1,3-Diene Polymerization Mediated by Homoleptic Tetramethylaluminates of the Rare-Earth Metals


Abstract
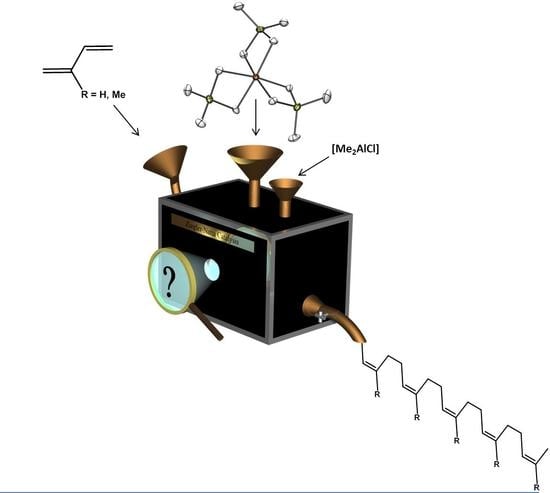
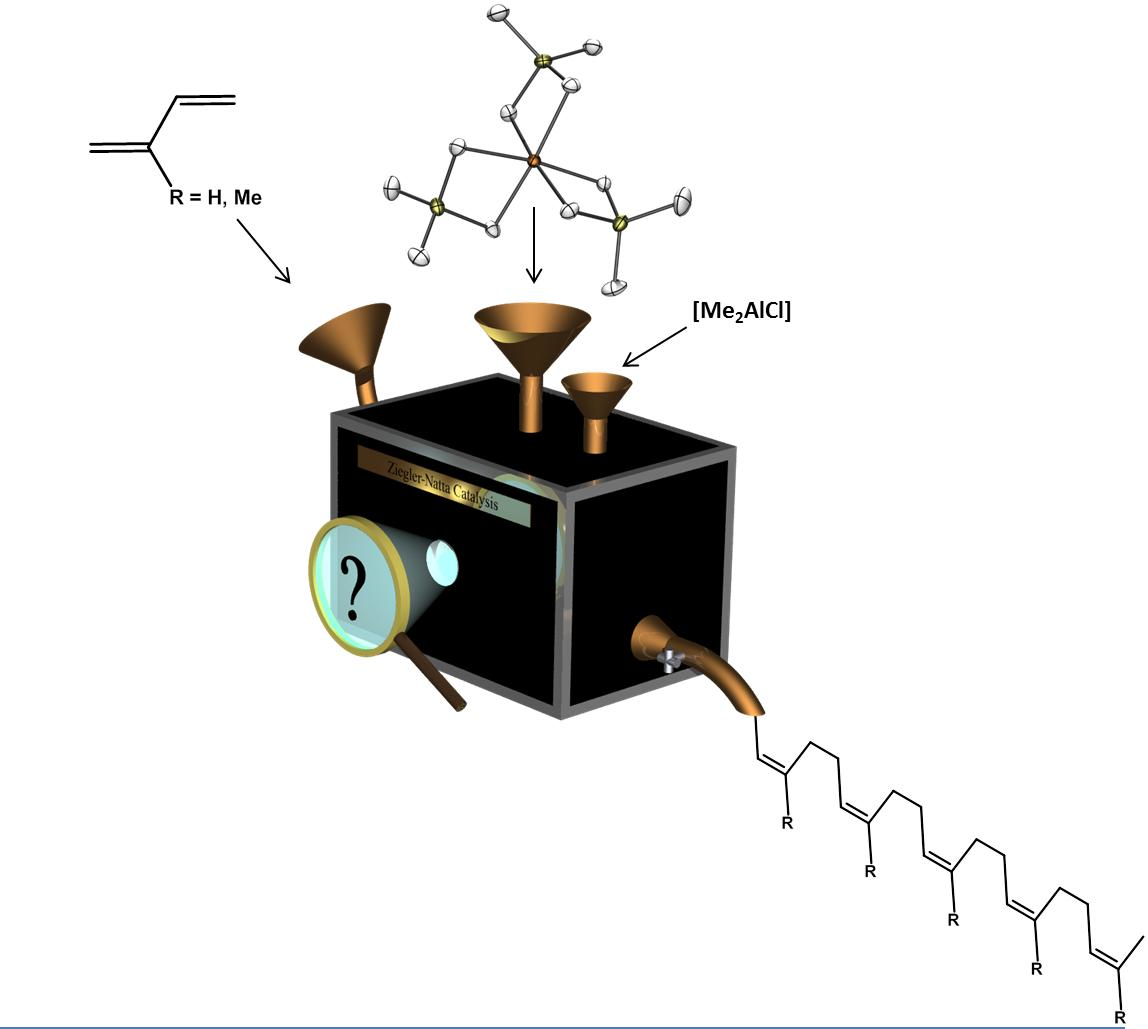
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
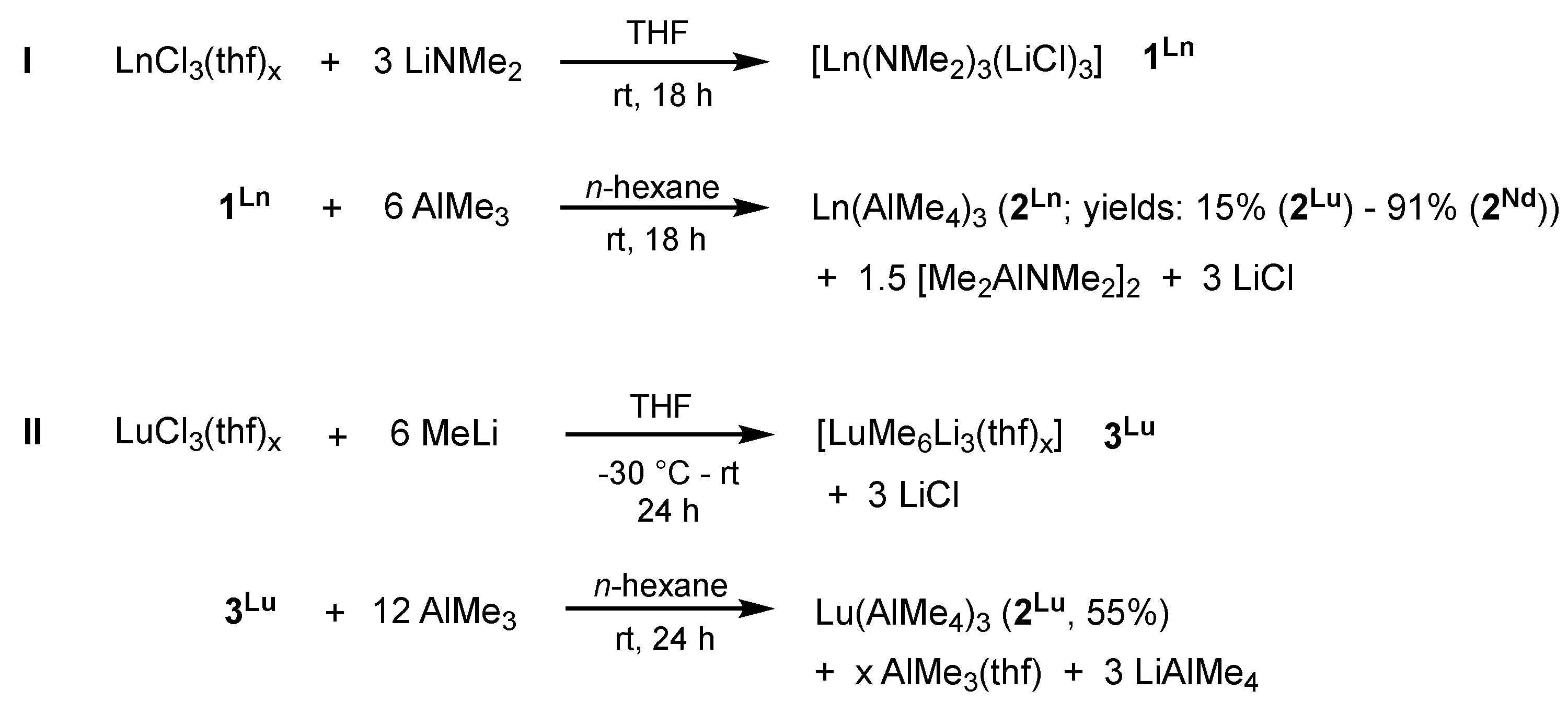{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Catalyst Systems
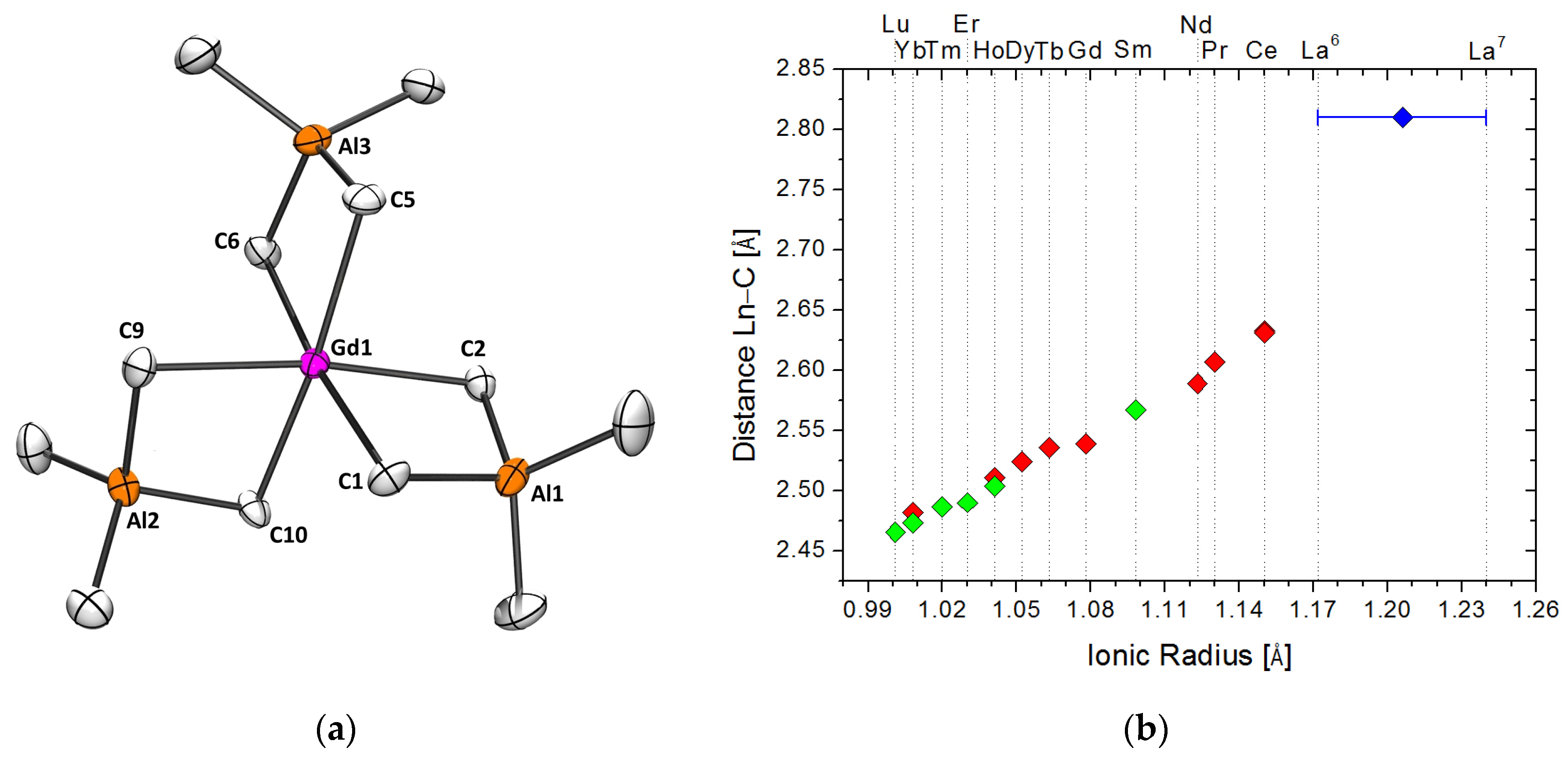
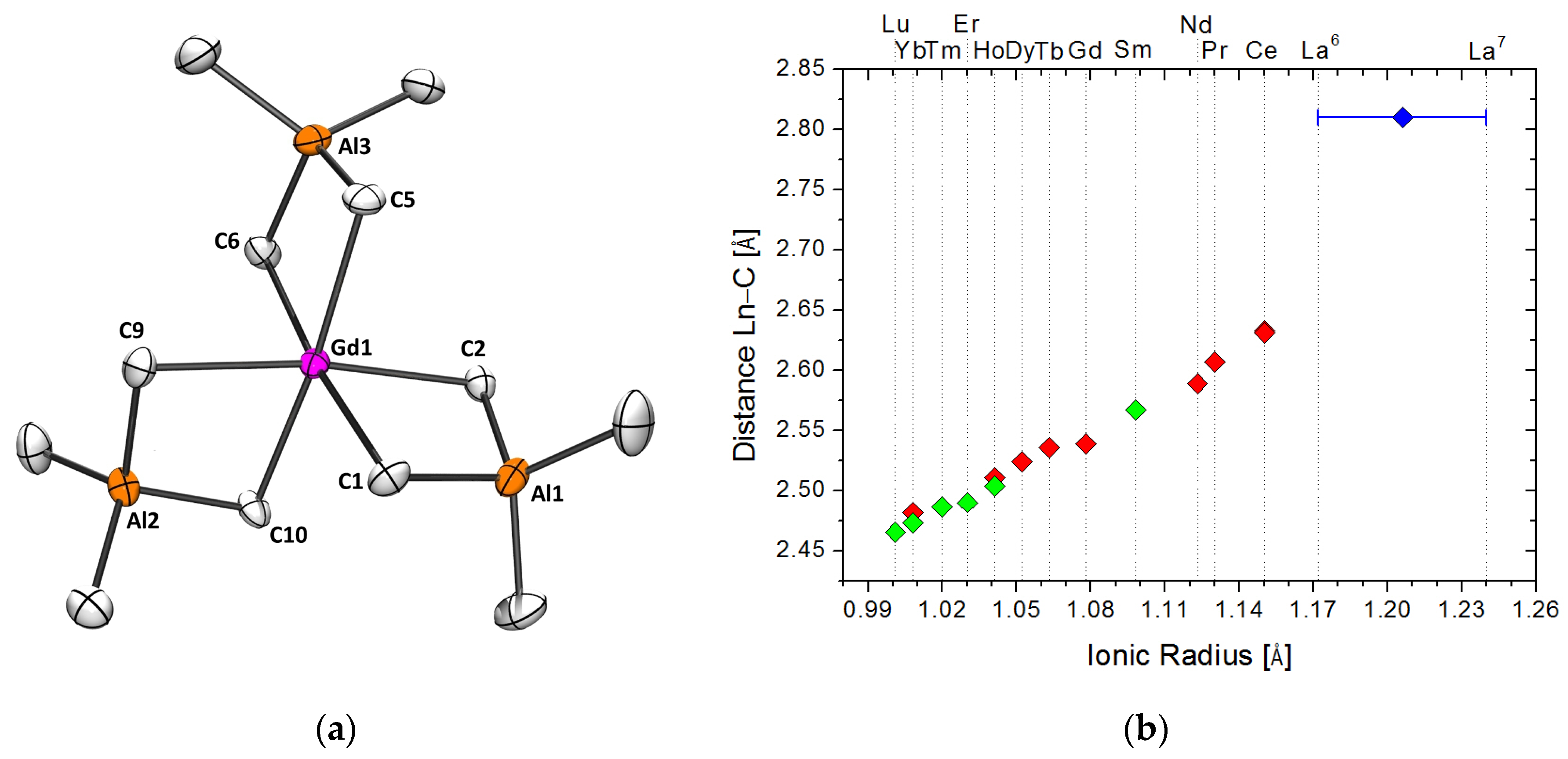
2.1.1. Precatalyst Synthesis and Structural Characterization
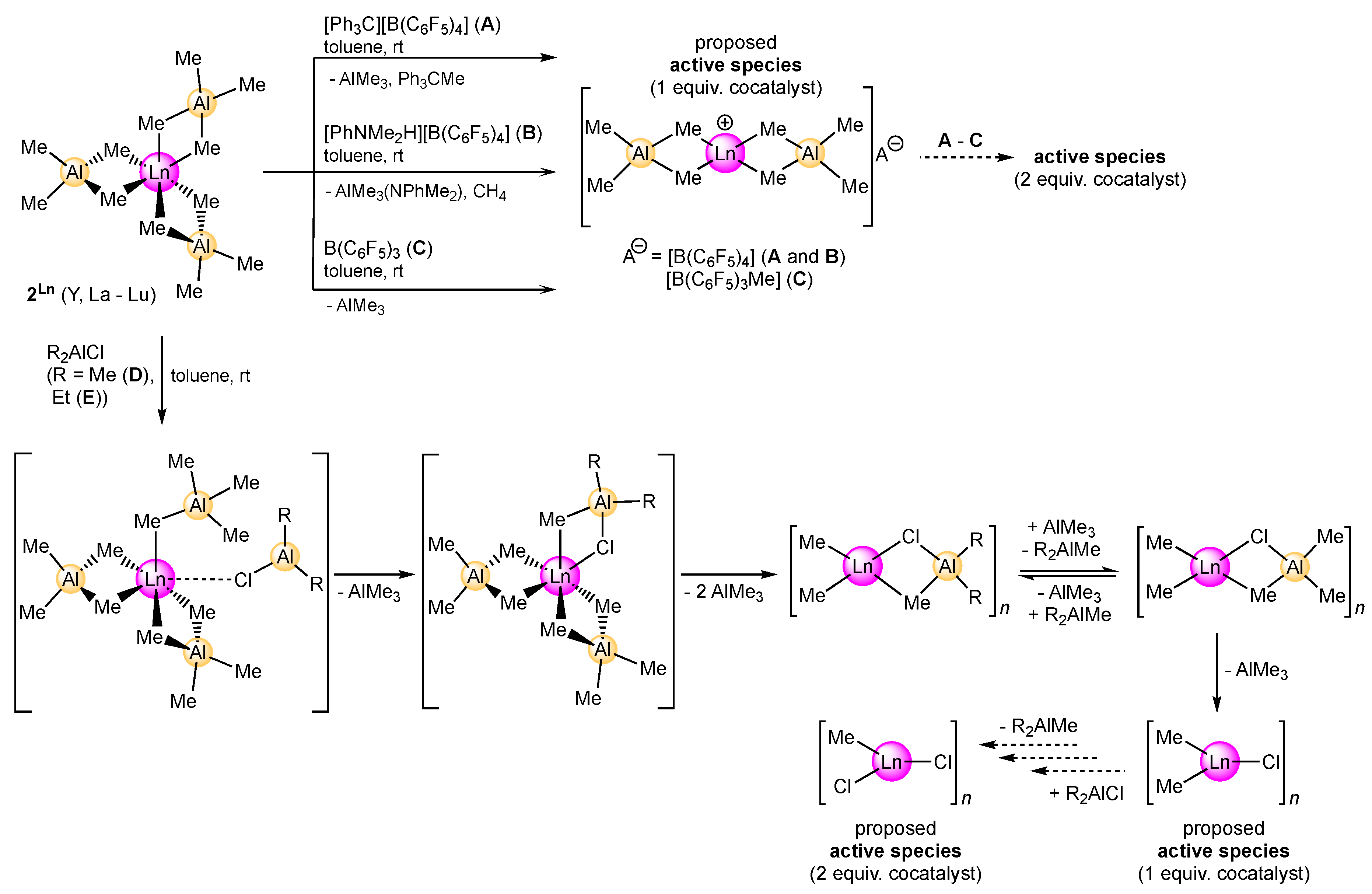
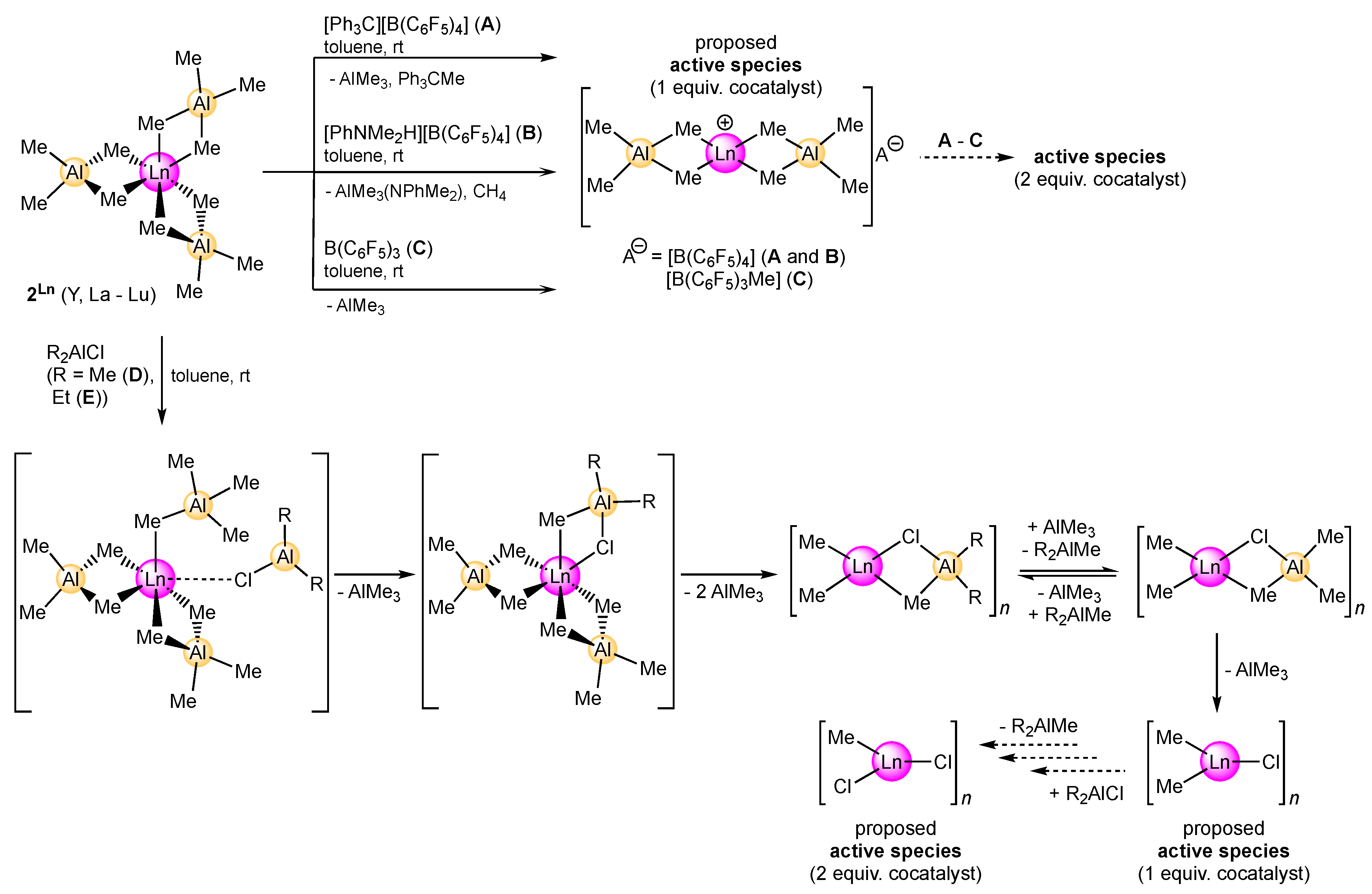
2.1.2. Activation by Cationizing Cocatalysts
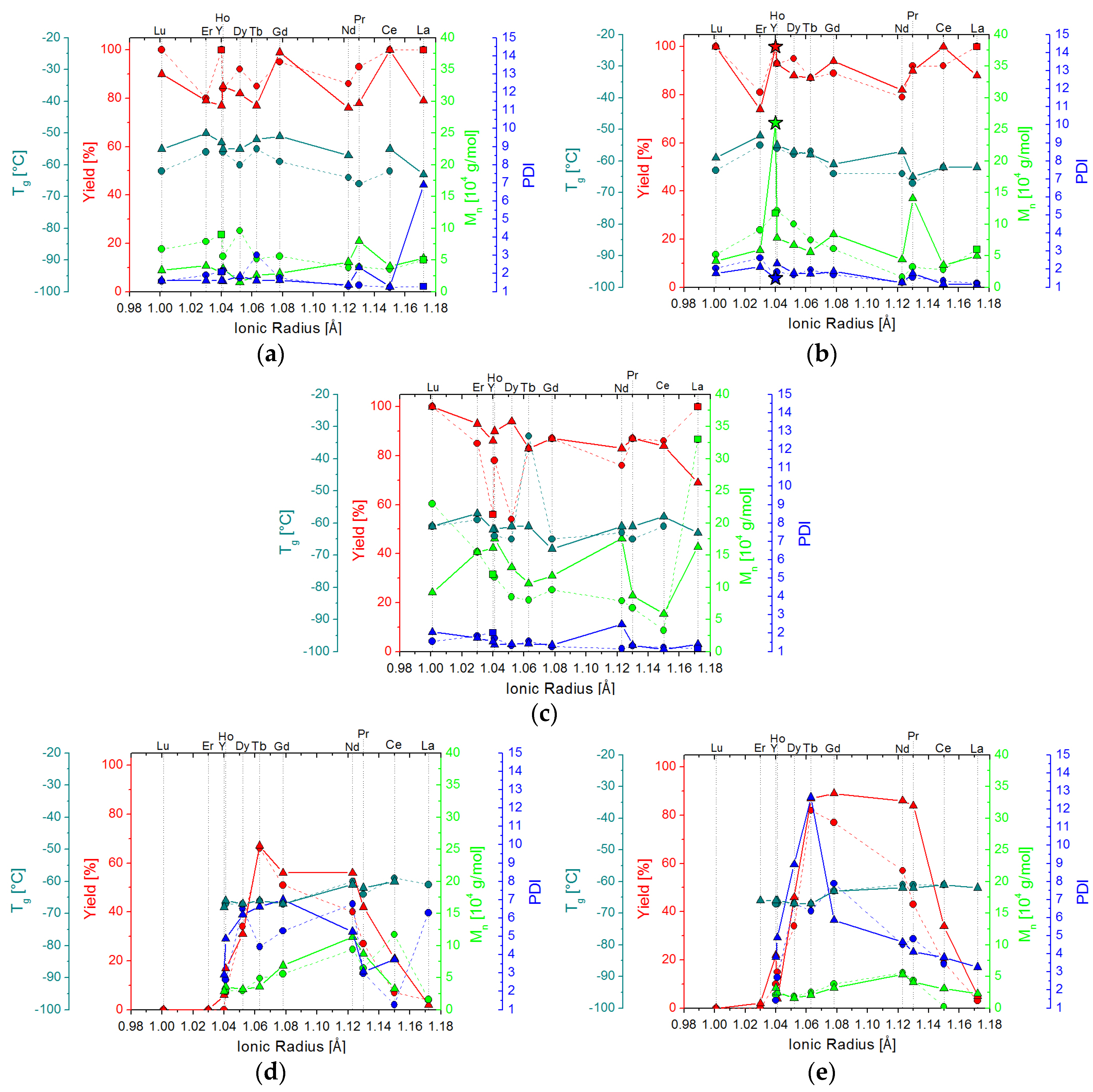
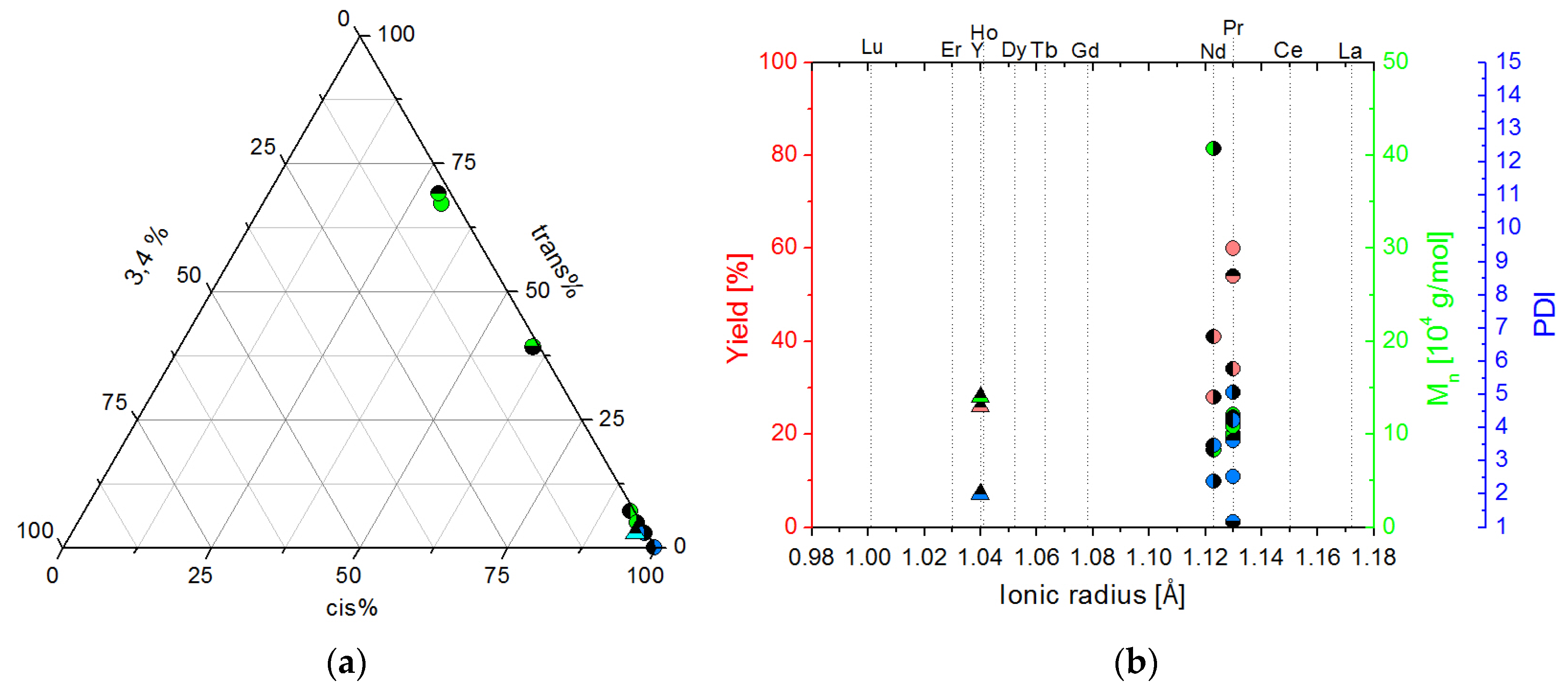
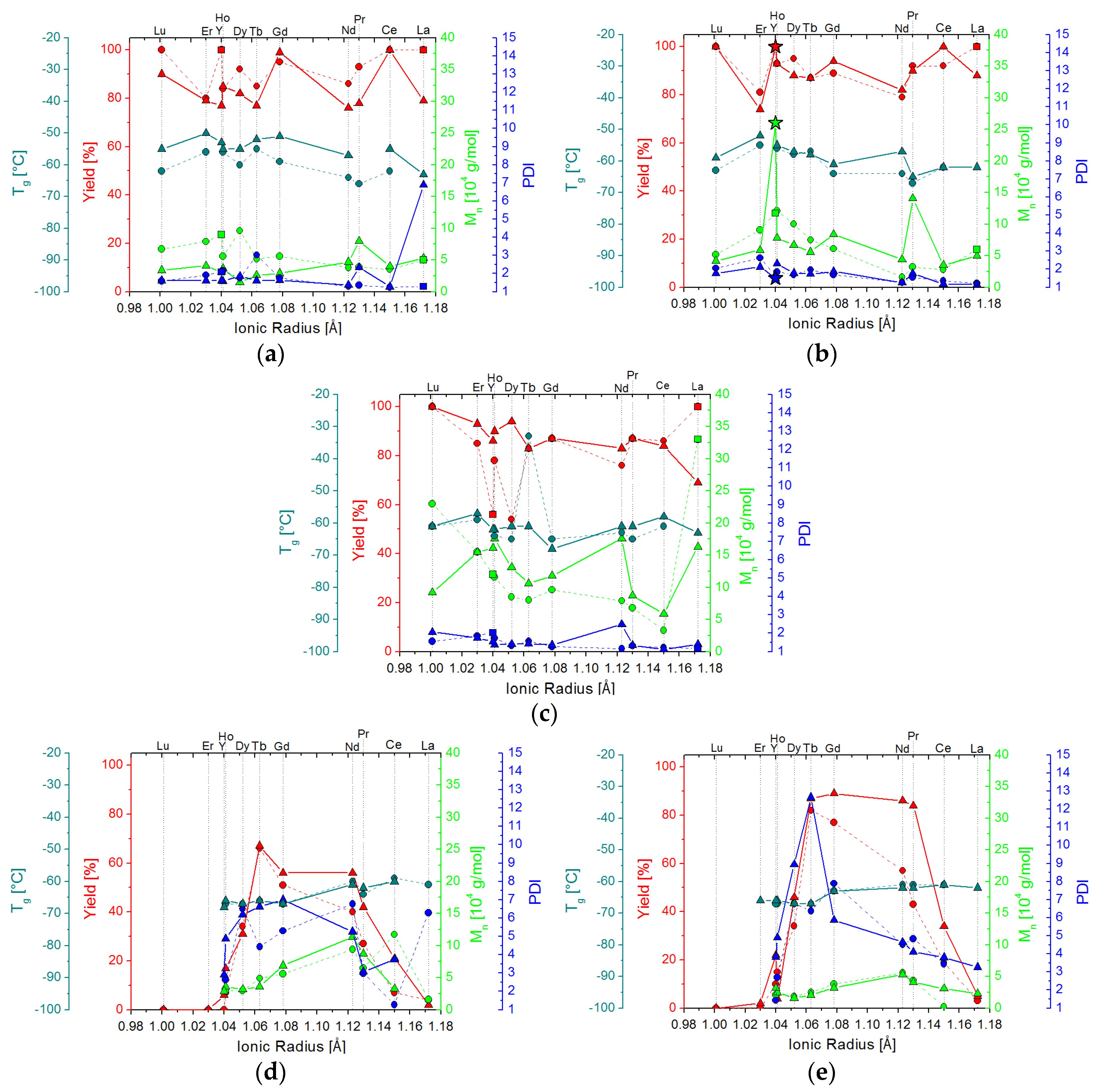
2.2. Isoprene Polymerization Catalysis
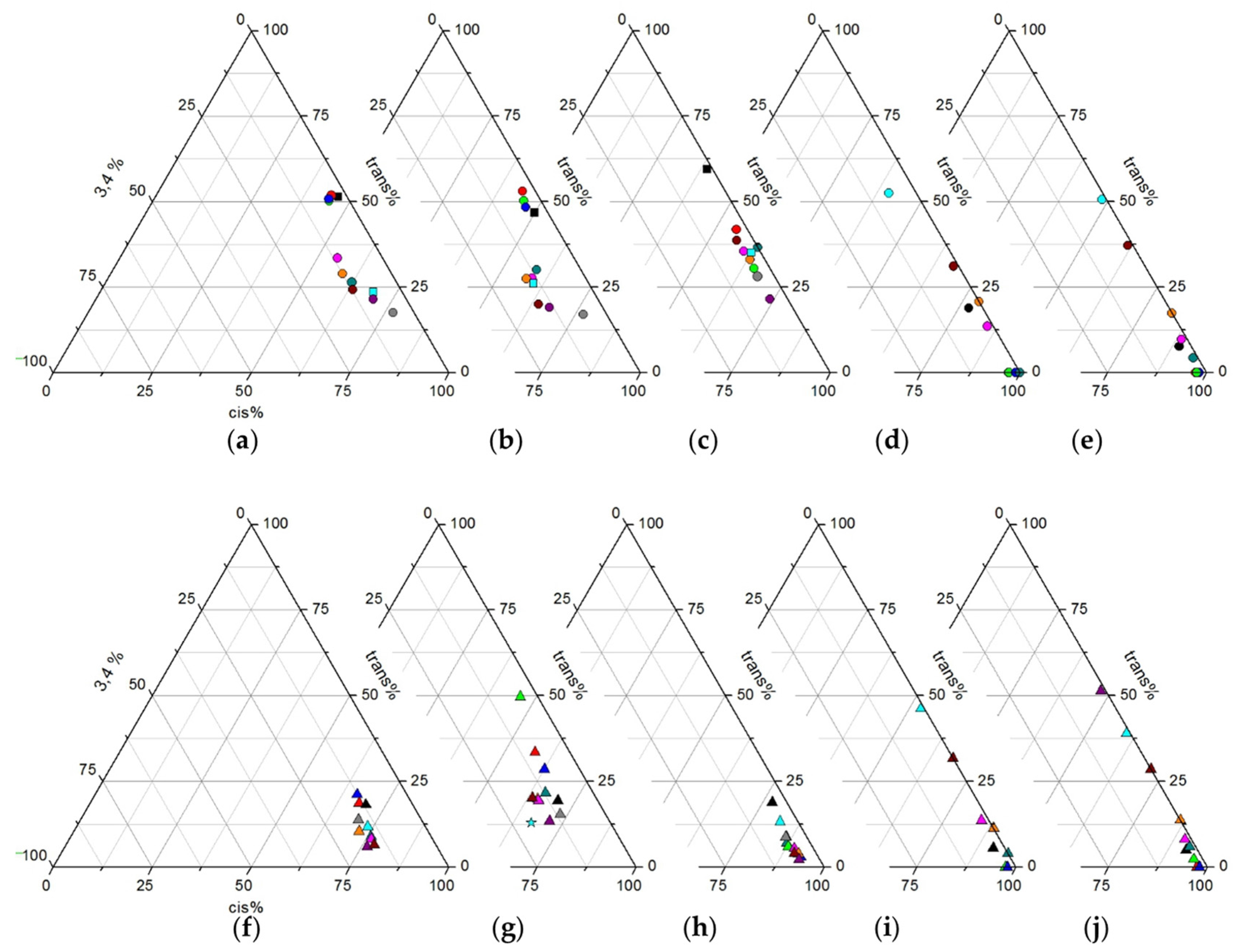
2.2.1. Polymers Obtained at Standard Conditions
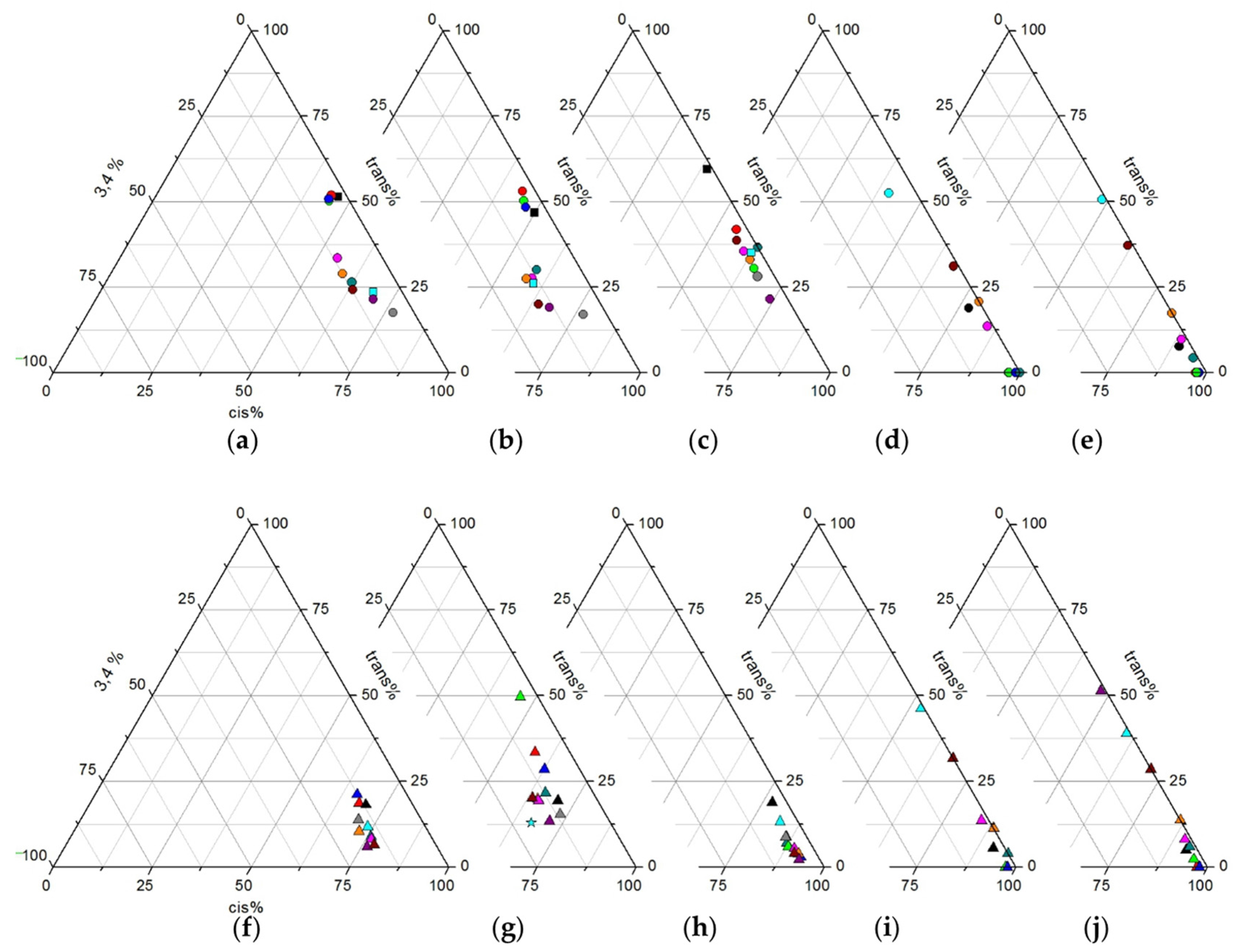
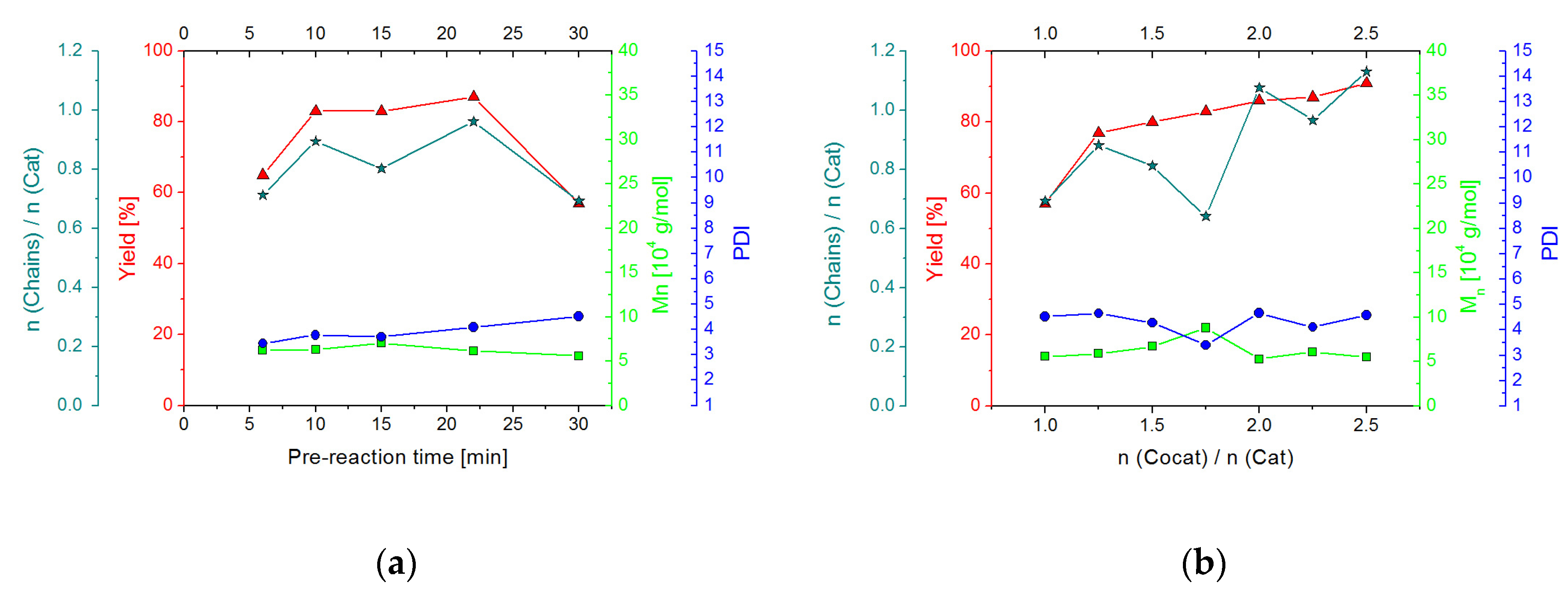
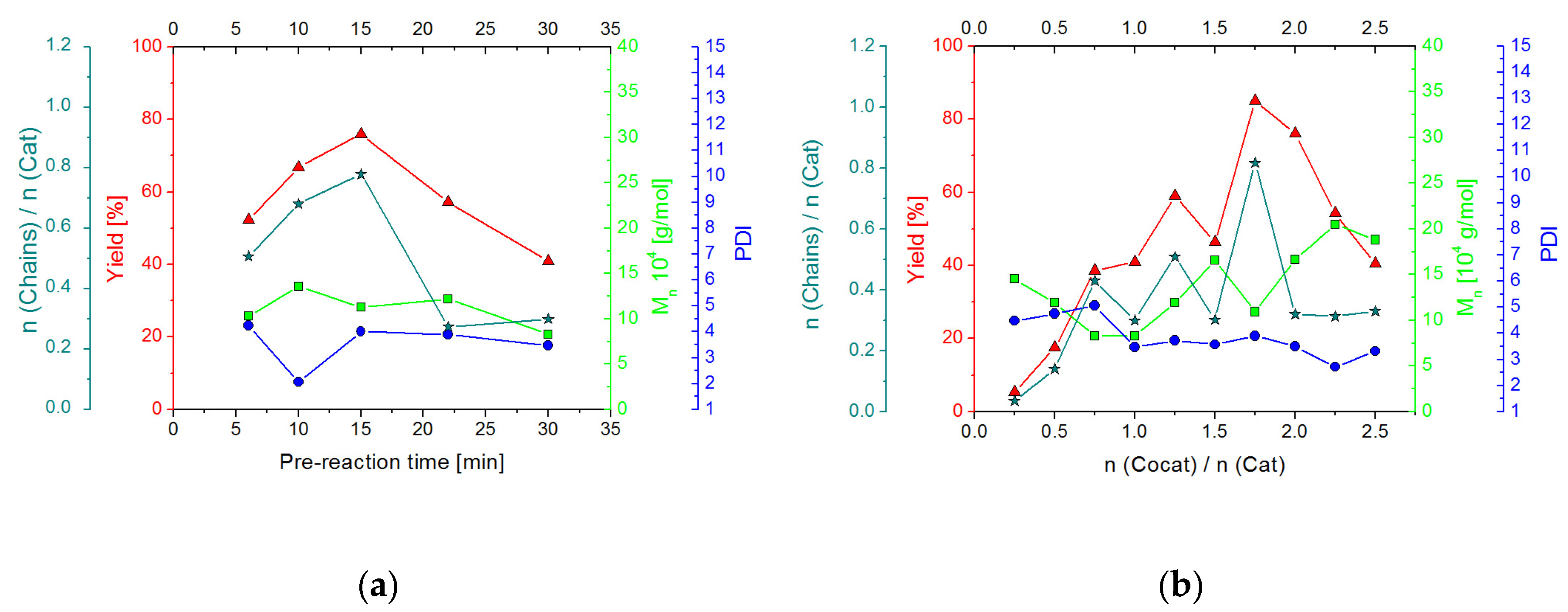
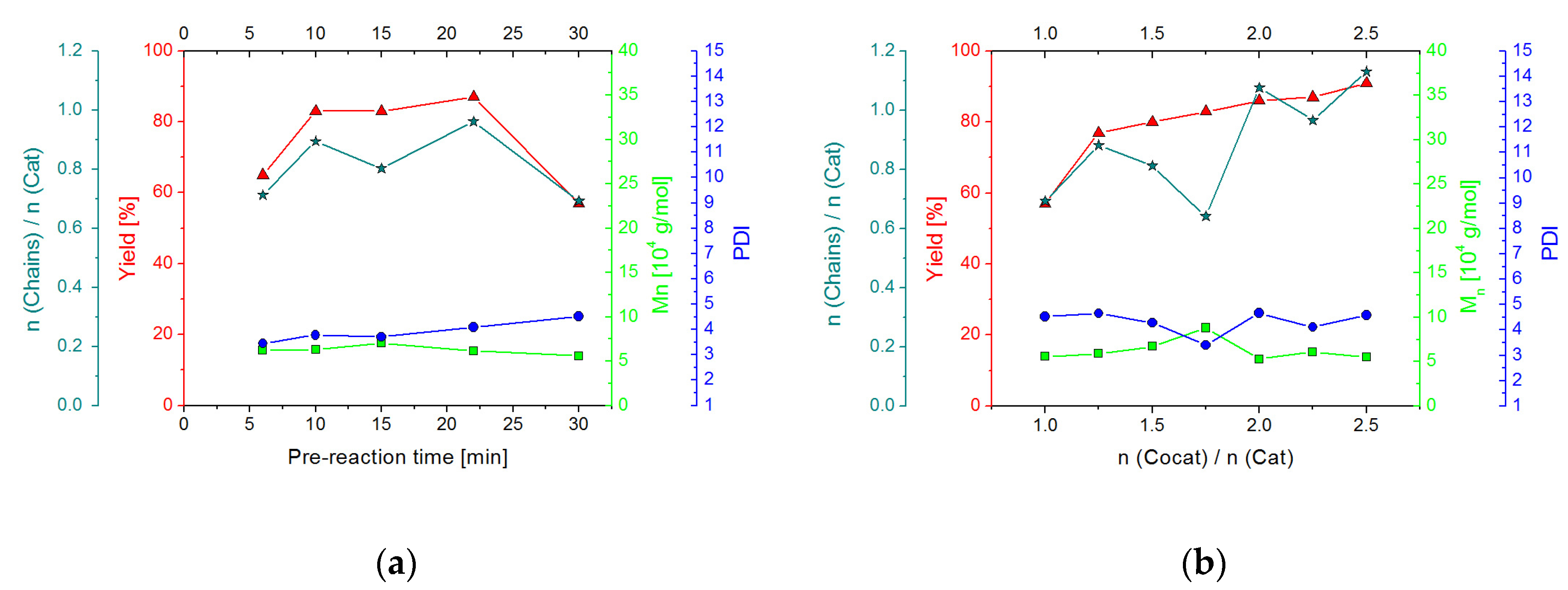
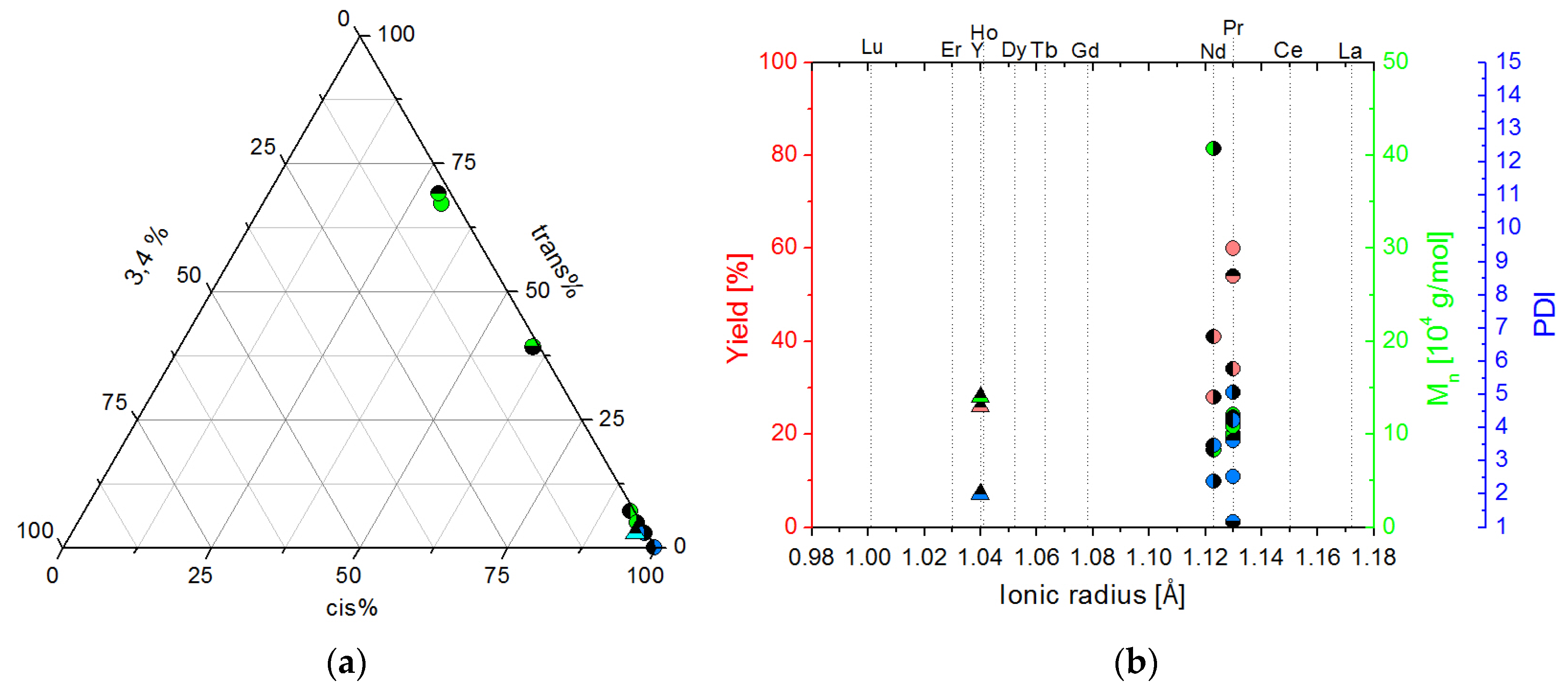
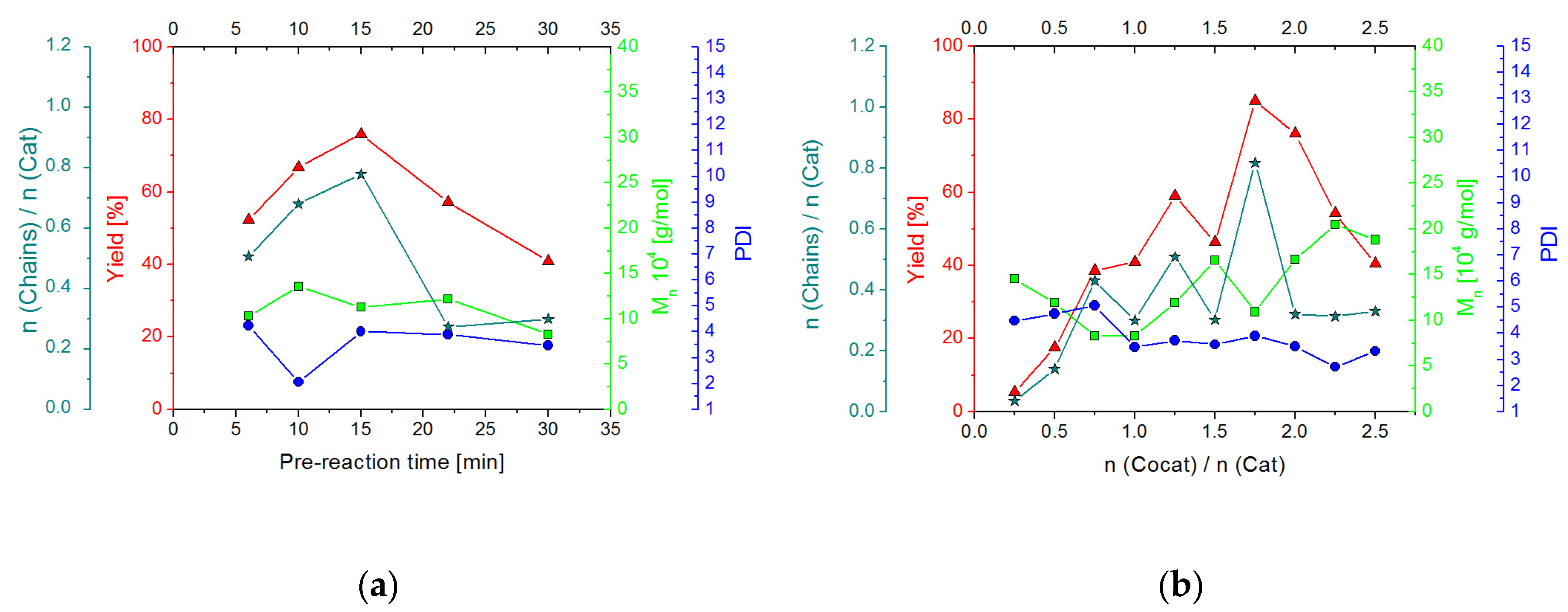
2.2.2. Screening Studies
2.3. 1,3-Butadiene Polymerization Catalysis
2.3.1. Polymers Obtained at Standard Conditions
2.3.2. Screening Studies
2.4. Ethylene Polymerization
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis of Tb(AlMe4)3 (2Tb)
3.3. New Protocol for the Synthesis of Lu(AlMe4)3 (2Lu)
3.4. Polymerization of Isoprene
3.5. Polymerization of 1,3-Butadiene
3.6. Polymerization of Ethylene
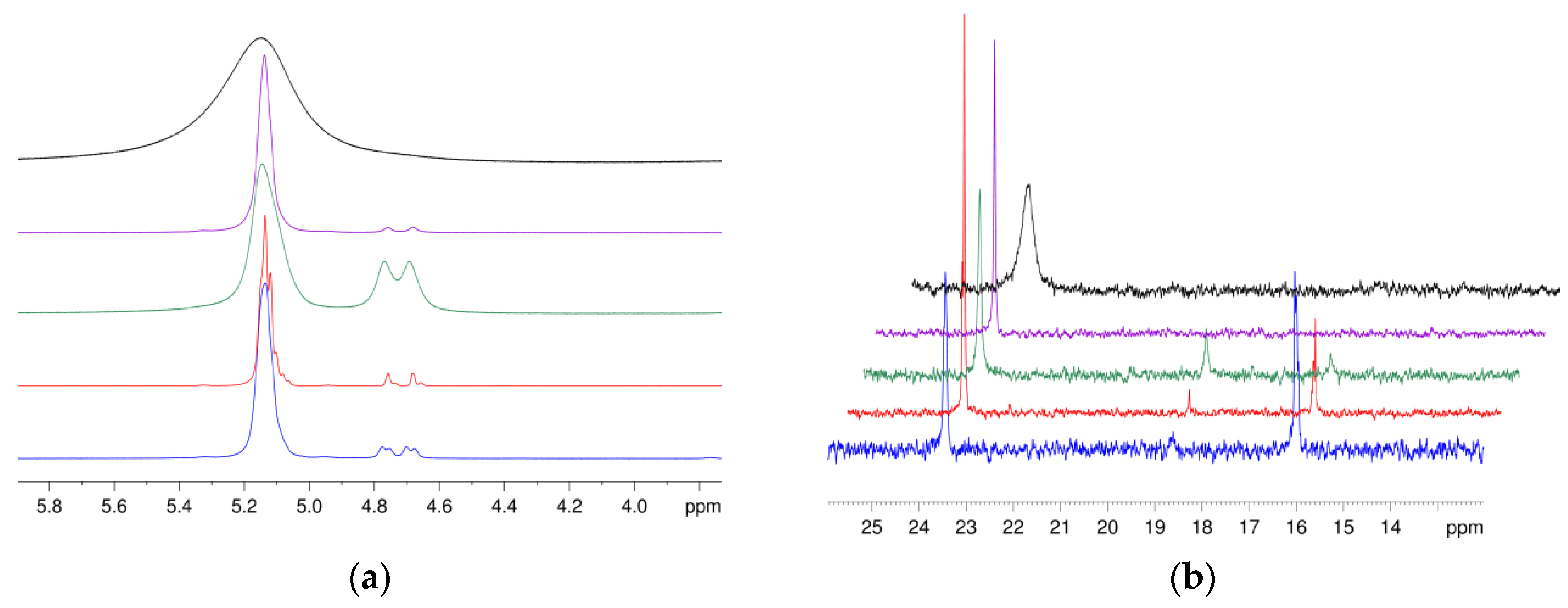
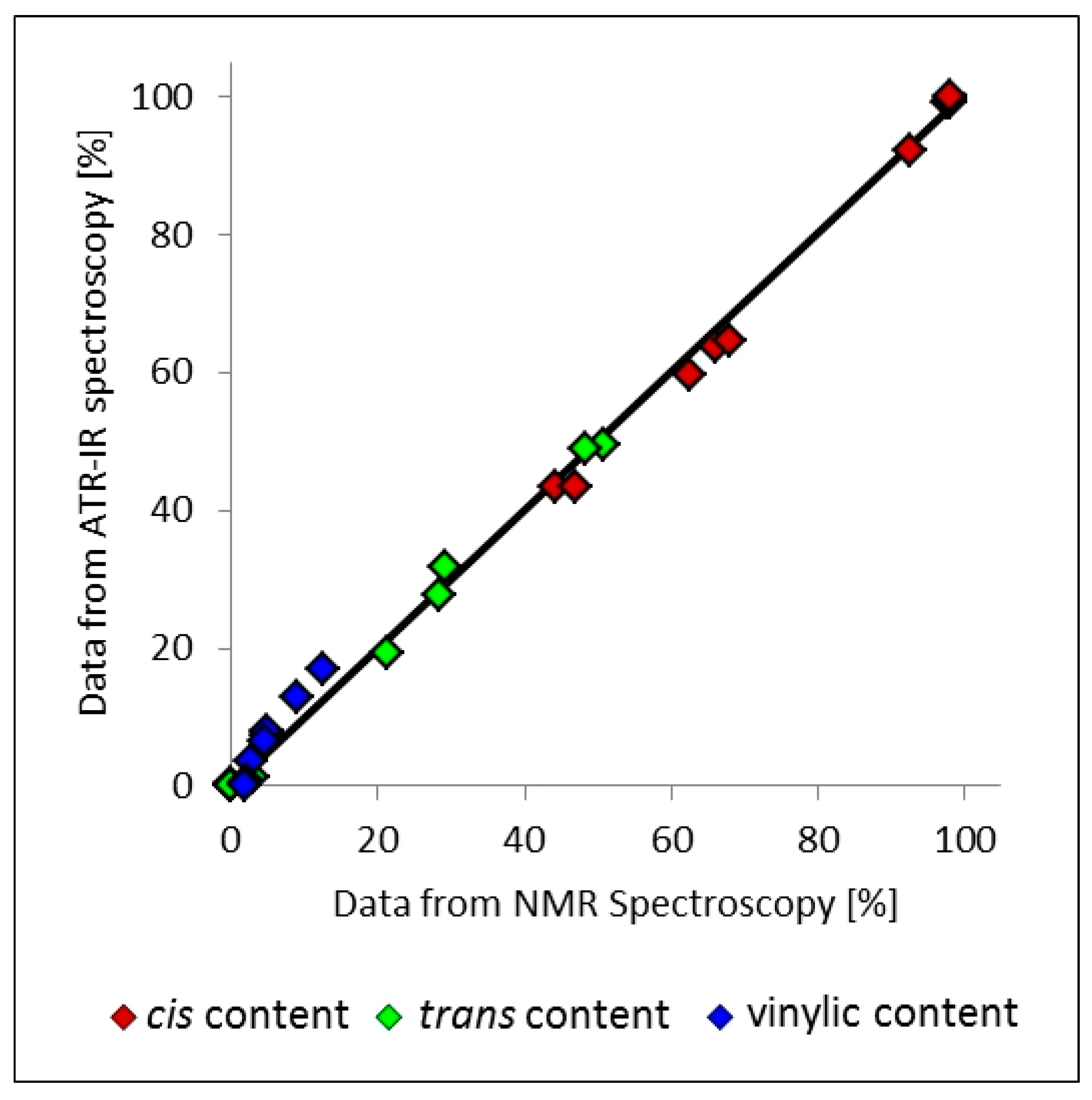
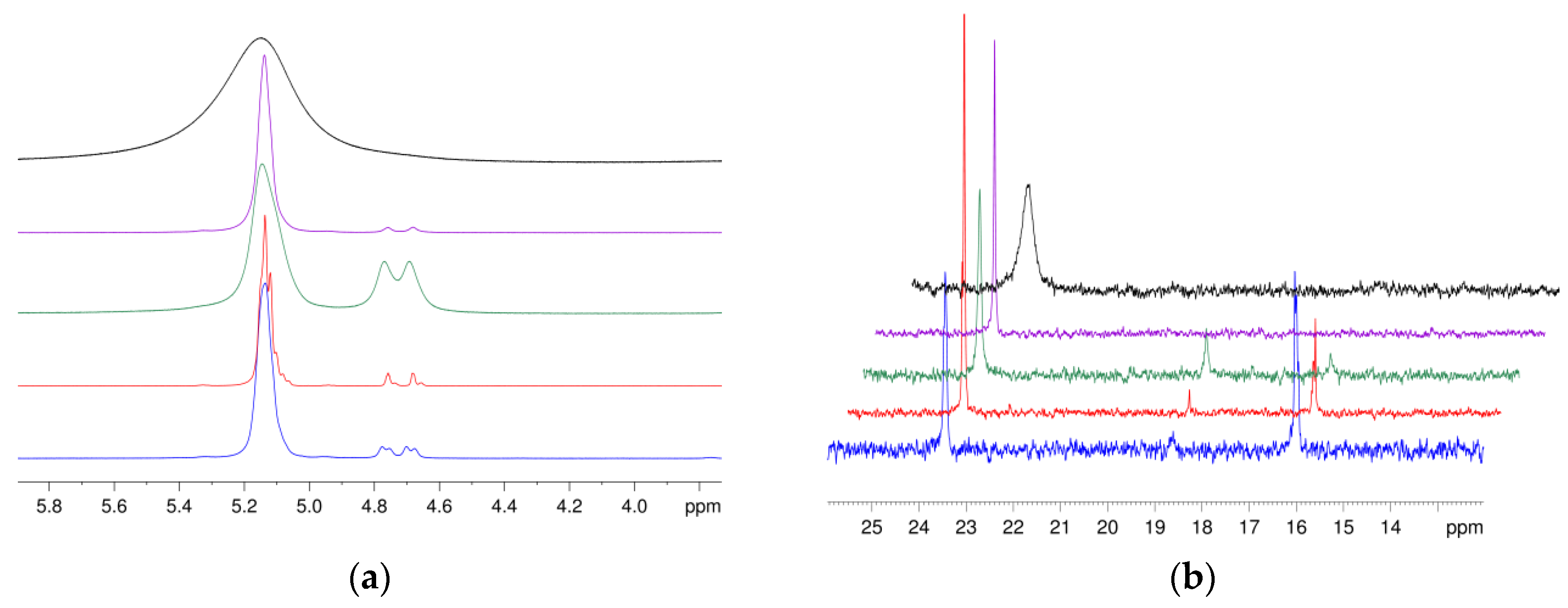
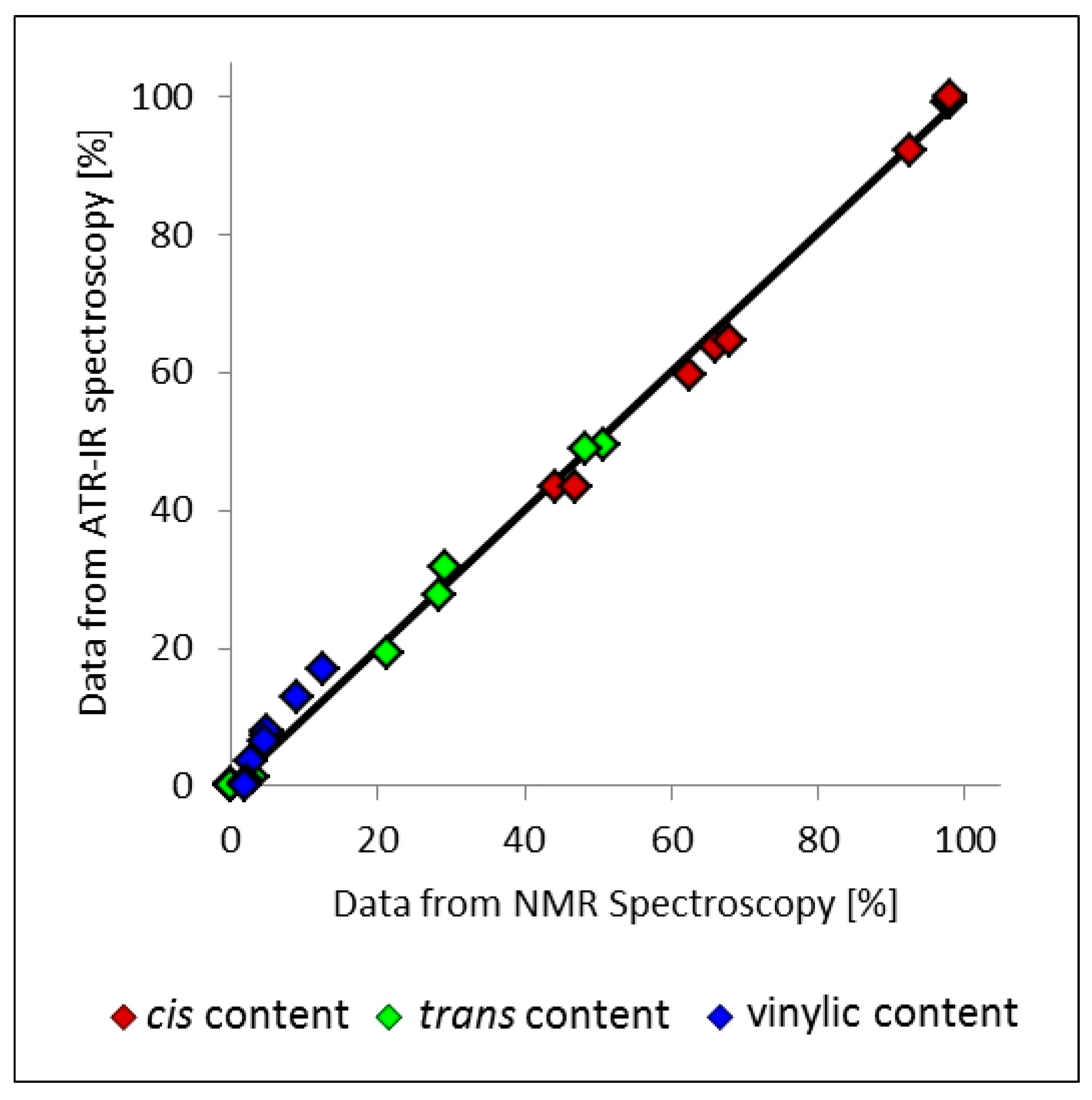
3.7. Application of NMR Spectroscopic Techniques for Microstructure Determination
3.8. Application of ATR-Infrared Spectroscopic Techniques for Microstructure Detection
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boor, J.J. Ziegler-Natta Catalysts and Polymerizations; Academic Press: New York, NY, USA; San Francisco, CA, USA; London, UK, 1979. [Google Scholar]
- Mülhaupt, R. Catalytic Polymerization and Post Polymerization Catalysis Fifty Years After the Discovery of Ziegler’s Catalysts. Macromol. Chem. Phys. 2003, 204, 289–327. [Google Scholar] [CrossRef]
- Eisch, J.J. Fifty Years of Ziegler–Natta Polymerization: From Serendipity to Science. A Personal Account. Organometallics 2012, 31, 4917–4932. [Google Scholar] [CrossRef]
- Fischbach, A.; Anwander, R. Rare-Earth Metals and Aluminum getting Close in Ziegler Type Organometallics. Adv. Polym. Sci. 2006, 204, 155–281. [Google Scholar]
- Friebe, L.; Nuyken, O.; Obrecht, W. Neodymium-Based Ziegler/Natta Catalysts and their Application in Diene Polymerization. Adv. Polym. Sci. 2006, 204, 1–154. [Google Scholar]
- Porri, L.; Ricci, G.; Giarrusso, A.; Shubin, N.; Lu, Z. Recent Developments in Lanthanide Catalysts for 1,3-Diene Polymerization. In ACS Symposium Series 749—Olefin Polymerization: Emerging Frontiers; Arjunan, P., McGrath, J.C., Hanlon, T., Eds.; Oxford University Press: New York, NY, USA, 2000; pp. 15–30. [Google Scholar]
- Osakada, K.; Takeuchi, D. Coordination Polymerization of Dienes, Allenes, and Methylenecycloalkanes. In Polymer Synthesis; Springer: Berlin/Heidelberg, UK, 2004; pp. 137–194. [Google Scholar]
- Shen, Z.; Ouyang, J. Handbook on the Physics and Chemistry of Rare Earths; Gschneidner, K.A., Jr., Fleming, L., Eds.; Elsevier Science Publishers: Amsterdam, The Netherlands, 1987; Chapter 61. [Google Scholar]
- Taube, R.; Sylvester, G. Applied Homogeneous Catalysis with Organometallic Compounds; Cornils, B., Herrmann, W.A., Eds.; Wiley-VCH: Weinheim, Germany, 2002; pp. 280–318. [Google Scholar]
- Zimmermann, M.; Anwander, R. Homoleptic Rare-Earth Metal Complexes Containing Ln−C σ-Bonds. Chem. Rev. 2010, 110, 6194–6259. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, A.; Klimpel, M.G.; Widenmeyer, M.; Herdtweck, E.; Scherer, W.; Anwander, R. Stereospecific Polymerization of Isoprene with Molecular and MCM-48-Grafted Lanthanide(III) Tetraalkylaluminates. Angew. Chem. Int. Ed. 2004, 43, 2234–2239. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Frøystein, N.Å.; Fischbach, A.; Sirsch, P.; Dietrich, H.M.; Törnroos, K.W.; Herdtweck, E.; Anwander, R. Homoleptic Rare-Earth Metal(III) Tetramethylaluminates: Structural Chemistry, Reactivity, and Performance in Isoprene Polymerization. Chem. Eur. J. 2007, 13, 8784–8800. [Google Scholar] [CrossRef] [PubMed]
- Meermann, C.; Törnroos, K.W.; Nerdal, W.; Anwander, R. Rare-Earth Metal Mixed Chloro/Methyl Compounds: Heterogeneous–Homogeneous Borderline Catalysts in 1,3-Diene Polymerization. Angew. Chem. Int. Ed. 2007, 46, 6508–6513. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, A.; Perdih, F.; Herdtweck, E.; Anwander, R. Structure−Reactivity Relationships in Rare-Earth Metal Carboxylate-Based Binary Ziegler-Type Catalysts. Organometallics 2006, 25, 1626–1642. [Google Scholar] [CrossRef]
- Fischbach, A.; Perdih, F.; Sirsch, P.; Scherer, W.; Anwander, R. Rare-Earth Ziegler−Natta Catalysts: Carboxylate−Alkyl Interchange. Organometallics 2002, 21, 4569–4571. [Google Scholar] [CrossRef]
- Arndt, S.; Beckerle, K.; Zeimentz, P.M.; Spaniol, T.P.; Okuda, J. Cationic Yttrium Methyl Complexes as Functional Models for Polymerization Catalysts of 1,3-Dienes. Angew. Chem. Int. Ed. 2005, 44, 7473–7477. [Google Scholar] [CrossRef] [PubMed]
- Robert, D.; Spaniol, T.P.; Okuda, J. Neutral and Monocationic Half-Sandwich Methyl Rare-Earth Metal Complexes: Synthesis, Structure, and 1,3-Butadiene Polymerization Catalysis. Eur. J. Inorg. Chem. 2008, 2801–2809. [Google Scholar] [CrossRef]
- Litlabø, R.; Lee, H.S.; Niemeyer, M.; Törnroos, K.W.; Anwander, R. Rare-Earth metal bis(tetramethylaluminate) complexes supported by a sterically crowded triazenido ligand. Dalton Trans. 2010, 39, 6815–6825. [Google Scholar] [CrossRef] [PubMed]
- Occhipinti, G.; Meermann, C.; Dietrich, H.M.; Litlabø, R.; Auras, F.; Törnroos, K.W.; Maichle-Mössmer, C.; Jensen, V.R.; Anwander, R. Synthesis and Stability of Homoleptic Metal(III) Tetramethylaluminates. J. Am. Chem. Soc. 2011, 133, 6323–6337. [Google Scholar] [CrossRef] [PubMed]
- Nieland, A.; Mix, A.; Neumann, B.; Stammler, H.G.; Mitzel, N.W. Lanthanoid Tetramethylaluminates and Their Paramagnetic NMR Parameters. Eur. J. Inorg. Chem. 2014, 51–57. [Google Scholar] [CrossRef]
- Evans, W.J.; Anwander, R.; Ziller, J.W. Inclusion of Al2Me6 in the Crystalline Lattice of the Organometallic Complexes LnAl3Me12. Organometallics 1995, 14, 1107–1109. [Google Scholar] [CrossRef]
- König, S.N.; Chilton, N.F.; Maichle-Mössmer, C.; Pineda, E.M.; Pugh, T.; Anwander, R.; Layfield, R.A. Fast magnetic relaxation in an octahedral dysprosium tetramethyl-aluminate complex. Dalton Trans. 2014, 43, 3035–3038. [Google Scholar] [CrossRef] [PubMed]
- Klooster, W.T.; Lu, R.S.; Anwander, R.; Evans, W.J.; Koetzle, T.F.; Bau, R. Neutron Diffraction Study of [Nd(AlMe4)3] 0.5 Al2Me6 at 100 K: The First Detailed Look at a Bridging Methyl Group with a Trigonal-Bipyramidal Carbon Atom. Angew. Chem. Int. Ed. 1998, 37, 1268–1270. [Google Scholar] [CrossRef]
- Dietrich, H.M.; Raudaschl-Sieber, G.; Anwander, R. Trimethylyttrium and Trimethyllutetium. Angew. Chem. Int. Ed. 2005, 44, 5303–5306. [Google Scholar] [CrossRef] [PubMed]
- Schumann, H.; Mueller, J.; Bruncks, N.; Lauke, H.; Pickardt, J.; Schwarz, H.; Eckart, K. Organometallic compounds of the lanthanides. Part 17. Tris[(tetramethylethylenediamine)lithium] hexamethyl derivatives of the rare earths. Organometallics 1984, 3, 69–74. [Google Scholar] [CrossRef]
- Schumann, H.; Müller, J. Tris[(N,N,N′,N′-tetramethylethylenediamine)lithium]hexamethylerbate(III) and -lutetate(III). Angew. Chem. Int. Ed. 1978, 17, 276. [Google Scholar] [CrossRef]
- Schumann, H.; Pickardt, J.; Bruncks, N. Crystal and Molecular Structure of[Li(tmen)]3[Er(CH3)6]. Angew. Chem. Int. Ed. 1981, 20, 120–121. [Google Scholar] [CrossRef]
- Shannon, R.D.; Prewitt, C.T. Effective Ionic Radii in Oxides and Fluorides. Acta Cryst. 1969, B25, 925–946. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Cryst. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Chen, E.Y.-X.; Marks, T.J. Cocatalysts for Metal-Catalyzed Olefin Polymerization: Activators, Activation Processes, and Structure−Activity Relationships. Chem. Rev. 2000, 100, 1391–1434. [Google Scholar] [CrossRef] [PubMed]
- Bonath, M.; Hollfelder, C.O.; Schädle, D.; Maichle-Mössmer, C.; Sirsch, P.; Anwander, R. C-H Bond Activation and Isoprene Polymerization by Lutetium Alkylaluminate/gallate Complexes Bearing a Peripheral Boryl and a Bulky Hydridotris(pyrazolyl)borato Ligand. Eur. J. Inorg. Chem. 2017, 4683–4692. [Google Scholar] [CrossRef]
- Diether, D.; Tyulyunov, K.; Maichle-Mössmer, C.; Anwander, R. Fluorenyl Half-Sandwich Bis(tetramethylaluminate) Complexes of the Rare-Earth Metals: Synthesis, Structure, and Isoprene Polymerization. Organometallics 2017, 36, 4649–4659. [Google Scholar] [CrossRef]
- Monteil, V.; Spitz, R.; Boisson, C. Polymerization of butadiene and copolymerization of butadiene with styrene using neodymium amide catalysts. Polym. Int. 2004, 53, 576–581. [Google Scholar] [CrossRef]
- Dietrich, H.M.; Schuster, O.; Törnroos, K.W.; Anwander, R. Heterobimetallic Half-Lanthanidocene Clusters: Novel Mixed Tetramethylaluminato/Chloro Coordination. Angew. Chem. Int. Ed. 2006, 45, 4858–4863. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Champagne, T.M.; Giarikos, D.G.; Ziller, J.W. Lanthanide Metallocene Reactivity with Dialkyl Aluminum Chlorides: Modeling Reactions Used to Generate Isoprene Polymerization Catalysts. Organometallics 2005, 24, 570–579. [Google Scholar] [CrossRef]
- Evans, W.J.; Champagne, T.M.; Ziller, J.W. An Ethyl Aluminum Oxide (EAO) Complex with μ-η1:η2-Ethyl Coordination Derived from a Samarocene Carboxylate and Triethylaluminum. Organometallics 2005, 24, 4882–4885. [Google Scholar] [CrossRef]
- Hayes, P.G.; Piers, W.E.; Parvez, M. Cationic Organoscandium β-Diketiminato Chemistry: Arene Exchange Kinetics in Solvent Separated Ion Pairs. J. Am. Chem. Soc. 2003, 125, 5622–5623. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Nishiura, M.; Li, X.; Xi, Z.; Hou, Z. Cationic Scandium Allyl Complexes Bearing Mono(cyclopentadienyl) Ligands: Synthesis, Novel Structural Variety, and Olefin Polymerization Catalysis. Chem. Asian J. 2008, 3, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Tardif, O.; Kaita, S. Generation of cationic indenyl silylamide gadolinium and scandium complexes [(Ind)Ln{N(SiMe3)2}]+[B(C6F5)4]− and their reactivity for 1,3-butadiene polymerization. Dalton Trans. 2008, 2531–2533. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Su, Q.; Chen, J.; Luo, Y. Synthesis, characterization of cationic half-sandwich scandium mono(silylamide) complexes and their unexpected reactivity toward C–Cl σ bond activation of chlorobenzene. J. Organomet. Chem. 2014, 769, 119–123. [Google Scholar] [CrossRef]
- Zimmermann, M.; Törnroos, K.W.; Anwander, R. Cationic Rare-Earth-Metal Half-Sandwich Complexes for the Living trans-1,4-Isoprene Polymerization. Angew. Chem. Int. Ed. 2008, 47, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Tobisch, S.; Bögel, H.; Taube, R. Mechanistic Studies of the 1,4-Cis Polymerization of Butadiene According to the π-Allyl Insertion Mechanism. 1. Density Functional Study of the C−C Bond Formation Reaction in Cationic (η3-Allyl)(η2-/η4-butadiene)nickel(II) Complexes [Ni(C3H5)(C4H6)]+ and [Ni(C3H5)(C4H6)(C2H4)]+. Organometallics 1996, 15, 3563–3571. [Google Scholar]
- Taube, R.; Windisch, H.; Maiwald, S. The catalysis of the stereospecific butadiene polymerization by Allyl Nickel and Allyl Lanthanide complexes—A mechanistic comparison. Macromol. Symp. 1995, 89, 393–409. [Google Scholar] [CrossRef]
- Ward, B.D.; Bellemin-Laponnaz, S.; Gade, L.H. C3 Chirality in Polymerization Catalysis: A Highly Active Dicationic Scandium(III) Catalyst for the Isoselective Polymerization of 1-Hexene. Angew. Chem. Int. Ed. 2005, 44, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Arndt, S.; Spaniol, T.P.; Okuda, J. Homogeneous Ethylene-Polymerization Catalysts Based on Alkyl Cations of the Rare-Earth Metals: Are Dicationic Mono(alkyl) Complexes the Active Species? Angew. Chem. Int. Ed. 2003, 42, 5075–5079. [Google Scholar] [CrossRef] [PubMed]
- Dettenrieder, N.; Hollfelder, C.O.; Jende, L.N.; Maichle-Mössmer, C.; Anwander, R. Half-Sandwich Rare-Earth-Metal Alkylaluminate Complexes Bearing Peripheral Boryl Ligands. Organometallics 2014, 33, 1528–1531. [Google Scholar] [CrossRef]
- Valente, A.; Mortreux, A.; Visseaux, M.; Zinck, P. Coordinative Chain Transfer Polymerization. Chem. Rev. 2013, 113, 3836–3857. [Google Scholar] [CrossRef] [PubMed]
- Brintzinger, H.H.; Fischer, D.; Mülhaupt, R.; Rieger, B.; Waymouth, R.M. Stereospecific Olefin Polymerization with Chiral Metallocene Catalysts. Angew. Chem. Int. Ed. 1995, 34, 1143–1170. [Google Scholar] [CrossRef]
- Rocha, T.C.J.; Coutinho, F.M.B.; Soares, B.G. Effect of alkylaluminum structure on Ziegler-Natta catalyst systems based on neodymium for producing high-cis polybutadiene. Polym. Bull. 2009, 62, 1–10. [Google Scholar] [CrossRef]
- Hagihara, H.; Shiono, T.; Ikeda, T. Stereospecificity of propene polymerization with achiral titanocene-based catalysts. Macromol. Chem. Phys. 1998, 199, 2439–2444. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with Shelxl. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Hubschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Elgert, K.-F.; Quack, G.; Stützel, B. Zur Struktur des Polybutadiens, 3. Das 13C-NMR-Spektrum des cis-1,4-1,2-polybutadiens. Makromol. Chem. 1975, 176, 759–765. [Google Scholar] [CrossRef]
- Elgert, K.-F.; Quack, G.; Stützel, B. Zur Struktur des Polybutadiens, 2. Das 13C-NMR-Spektrum des 1,2-polybutadiens. Makromol. Chem. 1974, 175, 1955–1960. [Google Scholar] [CrossRef]
- Elgert, K.-F.; Stützel, B.; Frenzel, P.; Cantow, H.-J.; Streck, R. Zur Struktur des Polybutadiens, 1. Das 13C-NMR-Spektrum der 1,4-Polybutadiene. Makromol. Chem. 1973, 170, 257–260. [Google Scholar] [CrossRef]
- Santee, E.R.; Chang, R.; Morton, M. 300 MHz proton NMR of polybutadiene: Measurement of cis-trans isomeric content. J. Polym. Sci. Polym. Lett. Ed. 1973, 11, 449–452. [Google Scholar] [CrossRef]
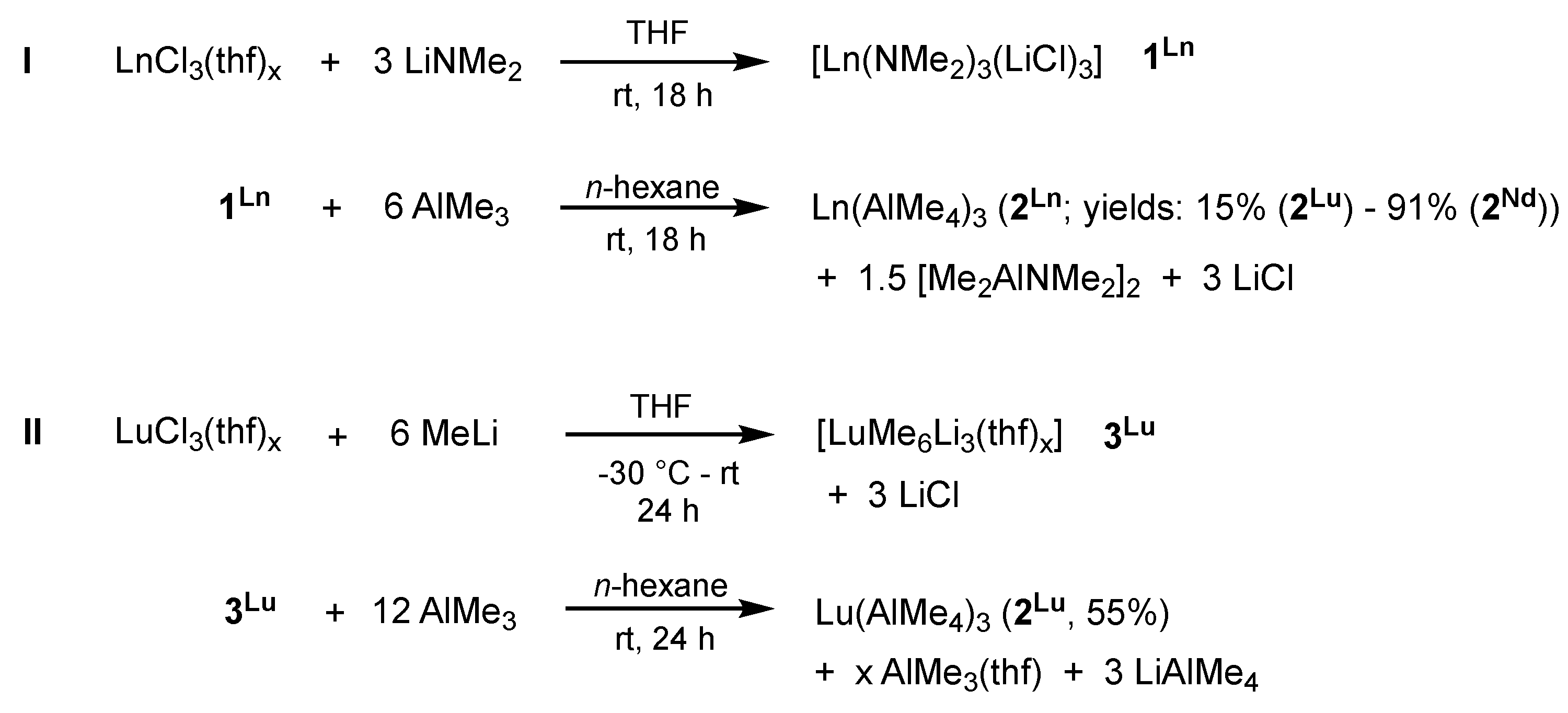
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hollfelder, C.O.; Jende, L.N.; Diether, D.; Zelger, T.; Stauder, R.; Maichle-Mössmer, C.; Anwander, R. 1,3-Diene Polymerization Mediated by Homoleptic Tetramethylaluminates of the Rare-Earth Metals. Catalysts 2018, 8, 61. https://doi.org/10.3390/catal8020061
Hollfelder CO, Jende LN, Diether D, Zelger T, Stauder R, Maichle-Mössmer C, Anwander R. 1,3-Diene Polymerization Mediated by Homoleptic Tetramethylaluminates of the Rare-Earth Metals. Catalysts. 2018; 8(2):61. https://doi.org/10.3390/catal8020061
Chicago/Turabian StyleHollfelder, Christoph O., Lars N. Jende, Dominic Diether, Theresa Zelger, Rita Stauder, Cäcilia Maichle-Mössmer, and Reiner Anwander. 2018. "1,3-Diene Polymerization Mediated by Homoleptic Tetramethylaluminates of the Rare-Earth Metals" Catalysts 8, no. 2: 61. https://doi.org/10.3390/catal8020061
APA StyleHollfelder, C. O., Jende, L. N., Diether, D., Zelger, T., Stauder, R., Maichle-Mössmer, C., & Anwander, R. (2018). 1,3-Diene Polymerization Mediated by Homoleptic Tetramethylaluminates of the Rare-Earth Metals. Catalysts, 8(2), 61. https://doi.org/10.3390/catal8020061