Perovskite Structure Associated with Precious Metals: Influence on Heterogenous Catalytic Process
by

Guilhermina Ferreira Teixeira
1, 
Euripedes Silva Junior
2,
Ramon Vilela
1,
Maria Aparecida Zaghete
2 and
Flávio Colmati
1,* 1
Laboratório de Bio-eletrocatálise e Células Combustíveis (LABEL-FC)-Instituto de Química, Universidade Federal de Goiás (UFG), 74690-900 Goiânia-Goiás, Brazil
2
Laboratório Interdisciplinar de Eletroquímica e Cerâmica (LIEC)-Centro de Desenvolvimento de Materiais Funcionais (CDMF), Instituto de Química-UNESP, 14800-060 Araraquara-SP, Brazil
*
Author to whom correspondence should be addressed.
Catalysts 2019, 9(9), 721; https://doi.org/10.3390/catal9090721
Submission received: 29 July 2019
/
Revised: 21 August 2019
/
Accepted: 23 August 2019
/
Published: 27 August 2019
(This article belongs to the Special Issue Catalysis by Precious Metals, Past and Future)
Abstract
:The use of perovskite-based materials and their derivatives can have an important role in the heterogeneous catalytic field based on photochemical processes. Photochemical reactions have a great potential to solve environmental damage issues. The presence of precious metals in the perovskite structure (i.e., Ag, Au, or Pt) may improve its efficiency significantly. The precious metal may comprise the perovskite lattice as well as form a heterostructure with it. The efficiency of catalytic materials is directly related to processing conditions. Based on this, this review will address the use of perovskite materials combined with precious metal as well as their processing methods for the use in catalyzed reactions.
1. Introduction
Perovskite-based materials have been extensively used in photocatalytic systems. The incorporation of a precious metal on perovskite lattice or the use of these elements to modify the surface of perovskite particles result in better light absorptions, which promotes the photochemical behavior required for the catalytic process, such as photocatalysis and electrocatalysis; silver (Ag) and gold (Au) are the most used precious metals for this purpose. In order to understand the advantages of using perovskite structures associated with precious metals as photocatalysis in the catalytic process, this review addresses considerations on photocatalysis principles, perovskite structure, and synthesis methods to obtain a catalyst.
2. General Considerations on Photocatalysis
According to the IUPAC Gold Book [1], photocatalysis is defined as a change in the rate of a chemical reaction or its initiation under the action of ultraviolet, visible or infrared radiation in the presence of a substance—the photocatalyst—that absorbs light and is involved in the chemical transformation of the reaction partners. Thereby, the photo term comprises the light (UV-vis, IR, etc.), while the catalysis ones represent the process through which a catalyst compound or a substance changes the reaction rate of chemical transformation by increasing the reaction kinetics without modifying the overall standard Gibbs energy (ΔG0) alteration in the reaction [1]. Considering the catalyst aspect, a photocatalyst is defined according to the IUPAC as a compound or a substance able to produce, upon absorption of light, chemical transformations of the reaction partners. The excited state of the photocatalyst repeatedly interacts with the reaction partners forming reaction intermediates and regenerates itself after each cycle of such interactions [1]. Therefore, a photocatalytic process can be understood as a synergic process in which light radiation and the catalyst act jointly to support and speed up a chemical reaction.
The photocatalytic processes are included in a broader and general category of chemical and photochemical reactions known as advanced oxidation processes (AOPs), which apply powerful oxidizing agents in order to degrade persistent organic pollutants as well as remove certain inorganic pollutants. Through such process, the pollutants are converted partially or totally into simpler and less toxic species resulting in easily degraded substances through the application of common technologies [2,3,4,5]. The development of the AOPs started in the 1980s from new approaches for potable water treatment that dispose of hydroxyl radicals (●OH) as major oxidizing agent [3,4,5]
Because of their non-selective nature, hydroxyl radicals may easily lead to the mineralization of a wide range of potentially toxic organic species and depending on the operating conditions (temperature, pH, pollutant concentration, etc.) transform them into less complex and harmful intermediate products [5,6,7]. Although molecular ozone is a selective chemical oxidant and has a good oxidizing capability, hydroxyl radicals can reach reaction rates of 1 million to 1 billion times faster than those found in chemical oxidants and consequently display rate constants ranging from 106 to 1010 (M−1 s−1) [2,4,7]. Even though fluorine is a strong oxidizing agent, its use in drink water treatment or to degrade organic pollutants is constantly intensively discussed due to the risk that fluoride derivates may represent to the human health and biological systems [8,9], in addition to being an expensive treatment technology on a large scale [10,11].
Most industrial waste and wastewater treatment processes employ classical treatment methods from different physical, chemical, and biological processes, which only carry over organic compounds from water to another phase generating hazardous sludge that results in secondary pollution [12,13,14,15,16]. Despite usually being high-cost technologies, AOPs emerge as an interesting solution to wastewater treatment for presenting attractive characteristics such as versatile treatment —enabled before or after conventional treatment or even during the main stage; mineralization of several chemical pollutants, as well as intermediates; no generation of sludge hazardous (eco-friendliness technologies) as it occurs in the physical, chemical, and/or biological processes; high reaction rate, and satisfactory cost-effectiveness [13,14,17,18].
From the point of view of a chemical reaction, applying AOPs in wastewater treatments comprises basically the following steps: (i) firstly, the generation of strong oxidizing agents (, , , etc.) from the species into medium reactional (ions, molecules, catalyst, etc.); (ii) followed by the oxidizing species reacting with organic contaminants molecules in solution, generally converting them into easily degradable intermediary compounds; (iii) finally, the oxidation of these intermediates leading to complete mineralization in water, carbon dioxide, and inorganic salts [3,4,18,19,20]. In contrast, AOPs exhibit complex oxidation mechanisms due to the very large number of reactions that may occur; therefore, it is laborious to predict all reaction pathways of products formed [5,18,19,20]. In this regard, although AOPs display fast oxidation rates for many persistent organic pollutants, controlling the formation of reaction products becomes impossible. In particular cases, depending on medium conditions of the supports reaction, these products can be more toxic than parent molecules due to a fast attack to the water contaminant molecules by some free radicals, just as other intermediate species available in solution, such as short-lived species like hydroxyl radicals [5,19,20].
Although AOPs show many advantages in relation to other conventional oxidation processes, an important consequence of most AOPs—regarded as a remarkable disadvantage to any oxidation process based on hydroxyl radical attack—consists of the scavenging of radicals by scavenger agents presents in the solution (, , excess H2O2 and pH conditions) [2,5,18,21,22,23]. In this regard, the scavengers trap the radicals decreasing their availability in the reactive medium and consequently compromising the degradation efficiency, which lead others species to emerge with much lower oxidizing power than the hydroxyl radical. It is worth highlighting that scavenger species (ions, radicals, charge carriers, etc.) can act either in a favorable or harmful manner during the degradation process.
AOPs are classified as homogeneous and heterogeneous systems; in both cases, the hydroxyl radicals are generated with or without the presence of a stimulus from an external source (UV, ultrasound, microwave, etc.) [17,19,24]. In the homogeneous systems, a single phase is formed between the catalyst and the substrate, whereas in the heterogeneous systems the substrate and the catalyst present more than one phase, in which the catalyst can be found as a solid phase [19].
2.1. Heterogeneous Photocatalysis: TiO2 vs. Perovskite-based Materials
In the context of the AOPs systems, heterogeneous photocatalysis based on a semiconductor photocatalyst is among the most explored procedures in the area of water and wastewater treatment. The first experiments regarding AOPs were performed in the 1930s and used TiO2 in aqueous suspension under UV irradiation in order to degrade dyes [25,26,27]. Nonetheless, the applications of semiconductor photocatalysts are not limited to degradation of dyes and other potential pollutants that are harmful to human health and the environment. Its application also involves the production of hydrogen, purification of air, and antibacterial activity [12,26,27,28,29,30]. The evolution of photocatalysis progressed in parallel to advances in researches on the investigation of TiO2 properties applied to photoelectrochemical reactions. The advantages of using TiO2 include higher photoreactivity and physical-chemical stability as well as lower cost and toxicity in relation to other photocatalyst candidates [25,26,29,30]. Researchers investigating the use of TiO2 in photocatalytic processes were boosted in the year of 1972 when Honda and Fujishima pioneered and reported the decomposition of water into H2 and O2 through means of a photochemical experiment under sunlight irradiation [26,30].
In the photoactivation of the semiconductor, electrons located in the valence band maximum (VBM) are photoexcited under hν ≥ Eg toward conduction band minimum (CBM) leaving positive holes in the valence band. Subsequently, these charge carriers may have different destinations in the photocatalytic process [23,25,31,32,33,34,35]. A photocatalyst will oxidize a species upon a less positive oxidation potential (Eox) than the potential edge conduction band (CBM) and analogously reduces it upon a more positive reduction potential (Ered) than the potential edge valence band (VBM) [36]. Thus, the positions of the electronic band-edge structures of the semiconductor can easily promote both oxidation and reduction of the species presents in solution since the valence/conduction band gap (Eg) is sufficiently large for encompassing the redox potentials of some species that are essential to the photocatalytic process [25,33,34,35,36,37].
TiO2-based photocatalysts are undeniable the most studied material in the context of photocatalytic applications. Nevertheless, its use as pristine TiO2 bulk is restricted to the UV region since the TiO2 anatase electronic structure absorbs at ~3.2 eV and [25,26,38]. Complex oxides systems—such as perovskite-based photocatalysts—emerge as an interesting alternative to photocatalytic processes [39,40,41]. Among the different advantages of the photophysical properties of perovskite, we can highlight: (i) their structures can be formed from a wide variety of elements, although their basic structures are similar; (ii) their valency, stoichiometry, and vacancy can be easily changed from the simple and non-laborious proceedings; (iii) possibility of a satisfactory prediction of their surface properties since their bulk structures are very well characterized; (iv) their crystal structures generally provide an appropriate electronic structure that shifts the band gap energy to visible-light absorption; (v) their crystal structural arrangements allow lattice distortions, strongly affecting the separation of photogenerated charge carriers and avoiding the recombination processes [41,42]. Therefore, the possibility of controlling the physical-chemical properties of perovskite structures allows unraveling the relationship between structural properties and photocatalytic activity, which makes this material a good photocatalyst alternative to TiO2 [42].
2.2. Perovskite Structure
The perovskite-type structure term comes from mineral calcium titanate (CaTiO3), also called perovskite. Materials under perovskite-type structure have a general chemical formula of type ABX3, in which atoms A and B are metal cations and X atoms are non-metallic anions, so that upon the occupation of the site X with oxygen, this structure is called perovskite oxides (ABO3), as illustrated in Figure 1. In addition, the A atom can be monovalent, divalent or trivalent, while the B atom can be trivalent, tetravalent or pentavalent [43,44]. In a typical perovskite oxide structure, the lattice is formed by small B cations that are six-fold coordinated to oxygen (BO6), while larger A cations are twelve-fold coordinated by oxygen (AO12), so that BO6 octahedra are corner connected and AO12 clusters are arranged between the eight BO6 octahedra, resulting in a crystal structure that extends in three dimensions and has excellent flexibility depending on the valence and ionic radii of the A and B cations [43,44,45].
An ideal, fully ordered perovskite oxide structure shows a cubic crystal structure with space group ; however, there are several cases in which the ideal structure is distorted. The distortions observed in ABO3 structures can derive from (i) distortions in the BO6 octahedra, (ii) displacement of the B-site cation within the octahedron, (iii) displacement of the A-site cation within the lattice, and (iv) tilting of the corner-sharing octahedrons [47,48]. Therefore, the A and B cations can promote strain, stress, and/or distortions under the perovskite structure according to the distortion degree, also known as tolerance factor (t), depending on the ionic radii of the A and B cations [49,50].
Nevertheless, in general terms, the tolerance factor is not solely the unique variable able to determine the formability and stability of a general perovskite structure since other non-geometric factors, such as bond valence and chemical stability, should also be considered [51,52]. Thereby, the size of the atoms constituting the structure must be adequate to make the perovskite structure geometrically robust as well as less reactive under ambient conditions, for example, in the presence of moisture and oxygen [51].
Figure 2 shows two correlated metals with similar perovskite structures, whereby the difference between the ionic radii of cations Sr2+ (118 pm) and Ca2+ (100 pm) promoted distinct degrees of BO6 octahedral distortions, resulting in a more “distorted” crystal structure of CaVO3 in relation to the SrVO3. Despite being similar perovskite structures, because of the variation in cation radii, the crystal structure of SrVO3 is cubic (c = 3.842 Å), whereas CaVO3 is orthorhombically distorted (c = 3.770 Å) [53].
In terms of photocatalytic process, there are several factors assigned to structural properties that affect the photocatalytic performance of ABO3-based photocatalysts. The main factors are related to electron-hole separation as well as the carrier transport mechanisms within the crystal lattice, such as exciton lifetime and diffusion length, exciton binding energy, and electron/hole effective mass [39,54]. Thereby, the lattice distortion and structural defects may change the electronic structure of perovskite oxide materials remarkably, consequently affecting the carrier transport mechanisms [39,44,47,48].
The electronic structure in perovskite oxides is defined by a lattice of BO6 octahedra clusters, whereby their B-O-B bonds build up an electrical conduce way along the crystal structure through which the electrons and/or holes “flow” [43,45,55]. The lattice distortion degree of the pristine ABO3 induced by ion doping may induce structural defects on the crystal structure generating and/or rearranging energy state levels that change the electronic structure strongly. These lattice distortions affect the separation of charge carriers avoiding the recombination processes and/or shifting the band gap energy to enable visible-light absorption and tuning the band edge potentials to meet the requirement of specific photocatalytic reactions [39,56,57].
2.3. Perovskite-based Photocatalysts Supported by Precious Metals: A Synergistic Effect on Photocatalytic Performance
Aiming at achieving the best of the photocatalytic process, innumerous strategies can be applied. Although other challenges are yet to be overcome in the photocatalytic universe, hampering the electron-hole pair recombination is an essential strategy to ensure optimum photocatalytic efficiency. In this regard, our review addresses one of the most interesting strategies to improve the photocatalytic activity of perovskite-based semiconductor. This strategy is based on the modification of perovskite structure by inserting a precious metal into its crystalline lattice through doping process or obtaining precious metal/perovskite composite through modification of perovskite particles surface with a precious metal in order to decrease and/or retard the recombination process of charge carriers, apart from shifting the band gap values to visible-light absorption region.
Precious metals integrate a class of dopants widely used in the development of new photocatalysts since their optical properties may allow the activity in visible light as well as decrease the electron-hole pair recombination promoting a synergistic effect on the photocatalytic activity [58,59]. On the whole, individual precious metals nanoparticles (Ag, Au, Pt, Rh, Ir, etc.) or precious metals supported on the photocatalyst surface exhibit a specific behavior when interacting with light at plasma frequency, which enables the visible light absorption by the surface plasmon resonance (SPR) effect [60,61]. The plasmon term can be understood as a wave coming from the collective oscillation of electrons on the surface of metals. Basically, the electric field associated with light exerts a force on the outermost electrons of the conduction band of metal redistributing the spatial electron density and generating intense electric fields on the surface metal. In a given oscillation pattern at plasma frequency, when the depth of light penetration is approximately equal to the size of the metallic nanoparticles, such oscillation resonates with incident light resulting in a strong oscillation of the surface electrons that change the absorption profile of metal [61,62].
The key point of the SPR is the frequency of incident radiation, which must be resonant with the frequency of oscillation of the electrons on the metal surface. However, if the incident radiation is lower than plasma frequency, it will be reflected because the electrons of metal filter the electric field incident. In contrast, if the incident radiation is higher than the plasma frequency, the light will be transmitted because the electrons are incapable of responding quickly to the electric field oscillation to filter it [61].
The oscillations of electrons and electromagnetic fields under resonance frequency are defined as located at the surface plasmonic resonance (LSPR), whereby electrons would oscillate with the maximum amplitude. Nevertheless, for a same noble metal, the resonance frequency can be attuned by means of change in size, shape, and nature of the surrounding medium and type of matrix (photocatalyst) when the precious metal is a dopant on the photocatalyst surface [58,59,63]. In this respect, silver and gold nanoparticles are good examples of metals that present an SPR effect due to decreased particles size. A smaller particle size allows silver to absorb light at a visible frequency and gold to absorb light at IR frequency [61].
Thus, the great interest in precious metals nanostructured for photocatalytic applications arise from the localized surface plasmon resonance mechanisms since it is well-known that such plasmonic effect can increase the cross-section absorption in photocatalyst through the process of strong field enhancement, consequently shifting the light absorption to larger wavelengths, in addition to enhancing the separation of charge carriers photogenerated in photocatalyst [58,60].
In this regard, for metallic nanoparticles supported on the semiconductor surface, the energy generates by the electron oscillation via LSPR effect can be transferred to the semiconductor as electromagnetic energy or as excited electrons. In the first case, the energy absorbed in the excitation of the plastron band is quickly converted to heating, and then this energy is transferred to the semiconductor via photothermal effect [58,59,60,62]. The second case involves a complex charge of transfer mechanisms in which the surface plasmon resonance acts by amplifying the light absorption of the metallic nanoparticles directly injecting photogenerated electrons from the metallic nanoparticles to the semiconductor conduction band via direct transfer mechanism and/or through the resonance electron transfer mechanism. The local electromagnetic field generated by LSPR, in turn, facilitates the generation of electron-hole pairs close to semiconductor surface, in addition to creating a Schottky barrier suppressing the recombination processes since the photogenerated electrons are quickly transferred to the semiconductor conduction band [58,59,62,63]. In the case of our work, the semiconductors are the perovskite compounds used as the catalyst.
Figure 3 illustrates a scheme presenting both direct electron transfer and resonance electron transfer which have been proposed to explain the visible light activity of metallic nanoparticles on the photocatalyst. In the scheme, Au is the metallic nanoparticles and TiO2 is the photocatalyst semiconductor. However, this scheme can be applied to illustrate these electronic transfer effects whenever a perovskite structure is used as a semiconductor. Details on the species formed during the photocatalytic process can be found further in the reference section [63].
On the plasmonic point of view, the decision on the noble metals to be used should consider those able to support a strong surface plasmon resonance effect at the desired resonance wavelength. The ability of a metal to support an SPR depends on their dielectric properties, i.e., dielectric function (ε), including its real (εr) and an imaginary (εi) part, as both of which vary according to the excitation frequency [60]. Therefore, a resonance in the absorption occurs at the plasmon frequency when the real part of the dielectric function reaches zero [61]. Thereby, among the noble metals, silver is undoubtedly the most employed one in plasmonic applications for its unique electrical and optical properties, in addition to low cost in relation to Au and Pt [60], for example.
In a hybrid structure applied in plasmonic photocatalysis, the charge-transfer mechanism between metallic nanoparticles and semiconductors displays a fundamental role for noble metals to present an intense SPR effect in the range of energy level states of the semiconductor conduction band in order to enable the electron transfer between metal and semiconductor [64,65,66]. Generally, interband transitions between Ag/semiconductor hybrid structures occur at high frequencies, therefore, silver nanoparticles present an intense SPR effect in the range of 400–450 nm [65], whereas, for Au and Cu, these transitions limit their SPR excitation to wavelengths above 500 and 600 nm, respectively [67]. It is worth mentioning that these interband transitions from the SPR effect in metal/semiconductor hybrid structures strongly depend on the electronic band structure and mainly on the minimum conduction band.
Therefore, plasmonic photocatalysis applications emerge as new and upgraded approaches on the photocatalytic universe and combined with the structure-property relationship of perovskite materials generate new structures of photocatalysts by blending the unique properties of perovskite compounds and the optical and electrical properties of precious metals nanostructured highlighted by the SPR effect.
The plasmonic effect caused by precious metals on the perovskite structure not only improves the photocatalytic behavior of the material, the incorporation of metals such as Pt, Pd, and Rh into a perovskite structure, also stabilizes the metal against sintering, metal-support interaction, and volatilization. The catalytic activity and selectivity of perovskite in the exhaust cleaning process are enhanced even if small amounts of precious metals are combined with the perovskite structure [68]. As aforementioned and illustrated in Figure 4, the distortion of BO6 octahedral of perovskite is promoted by ionic radii of cations present in the crystalline lattice resulting in a more distorted structure. Thus, the same precious-doped perovskite matrix may suffer different distortion degrees according to the kind of precious metal present in its structure. Thereby, the lattice distortion may change the electronic structure of perovskite oxide materials remarkably and consequently affect the carrier transport mechanisms [39,44,47,48,55].
Based-perovskite plasmonic photocatalytic materials can be processed as powder or film. Our work approaches the chemical synthesis method (bottom-up method) most commonly used to produce perovskite powders. Through this method, it is possible to control the stoichiometry, shape, surface area, and size of the particles, among other relevant features.
3. Chemical Synthesis for Perovskite Photocatalyst Obtaining
3.1. Solid-State Reaction
The solid-state reaction method is the oldest method to produce inorganic powders. It is the simplest and most widely used method in the synthesis of compounds with perovskite structure. This reaction consists of the combined heating of two or more non-volatile solids that react to form the desired product. The reactants in the stoichiometric ratios should be very well mixed at the level of individual particles on a scale around 1 μm or 10−3 mm [69]. An efficient mix process ensures the maximum contact among the reactants minimizing the diffusion distance among them. The mix is performed using a mortar or a mill for the precursors to be subsequently heated at high temperature in a furnace for several hours in an alumina crucible [70]. The reaction can be slow and its speed increases upon a higher temperature promoting the faster diffusion rate of the ions that compose the final product. The reaction occurs at the interface of the solids (reactants); after the surface layer reacts, the reaction continues as the reactant diffuse from the bulk to the interface [69,70].
Iwashina and Kudo obtained SrTiO3 powders doped with Rhodium at Ti sites (SrTiO3:Rh). We performed the airborne calcination of precursor mixture according to the following two steps: firstly, at 1173 K for 1 hour and then at 1373 K for 10 hours. X-ray diffraction patterns revealed that the single phase of SrTiO3: Rh is obtained until 4 atom% of dopant. Small amounts of impurity phase are obtained for 7 and 10% of atom dopant. After powder preparation, this one was employed in photoelectrochemical water splitting by covering of an ITO electrode. Undoped SrTiO3 is an n-type semiconductor and the doping process transforms it in a p-type semiconductor. The optimum material was the one that doped with 7 atom% of Rhodium. The dopant contributes to the visible light absorber and a recombination center for photogenerated electrons and holes because these factors are responsible for the device efficiency in the photocatalysis process. Through diffuse reflectance spectra, we found that SrTiO3:Rh (7 atom%) had visible light absorption bands that did not occur for the non-doped material. The Faradaic efficiency for H2 and O2 evolution were 100% within experimental error and stable cathodic photocurrent could be observed for a long period of time [71].
The (AgySr1−y)(Ti1−yNby)O3 was produced seeking to evaluate its efficiency in the photocatalytic oxidation through the decomposition of gaseous 2-propanol under UV and visible light irradiations. The products present a cubic crystalline structure with homogeneous SrTiO3 crystalline phase at least up to y = 0.1. The band gap energy value (3.18 eV) was similar to (AgySr1−y)(Ti1−yNby)O3 and pure SrTiO3. The density of states calculations for (Ag0.25Sr0.75)(Ti0.75Nb0.25)O3, Nb 4d contributes to the conduction band, but did not contribute to the valence band. In contrast, Ag 4d contributed only to the valence band, meanwhile, we may consider that Ag 4d did not mix with valence band composed of O 2p, but Ag 4d forms an isolated band in the forbidden above the valence band. We evaluated the photocatalytic activity in the decomposition of gaseous 2-propanol by monitoring the acetone and CO2 concentration on the subjection of the photocatalyst under UV and visible lights. After adsorbed into the semiconductor surface, the 2-propanol concentrations in presence of (Ag0.25Sr0.75)(Ti0.75Nb0.25)O3 were 18 and 2.5 ppmv, respectively, showing that acetone and CO2 are generated when absorbing either UV or visible light. In addition, the color of (Ag0.25Sr0.75)(Ti0.75Nb0.25)O3 did not change at all after photocatalytic test indicating the stability of the compound under light irradiation. The effective work of photocatalyst at decomposing organic compounds occurs if photoexcited electrons are consumed in the oxygen reduction. In the case of (Ag0.25Sr0.75)(Ti0.75Nb0.25)O3, the Ag+ was attributed to the electronic structure of matrix valence band and not to that of its conduction band. If the Ag+ were attributed to the electronic structure of the matrix conduction band, photoexcited electrons could not be consumed in the oxygen reduction. Despite (Ag0.25Sr0.75)(Ti0.75Nb0.25)O3 presenting photocatalytic activity under both UV and visible light irradiation, the formation of the Ag 4d isolated band above the valence band composed of O 2p is responsible for making the efficiency of the compound under UV light superior to that under visible light [72].
Saadetnejad and Yildirim studied the photocatalytic hydrogen production by water splitting over Au/Al-SrTiO3. The Al-doped SrTiO3 was obtained from solid-state reaction and loaded with Au via the homogeneous deposition-precipitation method. XRD patterns indicated that samples were indeed SrTiO3 with good crystallinity, which contributes to an efficient migration of charge carriers in the water-splitting process. The best performing catalyst was obtained by using a material with composition equal to 1% Al doped, 0.25% Au loaded SrTiO3, in the presence of methanol as a sacrificial agent, while 1% Al doped 0.50% Au was the best for the isopropyl medium. The relative performance of methanol and isopropyl medium seems to depend on the alcohol concentration since the isopropyl performs better at 10% and 30%, while methanol is better at 20%. Furthermore, the distinguished results obtained from differences among concentrations and different alcohol types may be related to the carbon number present in the alcohol molecules, which may result in different byproducts, the redox potential of alcohol, and their OH scavenging behavior [73].
AgTaO3/AgBr heterojunction presents intense visible light absorption and consequently better photocatalyst perform than individual AgTaO3 and AgBr in the methyl orange degradation. AgTaO3 showed no efficiency at dye degradation under visible light irradiation. After the heterojunction formation, the photocatalytic activity efficiency significantly improves due to the enhanced interfacial charge separation efficiency between AgTaO3 and AgBr. The heterojunction induced an efficient transfer of photogenerated electrons from AgBr conduction band to AgTaO3 conduction band leading to a better charge separation improving photocatalytic behavior. We applied solid-state reaction to obtain Perovskite AgTaO3 and solution method to produce AgBr. Composites were processed by mixing individual AgTaO3 and AgBr suspensions containing poly(ethyleneglycol)-block-poly(ethylene glycol) NH3.H2O. After a photocatalytic test, an additional weak diffraction peak regarding cubic Ag was observed in AgTaO3/AgBr heterostructure indicating the formation of the ternary AgTaO3/AgBr/Ag. XPS results indicated that partial Ag+ ions present in the heterojunction were reduced to metallic Ag nanoparticles to form the ternary AgTaO3/AgBr/Ag during the photocatalytic experiment. It was evidenced that the photogenerated electrons may migrate from the conduction band of AgBr to the conduction band of AgTaO3 [74].
Konta and co-workers investigated the photocatalytic application of SrTiO3:M(0.5%) (M = Mn, Ru, Rh, Pd, Ir, and Pt ions) powders in H2 evolution from methanol solution and O2 evolution from an aqueous solution nitrate solution under visible light irradiation. All material exhibited pure phase of SrTiO3, except for SrTiO3:Pd(0.5%), since its Pd2+ ion radius are much larger than Ti4+ ion radius. The diffuse reflectance spectra of materials revealed that all samples presented visible light absorption. The spectra of the sample doped with Mn, Ru, Rh, and Ir, we found an absorption band as a shoulder in addition to the absorption band regarding SrTiO3 indicating that the dopants had created discontinuous levels in the forbidden band. The presence of two absorption bands as shoulders in the diffuse reflectance spectra of SrTiO3:Rh powders result from some doping levels formed in the forbidden band due to different oxidation numbers (Rh3+ and Rh5+). SrTiO3:Rh showed the best photocatalytic activity in H2 evolution, while SrTiO3:Ru proved the best for O2 evolution. In contrast to the higher O2 evolution for SrTiO3:Mn and SrTiO3:Ru, their low photocatalytic activity for H2 evolution would occur due to the surface states generated by Mn and Ru dopants below the conduction bands, resulting in difficulty for the H2 evolution. Because SrTiO3:Rh showed the highest activity for H2 evolution, further investigations should be carried out to approach this compound. We found the occurrence of at least two different species of doped Rh (Rh3+ and probably Rh5+). Rh with higher oxidation number acts as an electron acceptor because they can be easily reduced to Rh3+ at an early stage of the photocatalytic reaction; in addition, during the photocatalytic process, the Rh3+ acts as an electron donor. The SrTiO3:Rh visible light response resulted from the electronic transition from the electron donor level formed by Rh ions to the Ti 3d orbitals present in the conduction band of SrTiO3 matrix. Despite its excellent performance in H2 evolution, the SrTiO3:Rh presented no activity to O2 evolution. One of the reasons for no O2 evolution using SrTiO3:Rh as photocatalyst is the kinetic limitation of the lack of active sites. The photocatalytic reaction may be efficient after reducing the Rh with high oxidation number. However, these species cannot be reduced in an aqueous silver nitrate solution medium because they act as recombination center in the absence of a hole scavenger, which is another reason for the absence of photocatalytic activity in O2 evolution [75]. Table 1 shows both H2 and O2 evolution when using SrTiO3:M(0.5%) under visible light absorption as well as the respective band-gap values of semiconductors.
3.2. Sol–Gel Method
Sol–gel method also represents a widely used approach to produce perovskite particles. This process consists in the transition of a solution system from a liquid known as “sol”, most often a colloid liquid, into a solid phase named “gel”. Inorganic metal salts or metalorganic compounds are usually employed as starting materials for the sol–gel method. To form the desired product, it initially occurs the hydrolysis of a precursor metal to produce the metal hydroxide solution. Subsequently, an immediate condensation occurs providing a three-dimensional gel that is dried by removing the solvent. Depending on the way the solvent is removed, the formation of the Xerogel or Aerogel occurs. If the solvent is removed slowly at room temperature, a Xerogel is formed. In contrast, if the drying process is performed under supercritical conditions, the Aerogel is obtained [76,77,78].
Since the ion–codoped semicondutor has been improving photocatalytic activities under visible-light irradiation, Zhang and co-workers studied the effect of monovalence silver and trivalence lanthanum codoping CaTiO3 in the water-splitting process. The sol–gel method was chosen to provide the perovskite material and the ultrasonic dispersing technique was used to enhance the properties of the materials. XRD patterns showed the pure CaTiO3 orthorhombic phase even for doped samples. Diffuse reflection spectra showed a shift to a longer wavelength when replacing Ca2+ by Ag+ with La3+. Such behavior may result from the electronic transition between the electrons present in the Ag 4d5s orbital to the O 2p + Ti 3d hybrid orbital. The Ag–La codoped CaTiO3 introduces a band in the visible light region due to the transition of Ag 4d5s electrons to the conduction band. The photocatalytic efficiency increased until the amount of dopant equal to 3% and is much higher than the value obtained from pure CaTiO3. Upon a doped concentration lower than the optimum value, the photocatalytic activity increased because of the absence of enough capture traps of charge carriers in the photocatalyst particles. In contrast, the recombination rate increases when the doped concentration is higher than the optimal value, which promotes lower photocatalyst activity [79].
ZnTiO3 is UV light active photocatalyst and the formation of a composite between this perovskite and plasmonic metals (e.g., Au) represent satisfactory applications of this perovskite in visible light assisted photocatalytic reactions. Gold is considered an efficient metal for this purpose due to its great properties, such as surface plasmon resonance characteristics, electron storage effect, chemical stability, and high catalytic activity. In this sense, Hemalata Reddy and co-workers obtained ZnTiO3 through sol-gel auto combustion method and subsequently loaded the particles with Au nanoparticles using the precipitation-deposition method forming Au/ZnTiO3 nanocomposites. The material was applied in H2 generation from methanol solution under UV and visible light irradiation. Pure ZnTiO3 has absorption in UV region and the addition of Au on ZnTiO3 surface promotes an additional absorption in the visible region due to the surface plasmon absorption corresponding to the gold nanoparticles. The photocatalytic tests were performed by using Au/ZnTiO3 nanocomposite with Au concentration varying from 0.5 to .5 wt%. We achieved the best results for H2 evolution using the nanocomposites with 1 wt% of Au under visible light irradiation, while pure ZnTiO3 efficiency was practically insignificant. In contrast, through UV as the radiation source, the photocatalytic efficiency increases with a higher Au content up to 1.5% wt due to the separation of charge carriers in the interface between Au nanoparticles and perovskite particles through the Schottky barrier. In addition, the interfacial defect sites in the nanocomposites act as Au-induced charge separation effect that balances the recombination effect of Au/ZnTiO3 in photocatalytic reaction from UV light irradiation [80].
The physicochemical properties and photocatalytic activity for the degradation of methylene blue under simulated solar light were investigated in novel Au-induced nanostructured BiFeO3 homojunctions (Aux-BFO, x = 0, 0. 6, 1.2, 1.8, 2.4 wt%) obtained through in situ synthesis, despite of a simple reduction method using spinning techniques and post-thermal processing. Among the Aux-BFO structures, the Au1.2-BFO sample proved the best photocatalytic activity (85.76%) after 3h of irritation, being much higher than pristine BFO samples (49.49%) considering the same experimental conditions. The authors attributed the notorious improvement of photocatalytic activity primarily to the SPR effect of Au NPs in the hierarchical of Au-BFO nanostructured homojunction and secondly to the structural defects (Fe2+/Fe3+ pairs and oxygen vacancy) [81].
3.3. Hydrothermal Method
Another widely used synthesis route to grow perovskite structures is the hydrothermal method. This reaction occurs in an aqueous solution into a close system under high pressure and temperature above the boiling point. When the materials synthesis is performed in a nonaqueous solvent at relatively high temperature, the process is known as the solvothermal method. In the hydrothermal/solvothermal method, the temperature is relatively high, but lower than the temperature employed in the methods explained above. In these methods, the crystallization process occurs directly in the solution, which excludes the calcination step and contributes to the growth of particles with very well-controlled morphology [82].
CaTiO3 orthorhombic nanocuboids with a size around 0.3–0.5 µm in width and 0.8–1.1 µm in length were produced using the conventional hydrothermal method. After the CaTiO3 synthesis, the hybridized Au nanoparticles were deposited on perovskite surface obtaining Au@CaTiO3 nanocomposite with an Au mass fraction of 4.3%. XPS spectrum indicated that Au species exist in the metallic state on perovskite surface. We performed photocatalytic tests under simulated-sunlight, ultraviolet, and visible light using Rhodamina B as pollutant model. After 120 minutes of simulated-sunlight irradiation, the degradation of Rhodamine reached 76.4% using pure CaTiO3 and 99.9% when the photocatalyst was the composite. Under UV light irradiation, the efficiency of CaTiO3 reached 95.7%, while the efficiency of Au@CaTiO3 was 99.9%. By irradiating the system with a visible light source, the CaTiO3 efficiency had the worst result (15.1%), whereas the composite presented 46.1% of efficiency. The presence of Au onto CaTiO3 enhanced visible-light absorption; decreased charge-transfer resistance enhanced the photocatalytic performance of the material. CaTiO3 is excited under UV irradiation producing electrons in its conduction band and holes in its valence band. Even though Au nanoparticles could not be excited under UV irradiation, they can act as electron sinks to capture the photogenerated electrons in CaTiO3. The electronic transfer between the conduction band of CaTiO3 to Au nanoparticles promotes the spatial separation of the electron/hole pair in perovskite. For this reason, more holes in the valence band of CaTiO3 are available in the photocatalytic process. CaTiO3 cannot be directly excited to produce electron/hole pairs under visible light irradiation. However, the located surface plasmon resonance (LSPR) of Au nanoparticles is induced by visible-light absorption stimulating the generation of electron/hole pairs in CaTiO3 due to electromagnetic field caused by LSPR effect. This is why Au@CaTiO3 nanocomposites also present visible-light photocatalytic degradation of Rhodamine B. When simulated sunlight is used as irradiation source, Au nanoparticles act as electron sinks and are responsible for the LSPR effect in the Au@CaTiO3 composites, improving the photocatalytic performance of nanocomposite under simulated sunlight irradiation [83].
Malkhasian studied the photocatalytic performance of Pt/AgVO3 nanowires under visible light irradiation in the photooxidation of atrazine. The author used the hydrothermal method to synthesize β-AgVO3 nanowires posteriorly used to obtain the Pt/AgVO3 compound. For this purpose, Pt metal was deposited on β-AgVO3 surface under UV-light irradiation using a photo-assisted deposition (PAD) method. XRD patterns reveal that only the β-AgVO3 peaks are present in the samples indicating that Pt content was well dispersed on the perovskite surface, which was confirmed through TEM analyses. In contrast, higher Pt percentage decreases the crystalline size of β-AgVO3. Higher Pt weight percentage of Pt from 0.2% to 0.6% also leads to improved photocatalytic efficiency. When using 0.6 g/L of the 0.6%wt Pt/β-AgVO3, the oxidation of atrazine was 99% after a 60-minute reaction time. The oxidation time of atrazine decreases from 60 minutes to 40 minutes upon the use of 0.9 g/L of photocatalyst. This behavior results from the higher number of available active sites to oxidize atrazine with higher photocatalyst concentration. In addition, the photocatalyst can be used five times without losing its stability [84].
Me-loaded NaNbO3 catalysts (Me = Fe, Ni, Co, and Ag) were prepared, aiming at producing H2 through the water-splitting process. The material was obtained by impregnating NaNbO3 (previously synthesized through hydrothermal method) in an aqueous solution of cation nitrates followed calcination. The formation of colored powders indicated the formation of metal oxidized on the surface of perovskite particles. The samples presented similar morphologies composed by irregular structures without significant modification caused by metal loading on NaNbO3. Photocatalyst results showed that different metal oxides loaded with NaNbO3 have a different effect in the photocatalysis for H2 evolution. Interestingly, only Ag/NaNbO3 proved superior efficiency than pristine NaNbO3 in the performed tests. This behavior was attributed to the fact that the difference between the electronegativity of Ag and Nb is higher than the difference between the electronegativity of Nb and the other metals employed on the surface of NaNbO3. Additionally, the Ag species on the perovskite surface acts as active site for proton reduction, which can explain the highest photocatalytic activity of Ag/NaNbO3 [85]. Ruthenium particles supported on hierarchical nanoflowers assembled by cubic phase NaNbO3 have been applied to degrade Rhodamine B. The surface modification of NaNbO3 with Ruthenium promotes the photosensitized degradation of dye, while Ru-dopping NaNbO3 inhibits the photocatalytic effect. The photocatalyst activity improved with surface decoration due to efficienct electron transfer and charge separation achieved by composite. The best results occurred for NaNbO3 with 0.5% of Ru species and the degradation of Rhodamine B follows pseudo-first order kinetics. In addition, the photocatalyst can be used at least five times successively without losing its photocatalytic efficiency. Interestingly, the degradation time reduces after reusing photocatalyst. After the catalyst process, both crystalline structure and morphology of the perovskite do not change, which may indicate the stability of photocatalyst posteriorly confirmed through the XPS analysis [86].
The presence of Ag nanoparticles onto KNbO3 nanowires drastically improved the photoreactivity of the perovskite and the photodecomposition of Rhodamine B under UV and visible light irradiation. The material begins to lose its activity after three cycles of aqueous organic compound degradation under UV or visible light irradiation. The use of p-benzoquinone, t-butyl alcohol, and ammonium oxalate scavengers revealed that under UV illumination, ·O2− and ·OH are the major active species in the reaction. When visible light is used as an irradiation source, the major active species are·O2− and H+. During the photocatalytic process under UV irradiation, electrons present in the valence band of KNbO3 are excited to the conduction band with hole generation in the valence band. Due to the energy gap of Ag/KNbO3 composites, the photoexcited electrons could transfer from KNbO3 perovskite to Ag nanoparticles, enabling the separation between photogenerated electrons and holes in KNbO3 to promote the photocatalytic reaction. However, the excess of Ag on perovskite surface can act as a recombination center and partly avoid the UV absorption of perovskite decreasing the photocatalytic activity of the material. Visible-light irradiation promotes the photoexcitation of Ag nanoparticles electrons through intraband excitations within the sp band. Furthermore, the photocatalytic process under visible-light is driven by plasmonic sensitization due to an electron electron-transfer process from Ag nanoparticles to KNbO3 via localized surface plasmonic resonance. In addition, researchers revealed that the Ag/KNbO3 composites proved efficient not only for organic dye decomposition, but also for application in other areas, like water splitting [87]. More recently, Xing et al. studied the new application of Ag/KNbO3 nanocomposites in photocatalytic NH3 synthesis by converting N2 into NH3. The presence of Ag on KNbO3 surface significantly enhances the photocatalytic behavior of perovskite. The highest performance for NH3 generation is achieved when the Ag content in the sample is 0.5%, which reaches about four times more than pure KNbO3. NH3 generation is related to Ag content since the ammonium production increases with higher Ag and then reduces. Pristine KNbO3 has a poor response on visible light irradiation due to its high band gap value and the formation of composite with Ag nanoparticles significantly improves the performance of the material under visible light exposure due to the photosensitization effect of Ag nanoparticles. In addition, the composites also proved efficient at H2 generation under simulated sunlight radiation and photocatalytic Rhodamine B degradation [88].
Ag-decorated ATaO3 nanocubes (A = K, Na) were prepared using the hydrothermal method in order to evaluate their photocatalytic water-splitting activity under simulated and pure sunlight. The photocatalytic water-splitting results revealed that both Ag-decorated KTaO3 and NaTaO3 nanocubes exhibited a rate for H2 evolution from aqueous CH3OH solutions up to 185.60 and 3.54 μmol/hg under simulated sunlight, receptively, which were more than two times of pristine NaTaO3 and KTaO3 in the same experimental conditions. In contrast, under purely visible light illumination, the photocatalytic performance of both Ag-decorated ATaO3 materials was lower, therefore, the highest H2 evolution of 25.94 and 0.83 μmol/h g for Ag-decorated KTaO3 and NaTaO3, respectively. The authors conclude that the surface plasmonic effect of Ag nanoparticles was responsible for increasing appreciably the quantum efficiency photocatalytic water-splitting activity of the Ag-decorated K, NaTaO3 materials [89].
As observed, several perovskites structures applied in photocatalysis are obtained from hydrothermal synthesis. One of the most interesting advantages in the use of this method to obtain semiconductor materials is the possibility of producing the same material with different morphologies. Spherical shaped SrTiO3 with sponge-like mesoporous morphology [90], cubic-like SrTiO3 [91,92], SrTiO3 dendritic particles, among other SrTiO3 morphologies, can be obtained from hydrothermal method [93] (Figure 4). In this sense, the syntheses parameters can be controlled in order to produce a material with different morphology. The work conducted by Kalyani et al. is a good example of how synthesis parameters influence the product obtained since the authors developed a comprehensive study on the SrTiO3 growth mechanism as a function of the synthesis conditions [93]. After the synthesis, the surface of the particles can be easily decorated with a precious metal through a chemical solution process; in some cases, the metal deposition process may be photo-assisted [83,84]. Figure 5 from the work published by Kumar et al. exemplifies an easy chemical solution route to deposit precious metal nanoparticles on the perovskite surface [93]. Kumar addressed the fabrication of photoanodes of NaNbO3 nanorods and Ag decorated NaNbO3 nanorods across hydrothermal method and chemical solution method. Due to the visible plasmonic effect, Ag nanoparticles are responsible for reducing the material optical band gap. Additionally, the Ag decorated NaNbO3 nanorods presented the best photocurrent density because the surface plasmon mediated electron transfer from metallic nanoparticles decorated NaNbO3 nanorods [94].
Based on the processing routes discussed here, any pure perovskite structure composed of precious metal or precious metal/perovskite hybrid structure can be obtained aiming at the applicability in a catalyst test.
These materials can be also processed as film. Jeong and co-workers [91] developed a fully solution-deposited nanocomposite photoanode from the impregnation of Ag NPs in the BiVO4 films prepared through spin coating and post-annealing (500 °C for 2h) to evaluate the photocatalytic water-splitting activity under simulated sunlight of 1 sun (100 mW/cm2) with Xe lamp. Ag nanoparticles-incorporated BiVO4 nanocomposite, for optimum Ag concentration of 4.0 mM, exhibited an appreciable improvement of photocatalytic performance at low potential (0.4 V), in addition to generating a saturated photocatalytic current density 3.3 times higher than the pristine BiVO4 film. Jeong et al. explain that these results can be associated with the SPR-induced enhancement mechanism, once Ag NPs impregnated in BiVO4 films are responsible for promoting enhanced carrier generation and separation. The performance of the films enhances the current density at low potentials and improves the kinetics of the carrier generation and separation due to the efficient charge carrier generation [95].
Zhang et al. [96] was approached by the role of charge transfer (CT) on the degradation of Rhodamine B under UV (265 nm) and UV–Vis light (Xe lamp 500 W with a 420 nm optical filter) using Ag-nanoparticle-dispersed BaTiO3 (Ag/BTO) composite films prepared through the sol-gel and spin-coating methods. Among the BTO materials obtained, only the monolithic BTO did not exhibit the absorption peak in the visible wavelength region of 450–600 nm, which is the peak associated with the SPR effect of the Ag NPs. The optical absorption spectra revealed that as Ag content increases until 25 mol%, a red-shift was observed, whereas a blue shift occurs with Ag content of 30 mol%. The photocatalytic results showed that Ag25/BTO film was the best Ag/BTO composite films; therefore, its photocatalytic efficiency was better under visible (62%) rather than UV–Vis (42%) light irradiation. The authors attributed this improvement in photocatalytic activity of the Ag25/BTO film to the SPR effect of the Ag nanoparticles derived from the maximum absorption profile of the band with slightly red-shift trend from 517 to 523 nm. XPS and PL measurements provide great evidence on the synergetic effect between the charge transfer from perovskite to Ag nanoparticles under UV light and charge transfer from Ag nanoparticles to BaTiO3 under visible light irradiation [96].
Until now, we approached the photocatalysis as environmental contamination solution, however, the electrocatalysis also represents an alternative to this purpose. Both processes involve oxidation and reduction mechanisms from photochemical reactions. As explained, in the photocatalysis the redox reaction is promoted by light absorption, while in the case of electrocatalysis, the redox process occurs through the flow of an electric current through an external circuit in the reaction system, as in further explanations below.
4. Application of Perovskites in Electrocatalysis
Electrocatalysis is the study area concerning chemical reactions that occur on electrode surfaces. It is strategical to develop devices and methodologies applied to produce clean energy and benefit a response to the increasing worldwide energetic demand [97]. The most efficient electrocatalysts derive from noble metals, e.g. Pt, Ru, Rh, and Ir based nanomaterials, but the high cost of these materials makes the large-scale production of technologies aiming at sustainable energy production, conversion, and storage a goal difficult to be achieved [98,99]. To overcome such issue, perovskites have been gaining attention as promising materials to act as electrocatalysts for their exceptional thermal stability, ionic conductivity, electron mobility, and redox behavior [100]. The perovskite electrocatalysts can be applied to plenty of electrochemical reactions, such as oxygen reduction, oxygen evaluation, hydrogen evaluation, alcohol oxidation, carbon dioxide reduction, water splitting, among others [101]. In this section, we present some insights on the perovskites electrocatalysts containing precious metals and their applications aiming at the development of energy generation devices.
The oxygen reduction reaction (ORR) and oxygen evaluation reaction (OER) have great importance in the development of electrochemical energy-conversion devices such metal-air batteries, polymer electrolyte membrane fuel cells (PEMFC), and other devices, e.g. in oxygen sensors. The large overpotential associated with these processes, due their slow reactions kinetics, is one of the major challenges to be overcome to develop high-performance catalysts [102]. Taking as an example the PEMFC, which accounts for one of the most representative applications of oxygen electrocatalysis, the oxygen could react through 2-electron or 4-electron pathways, as shown in the chemical equations below [103]:
| ORR in alkaline medium | |
| O2 + 2H2O + 4e− → 4OH− (4-electrons process) | −1 |
| O2 + H2O + 2e− → HO2− + OH− | (2a) |
| H2O + HO2− + 2e− → 3OH− (2-electrons process) | (2b) |
| ORR in acid medium | |
| O2 + 4H+ + 4e− → 2H2O (4-electrons process) | −3 |
| O2 + 2H+ + 2e− → H2O2 | (4a) |
| H2O2 + 2H+ + 2e− → 2H2O (2-electrons process) | (4b) |
Currently, the most efficient catalysts for both anodic and cathodic electrochemical reactions in PEMFC are the Pt-based electrocatalysts. However, the high costs involved in large scale production represent an enormous problem. In this scenario, the perovskites structures arise as one alternative to obtain cheaper electrocatalysts that exhibit the same features of the Pt electrocatalysts, like outstanding catalytic and electrical properties and superior resistant characteristics to corrosion [104]. Recently, perovskite-type oxides have gained noticeable popularity as cost-effective bifunctional oxygen catalysts with promising catalytic activity and stability for the ORR and OER in an alkaline medium for their excellent electrical properties and abundant active sites [105].
Zhang et al. [106] provide an example of a precious metal-containing perovskite catalyst applied to ORR by using thermal treatments incorporated Ag-decorating nanoparticles into PBMO5 perovskites nanofibers matrix, followed by a second thermal treatment, which allowed the Ag nanoparticles to be exsolved from the crystal structure, establishing a strong interaction with the perovskite matrix and generating the Ag-(PrBa)0.95Mn2O5+δ (Ag-PBMO5) catalyst, as illustrated in Figure 6a. This catalyst has favorable ORR activity enhanced in relation to the PBMO5 and higher durability than the state-of-art Pt/C catalyst in alkaline solution. The ORR electrocatalytic tests were performed using linear sweep voltammetry (LSV) technique and the results revealed that Ag-PBMO5 catalyst demonstrated considerably higher catalytic activity in comparison to the its predecessors with ORR Eonset and E1/2 of Ag-PBMO5 catalyst of 0.92 V and 0.81 V vs. RHE, and the ORR Eonset and E1/2 of PBMO5 catalyst of 0.84 V and 0.74 V vs. RHE, respectively. The use of a Koutecky–Levich analysis based on the LSV experiments performed with a rotating electrode revealed that the Ag-PBMO5 electrocatalyst reduces the oxygen completely and directly to OH− though a 4-electron reaction pathway, achieving the highest energy efficiency in theory. The outstanding catalytic features of Ag-PBMO5 arises from a sum of factors, like crystal structure of reconstructed perovskite with oxygen defects internally ordered, which facilitates the charge transfer during the ORR through the perovskite structure and the resulting Mn3+ valence with eg occupancy, which moderates the intermediate binding strength on Mn sites and contributes to electrocatalytic activity of PBMO5. The contribution of exsolved Ag nanoparticles was investigated using DFT calculations, which revealed a significant ligand effect between the nanoparticles and PBMO5 and an increase in electron occupancy in s, p, and d orbitals on Mn sites, which results in a charge transfer from Ag to Mn sites. In this case, the d-band orbitals of Ag atoms are not completely filled and vacancies are generated, which consequently narrows the d-orbital of Ag and up-shifts the d-band center of Ag atom. The Ag-atom with and up-shifted d-band center is expected to adsorb O2 more strongly and split the O–O bond more efficiently. Therefore, charge transfer plays a crucial role in the enhancement of the ORR activity.
Zhu et al. [107] reported another example of perovskite electrocatalyst with exsolved Ag nanoparticles. The heterostructured Ag nanoparticle-decorated perovskite (denoted by e-SANC) was obtained from the precursor Sr0.95Ag0.05Nb0.1Co0.9O3−δ (SANC) through an easy exsolving process. The ORR activity was measured using the electrochemical impedance spectroscopy (EIS) technique and data were calculated from impedance loops the area-specific resistance (ASR). Electrocatalysts with lower ASR values show higher ORR activity. The e-SANC electrocatalyst exhibit very low area-specific resistance (0.214 Ω cm2 at 500 °C), which is much lower than its precursor (0.363 Ω cm2 at 500 °C). To illustrate the effect of Ag atoms in the perovskite structure, the results were compared to an electrode prepared without Ag (SNC0.95). In this case, the e-SANC electrocatalyst also showed ASR values at least 50% lower in addition to lower energy activation for ORR for the entire temperature range studied. The analysis of the circuit model used for EIS experiments can offer some insights on the ORR mechanism. The high-frequency resistance (RE1) is associated with the charge-transfer process and the results indicate that this feature is not affected in a significant way when comparing the catalysts e-SANC and SNC0.95. The low-frequency resistance (RE2) is related to oxygen surface process, like adsorption, desorption, dissociation at gas–cathode interface, and surface diffusion of intermediate species. The RE2 value for e-SANC decreased greatly when compared to SNC0.95, indicating that the Ag nanoparticles increase the velocity of the oxygen surface processes, an effect also found in kex (surface exchange coefficient) measurements using electrical conductivity relaxation (ECR) technique. The e-SANC electrocatalyst performance was evaluated in a solid oxide fuel cell (SOFC), in which the material was used as a cathode; the system showed the power-density peak of 1116 mW cm−2, having remained stable for 140 h at 500 °C. Furthermore, the cathode tolerance to CO2 was improved in relation to the precursor. It is possible to find that the exsolving methods have great potential to prepare precious metal nanoparticle-modified perovskites oxides as efficient catalysts towards fuel cells applications [108].
Taking into account oxygen electrocatalysis, there are some cases of bifunctional catalysts, which means that they act in ORR and OER. The bifunctional electrocatalysts are fundamental to the development of regenerative fuel cells, which are devices that produce energy and electrolytically regenerate their reactants by using stacks of electrochemical cells [109]. Retuerto and co-workers [110] prepared the functional perovskite electrocatalyst La1.5Sr0.5NiMn0.5Ru0.5O6 (denoted as LSNMR) from the mixture of La2O3, the Ru, Mn and Ni oxides, and SrCO3 at stoichiometric proportions. The efficiency of LSNMR electrocatalyst towards ORR and OER was measured in alkaline medium using the bifunctional index (BI) proposed by Schuhmann et al. [111]. The BI value determined to LSNMR bifunctional electrocatalyst was approximately 0.83 V, while an ideal ORR-OER electrocatalyst must show a BI value close to 0 V and the state-of-art bifunctional electrocatalysts shows values around 0.9 V. To the best of our knowledge, the LSNMR electrocatalyst appears to be the unique example of bifunctional catalyst for ORR and OER containing a precious metal into a perovskite structure. For comparison purposes, we can highlight the BI values of some bifunctional electrocatalysts, such as La0.58Sr0.4Co0.2Fe0.8O3/FeNx−C (0.86 V) [112], La0.7(Ba0.5Sr0.5)0.3Co0.8Fe0.2O3−δ/C (0.88 V) [113], La0.58Sr0.4Co0.2Fe0.8O3/N-CNT (0.826 V) [111], LaNi0.8Fe0.2O3 (1.02 V) [114], LaNi0.85Mg0.15O3 (1.15 V) [115], and the state-of-art catalysts RuO2 (0.8 V) and IrO2 (1.32 V) [112].
Water splitting has been arising interest for its possibility to generate hydrogen in a carbon-neutral manner. This process can be achieved by using electrodes under a light source, converting H2O to H2 and O2 through a photoelectrochemical (PEC) process [116]. Several transition metal oxides, and oxygen and halide perovskites have been used in the PEC evaluation of H2 [117]. Kumar and co-workers [94] provide an example after having developed an anode of NaNbO3 nanorods decorated with plasmonic Ag nanoparticles applied to PEC H2 evaluation. The results showed that both bare NaNbO3 and the Ag-decorated nanorods exhibit low current density due to H2 generation in dark conditions, but when the system was irradiated with a halogen lamp, the Ag-decorated nanorods anode exhibit 4-fold current density in relation to bare NaNbO3 catalyst. The origins of such an enhancement were investigated using the EIS technique, which revealed that the decoration of NaNbO3 increases its donor density, indicating the transfer of majority carriers from plasmonic Ag nanoparticles to perovskite matrix resulting in a higher concentration of charge carriers.
5. Final Considerations
Photocatalysis and electrocatalysis have been great routes to degrade persistent organic pollutants based on advanced oxidative processes. Because of their electrical and optical properties, perovskite structured materials represent a good alternative to replace TiO2-based materials in environmental treatment using AOPs. The perovskite properties are improved with the presence of precious metals in its crystalline structure and/or the formation of a precious metal/perovskite structure hybrid material. The most used precious metals for this purpose are silver and gold. This occurs due to the surface plasmon resonance phenom presented by metals. These semiconductors can be processed as powder and/or films. Chemical syntheses are efficient routes to produce these catalysts, especially hydrothermal synthesis. Based on the processing route discussed here, it is possible to obtain a catalyst with appropriate characteristics to be applied in both the dye decomposition and water-splitting process.
Author Contributions
Bibliographical review about photocatalysis and the use of perovskites in this issue, chemical synthesis survey and eletrocathalysis approach were done by G.F.T.; E.S.J. and R.V. under supervision of M.A.Z. and F.C. All authors contributed to the manuscript preparation, and as the same way, all authors have given approval for the final version of the review.
Funding
This research was funded by Chemistry Postgraduate Program of Federal University of Goiás, CAPES (Process number 88882.306480/2018-1) and FAPESP-CEPID/CDMF 2013/07296-2
Acknowledgments
The authors would like to thank the Brazilian research agencies CAPES (Process: 88882.306480/2018-1), CNPq, (Process 554569/2010-8 and 475609/2008-5) and, FAPESP-CEPID/CDMF 2013/07296-2 for granting the support to research groups.
Conflicts of Interest
The authors declare no conflict of interest.
References
- IUPAC. Compendium of chemical terminology. In The Gold Book, 2nd ed.; McNaught, A.D., Wilkinson, A., Eds.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar]
- Deng, Y.; Zhao, R. Advanced Oxidation Processes (AOPs) in Wastewater Treatment. Curr. Pollut. Rep. 2015, 1, 167–176. [Google Scholar] [CrossRef]
- Yang, L.; Yang, L.; Ding, L.; Deng, F.; Luo, X.-B.; Luo, S.-L. Principle for the application of nanomaterials in environmental pollution control and resource reutilization. In Nanomaterial for the Removal of Pollutants and Resource Reutilization; Luo, X., Deng, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–23. [Google Scholar]
- Glaze, W.H.; Kang, J.-W.; Chapin, D.H. The chemistry of water treatment processes involving ozone, hydrogen peroxide and ultraviolet radiation. Ozone Sci. Eng. 1987, 9, 335–352. [Google Scholar] [CrossRef]
- Parsons, S. Advanced Oxidation Processes for Water and Wastewater Treatment; IWA Publishing: London, UK, 2004; pp. 1–347. [Google Scholar]
- Fan, X.; Hao, H.; Shen, X.; Chen, F.; Zhang, J. Removal and degradation pathway study of sulfasalazine with Fenton-like reaction. J. Hazard. Mater. 2001, 190, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.; Ullah, R.; Butt, A.M.; Gohar, N.D. Strategies of making TiO2 and ZnO visible light active. J. Hazard. Mater. 2009, 170, 560–569. [Google Scholar]
- WHO. Fluoride in drinking-water. In Guidelines for Drinking-Water Quality, 3rd ed.; WHO Press: Geneva, Switzerland, 2006; Volume 1, pp. 375–377. [Google Scholar]
- Ibrahim, M.; Asimrasheed, M.; Sumalatha, M.; Prabhakar, P. Effects of fluoride contents in ground water: A review. Inter. J. Pharm. Appl. 2011, 2, 128–134. [Google Scholar]
- Singh, J.; Singh, P.; Singh, A. Fluoride ions vs. removal technologies: A study. Arab. J. Chem. 2016, 9, 815–824. [Google Scholar] [CrossRef]
- Modi, S.; Soni, R. Merits and Demerits of different technologies of defluoridation for drinking water. IOSR J. Environ. Sci. Toxicol. Food. Technol. 2013, 3, 24–27. [Google Scholar] [CrossRef]
- Gupta, V.K.; Ali, I.; Saleh, T.A.; Nayak, A.; Agarwal, S. Chemical treatment technologies for waste-water recycling—An overview. RSC Adv. 2012, 2, 6380–6388. [Google Scholar] [CrossRef]
- Gogate, P.R.; Pandit, A.B. A review of imperative technologies for wastewater treatment II: Hybrid methods. Adv. Environ. Res. 2004, 8, 553–597. [Google Scholar] [CrossRef]
- Oller, I.; Malato, S.; Sanchez-Perez, J.A. Combination of advanced oxidation processes and biological treatments for wastewater decontamination—A review. Sci. Total Environ. 2011, 409, 4141–4166. [Google Scholar] [CrossRef]
- Esplugas, S.; Bila, D.M.; Krause, L.G.T.; Dezotti, M. Ozonation and advanced oxidation technologies to remove endocrine disrupting chemicals (EDCs) and pharmaceuticals and personal care products (PPCPs) in water effluents. J. Hazard. Mater. 2007, 149, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, R.; Gracia, F.; Stephen, A. Basic principles, mechanism, and challenges of photocatalysis. In Nanocomposites for Visible Light-Induced Photocatalysis; Khan, M.M., Pradhan, D., Shon, Y., Eds.; Springer: Cham, Switzerland, 2018; pp. 19–40. [Google Scholar]
- Serpone, N.; Horikoshi, S.; Emeline, A.V. Microwaves in advanced oxidation processes for environmental applications. A brief review. J. Photochem. Photobiol. C 2010, 11, 114–131. [Google Scholar] [CrossRef]
- Legrini, O.; Oliveros, E.; Braun, A.M. Photochemical processes for water treatment. Chem. Rev. 1993, 93, 671–698. [Google Scholar] [CrossRef]
- Huang, C.P.; Dong, C.; Tang, Z. Advanced chemical oxidation: Its present role and potential future in hazardous waste treatment. Waste Manag. 1993, 13, 361–377. [Google Scholar] [CrossRef]
- Pignatello, J.J.; Oliveros, S.E.; Mackay, A. Advanced oxidation processes of organic contaminant destruction based of the Fenton reaction and related chemistry. Crit. Rev. Environ. Sci. Technol. 2006, 36, 1–84. [Google Scholar] [CrossRef]
- Carraway, E.R.; Hoffman, A.J.; Hoffmann, M.R. Photocatalytic oxidation of organic acids on quantum-sized semiconductor colloids. Environ. Sci. Technol. 1994, 28, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Neppolian, B.; Choi, H.S.; Sakthivel, S.; Arabindoo, B.; Murugesan, V. Solar light induced and TiO2 assisted degradation of textile dye reactive blue 4. Chemosphere 2002, 46, 1173–1181. [Google Scholar] [CrossRef]
- Reza, K.M.; Kurny, A.S.; Gulshan, F. Parameters affecting the photocatalytic degradation of dyes using TiO2: A review. Appl. Water Sci. 2015, 4, 1569–1578. [Google Scholar] [CrossRef]
- Lim, M.; Son, Y.; Khim, J. Frequency effects on the sonochemical degradation of chlorinated compounds. Ultrason. Sonochem. 2011, 8, 460–465. [Google Scholar] [CrossRef]
- Doodeve, C.F.; Kitchener, J.A. Photosensitisation by titanium dioxide. Trans. Faraday Soc. 1938, 34, 570–579. [Google Scholar]
- Hashimoto, K.; Irie, H.; Fujishima, A. TiO2 photocatalysis: A historical overview and future prospects. Jpn. J. Appl. Phys. 2005, 44, 8269–8285. [Google Scholar] [CrossRef]
- Ameta, S.C.; Ameta, R. Introduction. In Advanced Oxidation Processes for Wastewater Treatment; Ameta, S.C., Ameta, R., Eds.; Academic Press: Oxford, UK, 2018; pp. 1–12. [Google Scholar]
- Gaya, U.I. Principles of heterogeneous photocatalysis. In Heterogeneous Photocatalysis Using Inorganic Semiconductor Solids; Gaya, U.I., Ed.; Springer Science: Dordrecht, The Netherlands, 2014; pp. 1–34. [Google Scholar]
- Gratzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Cowan, A.J.; Durrant, J. Long-lived charge separated states in nanostructured semiconductor photoelectrodes for the production of solar fuels. Chem. Soc. Rev. 2013, 42, 2281–2293. [Google Scholar] [CrossRef]
- Al-Ekabi, H.; Serpone, M. Kinetic studies in heterogeneous photocatalysis. 1. Photocatalytic degradation of chlorinated phenols in aerated aqueous solutions over TiO2 supported on a glass matrix. J. Phys. Chem. 1988, 92, 5726–5731. [Google Scholar] [CrossRef]
- Serpone, N. Brief introductory remarks on heterogeneous photocatalysis. Sol. Energy Mater. Sol. Cells 1995, 38, 369–379. [Google Scholar] [CrossRef]
- Paramasivam, I.; Jha, H.; Liu, N.; Schmuki, P. A review of photocatalysis using self-organized TiO2 nanotubes and other ordered oxide nanostructures. Small 2012, 8, 3073–3103. [Google Scholar] [CrossRef]
- Shen, Y.; Guo, X.; Bo, X.; Wang, T.; Guo, X.; Xie, M.; Guo, X. Effect of template-induced surface species on electronic structure and photocatalytic activity of g-C3N4. Appl. Surf. Sci. 2017, 396, 933–938. [Google Scholar] [CrossRef]
- Ravelli, D.; Dondi, D.; Fagnoni, M.; Albini, A. Photocatalysis. A multi-faceted concept for green chemistry. Chem. Soc. Rev. 2009, 38, 1999–2011. [Google Scholar] [CrossRef]
- Liu, B.; Zhao, X.; Terashima, C.; Fujishima, A.; Nakata, K. Thermodynamic and kinetic analysis of heterogeneous photocatalysis for semiconductor systems. Phys. Chem. Chem. Phys. 2014, 16, 8751–8760. [Google Scholar] [CrossRef]
- Henderson, M.A. A surface science perspective on TiO2 photocatalysis. Surf. Sci. Rep. 2011, 66, 185–297. [Google Scholar] [CrossRef]
- Grabowska, E. Selected perovskite oxides: Characterization, preparation and photocatalytic properties—A review. Appl. Catal. B Environ. 2016, 186, 97–126. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, G.; Wang, L.; Irvine, J.T.S. Inorganic perovskite photocatalysts for solar energy utilization. Chem. Soc. Rev. 2016, 45, 5951–5984. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Cervantes, M.L.; Castillejos, E. Perovskites as catalysts in advanced oxidation processes for wastewater treatment. Catalysts 2019, 9, 230. [Google Scholar] [CrossRef]
- Shi, J.; Guo, L. ABO3-based photocatalysts for water splitting. Prog. Nat. Sci. Mater. Inter. 2012, 22, 592–615. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, R.H. Perovskites: Modern and Ancient; Almaz Press: Thunder Bay, ON, Canada, 2002; pp. 1–318. [Google Scholar]
- Glazer, A.M. The classification of tilted octahedra in perovskites. Acta Crystallogr. Sect. B 1972, 28, 3384–3392. [Google Scholar] [CrossRef]
- Tilley, R.J.D. Perovskites. Structure-Property Relationships; John Wiley & Sons Ltd.: Chichester, UK, 2016; pp. 1–315. [Google Scholar]
- Mishra, A.; Prasad, R. Preparation and application of perovskite catalyst for diesel soot emission control: An overview. Catal. Rev. 2014, 56, 57–81. [Google Scholar] [CrossRef]
- Knight, K.S. Structural phase transitions, oxygen vacancy ordering and protonation in doped BaCeO3: Results from time-of-flight neutron powder diffraction investigations. Solid State Ion. 2001, 145, 275–294. [Google Scholar] [CrossRef]
- Woodward, P.M. Octahedral tilting in perovskites. I. geometrical considerations. Acta Crystallogr. Sect. B Struct. Sci. 1997, 53, 32–43. [Google Scholar] [CrossRef]
- Goldschmidt, V.M. Die Gesetze der Krystallochemie. Naturwissenschaften 1926, 14, 477–485. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomie distances in halides and chaleogenides. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Fan, Z.; Sun, K.; Wang, J. Perovskites for photovoltaics: A combined review of organic–inorganic halide perovskites and ferroelectric oxide perovskites. J. Mater. Chem. A 2015, 3, 18809–18828. [Google Scholar] [CrossRef]
- Yamada, I.; Takamatsu, A.; Ikeno, H. Complementary evaluation of structure stability of perovskite oxides using bond-valence and density functional-theory calculations. Sci. Technol. Adv. Mater. 2018, 19, 102–107. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, Y.; Guo, L.; Zhao, W.W.; Barnes, A.; Zhang, H.-T.; Eatom, C.; Zheng, Y.; Brahlek, M.; Haneef, H.F.; et al. Correlated metals as transparent conductor. Nat. Mater. 2016, 15, 204–210. [Google Scholar] [CrossRef]
- Imran, Z.; Rafiq, M.A.; Hasan, M.M. Charge carrier transport mechanisms in perovskite CdTiO3 fibers. AIP Adv. 2014, 4, 067137. [Google Scholar] [CrossRef]
- Nikonov, A.V.; Kuterbekov, K.A.; Bekmyrza, K.Z.; Pavzderin, N.B. A brief review of conductivity and thermal expansion of perovskite-related oxides for SOFC cathode. Eurasian J. Phys. Funct. Mater. 2018, 2, 274–292. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Yang, L.; Ai, C.; Xie, P.; Lin, S.; Wang, C.-Z.; Lu, X. Tailoring band gap of perovskite BaTiO3 by Transition metals co-doping for visible-light photoelectrical applications: A first-principles study. Nanomaterials 2018, 8, 455. [Google Scholar] [CrossRef]
- Hwang, S.W.; Noh, T.H.; Cho, I.S. Optical properties, electronic structures, and photocatalytic performances of bandgap-tailored SrBi2Nb2−xVxO9 compounds. Catalysts 2019, 9, 393. [Google Scholar] [CrossRef]
- Linic, S.; Christopher, P.; Ingram, D.B. Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat. Mater. 2001, 10, 911–921. [Google Scholar] [CrossRef]
- Linic, S.; Aslam, U.; Boerigter, C.; Morabito, M. Photochemical transformations on plasmonic metal nanoparticles. Nat. Mater. 2015, 14, 567–576. [Google Scholar] [CrossRef]
- Nycenga, M.; Cobley, C.M.; Zeng, J.; Li, W.; Moran, C.H.; Zhang, Q.; Qin, D.; Xia, Y. Controlling the synthesis and assembly of silver nanostructures for plasmonic applications. Chem. Rev. 2011, 111, 3669–3712. [Google Scholar] [CrossRef]
- Wang, M.; Ye, M.; Iocozzia, J.; Lin, C.; Lin, Z. Plasmon-mediated solar energy conversion via photocatalysis in noble metal/semiconductor composites. Adv. Sci. 2016, 3, 1600024. [Google Scholar] [CrossRef]
- Hou, W.; Liu, Z.; Pavaskar, P.; Hung, W.H.; Cronin, S.B. Plasmonic enhancement of photocatalytic decomposition of methyl orange under visible light. J. Catal. 2011, 277, 149–153. [Google Scholar] [CrossRef]
- Nie, J.; Scheneider, J.; Sieland, F.; Zhou, L.; Xia, S.; Bahnemann. New insights into the surface plasmon resonance (SPR) driven photocatalytic H2 production of Au-TiO2. RSC Adv. 2018, 8, 25881–25887. [Google Scholar] [CrossRef]
- Liz-Marzán, L.M. Tailoring surface plasmons through the morphology and assembly of metal nanoparticles. Langmuir 2006, 22, 32–41. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, Y.; Yang, X.; Wang, S.; Shen, J.; Lin, B.; Li, C. Enhanced visible light photocatalytic activity of interlayer-isolated triplex Ag@SiO2@TiO2 core-shell nanoparticles. Nanoscale 2013, 5, 3359–3366. [Google Scholar] [CrossRef]
- Hou, W.; Cronin, S.B. A review of surface plasmon resonance-enhanced photocatalysis. Adv. Funct. Mater. 2013, 23, 1612–1619. [Google Scholar] [CrossRef]
- Wang, H.; Tam, F.; Grady, N.K.; Halas, N.J. Cu nanoshells: Effects of interband transitions on the nanoparticle plasmon resonance. J. Phys. Chem. B 2005, 109, 18218–18222. [Google Scholar] [CrossRef]
- Musialic-Piotrowwska, A.; Landmesser, H. Noble metal-doped perovskites for the oxidation of organic air pollutants. Catal. Today 2008, 137, 357–361. [Google Scholar] [CrossRef]
- Smart, L.E.; Moore, E.A. Solid State Chemistry, 3rd ed.; Taylor & Francis: Abingdon, UK, 2005; pp. 148–177. [Google Scholar]
- West, A.R. Solid State Chemistry and Its Applications, 2nd ed.; Wiley: Chichester, UK, 2014; pp. 187–228. [Google Scholar]
- Iwashina, K.; Kudo, A. Rh-doped SrTiO3 photocatalyst electrode showing cathodic photocurrent for water splitting under visible-light irradiation. J. Am. Chem. Soc. 2011, 133, 13272–13275. [Google Scholar] [CrossRef]
- Irie, H.; Maruyama, Y.; Hashimoto, K. Ag+ and Pb2+ doped SrTiO3 Photocatalysts. A correlation between band structure and photocatalytic activity. J. Phys. Chem. C 2007, 111, 1847–1852. [Google Scholar] [CrossRef]
- Saadetnejad, D.; Yıldırım, R. Photocatalytic hydrogen production by water splitting over Au/Al-SrTiO3. Int. J. Hydrog. Energy 2018, 43, 1116–1122. [Google Scholar] [CrossRef]
- Wang, F.; Wang, T.; Lang, J.; Su, Y.; Wang, X. Improved photocatalytic activity and durability of AgTaO3/AgBr heterojunction: The relevance of phase and electronic structure. J. Mol. Catal. A Chem. 2017, 426, 52–59. [Google Scholar] [CrossRef]
- Konta, R.; Ishii, T.; Kato, H.; Kudo, A. Photocatalytic activities of noble metal ion doped SrTiO3 under visible light irradiation. J. Phys. Chem. B 2004, 108, 8992–8995. [Google Scholar] [CrossRef]
- Sajjadi, S.P. Sol-gel process and its application in Nanotechnology. J. Polym. Eng. Technol. 2005, 13, 38–41. [Google Scholar]
- Rao, B.G.; Mukherjee, D.; Reddy, B.M. Nanostructures for novel therapy novel approaches for preparation of nanoparticles. In Nanostructures for Novel Therapy; Fricai, D., Grumezescu, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–36. [Google Scholar]
- Nayak, A.K.; Das, B. Introduction to polymeric gels. In Polymeric Gels. Characterization, Properties and Biomedical Applications; Pal, K., Banerjee, I., Eds.; Elsevier: Duxford, UK, 2018; pp. 3–27. [Google Scholar]
- Zhang, H.; Chen, G.; He, X.; Xu, J. Electronic structure and photocatalytic properties of Ag-La codoped CaTiO3. J. Alloy. Compd. 2012, 516, 91–95. [Google Scholar] [CrossRef]
- Reddy, K.H.; Martha, S.; Parida, K.M. Erratic charge transfer dynamics of Au/ZnTiO3 nanocomposites under UV and visible light irradiation and their related photocatalytic activities. Nanoscale 2018, 10, 18540–18554. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Chen, L.; Sun, H.; Zhang, H.; Guo, H.; Feng, L. In situ synthesis of au-induced hierarchical nanofibers/nanoflakes structured BiFeO3 homojunction photocatalyst with enhanced photocatalytic activity. Front. Chem. 2019, 6, 649–657. [Google Scholar] [CrossRef]
- Feng, S.; Li, G. Hydrothermal and solvothermal syntheses. In Modern Inorganic Synthetic Chemistry, 2nd ed.; Xu, R., Xu, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 73–104. [Google Scholar]
- Yan, Y.; Yang, H.; Yi, Z.; Li, R.; Wang, X. Enhanced photocatalytic performance and mechanism of Au@CaTiO3 composites with au nanoparticles assembled on CaTiO3 nanocuboids. Micromachines 2019, 10, 254. [Google Scholar] [CrossRef]
- Malkhasian, A.Y.S. Synthesis and characterization of Pt/AgVO3 nanowires for degradation of atrazine using visible light irradiation. J. Alloy. Compd. 2015, 649, 394–399. [Google Scholar] [CrossRef]
- Zielińska, B.; Borowiak-Palen, E.; Kalenczuk, R.J. Preparation, characterization and photocatalytic activity of metal-loaded NaNbO3. J. Phys. Chem. Solids 2011, 72, 117–123. [Google Scholar] [CrossRef]
- Chen, W.; Hu, Y.; Ba, M. Surface interaction between cubic phase NaNbO3 nanoflowers and Ru nanoparticles for enhancing visible-light driven photosensitized photocatalysis. Appl. Surf. Sci. 2018, 435, 483–493. [Google Scholar] [CrossRef]
- Zhang, T.; Lei, W.; Liu, P.; Rodriguez, J.A.; Yu, J.; Qi, Y.; Liu, G.; Liu, M. Organic Pollutant Photodecomposition by Ag/KNbO3 Nanocomposites: A combined experimental and theoretical study. J. Phys. Chem. C 2016, 120, 2777–2786. [Google Scholar] [CrossRef]
- Xing, P.; Wu, S.; Chen, Y.; Chen, P.; Hu, X.; Lin, H.; Zhao, L.; He, Y. New Application and Excellent Performance of Ag/KNbO3 Nanocomposite in Photocatalytic NH3 Synthesis. ACS Sustain. Chem. Eng. 2019, 7, 12408–124118. [Google Scholar] [CrossRef]
- Xu, D.; Yang, S.; Jin, Y.; Chen, M.; Fan, W.; Luo, B.; Shi, W. Ag-decorated ATaO3 (A = K, Na) nanocube plasmonic photocatalysts with enhanced photocatalytic water-splitting properties. Langmuir 2015, 31, 9694–9699. [Google Scholar] [CrossRef]
- Jayabal, P.; Sasirekha, V.; Mayandi, J.; Jeganathan, K.; Ramakrishnan, V. A facile hydrothermal synthesis of SrTiO3 for dye sensitized solar cell application. J. Alloy. Compd. 2014, 586, 456–461. [Google Scholar] [CrossRef]
- Gao, H.; Yang, H.; Wang, S. Hydrothermal synthesis, growth mechanism, optical properties and photocatalytic activity of cubic SrTiO3 particles for the degradation of cationic and anionic dyes. Optik 2018, 175, 237–249. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, Y.; Xu, L.; Cao, J.; Ho, W.; Lee, S.C. Visible-light-active plasmonic Ag-SrTiO3 nanocomposites for the degradation of NO in air with high selectivity. ACS Appl. Mater. Interfaces 2016, 8, 4165–4178. [Google Scholar] [CrossRef]
- Kalyani, V.; Vasile, B.S.; Ianculescu, A.; Testino, A.; Carino, A.; Buscaglia, M.T.; Buscaglia, V.; Nanni, P. Hydrothermal synthesis of SrTiO3: Role of interfaces. Cryst. Growth Des. 2015, 15, 5712–5725. [Google Scholar] [CrossRef]
- Kumar, D.; Singh, S.; Khare, N. Plasmonic Ag nanoparticles decorated NaNbO3 nanorods for efficient photoelectrochemical water splitting. Int. J. Hydrog. Energy 2018, 43, 1–8. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Shin, H.-M.; Jo, Y.-R.; Kin, Y.J.; Kim, S.; Lee, W.-J.; Lee, G.J.; Song, J.; Moon, B.J.; Seo, S.; et al. Plasmonic Silver nanoparticle-impregnated nanocomposite BiVO4 photoanode for plasmon-enhanced photocatalytic water splitting. J. Phys. Chem. C 2018, 122, 7088–7093. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, B.-P.; Li, S.; Huang, Z.; Yang, C.; Wang, H. Enhanced photocatalytic activity in Ag-nanoparticle-dispersed BaTiO3 composite thin films: Role of charge transfer. J. Adv. Ceram. 2017, 6, 1–10. [Google Scholar] [CrossRef]
- Gpel, W.; Schierbaum, K.D.; Vayenas, C.G.; Yentekakis, I.V.; Gerischer, H.; Vielstich, W.; Savage, P.E.; Moyes, R.B.; Bond, G.; Suslick, K.S. Special Catalytic Systems. In Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Weitkamp, J., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 1997; pp. 1283–1357. [Google Scholar]
- Wang, Y.-J.; Zhao, N.; Fang, B.; Li, H.; Bi, X.T.; Wang, H. Carbon-Supported Pt-Based Alloy Electrocatalysts for the Oxygen Reduction Reaction in Polymer Electrolyte Membrane Fuel Cells: Particle Size, Shape, and Composition Manipulation and Their Impact to Activity. Chem. Rev. 2015, 115, 3433–3467. [Google Scholar] [CrossRef] [Green Version]
- Shan, S.; Wu, J.; Kang, N.; Cronk, H.; Zhao, Y.; Zhao, W.; Skeete, Z.; Joseph, P.; Trimm, B.; Luo, J.; et al. Nanoscale Alloying in Electrocatalysts. Catalysts 2015, 5, 1465–1478. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Zhang, P.; Dai, S. Recent Advances of Lanthanum-Based Perovskite Oxides for Catalysis. ACS Catal. 2015, 5, 6370–6385. [Google Scholar] [CrossRef]
- Ghosh, S.; Basu, R.N. Multifunctional nanostructured electrocatalysts for energy conversion and storage: Current status and perspectives. Nanoscale 2018, 10, 11241–11280. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Rahaman, H. Noble metal-manganese oxide hybrid nanocatalysts. In Noble Metal-Metal Oxide Hybrid Nanoparticles; Elsevier: Amsterdam, The Netherlands, 2019; pp. 313–340. [Google Scholar]
- Zhang, J.; Xia, Z.; Dai, L. Carbon-based electrocatalysts for advanced energy conversion and storage. Sci. Adv. 2015, 1, e1500564. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, B. Recent advances in porous Pt-based nanostructures: Synthesis and electrochemical applications. Chem. Soc. Rev. 2014, 43, 2439–2444. [Google Scholar] [CrossRef]
- Shao-Horn, Y.; Hwang, J.; Yu, Y.; Katayama, Y.; Rao, R.R.; Giordano, L. Perovskites in catalysis and electrocatalysis. Science 2017, 358, 751–756. [Google Scholar] [Green Version]
- Zhang, Y.Q.; Tao, H.B.; Liu, J.; Sun, Y.F.; Chen, J.; Hua, B.; Thundat, T.; Luo, J.L. A rational design for enhanced oxygen reduction: Strongly coupled silver nanoparticles and engineered perovskite nanofibers. Nano Energy 2017, 38, 392–400. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, W.; Ran, R.; Chen, Y.; Shao, Z.; Liu, M. Promotion of Oxygen Reduction by Exsolved Silver Nanoparticles on a Perovskite Scaffold for Low-Temperature Solid Oxide Fuel Cells. Nano Lett. 2016, 16, 512–518. [Google Scholar] [CrossRef]
- Huang, K. An emerging platform for electrocatalysis: Perovskite exsolution. Sci. Bull. 2016, 61, 1783–1784. [Google Scholar] [CrossRef]
- Mitlitsky, F.; Myers, B.; Weisberg, A.H. Regenerative fuel cell systems. Energy Fuels 1998, 12, 56–71. [Google Scholar] [CrossRef]
- Retuerto, M.; Calle-Vallejo, F.; Pascual, L.; Lumbeeck, G.; Fernandez-Diaz, M.T.; Croft, M.; Gopalakrishnan, J.; Peña, M.A.; Hadermann, J.; Greenblatt, M. La1.5Sr0.5NiMn0.5Ru0.5O6 double perovskite with enhanced ORR/OER bifunctional catalytic activity. ACS Appl. Mater. Interfaces 2019, 11, 21454–21464. [Google Scholar] [CrossRef]
- Elumeeva, K.; Masa, J.; Tietz, F.; Yang, F.; Xia, W.; Muhler, M.; Schuhmann, W. A Simple Approach towards High-Performance Perovskite-Based Bifunctional Oxygen Electrocatalysts. ChemElectroChem 2016, 3, 138–143. [Google Scholar] [CrossRef]
- Rincón, R.A.; Masa, J.; Mehrpour, S.; Tietz, F.; Schuhmann, W. Activation of oxygen evolving perovskites for oxygen reduction by functionalization with Fe-Nx/C groups. Chem. Commun. 2014, 50, 14760–14762. [Google Scholar] [CrossRef]
- Elumeeva, K.; Masa, J.; Sierau, J.; Tietz, F.; Muhler, M.; Schuhmann, W. Perovskite-based bifunctional electrocatalysts for oxygen evolution and oxygen reduction in alkaline electrolytes. Electrochim. Acta 2016, 208, 25–32. [Google Scholar] [CrossRef]
- Zhang, D.; Song, Y.; Du, Z.; Wang, L.; Li, Y.; Goodenough, J.B. Active LaNi1−xFexO3 bifunctional catalysts for air cathodes in alkaline media. J. Mater. Chem. A 2015, 3, 9421–9426. [Google Scholar] [CrossRef]
- Du, Z.; Yang, P.; Wang, L.; Lu, Y.; Goodenough, J.B.; Zhang, J.; Zhang, D. Electrocatalytic performances of LaNi1−xMgxO3 perovskite oxides as bi-functional catalysts for lithium air batteries. J. Power Sources 2014, 265, 91–96. [Google Scholar] [CrossRef]
- Warwick, M.E.A.; Kaunisto, K.; Barreca, D.; Carraro, G.; Gasparotto, A.; Maccato, C.; Bontempi, E.; Sada, C.; Ruoko, T.-P.; Turner, S.; et al. Vapor Phase Processing of α-Fe2O3 Photoelectrodes for Water Splitting: An Insight into the Structure/Property Interplay. ACS Appl. Mater. Interfaces 2015, 7, 8667–8676. [Google Scholar] [CrossRef]
- Guerrero, A.; Bisquert, J. Perovskite semiconductors for photoelectrochemical water splitting applications. Curr. Opin. Electrochem. 2017, 2, 144–147. [Google Scholar] [CrossRef]
Figure 1.
(a) ABO3 ideal cubic perovskite structure and (b) the perovskite framework Mishra, A.; Prasad, R. Preparation and application of perovskite catalyst for diesel soot emission control: an overview. Catal Rev. 2014, 56, 57–81. Copyright 2014 Taylor & Francis [46].
Figure 1.
(a) ABO3 ideal cubic perovskite structure and (b) the perovskite framework Mishra, A.; Prasad, R. Preparation and application of perovskite catalyst for diesel soot emission control: an overview. Catal Rev. 2014, 56, 57–81. Copyright 2014 Taylor & Francis [46].
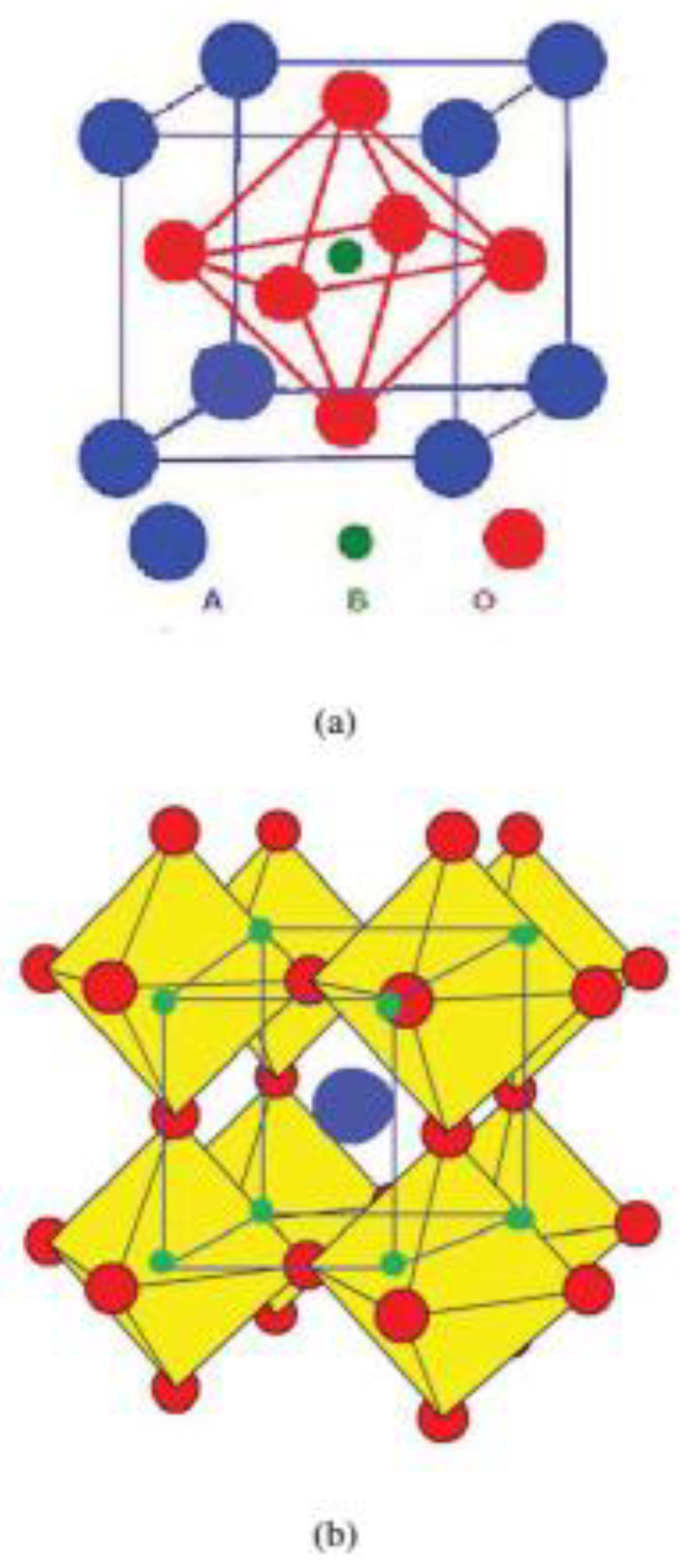
Figure 2.
Crystal structure of SrVO3 and CaVO3. Reprinted (adapted) with permission from Zhang, L.; Zhou, Y.; Guo, L.; Zhao, W.W.; Barnes, A.; Zhang, H.-T.; Eatom, C.; Zheng, Y.; Brahlek, M.; Haneef, H.F.; Podroza, N.J.; Chan, M.H.W.; Gopalan, V.; Rabe, K.M.; Engel-Herbert, R. Correlated metals as transparent conductor. Nat. Mater. 2016, 15, 204–210. Copyright 2015 Spring Nature [53].
Figure 2.
Crystal structure of SrVO3 and CaVO3. Reprinted (adapted) with permission from Zhang, L.; Zhou, Y.; Guo, L.; Zhao, W.W.; Barnes, A.; Zhang, H.-T.; Eatom, C.; Zheng, Y.; Brahlek, M.; Haneef, H.F.; Podroza, N.J.; Chan, M.H.W.; Gopalan, V.; Rabe, K.M.; Engel-Herbert, R. Correlated metals as transparent conductor. Nat. Mater. 2016, 15, 204–210. Copyright 2015 Spring Nature [53].
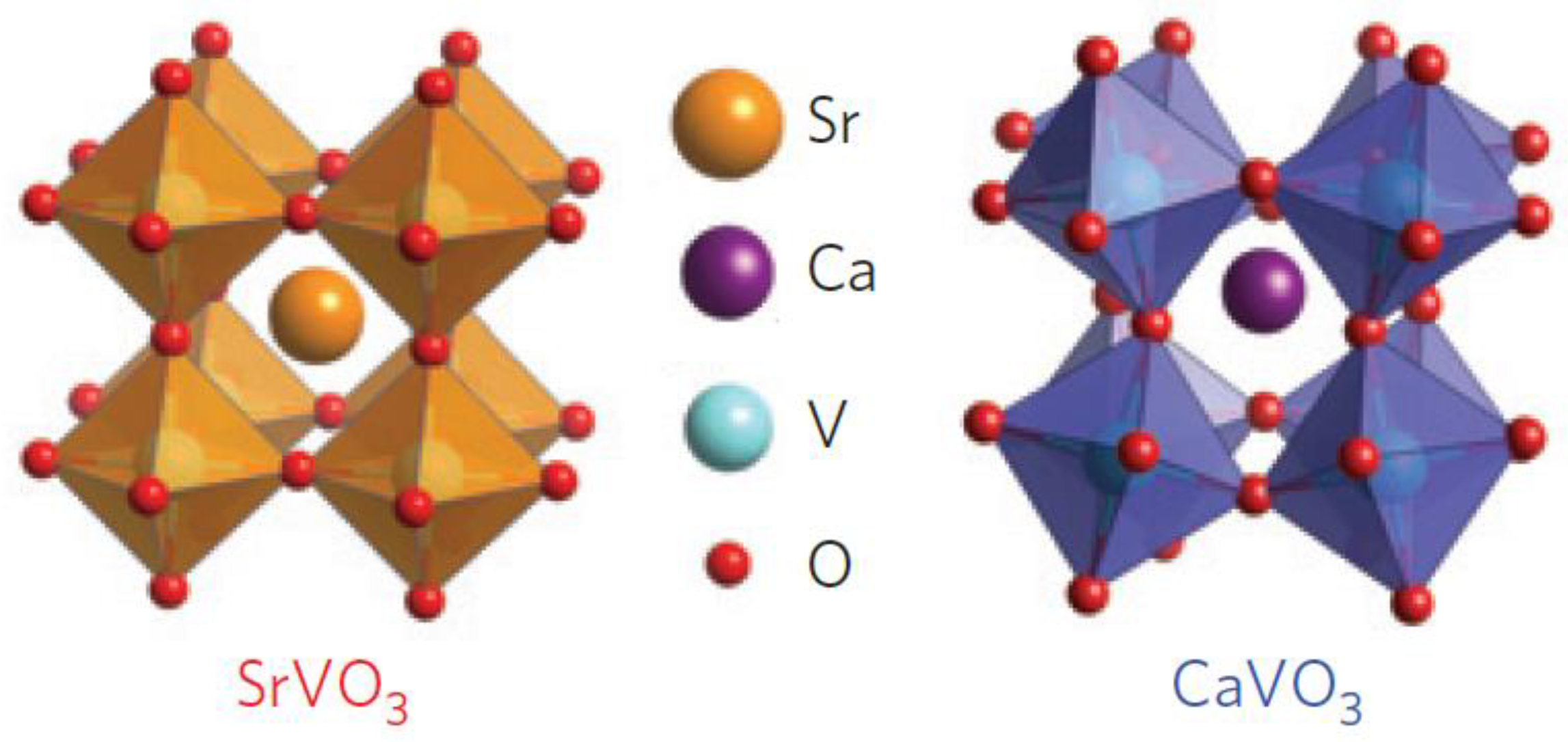
Figure 3.
The visible light activity of Au–TiO2 explained by two electron transfer mechanisms: resonance energy transfer process (right) and direct electron transfer process (left). Reprinted with permission from Nie, J.; Scheneider, J.; Sieland, F.; Zhou, L.; Xia, S.; Bahnemann. New insights into the surface plasmon resonance (SPR) driven photocatalytic H2 production of Au-TiO2. RSC Adv. 2018, 8, 25881–25887. Copyright 2018 The Royal Society of Chemistry [63].
Figure 3.
The visible light activity of Au–TiO2 explained by two electron transfer mechanisms: resonance energy transfer process (right) and direct electron transfer process (left). Reprinted with permission from Nie, J.; Scheneider, J.; Sieland, F.; Zhou, L.; Xia, S.; Bahnemann. New insights into the surface plasmon resonance (SPR) driven photocatalytic H2 production of Au-TiO2. RSC Adv. 2018, 8, 25881–25887. Copyright 2018 The Royal Society of Chemistry [63].
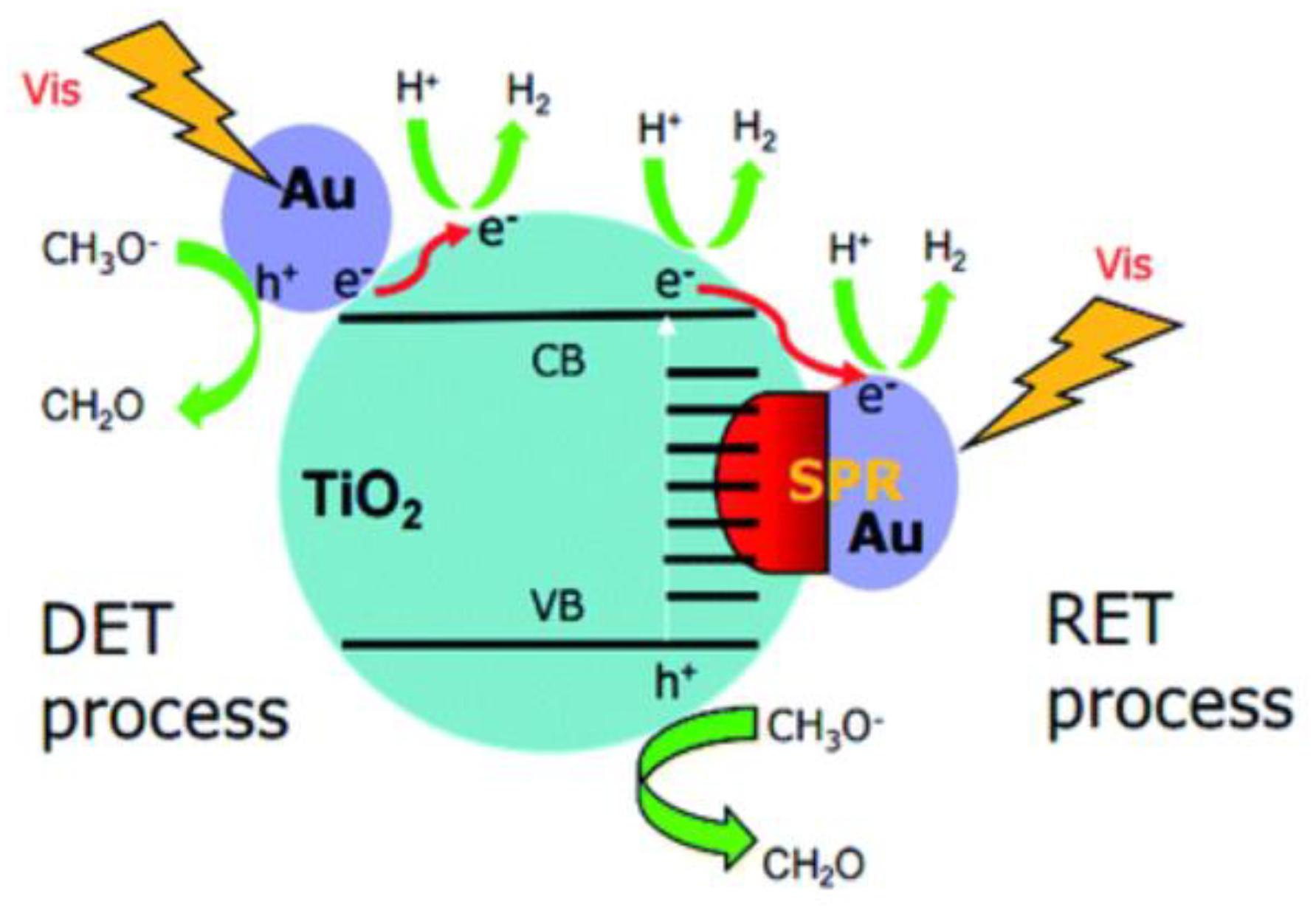
Figure 4.
(a) FE-SEM image of spherical shaped SrTiO3 with sponge-like mesoporous morphology. Reprinted (adapted) with permission from Jayabal, P.; Sasirekha, V.; Mayandi, J.; Jeganathan, K.; Ramakrishnan, V. A facile hydrothermal synthesis of SrTiO3 for dye-sensitized solar cell application. J. Alloys Compd. 2014, 586, 456–461. Copyright (2013) Elsevier B.V. [90]; (b) SEM image of cubic-shaped SrTiO3. Reprinted (adapted) with permission from Gao, H.; Yang, H.; Wang, S. Hydrothermal synthesis, growth mechanism, optical properties and photocatalytic activity of cubic SrTiO3 particles for the degradation of cationic and anionic dyes. Optik 2018, 175, 237–249. Copyright (2018) Elsevier GmbH [91]; (c) TEM image of 0.5%Ag-SrTiO3 nanocomposites (the arrows indicate Ag nanoparticles). Reprinted (adapted) with permission from Zhang, Q.; Huang, Y.; Xu, L.; Cao, J.; Ho, W.; Lee, S.C. Visible-light-active plasmonic Ag-SrTiO3 nanocomposites for the degradation of NO in air with high selectivity. ACS Appl. Mater. Interfaces 2016, 8, 4165–4178. Copyright (2016) American Chemical Society [92]; and (d) SEM image of SrTiO3 dendritic particles. Reprinted (adapted) with permission from Kalyani, V.; Vasile, B.S.; Ianculescu, A.; Testino, A.; Carino, A.; Buscaglia, M.T.; Buscaglia, V.; Nanni, P. Hydrothermal synthesis of SrTiO3: role of interfaces. Cryst Growth Des. 2015, 15, 5712–5725. Copyright (2015) American Chemical Society [93].
Figure 4.
(a) FE-SEM image of spherical shaped SrTiO3 with sponge-like mesoporous morphology. Reprinted (adapted) with permission from Jayabal, P.; Sasirekha, V.; Mayandi, J.; Jeganathan, K.; Ramakrishnan, V. A facile hydrothermal synthesis of SrTiO3 for dye-sensitized solar cell application. J. Alloys Compd. 2014, 586, 456–461. Copyright (2013) Elsevier B.V. [90]; (b) SEM image of cubic-shaped SrTiO3. Reprinted (adapted) with permission from Gao, H.; Yang, H.; Wang, S. Hydrothermal synthesis, growth mechanism, optical properties and photocatalytic activity of cubic SrTiO3 particles for the degradation of cationic and anionic dyes. Optik 2018, 175, 237–249. Copyright (2018) Elsevier GmbH [91]; (c) TEM image of 0.5%Ag-SrTiO3 nanocomposites (the arrows indicate Ag nanoparticles). Reprinted (adapted) with permission from Zhang, Q.; Huang, Y.; Xu, L.; Cao, J.; Ho, W.; Lee, S.C. Visible-light-active plasmonic Ag-SrTiO3 nanocomposites for the degradation of NO in air with high selectivity. ACS Appl. Mater. Interfaces 2016, 8, 4165–4178. Copyright (2016) American Chemical Society [92]; and (d) SEM image of SrTiO3 dendritic particles. Reprinted (adapted) with permission from Kalyani, V.; Vasile, B.S.; Ianculescu, A.; Testino, A.; Carino, A.; Buscaglia, M.T.; Buscaglia, V.; Nanni, P. Hydrothermal synthesis of SrTiO3: role of interfaces. Cryst Growth Des. 2015, 15, 5712–5725. Copyright (2015) American Chemical Society [93].

Figure 5.
Schematic representation of the different steps involved in the formation of perovskite particles from the hydrothermal method and Ag decoration pervoskite surface by the chemical solution. Reprinted with permission from Kumar, D.; Singh, S.; Khare, N. Plasmonic Ag nanoparticles decorated NaNbO3 nanorods for efficient photoelectrochemical water splitting. Int. J. Hydrogen Energ. 2018, 43, 1–8. Copyright (2018) Hydrogen Energy Publications LLC. Published by Elsevier Ltd [94].
Figure 5.
Schematic representation of the different steps involved in the formation of perovskite particles from the hydrothermal method and Ag decoration pervoskite surface by the chemical solution. Reprinted with permission from Kumar, D.; Singh, S.; Khare, N. Plasmonic Ag nanoparticles decorated NaNbO3 nanorods for efficient photoelectrochemical water splitting. Int. J. Hydrogen Energ. 2018, 43, 1–8. Copyright (2018) Hydrogen Energy Publications LLC. Published by Elsevier Ltd [94].
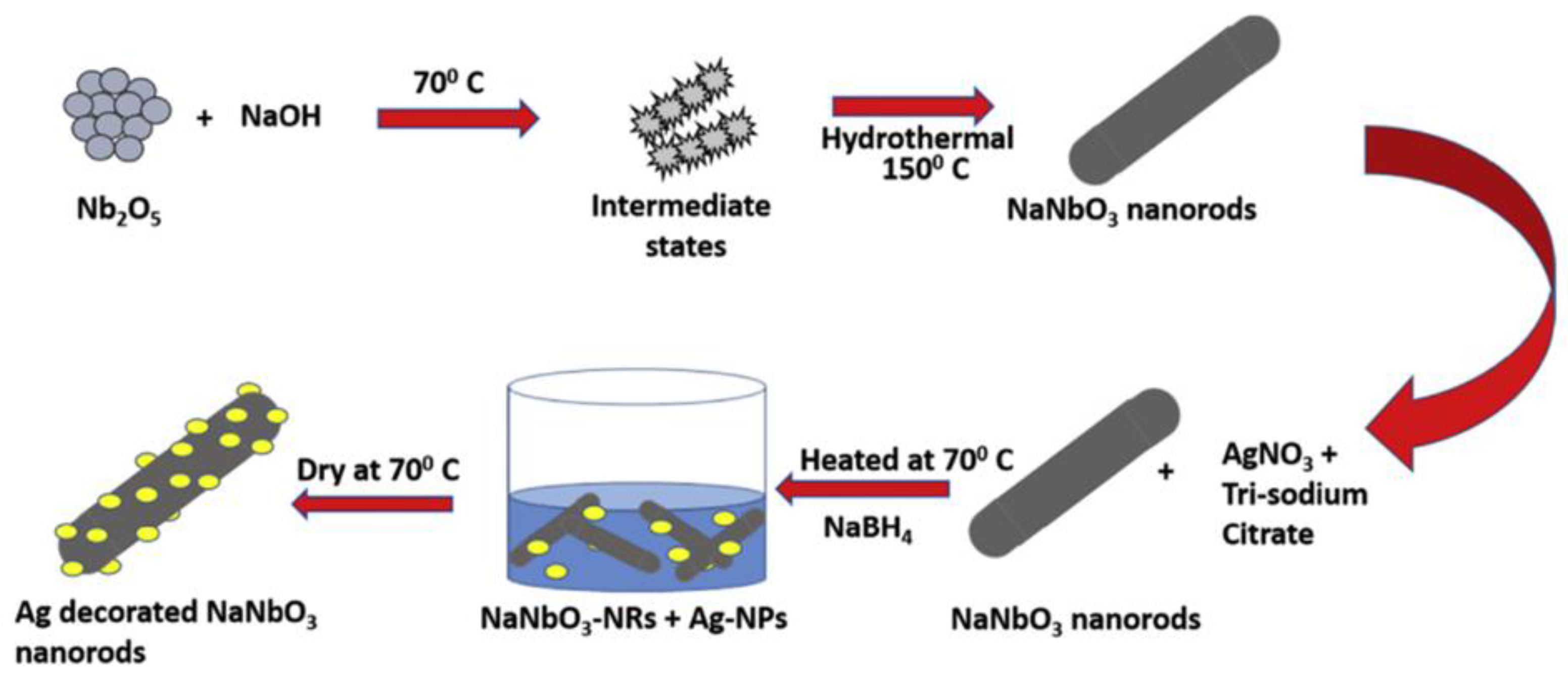
Figure 6.
(a) Synthesis steps for the obtention of Ag-PBMO5 electrocatalyst; (b) proposed scheme for ORR at the electrocatalyst surface; c) HRTEM images of exsolved Ag2O nanoparticles bonded to the perovskite surface and FESEM images of PBAMO3 nanofibers. Reprinted with permission from Zhang, Y.Q.; Tao, H.B.; Liu, J.; Sun, Y.F.; Chen, J.; Hua, B.; Thundat, T.; Luo, J.L. A rational design for enhanced oxygen reduction: Strongly coupled silver nanoparticles and engineered perovskite nanofibers. Nano Energy, 2017, 38, 392–400. Copyright (2017) Nano Energy Publications LLC. Published by Elsevier Ltd [106].
Figure 6.
(a) Synthesis steps for the obtention of Ag-PBMO5 electrocatalyst; (b) proposed scheme for ORR at the electrocatalyst surface; c) HRTEM images of exsolved Ag2O nanoparticles bonded to the perovskite surface and FESEM images of PBAMO3 nanofibers. Reprinted with permission from Zhang, Y.Q.; Tao, H.B.; Liu, J.; Sun, Y.F.; Chen, J.; Hua, B.; Thundat, T.; Luo, J.L. A rational design for enhanced oxygen reduction: Strongly coupled silver nanoparticles and engineered perovskite nanofibers. Nano Energy, 2017, 38, 392–400. Copyright (2017) Nano Energy Publications LLC. Published by Elsevier Ltd [106].
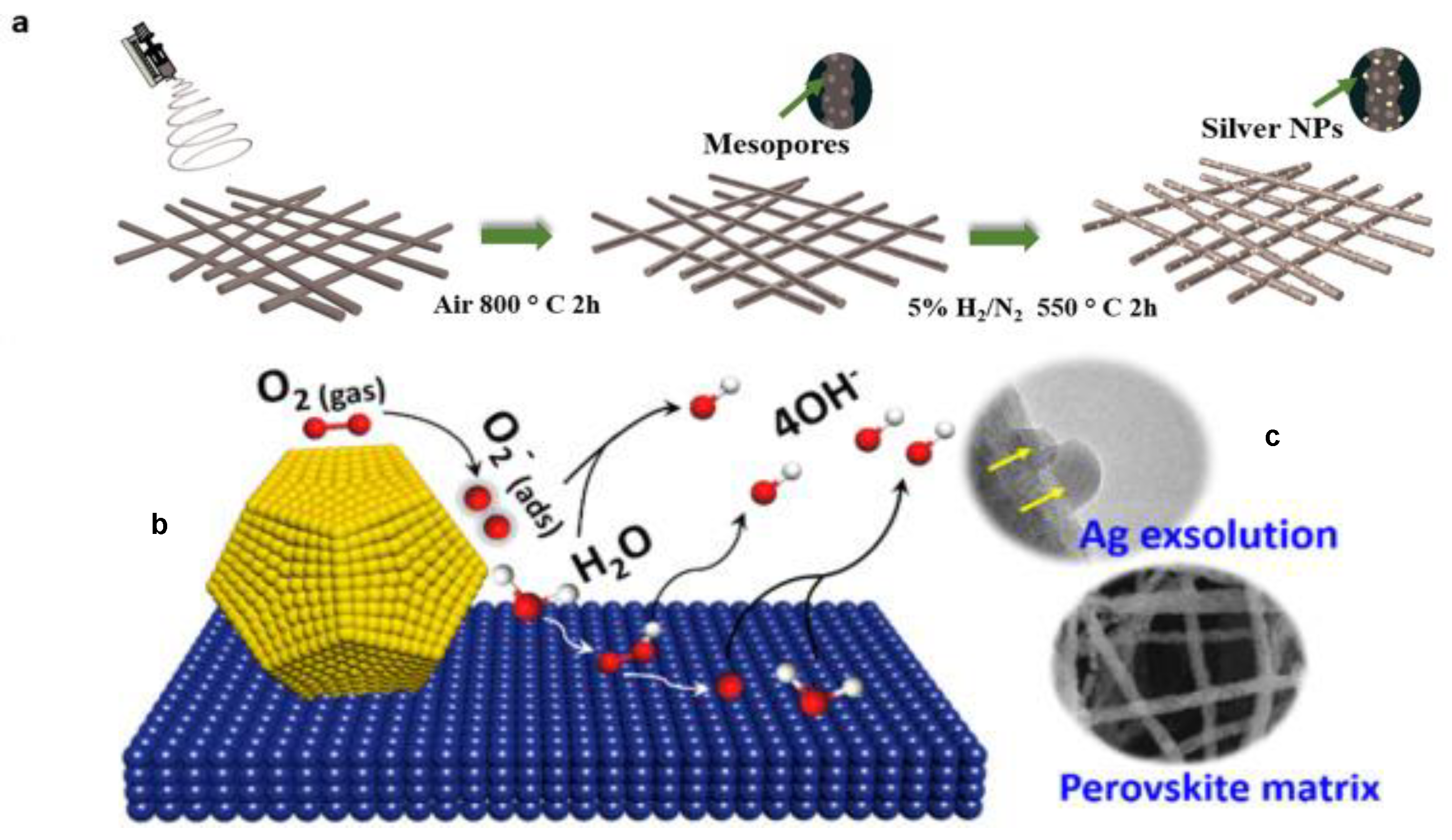

{kind=link}
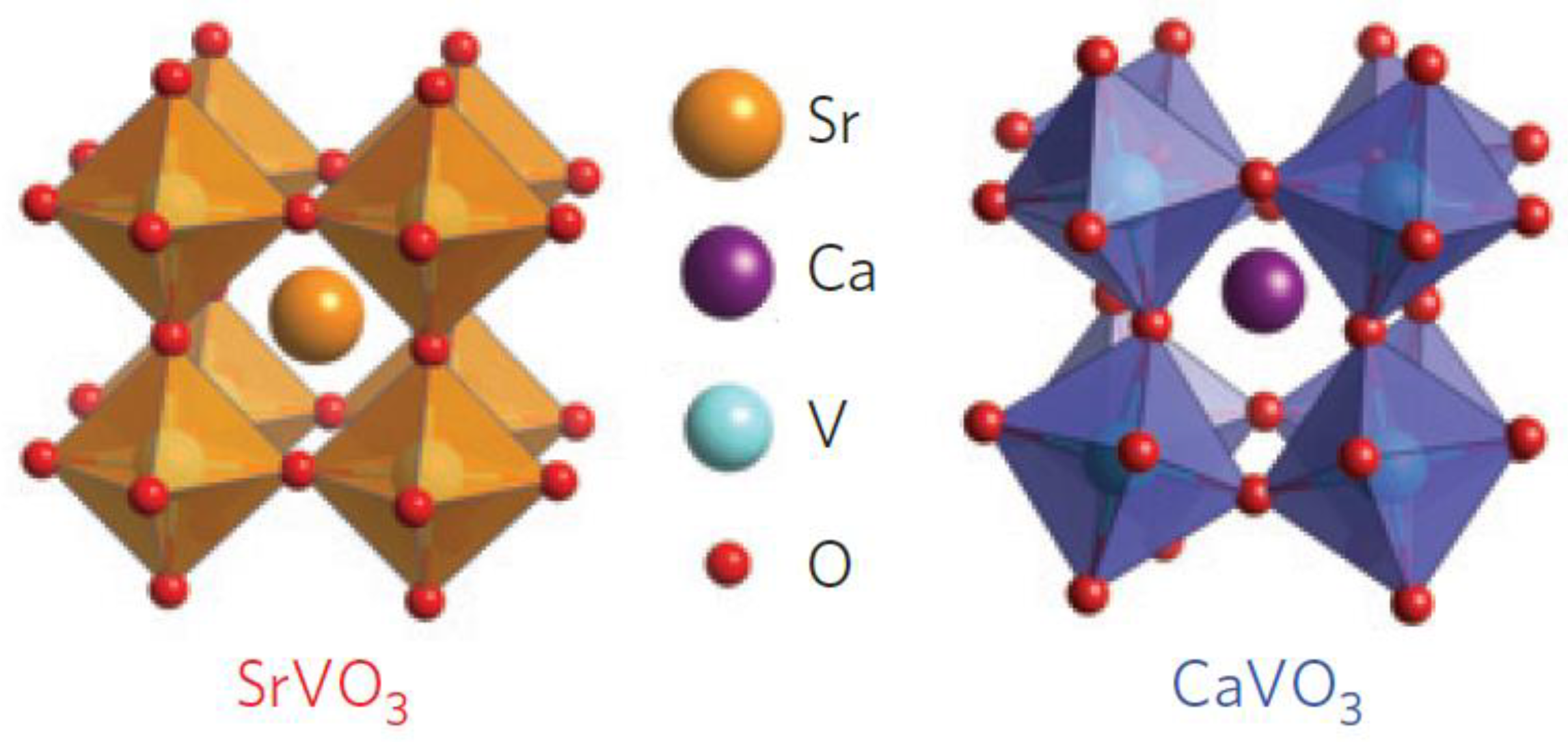{kind=link}
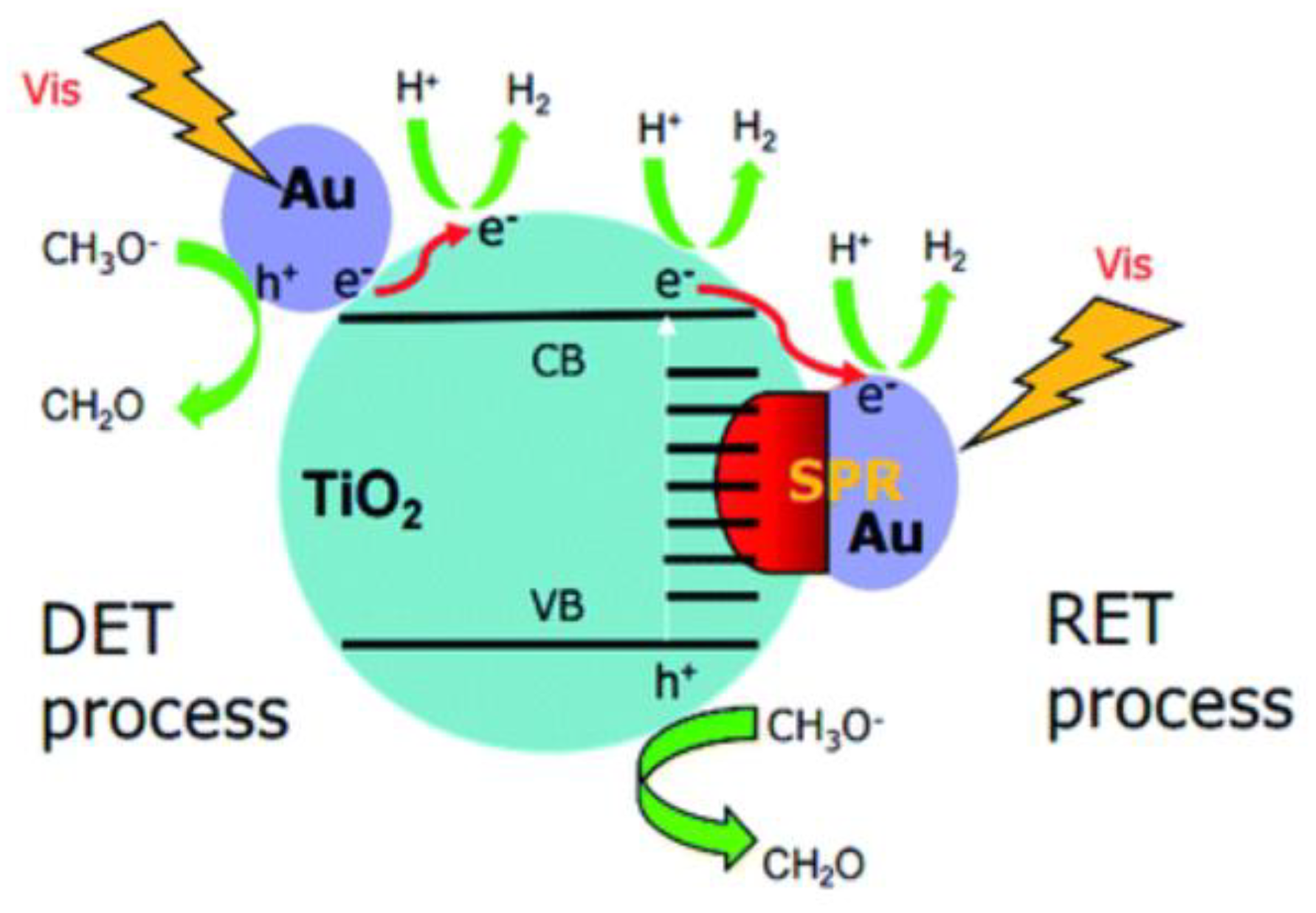{kind=link}
{kind=link}
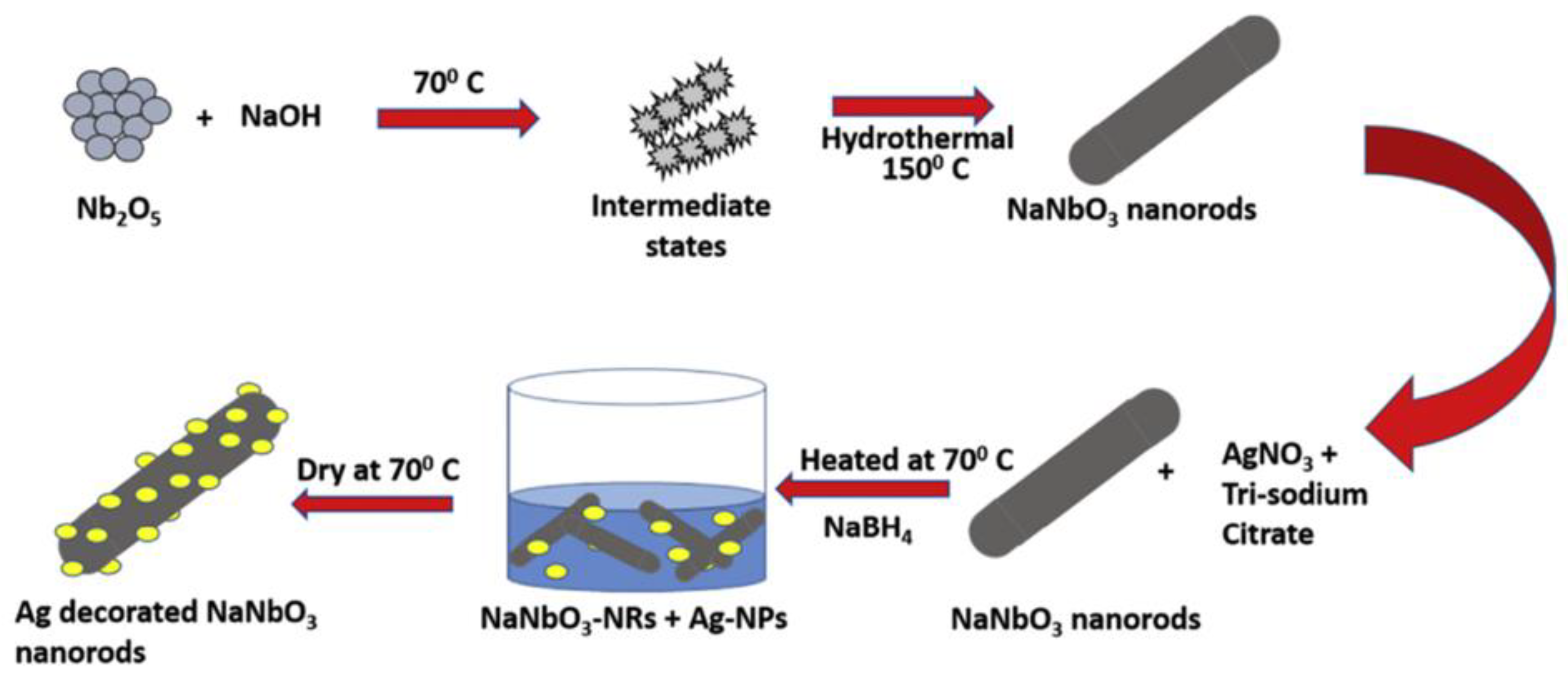{kind=link}
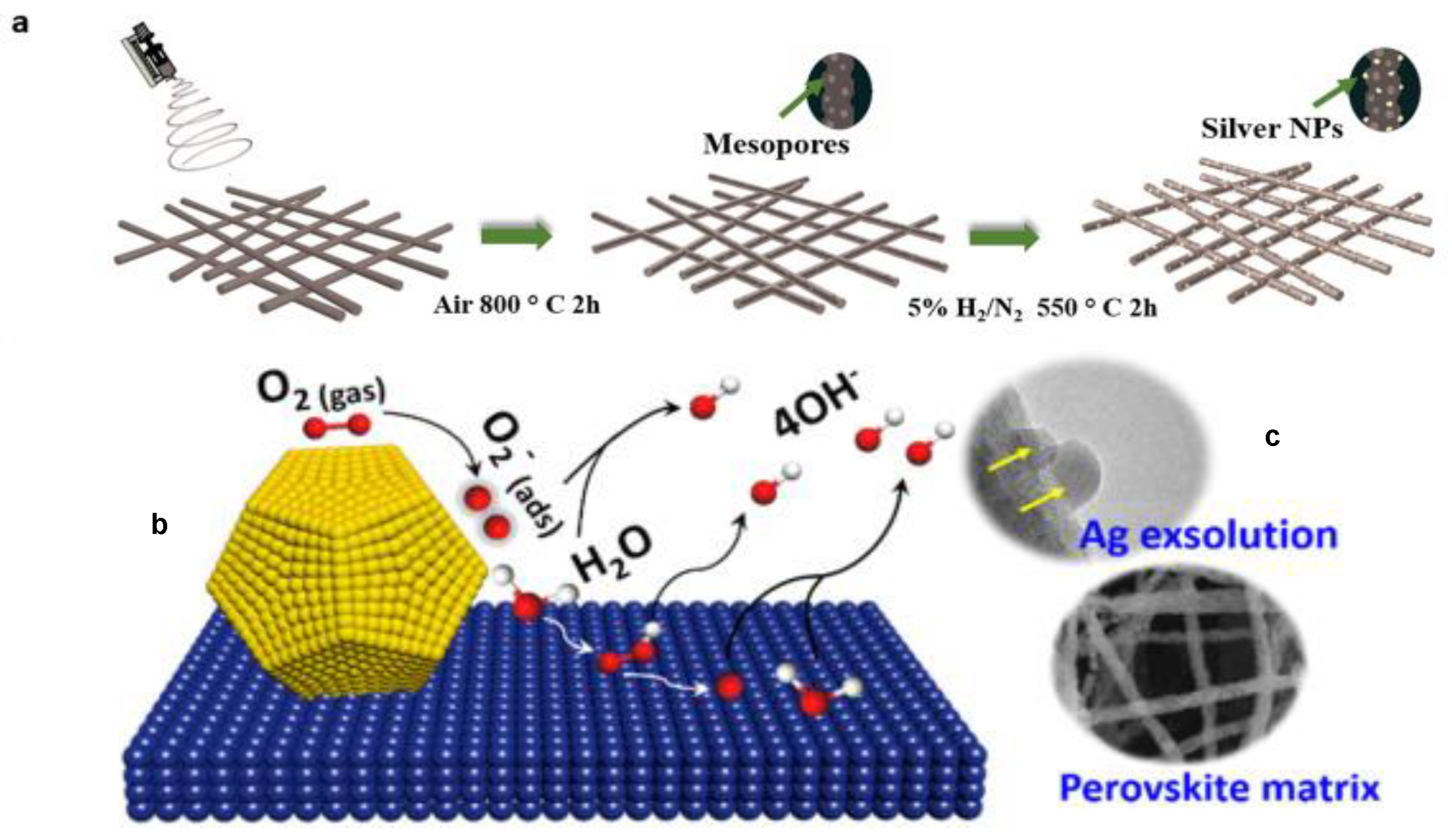{kind=link}
Table 1.
Photocatalytic activities of SrTiO3:M(0.5%) for H2 and O2 evolution from the aqueous solution containing sacrificial reagents under visible light irradiation a. Reprinted with permission from Konta, R.; Ishii, T.; Kato, H.; Kudo, A. Photocatalytic activities of noble metal ion doped SrTiO3 under visible light irradiation. J. Phys. Chem. B 2004, 108, 8992–8995. Copyright 2004 American Chemical Society [75].
Table 1.
Photocatalytic activities of SrTiO3:M(0.5%) for H2 and O2 evolution from the aqueous solution containing sacrificial reagents under visible light irradiation a. Reprinted with permission from Konta, R.; Ishii, T.; Kato, H.; Kudo, A. Photocatalytic activities of noble metal ion doped SrTiO3 under visible light irradiation. J. Phys. Chem. B 2004, 108, 8992–8995. Copyright 2004 American Chemical Society [75].
| M | Eg/eV | Activity/µmol h−1 | |
|---|---|---|---|
| H2 b | O2 c | ||
| Mn | 2.7 | 0.2 | 2.7 |
| Ru | 1.9 | 1.7 | 3.9 |
| Rh | 1.7 | 17.2 | 0 |
| Ir | 2.3 | 8.6 | 0.4 |
a Catalyst, 0.3 g; reactant solution, 150 mL; light source, 300 W Xe lamp with cutoff filter (λ > 440nm). b From 10 vol% aqueous MeOH; cocatalyst, Pt (0.5 wt %). c From 0.05 mol.L−1 aqueous AgNO3.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Teixeira, G.F.; Silva Junior, E.; Vilela, R.; Zaghete, M.A.; Colmati, F. Perovskite Structure Associated with Precious Metals: Influence on Heterogenous Catalytic Process. Catalysts 2019, 9, 721. https://doi.org/10.3390/catal9090721
AMA Style
Teixeira GF, Silva Junior E, Vilela R, Zaghete MA, Colmati F. Perovskite Structure Associated with Precious Metals: Influence on Heterogenous Catalytic Process. Catalysts. 2019; 9(9):721. https://doi.org/10.3390/catal9090721
Chicago/Turabian StyleTeixeira, Guilhermina Ferreira, Euripedes Silva Junior, Ramon Vilela, Maria Aparecida Zaghete, and Flávio Colmati. 2019. "Perovskite Structure Associated with Precious Metals: Influence on Heterogenous Catalytic Process" Catalysts 9, no. 9: 721. https://doi.org/10.3390/catal9090721
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.