytic Au(I)/Au(III) Arylation with the Hemilabile MeDalphos Ligand: Unusual Selectivity for Electron-Rich Iodoarenes and Efficient Application to Indoles. Chem. Sci. 2019, 10, 7183–7192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.; Adet, N.; Saffon-Merceron, N.; Bourissou, D. Au(I)/Au(III)-Catalyzed C-N Coupling. Chem. Commun. 2020, 56, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Akram, M.O.; Das, A.; Chakrabarty, I.; Patil, N.T. Ligand-Enabled Gold-Catalyzed C(Sp2))N Cross-Coupling Reactions of Aryl Iodides with Amines. Org. Lett. 2019, 21, 8101–8105. [Google Scholar] [CrossRef]
- Shaikh, A.C.; Shinde, D.R.; Patil, N.T. Gold vs Rhodium Catalysis: Tuning Reactivity through Catalyst Control in the C-H Alkynylation of Isoquinolones. Org. Lett. 2016, 18, 1056–1059. [Google Scholar] [CrossRef]
- Zhao, F.; Xu, B.; Ren, D.; Han, L.; Yu, Z.; Liu, T. C–H Alkynylation of N-Methylisoquinolone by Rhodium or Gold Catalysis: Theoretical Studies on the Mechanism, Regioselectivity, and Role of TIPS-EBX. Organometallics 2018, 37, 1026–1033. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, F.; Zhang, J. Catalytic Regioselective Control in the Diastereoselective 1,3-Dipolar Cycloaddition Reactions of 1-(1-Alkynyl)Cyclopropyl Ketones with Nitrones. Chem. –A Eur. J. 2010, 16, 6146–6150. [Google Scholar] [CrossRef]
- Zhang, J.; Shen, W.; Li, L.; Li, M. Reaction Mechanism and Chemoselectivity of Gold(I)-Catalyzed Cycloaddition of 1-(1-Alkynyl) Cyclopropyl Ketones with Nucleophiles to Yield Substituted Furans. Sci. China Chem. 2012, 55, 1413–1420. [Google Scholar]
- Zhang, Y.; Zhang, J. Kinetic Resolution of 1-(1-Alkynyl)Cyclopropyl Ketones by Gold(I)-Catalyzed Asymmetric [4+3]Cycloaddition with Nitrones: Scope, Mechanism and Applications. Chem. Commun. 2012, 48, 4710–4712. [Google Scholar] [CrossRef]
- Nakamura, I.; Kudo, Y.; Terada, M. Oxazepine Synthesis by Copper-Catalyzed Intermolecular Cascade Reactions between OPropargylic Oximes and Dipolarophiles. Angew. Chem. Int. Ed. 2013, 52, 7536–7539. [Google Scholar] [CrossRef]
- Nakamura, I.; Gima, S.; Kudo, Y.; Terada, M. Skeletal Rearrangement of O-Propargylic Formaldoximes by a Gold-Catalyzed Cyclization/Intermolecular Methylene Transfer Sequence. Angew. Chem. Int. Ed. 2015, 54, 7154–7157. [Google Scholar] [CrossRef] [PubMed]
- Solas, M.; Muñoz, M.A.; Suárez-Pantiga, S.; Sanz, R. Regiodivergent Hydration-Cyclization of Diynones under Gold Catalysis. Org. Lett. 2020, 22, 7681–7687. [Google Scholar] [CrossRef]
- Moran-Poladura, P.; Suárez-Pantiga, S.; Piedrafita, M.; Rubio, E.; Gonzalez, J.M. Regiocontrolled Gold(I)-Catalyzed Cyclization Reactions of N-(3-Iodoprop-2-Ynyl)-N-Tosylanilines. J. Organomet. Chem. 2011, 696, 12–15. [Google Scholar] [CrossRef]
- Wang, T.; Shi, S.; Vilhelmsen, H.M.; Zhang, T.; Rudolph, M.; Rominger, F.; Hashmi, A.S.K. Chemoselectivity Control: Goldcatalyzed Synthesis of 6,7-Dihydrobenzofuran-4(5H)-Ones and Benzofurans from 1-(Alkynyl)-7-Oxabycyclo[4.1.0]Heptan-2-Ones. Chem. –A Eur. J. 2013, 19, 12512–12516. [Google Scholar] [CrossRef]
- Fang, W.; Tang, X.-Y.; Shi, M. Gold(I)-Catalyzed Intramolecular Hydroarylation and the Subsequent Ring Enlargement of Methylenecyclopropanes to Cyclobutenes. RSC Adv. 2016, 6, 40474–40479. [Google Scholar] [CrossRef]
- Gimeno, A.; Cuenca, A.B.; Suárez-Pantiga, S.; de Arellano, C.R.; Medio-Simón, M.; Asensio, G. Competitive Gold-Activation Modes in Terminal Alkynes: An Experimental and Mechanistic Study. Chem. –A Eur. J. 2014, 20, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, P.D.; Lu, X.; Liu, R.-S. Gold-Catalyzed [5+2]- and [5+1]-Annulations between Ynamides and 1,2-Benzisoxazoles with Ligand-Controlled Chemoselectivity. ACS Catal. 2018, 8, 9697–9701. [Google Scholar] [CrossRef]
- Alonso, I.; Trillo, B.; López, F.; Montserrat, S.; Ujaque, G.; Castedo, L.; Lledós, A.; Mascareñas, J.L. Gold-Catalyzed [4C+2C] Cycloadditions of Allenedienes, Including an Enantioselective Version with New Phosphoramidite-Based Catalysts: Mechanistic Aspects of the Divergence between [4C+3C] and [4C+2C] Pathways. J. Am. Chem. Soc. 2009, 131, 13020–13030. [Google Scholar] [CrossRef]
- Mauleón, P.; Zeldin, R.M.; González, A.Z.; Toste, F.D. Ligand-Controlled Access to [4+2] and [4+3] Cycloadditions in Gold-Catalyzed Reactions of Allene-Dienes. J. Am. Chem. Soc. 2009, 131, 6348–6349. [Google Scholar] [CrossRef] [Green Version]
- Alcarazo, M.; Stork, T.; Anoop, A.; Thiel, W.; Fürstner, A. Steering the Surprisingly Modular π-Acceptor Properties of N-Heterocyclic Carbenes: Implications for Gold Catalysis. Angew. Chem. Int. Ed. 2010, 49, 2542–2546. [Google Scholar] [CrossRef] [Green Version]
- Sanz, R.; Miguel, D.; Rodríguez, F. Gold(I)-Catalyzed Tandem Reactions Initiated by 1,2-Indole Migrations. Angew. Chem. Int. Ed. 2008, 47, 7354–7357. [Google Scholar] [CrossRef]
- Álvarez, E.; Miguel, D.; García-García, P.; Fernández-Rodríguez, M.A.; Rodríguez, F.; Sanz, R. Solvent and Ligand Induced Switch of Selectivity in Gold(I)-Catalyzed Tandem Reactions of 3-Propargylindoles. Beilstein J. Org. Chem. 2011, 7, 786–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez, E.; Faza, O.N.; López, C.S.; Fernández-Rodríguez, M.A.; Brønsted, R.S. Acid-Catalyzed Cascade Reactions Involving 1,2-Indole Migration. Chem. –A Eur. J. 2015, 21, 12889–12893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, R.; Miguel, D.; Gohain, M.; García-García, P.; Fernández-Rodríguez, M.; González-Pérez, A.; Nieto-Faza, O.; de Lera, Á.; Rodríguez, F. Synthesis of Diverse Indole-Containing Scaffolds by Gold(I)-Catalyzed Tandem Reactions of 3-Propargylindoles Initiated by 1,2-Indole Migrations: Scope and Computational Studies. Chem. –A Eur. J. 2010, 16, 9818–9828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, M.; Bandini, M. Counterion Effects in Homogeneous Gold Catalysis. ACS Catal. 2015, 5, 1638–1652. [Google Scholar] [CrossRef]
- Macchioni, A. Ion Pairing in Transition-Metal Organometallic Chemistry. Chem. Rev. 2005, 105, 2039–2074. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Belpassi, L.; Zuccaccia, D.; Tarantelli, F.; Belanzoni, P. Counterion Effect in the Reaction Mechanism of NHC Gold(I)-Catalyzed Alkoxylation of Alkynes: Computational Insight into Experiment. ACS Catal. 2015, 5, 803–814. [Google Scholar] [CrossRef]
- Lu, Z.; Han, J.; Okoromoba, O.E.; Shimizu, N.; Amii, H.; Tormena, C.F.; Hammond, G.B.; Xu, B. Predicting Counterion Effects Using a Gold Affinity Index and a Hydrogen Bonding Basicity Index. Org. Lett. 2017, 19, 5848–5851. [Google Scholar] [CrossRef] [PubMed]
- Gatto, M.; Belanzoni, P.; Belpassi, L.; Biasiolo, L.; Del Zotto, A.; Tarantelli, F.; Zuccaccia, D. Solvent-, Silver-, and Acid-Free NHC-Au-X Catalyzed Hydration of Alkynes. The Pivotal Role of the Counterion. ACS Catal. 2016, 6, 7363–7376. [Google Scholar] [CrossRef]
- Hamilton, G.L.; Kang, E.J.; Mba, M.; Toste, F.D. A Powerful Chiral Counterion Strategy for Asymmetric Transition Metal Catalysis. Science 2007, 317, 496–499. [Google Scholar] [CrossRef] [Green Version]
- Dudnik, A.S.; Xia, Y.; Li, Y.; Gevorgyan, V. Computation-Guided Development of Au Catalyzed Cycloisomerizations Proceeding via 1,2-Si or 1,2-H Migrations: Regiodivergent Synthesis of Silylfurans. J. Am. Chem. Soc. 2010, 132, 7645–7655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, M.; Cera, G.; Perrotta, D.; Monari, M.; Bandini, M. Taming Gold(I)-Counterion Interplay in the Dearomatization of Indoles with Allenamides. Chem. –A Eur. J. 2014, 20, 9875–9878. [Google Scholar] [CrossRef] [PubMed]
- Rocchigiani, L.; Jia, M.; Bandini, M.; Macchioni, A. Assessing the Role of Counterion in Gold-Catalyzed Dearomatization of Indoles with Allenamides by NMR Studies. ACS Catal. 2015, 5, 3911–3915. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yan, W.; Akhmedov, N.G.; Shi, X. 1,2,3-Triazole as a Special “X-Factor” in Promoting Hashmi Phenol Synthesis. Org. Lett. 2010, 12, 344–347. [Google Scholar] [CrossRef]
- Weber, D.; Gagné, M.R. Dinuclear Gold-Silver Resting States May Explain Silver Effects in Gold(I)-Catalysis. Org. Lett. 2009, 11, 4962–4965. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.-Z.; Shi, M. Gold(I)- and Yb(OTf)3-Cocatalyzed Rearrangements of Epoxy Alkynes: Transfer of a Carbonyl Group in a Five-Membered Carbocycle. Chem. –A Eur. J. 2010, 16, 2496–2502. [Google Scholar] [CrossRef]
- Bhattacharjee, R.; Nijamudheen, A.; Datta, A. Mechanistic Insights into the Synergistic Catalysis by Au(I), Ga(III), and Counterions in the Nakamura Reaction. Org. Biomol. Chem. 2015, 13, 7412–7420. [Google Scholar] [CrossRef]
- Xi, Y.; Wang, D.; Ye, X.; Akhmedov, N.G.; Petersen, J.L.; Shi, X. Synergistic Au/Ga Catalysis in Ambient Nakamura Reaction. Org. Lett. 2014, 16, 306–309. [Google Scholar] [CrossRef]
Figure 1.
Overview of gold-catalytic reactions with π-systems studied in this article.

Scheme 1.
Nucleophilic functionalization of alkyne under gold catalysis.

Scheme 2.
Generic mechanism of a nucleophilic attack to an activated alkyne bearing an electron-donating group (EDG), e.g., a ynamide.
Scheme 2.
Generic mechanism of a nucleophilic attack to an activated alkyne bearing an electron-donating group (EDG), e.g., a ynamide.

Scheme 3.
(A,B) Formal [3+2] cycloaddition gold(I)-catalyzed between ynamides 3a and isoxazoles 3b, which, after deauration, leads to 2-amino pyrroles 3c/3d (Ye 2015) [39].
Scheme 3.
(A,B) Formal [3+2] cycloaddition gold(I)-catalyzed between ynamides 3a and isoxazoles 3b, which, after deauration, leads to 2-amino pyrroles 3c/3d (Ye 2015) [39].

Scheme 4.
Formal [3+2] cycloaddition between ynamides 4b and anthranils 4a catalyzed by IPrAuCl (4d)/AgNTf2 which affords indoles 4c (Hashmi 2016) [40].
Scheme 4.
Formal [3+2] cycloaddition between ynamides 4b and anthranils 4a catalyzed by IPrAuCl (4d)/AgNTf2 which affords indoles 4c (Hashmi 2016) [40].

Scheme 5.
The reaction of ynamides 5a with sulfoximines 5b catalyzed by Ph3PAuCl/AgOTf to afford the sulfoximinated products 5c (Wang and Chen 2016) [42].
Scheme 5.
The reaction of ynamides 5a with sulfoximines 5b catalyzed by Ph3PAuCl/AgOTf to afford the sulfoximinated products 5c (Wang and Chen 2016) [42].

Scheme 6.
Synthesis of 1,3-diaminopyrazoles 6c from 1-alkynyltriazenes 6a and imines 6b catalyzed by JohnPhosAuCl(6d)/AgNTf2 (Severin 2017) [41].
Scheme 6.
Synthesis of 1,3-diaminopyrazoles 6c from 1-alkynyltriazenes 6a and imines 6b catalyzed by JohnPhosAuCl(6d)/AgNTf2 (Severin 2017) [41].

Scheme 7.
Regioselectivity models description for the mechanism of the reaction of ynamide 7a with anthranil 7b, which afforded selectively 2-substituted 7-acyl-indole 7c compared with the 3-substituted 7-acyl-indole indole 7n (Lopez, Kolocouris) [43].
Scheme 7.
Regioselectivity models description for the mechanism of the reaction of ynamide 7a with anthranil 7b, which afforded selectively 2-substituted 7-acyl-indole 7c compared with the 3-substituted 7-acyl-indole indole 7n (Lopez, Kolocouris) [43].

Scheme 8.
Formation of α-oxo gold carbenes 8d from ynamide substrates 8a and X+-O− nucleophiles [4].
Scheme 8.
Formation of α-oxo gold carbenes 8d from ynamide substrates 8a and X+-O− nucleophiles [4].

Scheme 9.
Au(III)-catalyzed synthesis of α,β-unsaturated carbonyl derivatives 9c by oxidation of ynamides 9a (Martin 2011) [45].
Scheme 9.
Au(III)-catalyzed synthesis of α,β-unsaturated carbonyl derivatives 9c by oxidation of ynamides 9a (Martin 2011) [45].

Scheme 10.
Au(III)-catalyzed synthesis of O-heterocycles 10b from ynamides 10a via hydroalkoxylation (Kerwin 2009) [46].
Scheme 10.
Au(III)-catalyzed synthesis of O-heterocycles 10b from ynamides 10a via hydroalkoxylation (Kerwin 2009) [46].

Scheme 11.
Gold(I)-catalyzed synthesis of 2,5-diamino furans 11b from diynes 11a and water (Skrydstrup 2010) [47].
Scheme 11.
Gold(I)-catalyzed synthesis of 2,5-diamino furans 11b from diynes 11a and water (Skrydstrup 2010) [47].

Scheme 12.
Au(I)-catalyzed synthesis of dihydrobenzoxepines 12c by the formal [4+3] cycloaddition between ynamides 12a and epoxides 12b (Liu 2012) [48].
Scheme 12.
Au(I)-catalyzed synthesis of dihydrobenzoxepines 12c by the formal [4+3] cycloaddition between ynamides 12a and epoxides 12b (Liu 2012) [48].

Scheme 13.
Gold(I)-catalyzed cycloisomerization of tryptamine-derived ynamides 13a to afford fused indole N-heterocycles 13b or 13c (Liu 2019) [50].
Scheme 13.
Gold(I)-catalyzed cycloisomerization of tryptamine-derived ynamides 13a to afford fused indole N-heterocycles 13b or 13c (Liu 2019) [50].

Scheme 14.
Synthesis of sulfone-containing pyrrolo[2,1-a]isoquinolines 14d from the relevant diynamides 14a (Wang, Huang 2019) [51].
Scheme 14.
Synthesis of sulfone-containing pyrrolo[2,1-a]isoquinolines 14d from the relevant diynamides 14a (Wang, Huang 2019) [51].

Scheme 15.
Gold(I)-catalyzed formation of allenamide 15d from ynamide 15a through intramolecular [1,5]-hydride shift from benzyloxy group in keteniminium 15b (Gagosz 2017) [52].
Scheme 15.
Gold(I)-catalyzed formation of allenamide 15d from ynamide 15a through intramolecular [1,5]-hydride shift from benzyloxy group in keteniminium 15b (Gagosz 2017) [52].

Scheme 16.
Ynol derivatives employed in gold catalysis and their general reactivity profile [4].
Scheme 16.
Ynol derivatives employed in gold catalysis and their general reactivity profile [4].

Scheme 17.
Gold(I)-catalyzed reaction of alkynes 17a with furans 17b leading to phenols 17c (Hashmi 2015) [53].
Scheme 17.
Gold(I)-catalyzed reaction of alkynes 17a with furans 17b leading to phenols 17c (Hashmi 2015) [53].

Scheme 18.
Annulation of the anthranils 18b with aryl-ethynyl ethers 18a led to benzofuranoquinolines 18c using an Au(I) catalyst (Liu 2019) [54].
Scheme 18.
Annulation of the anthranils 18b with aryl-ethynyl ethers 18a led to benzofuranoquinolines 18c using an Au(I) catalyst (Liu 2019) [54].

Scheme 19.
General description of the Au-activated reactivity of allenes 19a substituted by an O-, S- or N-based nucleophilic group [55].
Scheme 19.
General description of the Au-activated reactivity of allenes 19a substituted by an O-, S- or N-based nucleophilic group [55].

Scheme 20.
Gold(I)-catalyzed synthesis of benzyloxybenzenes 20d from allene 20a (Zhang 2007) [56].
Scheme 20.
Gold(I)-catalyzed synthesis of benzyloxybenzenes 20d from allene 20a (Zhang 2007) [56].

Scheme 22.
Gold(I)-catalyzed synthesis of dihydrofuran derivatives 22c from allenols 22a (Hegedus 2006) [58].
Scheme 22.
Gold(I)-catalyzed synthesis of dihydrofuran derivatives 22c from allenols 22a (Hegedus 2006) [58].

Scheme 23.
Gold(I)-catalyzed synthesis of pyrrole derivatives 23c from allenamines 23a (Terada 2018) [59].
Scheme 23.
Gold(I)-catalyzed synthesis of pyrrole derivatives 23c from allenamines 23a (Terada 2018) [59].

Scheme 24.
Gold(I)-catalyzed hydroamination of N-, O- and S-allenyl derivatives 24a to afford 24c or 24d using co-catalyst PtCl2 (Muñoz 2019) [60].
Scheme 24.
Gold(I)-catalyzed hydroamination of N-, O- and S-allenyl derivatives 24a to afford 24c or 24d using co-catalyst PtCl2 (Muñoz 2019) [60].

Scheme 25.
Alkyne, allene and alkene derivatives with electron-withdrawing substituents [4].
Scheme 25.
Alkyne, allene and alkene derivatives with electron-withdrawing substituents [4].

Scheme 26.
General scheme of gold catalyst-activation of alkynyl carbonyl derivative 26a, and nucleophilic attack to the activated alkyne group of 26b to afford a variety of products 26c [4].
Scheme 26.
General scheme of gold catalyst-activation of alkynyl carbonyl derivative 26a, and nucleophilic attack to the activated alkyne group of 26b to afford a variety of products 26c [4].

Scheme 27.
Gold(I)-catalyzed cyclization of the 1,6-enynes 27a to form cyclobutane condensed bicyclic compounds 27d (Kang and Chung) [61].
Scheme 27.
Gold(I)-catalyzed cyclization of the 1,6-enynes 27a to form cyclobutane condensed bicyclic compounds 27d (Kang and Chung) [61].

Scheme 28.
Gold(I)-catalyzed reaction between propiolic acids 28b and alkenes 28a to form unsaturated δ-lactones 28d (Shin 2012) [62].
Scheme 28.
Gold(I)-catalyzed reaction between propiolic acids 28b and alkenes 28a to form unsaturated δ-lactones 28d (Shin 2012) [62].

Scheme 29.
Gold(Ι)-catalyzed synthesis of α-pyrones 29e from alkynyl-carboxylic acids 29a and alkynes 29b through enol esters 29c (Schreiber 2011) [63].
Scheme 29.
Gold(Ι)-catalyzed synthesis of α-pyrones 29e from alkynyl-carboxylic acids 29a and alkynes 29b through enol esters 29c (Schreiber 2011) [63].

Scheme 30.
Gold(I)-catalyzed synthesis of esters 30d or α-pyrones 30e,f from allenol esters 30a through allenol esters 30b (Schreiber 2007, 2011) [63,65].

Scheme 31.
Gold(I)-catalyzed synthesis of oxazepinones 31b by intramolecular hydroalkoxylation of alkynyl carboxamides 31a (Van der Eycken 2015) [66].
Scheme 31.
Gold(I)-catalyzed synthesis of oxazepinones 31b by intramolecular hydroalkoxylation of alkynyl carboxamides 31a (Van der Eycken 2015) [66].

Scheme 32.
Gold(III)-catalyzed synthesis of cyclic acetals 32e and exocyclic enol ethers 32b–32d via an intramolecular hydroalkoxylation of alkynyl esters 32a (Ley 2009) [67].
Scheme 32.
Gold(III)-catalyzed synthesis of cyclic acetals 32e and exocyclic enol ethers 32b–32d via an intramolecular hydroalkoxylation of alkynyl esters 32a (Ley 2009) [67].

Scheme 33.
Gold(III)-catalyzed synthesis of cyclic acetals 33b bearing enol ester groups via an intramolecular hydroalkoxylation of alkynyl esters 33a (Ley 2009) [67].
Scheme 33.
Gold(III)-catalyzed synthesis of cyclic acetals 33b bearing enol ester groups via an intramolecular hydroalkoxylation of alkynyl esters 33a (Ley 2009) [67].

Scheme 34.
Prevailing products 34c or 34d and intermediate 34b in gold-catalyzed oxidative functionalization of alkynyl carbonyl derivatives 34a [4].
Scheme 34.
Prevailing products 34c or 34d and intermediate 34b in gold-catalyzed oxidative functionalization of alkynyl carbonyl derivatives 34a [4].

Scheme 35.
Gold(I)-catalyzed intramolecular C-H functionalization of aryl groups, e.g., in 35a to afford 35d through intermediates 35b and 35c (Zhang 2012) [68].
Scheme 35.
Gold(I)-catalyzed intramolecular C-H functionalization of aryl groups, e.g., in 35a to afford 35d through intermediates 35b and 35c (Zhang 2012) [68].

Scheme 36.
Gold(Ι)-catalyzed synthesis of 1,4-dihydropyridines 36f from an aldehyde 36a and an aliphatic amine and alkynyl esters or alkynones 36b (Liu 2013) [70].
Scheme 36.
Gold(Ι)-catalyzed synthesis of 1,4-dihydropyridines 36f from an aldehyde 36a and an aliphatic amine and alkynyl esters or alkynones 36b (Liu 2013) [70].

Scheme 37.
Gold(I)-intermolecular condensation of ortho-acyl anilines 37a with alkynones 37b to afford 3-acyl-kinolines 37c (Liu 2012) [68].
Scheme 37.
Gold(I)-intermolecular condensation of ortho-acyl anilines 37a with alkynones 37b to afford 3-acyl-kinolines 37c (Liu 2012) [68].

Scheme 38.
General gold-catalyzed reaction models of allenyl carbonyl derivatives 38a that can afford furans 38d (R1 = alkyl group) or butenolides 38e (R1 = alkoxy group).
Scheme 38.
General gold-catalyzed reaction models of allenyl carbonyl derivatives 38a that can afford furans 38d (R1 = alkyl group) or butenolides 38e (R1 = alkoxy group).

Scheme 39.
Gold(III)-catalyzed synthesis of furans 39d from allenones 39a (Hashmi 2000) [71].
Scheme 39.
Gold(III)-catalyzed synthesis of furans 39d from allenones 39a (Hashmi 2000) [71].

Scheme 40.
Gold-catalyzed synthesis of furans 40e from the reaction of allenones 40a with TIPS-EBX reagent 40b via gold(I)/gold(III) redox cycle (Waser 2013, Ariafard 2017) [75,76].

Scheme 41.
Gold(I)-catalyzed synthesis of gold(I)-butenolides 41c from allenoates 41a (Hammond 2008, 2009) [77,78].


Scheme 43.
Palladium(II)-transmetallation studies of gold(I)-butenolides 43a (Rominger 2009, 2010, 2012) [81,82,83].

Scheme 44.
Gold(I)-catalyzed intramolecular additions of an O- and N-based nucleophilic group present into allenoates 44a to form dihydrofurans 44c or pyrroles 44d or tetrahydropyridines 44e (List 2016, Raven 2013, Xu 2015, Terada 2019, Sun 2015) [84,85,86,87,88].

Scheme 45.
Gold(I)-catalyzed C-H functionalization with TIPS-EBX reactions (Waser 2010, 2012, 2013, Li 2016, Patil 2017, Xia 2018) [91,92,93,94,95,96,97,98].

Scheme 46.
Resonance forms of a gold-carbene complex.

Scheme 47.
The reaction of IPrAuNTf2 47a with Mes2CN2 produced the first gold carbene derivative 47b without heteroatom donor substituents connected with the metal (Straub 2015) [107].
Scheme 47.
The reaction of IPrAuNTf2 47a with Mes2CN2 produced the first gold carbene derivative 47b without heteroatom donor substituents connected with the metal (Straub 2015) [107].

Scheme 48.
Synthesis of gold(III)-carbene derivative 48c from anion 48a and compound 48b (Mézailles 2006, Amgoune, Bourissou, Nebra, Fustier-Boutignon, Mézailles 2017) [108,109].


Scheme 50.
Cationic gold(I)–π-alkyne complexes, containing either an NHC ligand [118,119] or an alkyl phosphine ligand [120,121].

Scheme 51.
Generation of a C^N chelated alkyne complex 51b from 51a (Budzelaar 2017, 2018) [122,125].

Scheme 52.
Regiodivergent cycloisomerization of alkyne-tethered indoles 52a leading to azepino [4,5-b]indole 52c with Au(I) catalysis and the eight-member ring derivative 52e with Au(III) catalysis through spiranic intermediates 52b/52d (Echavarren 2006) [128].
Scheme 52.
Regiodivergent cycloisomerization of alkyne-tethered indoles 52a leading to azepino [4,5-b]indole 52c with Au(I) catalysis and the eight-member ring derivative 52e with Au(III) catalysis through spiranic intermediates 52b/52d (Echavarren 2006) [128].

Scheme 53.
Divergent catalysis in the cycloisomerization of allenic hydroxylamine ethers 53a with gold(I) or gold(III) complexes leading to dihydroisoxazoles 53b or 3,6-dihydro-1,2-oxazine 53c, respectively (Krause 2009) [129].
Scheme 53.
Divergent catalysis in the cycloisomerization of allenic hydroxylamine ethers 53a with gold(I) or gold(III) complexes leading to dihydroisoxazoles 53b or 3,6-dihydro-1,2-oxazine 53c, respectively (Krause 2009) [129].

Scheme 54.
Divergent cycloisomerization of 1,6-diyne esters 54a leading to cyclopenta[b]naphthalene 54b with Au(I) catalysis or cyclopentenyl diketone 54c with Au(III) catalysis (Chan 2014) [131].
Scheme 54.
Divergent cycloisomerization of 1,6-diyne esters 54a leading to cyclopenta[b]naphthalene 54b with Au(I) catalysis or cyclopentenyl diketone 54c with Au(III) catalysis (Chan 2014) [131].

Scheme 55.
Divergent reactivity for the reaction between a cinnamaldehyde and a ketene acetal with NHC gold(I) or gold(III) catalysts which affords the Mukaiyama reaction aldol product 55c or the Mukaiyama–Michael reaction product 55d, respectively (Toste 2015) [133].
Scheme 55.
Divergent reactivity for the reaction between a cinnamaldehyde and a ketene acetal with NHC gold(I) or gold(III) catalysts which affords the Mukaiyama reaction aldol product 55c or the Mukaiyama–Michael reaction product 55d, respectively (Toste 2015) [133].

Scheme 56.
Formal [3+2] versus [5+2] annulation in the Au(I) [138] versus Pt(II)-catalyzed [139] reaction between an isoxazole 56a and ynamide 56b, leading to 2-aminopyrrole 56g or 1,3-oxazepine 56i′, respectively (Lu and Ye 2015, 2017) [138,139].

Scheme 57.
Regiodivergent cycloisomerization of indolyl ynones 57a leading to the spyrocyclic product 57c′ with silver(I) catalysis or to carbazole 57e with gold(I) catalysis (Taylor and Unsworth 2016) [141].
Scheme 57.
Regiodivergent cycloisomerization of indolyl ynones 57a leading to the spyrocyclic product 57c′ with silver(I) catalysis or to carbazole 57e with gold(I) catalysis (Taylor and Unsworth 2016) [141].

Scheme 58.
C3- versus C2-arylation of N-phenyl substituted pyrrole 58a in the presence of palladium(II) versus gold(I) catalyst, leading to pyrrole derivatives 58c and 58d, respectively (Bourissou 2017, Yamaguchi 2014 [142,143]).

Scheme 59.
Activation of phenyliodide 59b by MeDalPhos gold(I) catalyst 59a. MeDalPhos ligand induces oscillation of oxidative state between Au(I) and Au(III) during the catalytic cycle (Bourissou 2019 [144], 2020 [145], Patil 2020 [146]).

Scheme 60.
Regioselective C-H alkynylation of isoquinolones in the presence of gold(I) or rhodium(II) catalysts leading to C4 or C8 C-H alkyne insertion in products 60e or 60e′, respectively (Patil 2016 [147]).
Scheme 60.
Regioselective C-H alkynylation of isoquinolones in the presence of gold(I) or rhodium(II) catalysts leading to C4 or C8 C-H alkyne insertion in products 60e or 60e′, respectively (Patil 2016 [147]).

Scheme 61.
Regioselective control in the diastereoselective 1,3-dipolar cycloaddition reactions of 1-(1-alkynyl)cyclopropyl ketones 61a with nitrones 61b using Sc(III) or Au(I) catalysts to afford a tetrahydro-1,2-oxazine derivative 61e′ or a 5,7-fused bicyclic furo[3,4-d][1,2]-oxazepine 61e (Zhang 2010 [149]).
Scheme 61.
Regioselective control in the diastereoselective 1,3-dipolar cycloaddition reactions of 1-(1-alkynyl)cyclopropyl ketones 61a with nitrones 61b using Sc(III) or Au(I) catalysts to afford a tetrahydro-1,2-oxazine derivative 61e′ or a 5,7-fused bicyclic furo[3,4-d][1,2]-oxazepine 61e (Zhang 2010 [149]).

Scheme 62.
The reaction between O-propargylic-oximes 62a and dipolarophiles 62b catalyzed by Cu(I) or Au(I) to afford an oxazepine derivative 62f′ or isoxazole 62e, respectively (Nakamura 2013, 2015 [152,153]).

Scheme 63.
Gold(I)-catalyzed regiodivergent hydration-cyclization of diynones 63a that can lead to the formation of 3(2H)-furanones 63b using Ph3PAuCl/AgSbF6 or 4-pyrones 63c using IPrAuNTf2 (Sanz 2020 [154]).
Scheme 63.
Gold(I)-catalyzed regiodivergent hydration-cyclization of diynones 63a that can lead to the formation of 3(2H)-furanones 63b using Ph3PAuCl/AgSbF6 or 4-pyrones 63c using IPrAuNTf2 (Sanz 2020 [154]).

Scheme 64.
Ligand controlled gold(I)-catalyzed regioselective intramolecular hydroarylation of N-(3-iodoprop-2-ynyl)-N-tosylanilines 64a leading to 4-iodide quinoline 64b with (2,4-tBu2C6H3O)3PAuCl/AgBF4 and 3-iodide quinoline 64c with the IPrAuNTf2; in the case of (2,4-tBu2C6H3O)3PAuCl, the AgBF4 is used as Cl− scavenger (González 2011 [155]).
Scheme 64.
Ligand controlled gold(I)-catalyzed regioselective intramolecular hydroarylation of N-(3-iodoprop-2-ynyl)-N-tosylanilines 64a leading to 4-iodide quinoline 64b with (2,4-tBu2C6H3O)3PAuCl/AgBF4 and 3-iodide quinoline 64c with the IPrAuNTf2; in the case of (2,4-tBu2C6H3O)3PAuCl, the AgBF4 is used as Cl− scavenger (González 2011 [155]).

Scheme 65.
Chemoselectivity control in gold(I)-catalyzed transformation of 1-(alkynyl)-7-oxabicyclo[0,1,4]heptan-2-ones 65a to benzofurans 65c with catalyst IPrAuCl/AgOTf and 6,7-dihydrobenzofunan-4(5H)-ones 65b with catalyst Ph3PAuCl/AgOTf (Hashmi 2013 [156]).
Scheme 65.
Chemoselectivity control in gold(I)-catalyzed transformation of 1-(alkynyl)-7-oxabicyclo[0,1,4]heptan-2-ones 65a to benzofurans 65c with catalyst IPrAuCl/AgOTf and 6,7-dihydrobenzofunan-4(5H)-ones 65b with catalyst Ph3PAuCl/AgOTf (Hashmi 2013 [156]).

Scheme 66.
Gold(I)-catalyzed transformation of ortho-(propargyloxy)aryl methylenecyclopropanes 66a to cyclobutene-substituted 2H-chromene 66b or methylenecyclopropane-2H-chromene 66c (Shi 2016 [157]).
Scheme 66.
Gold(I)-catalyzed transformation of ortho-(propargyloxy)aryl methylenecyclopropanes 66a to cyclobutene-substituted 2H-chromene 66b or methylenecyclopropane-2H-chromene 66c (Shi 2016 [157]).

Scheme 67.
Ligand-dependent competitive activation modes of terminal alkyne 67a to quinazolin-2-one 67b and indole 67c (Medio-Simón 2014 [158]).
Scheme 67.
Ligand-dependent competitive activation modes of terminal alkyne 67a to quinazolin-2-one 67b and indole 67c (Medio-Simón 2014 [158]).

Scheme 68.
Ligand-dependent [5+2] vs. [5+1] annulations between ynamides 68a and 1,2-benzisoxazoles 68b to heterocyclic derivatives 68c or 68d (Liu 2018 [159]).
Scheme 68.
Ligand-dependent [5+2] vs. [5+1] annulations between ynamides 68a and 1,2-benzisoxazoles 68b to heterocyclic derivatives 68c or 68d (Liu 2018 [159]).

Scheme 69.
Gold(I)-catalyzed [4+2] vs. [4+3] cycloaddition of allene-dienes 69a that can result in 69b or 69c (Toste 2009 [160]).
Scheme 69.
Gold(I)-catalyzed [4+2] vs. [4+3] cycloaddition of allene-dienes 69a that can result in 69b or 69c (Toste 2009 [160]).

Scheme 70.
Tuning π-acceptor ability of NHC ligands to control the reaction outcome of the gold(I)-catalyzed [3+2] versus [2+2] cycloaddition of allene-dienes 70a to 70b or 70c/70d, respectively (Fürstner 2010 [162]).
Scheme 70.
Tuning π-acceptor ability of NHC ligands to control the reaction outcome of the gold(I)-catalyzed [3+2] versus [2+2] cycloaddition of allene-dienes 70a to 70b or 70c/70d, respectively (Fürstner 2010 [162]).

Scheme 71.
Gold (I)-catalyzed tandem cyclization of 3-propargylindole 71a to 3-(inden-2-yl)indoles 71b or 71c initiated by 1,2-indole migration (Sanz 2008, 2011 [163,164]).

Scheme 72.
The counterion effect in the cationic gold(I) catalytic cycle; (a) no active proton is involved (e.g., NuH=alkene); (b) an active proton is involved (e.g., NuH=RO-H, R2N-H) [169].
Scheme 72.
The counterion effect in the cationic gold(I) catalytic cycle; (a) no active proton is involved (e.g., NuH=alkene); (b) an active proton is involved (e.g., NuH=RO-H, R2N-H) [169].

Scheme 73.
Gold(I)-catalyzed cycloisomerization reaction of 1,6-enyne 73a to 73b [170].
Scheme 73.
Gold(I)-catalyzed cycloisomerization reaction of 1,6-enyne 73a to 73b [170].

Scheme 74.
Gold(I)-catalyzed hydration reaction of alkyne 74a to ketone 74b and mechanistic details (Zuccaccia 2016 [171]).
Scheme 74.
Gold(I)-catalyzed hydration reaction of alkyne 74a to ketone 74b and mechanistic details (Zuccaccia 2016 [171]).

Scheme 75.
Chiral counterion (R)-TRIP can induce enantioselectivity [172].
Scheme 75.
Chiral counterion (R)-TRIP can induce enantioselectivity [172].

Scheme 76.
Chiral counterion strategy for enantioselective functionalization of allenes (ee = enantiomeric excess) [172].
Scheme 76.
Chiral counterion strategy for enantioselective functionalization of allenes (ee = enantiomeric excess) [172].

Scheme 77.
The counterion directing divergent cycloisomerization of homopropargyl ketones 77a to furans 77b or 77c (Li and Gevorgyan 2010 [173]).
Scheme 77.
The counterion directing divergent cycloisomerization of homopropargyl ketones 77a to furans 77b or 77c (Li and Gevorgyan 2010 [173]).

Scheme 78.
Counterion directing divergent dearomatization of indoles 78a with allenamides 78b through the formation of 78c or 78d (Bandini 2014, 2015 [174,175]).

Scheme 79.
Pyridine N-oxide acting as a hydrogen bonding acceptor additive in the gold(I)-catalyzed cyclization of propargyl amide 79a to form 79c (Hammond 2021 [126]).
Scheme 79.
Pyridine N-oxide acting as a hydrogen bonding acceptor additive in the gold(I)-catalyzed cyclization of propargyl amide 79a to form 79c (Hammond 2021 [126]).

Scheme 80.
Triazole-gold(I) complex is used as a gold(I) catalyst to convert compounds 80a to phenols 80b (Shi 2010) [176].
Scheme 80.
Triazole-gold(I) complex is used as a gold(I) catalyst to convert compounds 80a to phenols 80b (Shi 2010) [176].

Scheme 81.
Yb(III) activation of gold(I) in the rearrangement reaction of epoxy alkynes 81a to compounds 81b (Shi 2010 [178]).
Scheme 81.
Yb(III) activation of gold(I) in the rearrangement reaction of epoxy alkynes 81a to compounds 81b (Shi 2010 [178]).

Scheme 82.
Two possible ways of action of acidic co-catalyst as a Lewis acid or Brönsted–Lowry acid, see structures 82a or 82b, respectively [126].
Scheme 82.
Two possible ways of action of acidic co-catalyst as a Lewis acid or Brönsted–Lowry acid, see structures 82a or 82b, respectively [126].

Scheme 83.
(A) Au(I)/Ga(III) catalytic system in ambient Nakamura reaction that converts diketone 83a and alkyne 83b to α,α′-vinyl diketone 83c. (B) Ga(III) diketone complex 83d as an active intermediate formed during the catalytic cycle (Petersen 2014 [180]).
Scheme 83.
(A) Au(I)/Ga(III) catalytic system in ambient Nakamura reaction that converts diketone 83a and alkyne 83b to α,α′-vinyl diketone 83c. (B) Ga(III) diketone complex 83d as an active intermediate formed during the catalytic cycle (Petersen 2014 [180]).

Scheme 84.
Study of protodeauration using acids of different strengths.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
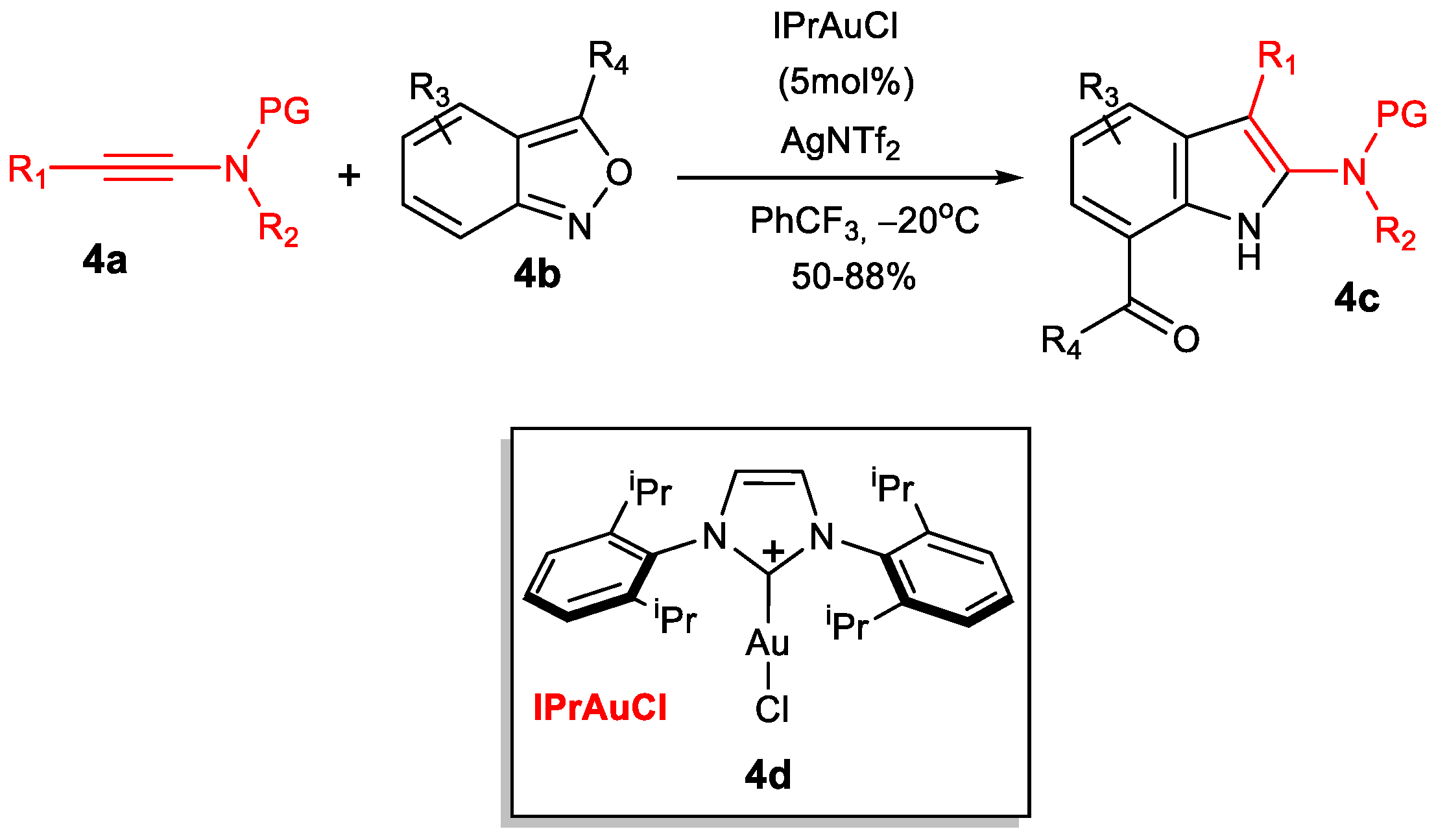{kind=link}
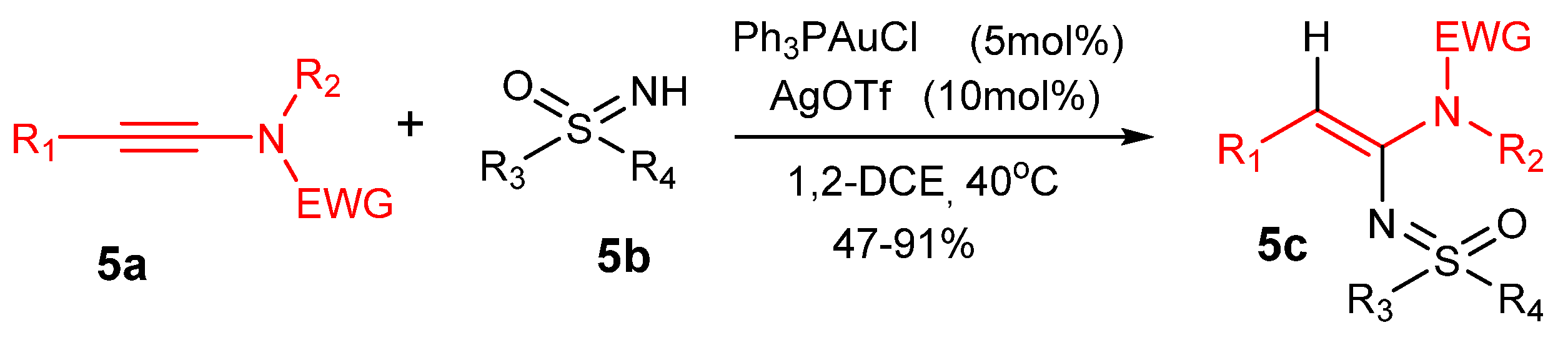{kind=link}
{kind=link}
{kind=link}
{kind=link}
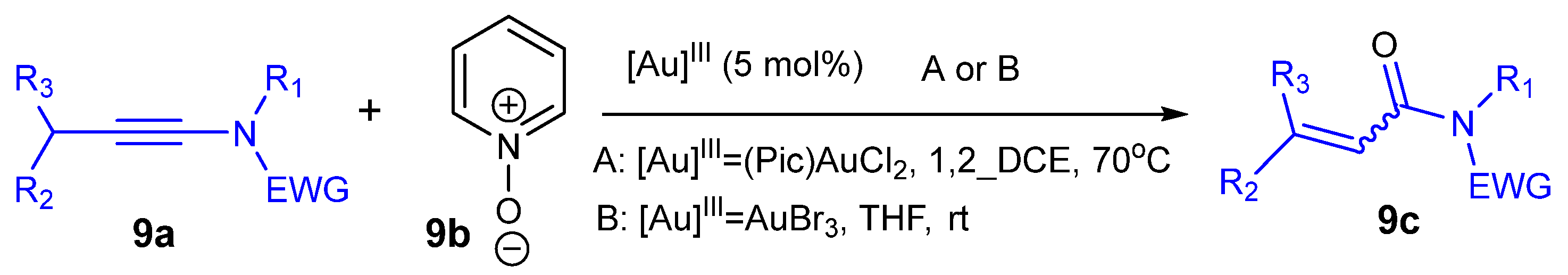{kind=link}
{kind=link}
{kind=link}
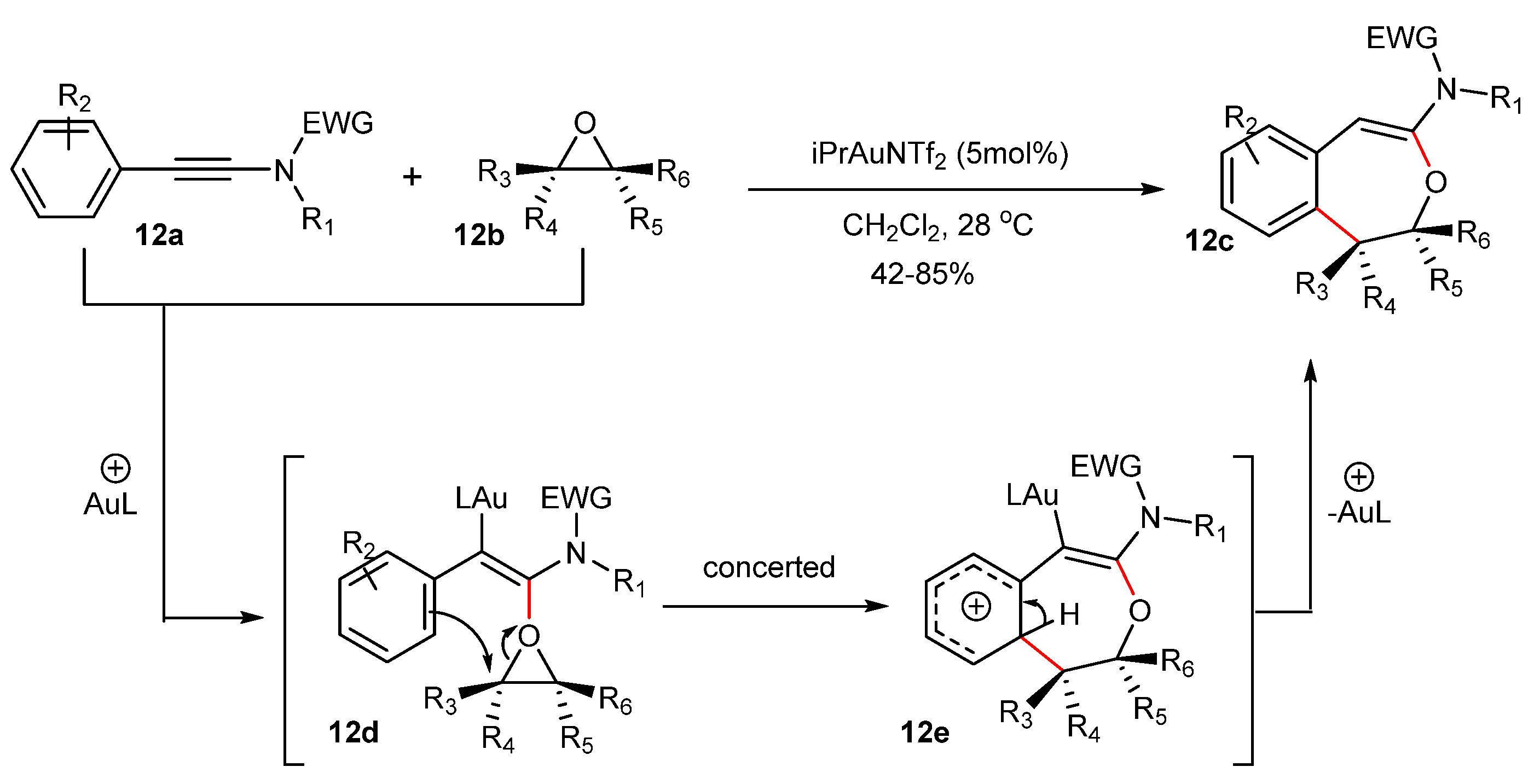{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
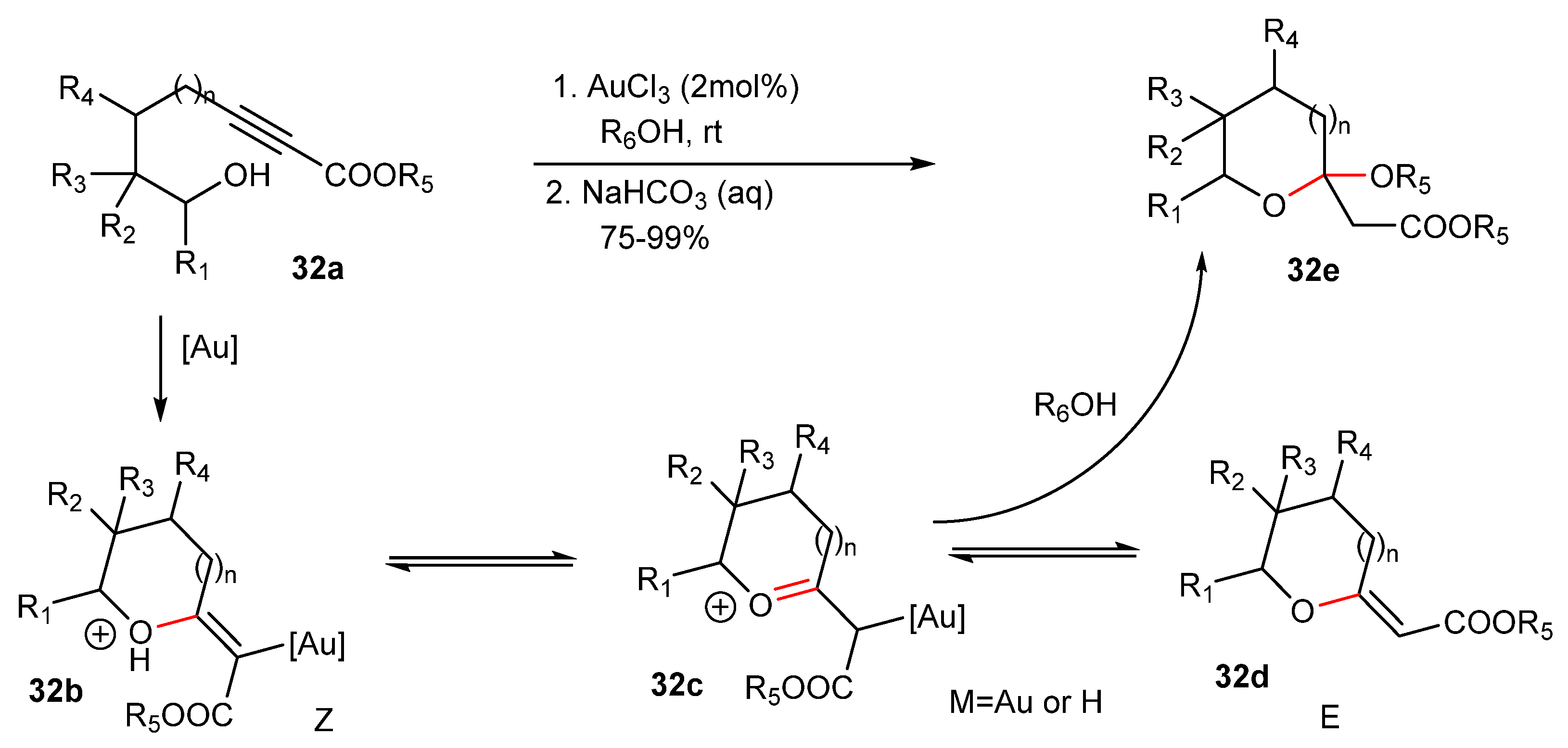{kind=link}
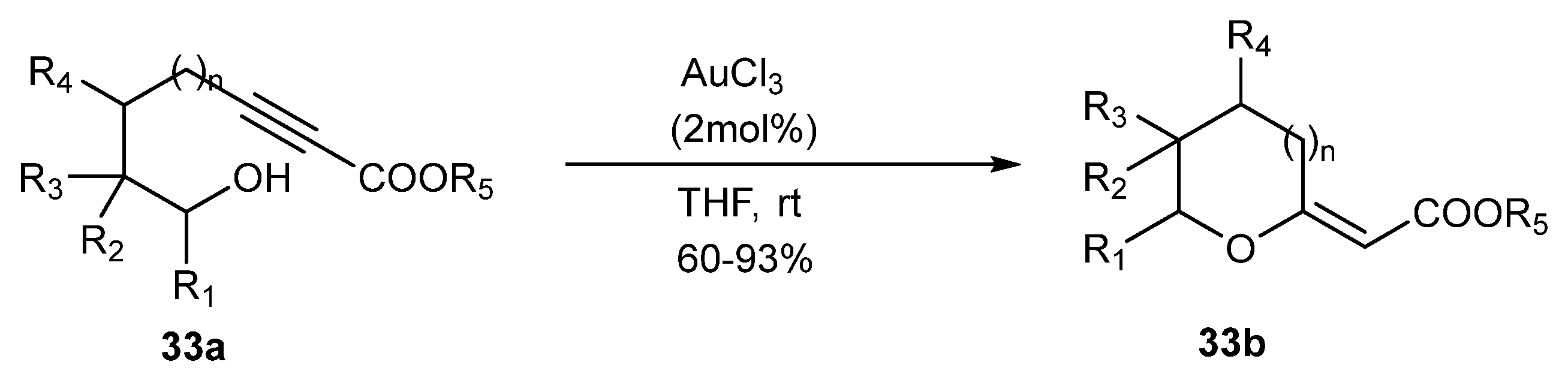{kind=link}
{kind=link}
{kind=link}
{kind=link}
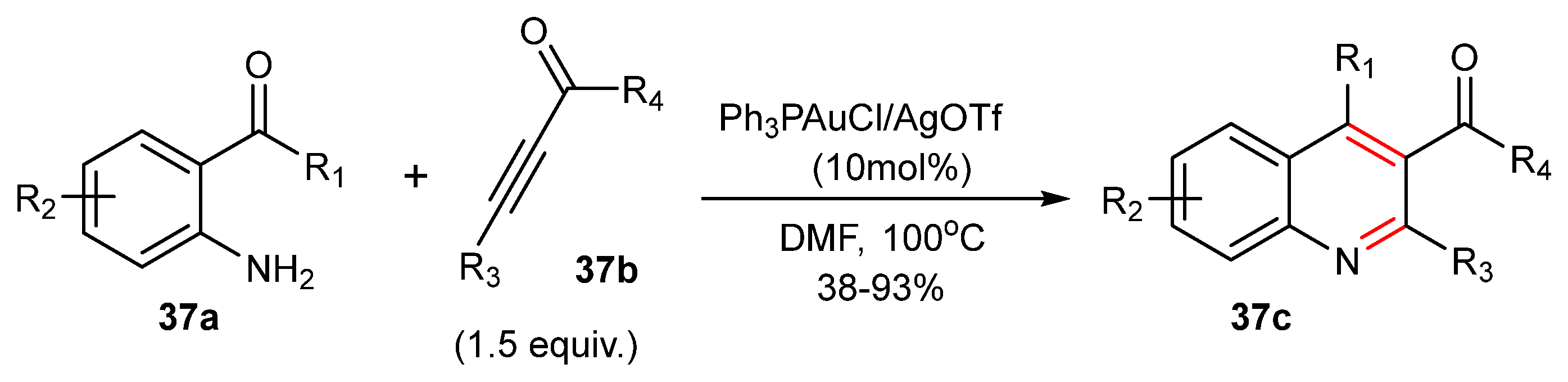{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
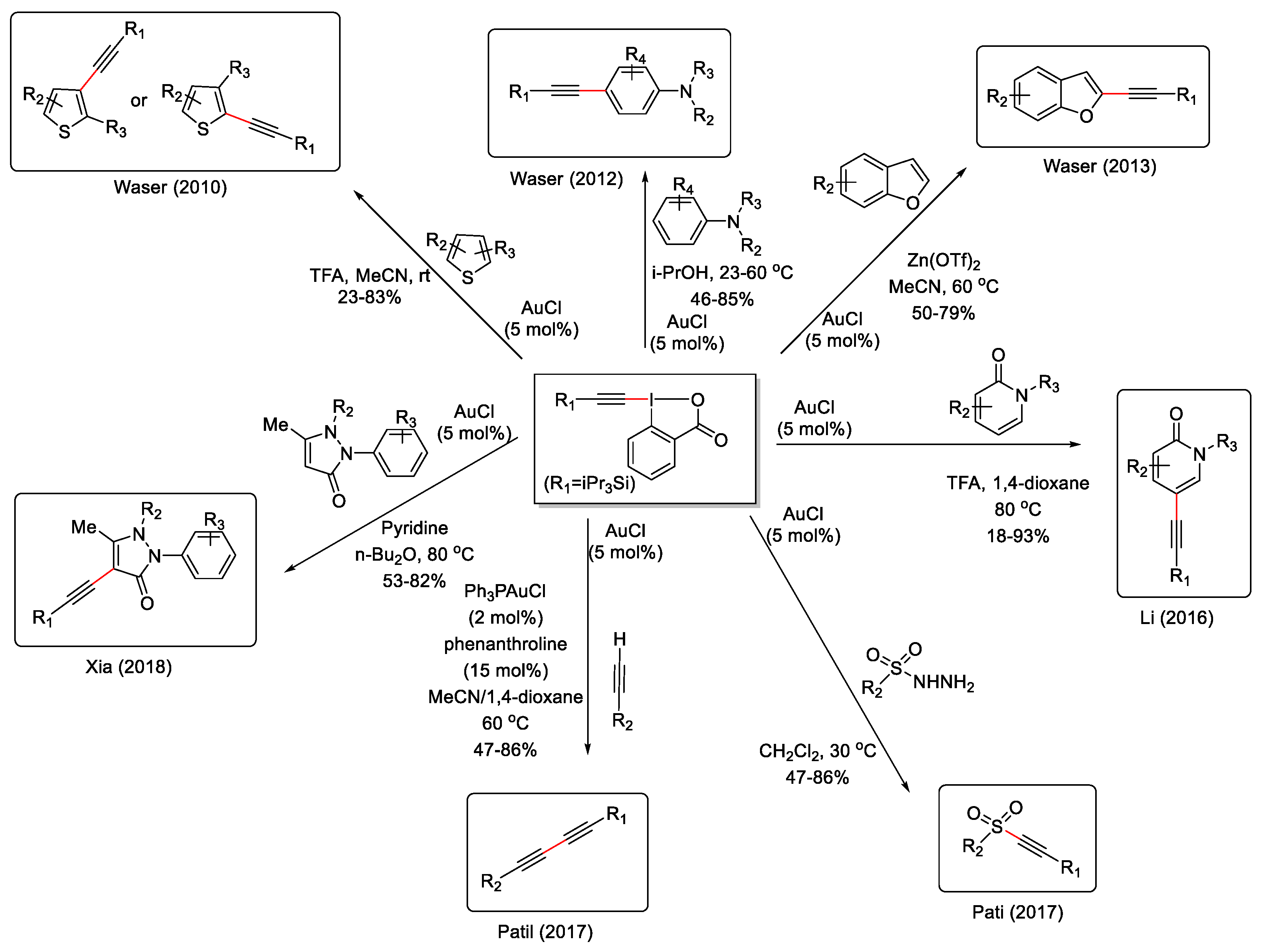{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
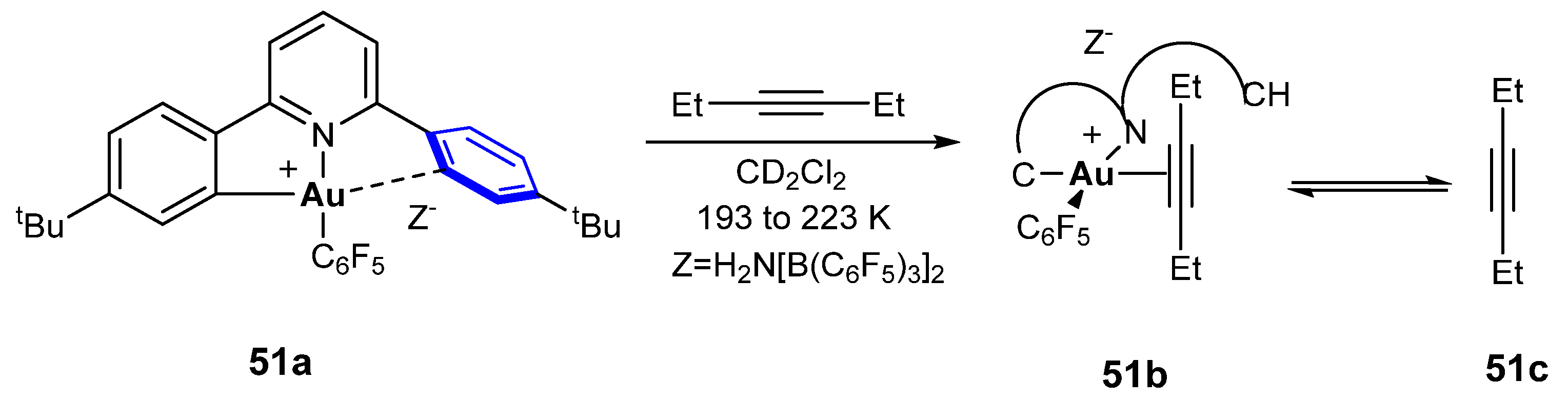{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
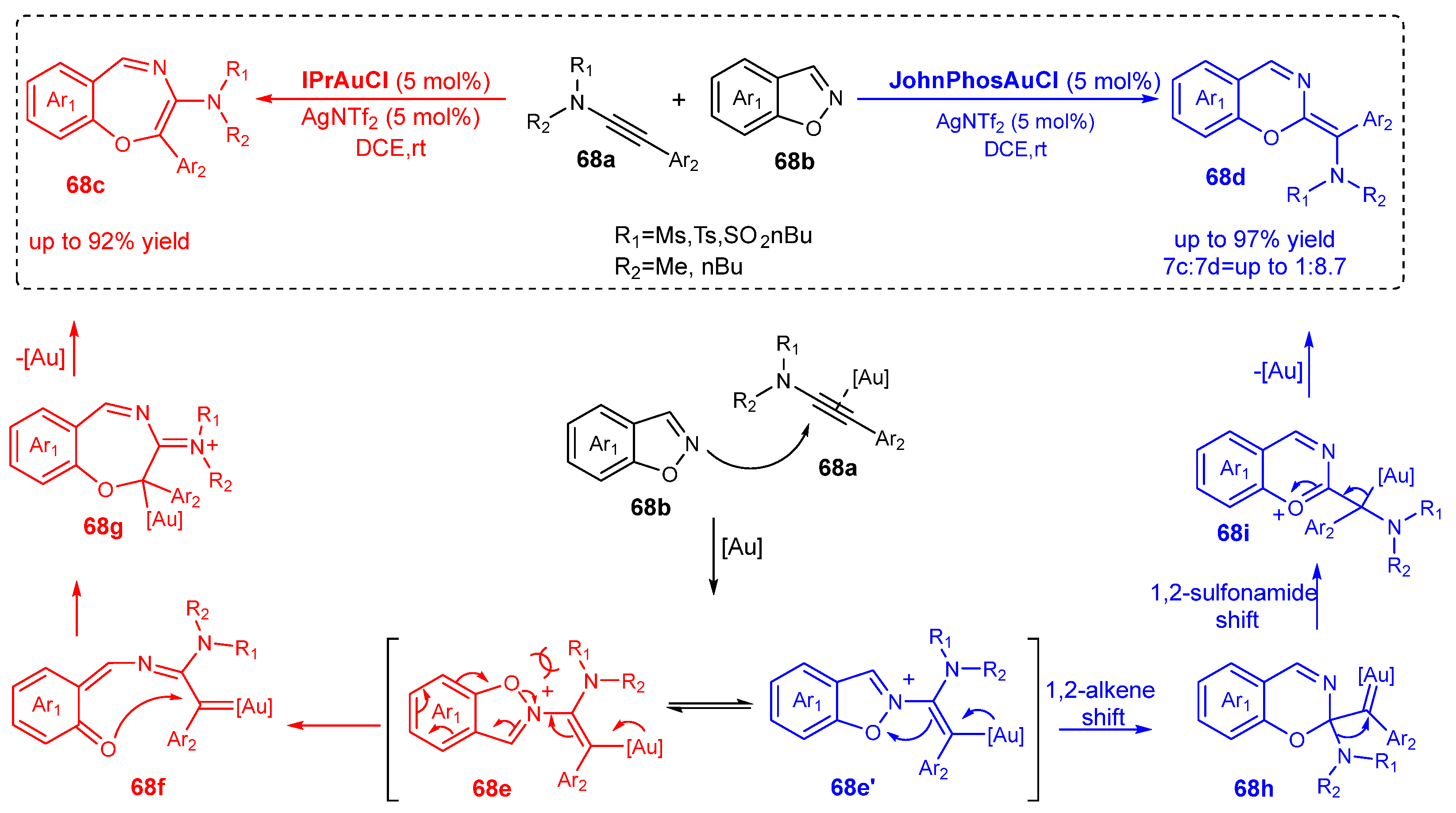{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Gold(I) affinity-related counterion effect on cycloisomerization of 1,6-enyne [170].
Table 1.
Gold(I) affinity-related counterion effect on cycloisomerization of 1,6-enyne [170].
| X− | AcO− | TfO− | BF4− | SbF6− | CTf3− | Al[OC(CF3)3]4− |
|---|---|---|---|---|---|---|
| Gold affinity index | 6.1 | 2.4 | 0.5 | 0 | 0.2 | ~0 |
| Relative initial rate | 0.0 | 1.0 | 7.1 | 21 | 32 | 42 |
Table 2.
Experimental data of the counterion effect on the gold(I)-catalyzed hydration reaction of alkyne [171].
Table 2.
Experimental data of the counterion effect on the gold(I)-catalyzed hydration reaction of alkyne [171].
| X | BF4 | SbF6 | BArF | OTf | NTf2 | OTs | TFA | NTf2 |
|---|---|---|---|---|---|---|---|---|
| Ketone yield/% | <1 | <1 | <1 | >99 | >99 | <1 | <1 | >99 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Stylianakis, I.; Kolocouris, A. Comprehensive Overview of Homogeneous Gold-Catalyzed Transformations of π-Systems for Application Scientists. Catalysts 2023, 13, 921. https://doi.org/10.3390/catal13060921
AMA Style
Stylianakis I, Kolocouris A. Comprehensive Overview of Homogeneous Gold-Catalyzed Transformations of π-Systems for Application Scientists. Catalysts. 2023; 13(6):921. https://doi.org/10.3390/catal13060921
Chicago/Turabian StyleStylianakis, Ioannis, and Antonios Kolocouris. 2023. "Comprehensive Overview of Homogeneous Gold-Catalyzed Transformations of π-Systems for Application Scientists" Catalysts 13, no. 6: 921. https://doi.org/10.3390/catal13060921
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.