Atomistic Molecular Dynamics Simulations of the Initial Crystallization Stage in an SWCNT-Polyetherimide Nanocomposite

 , ,
, ,  ,
,  and
and
Abstract
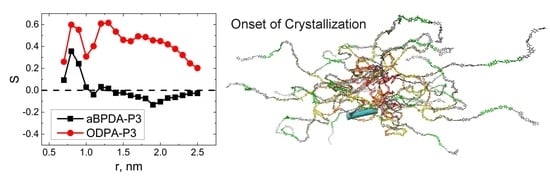
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
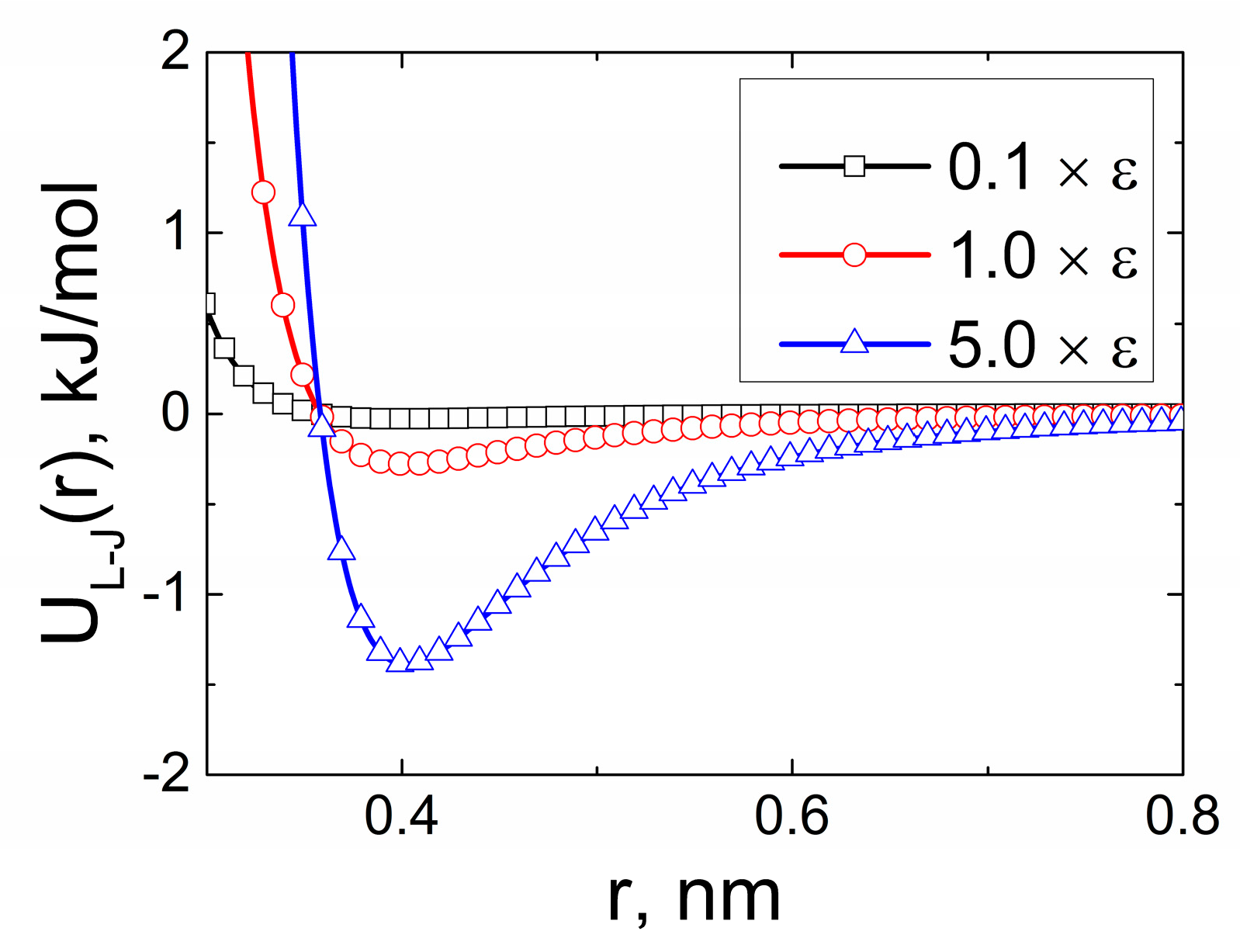
2. Computer Simulations
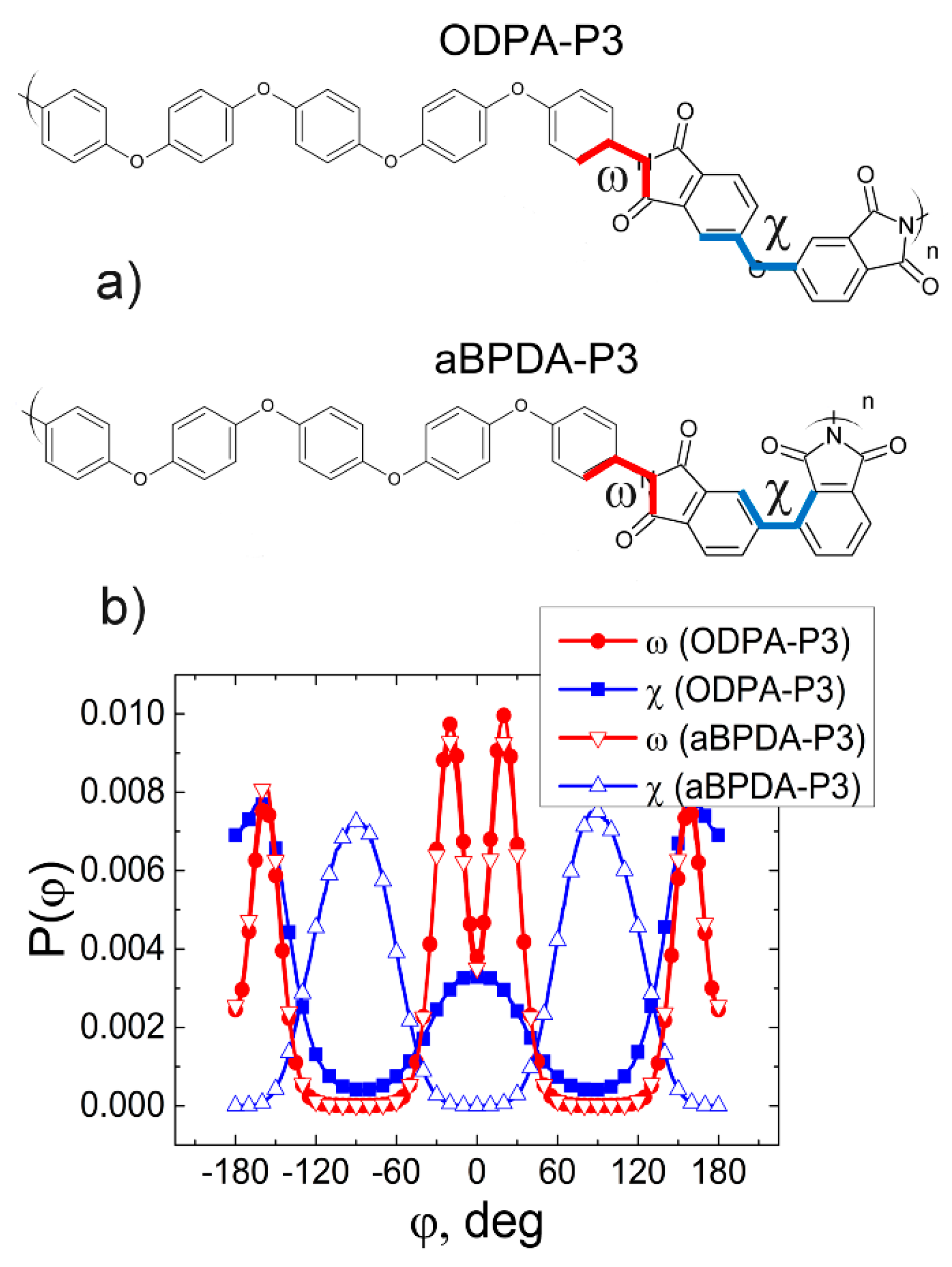
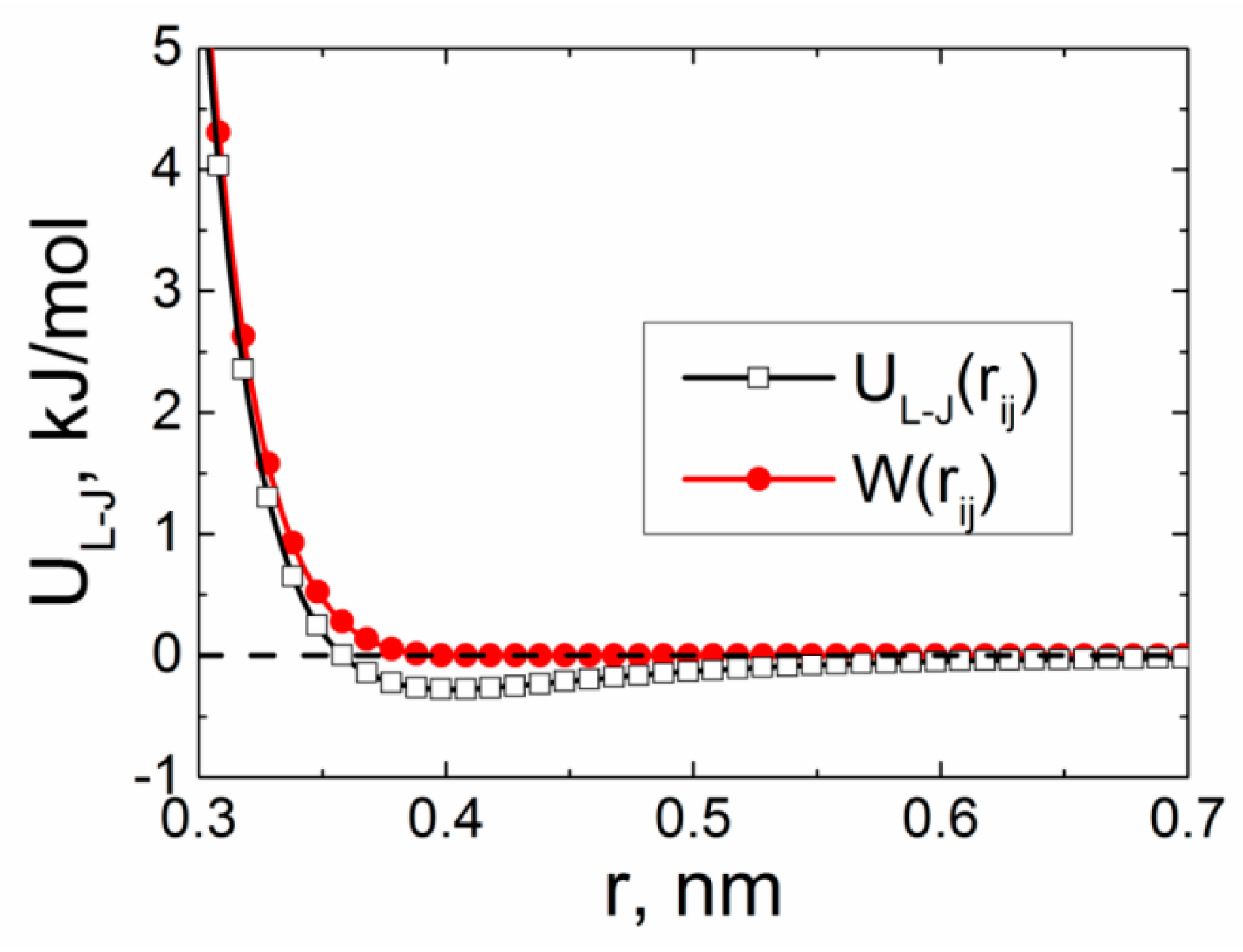
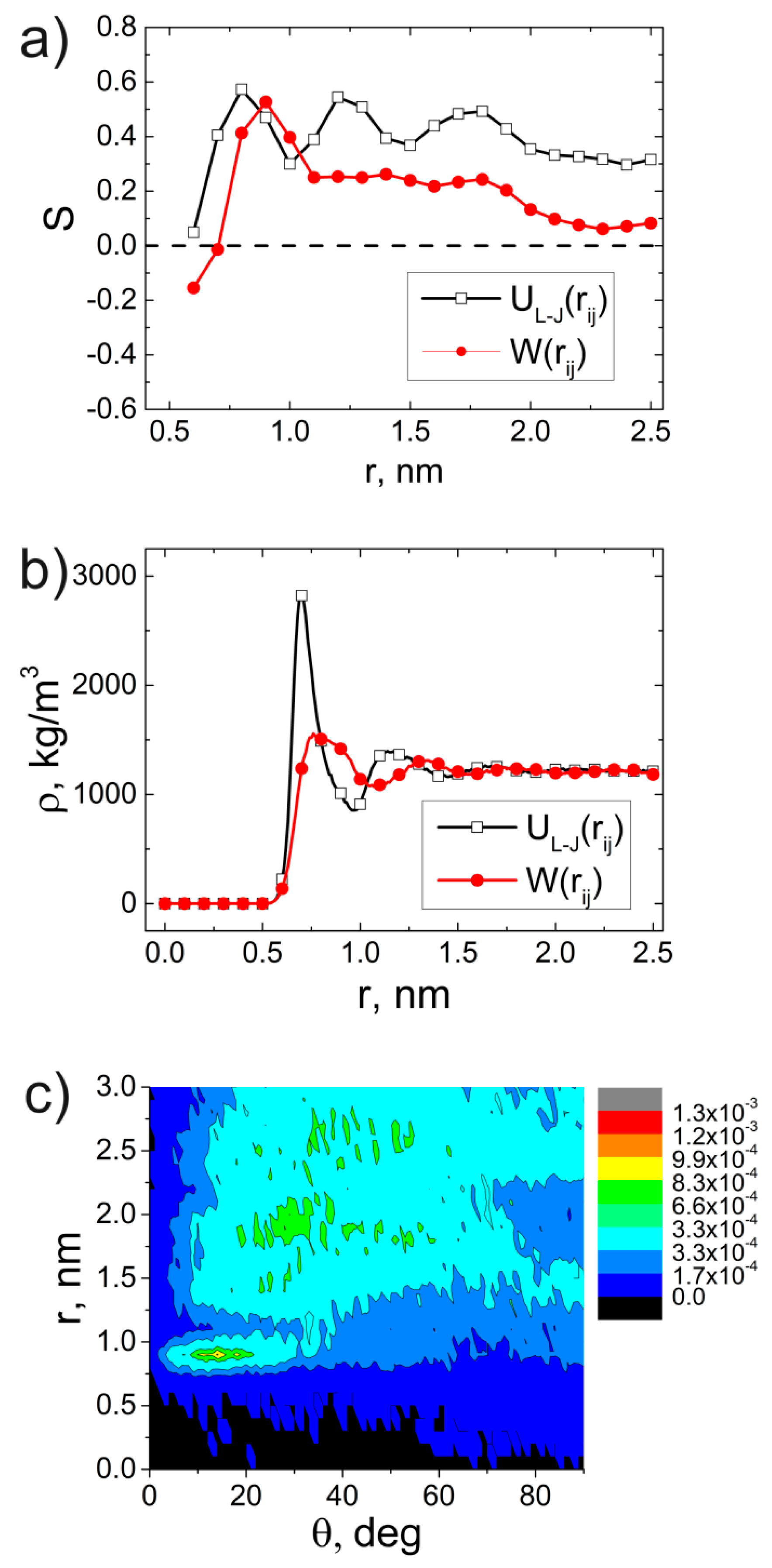
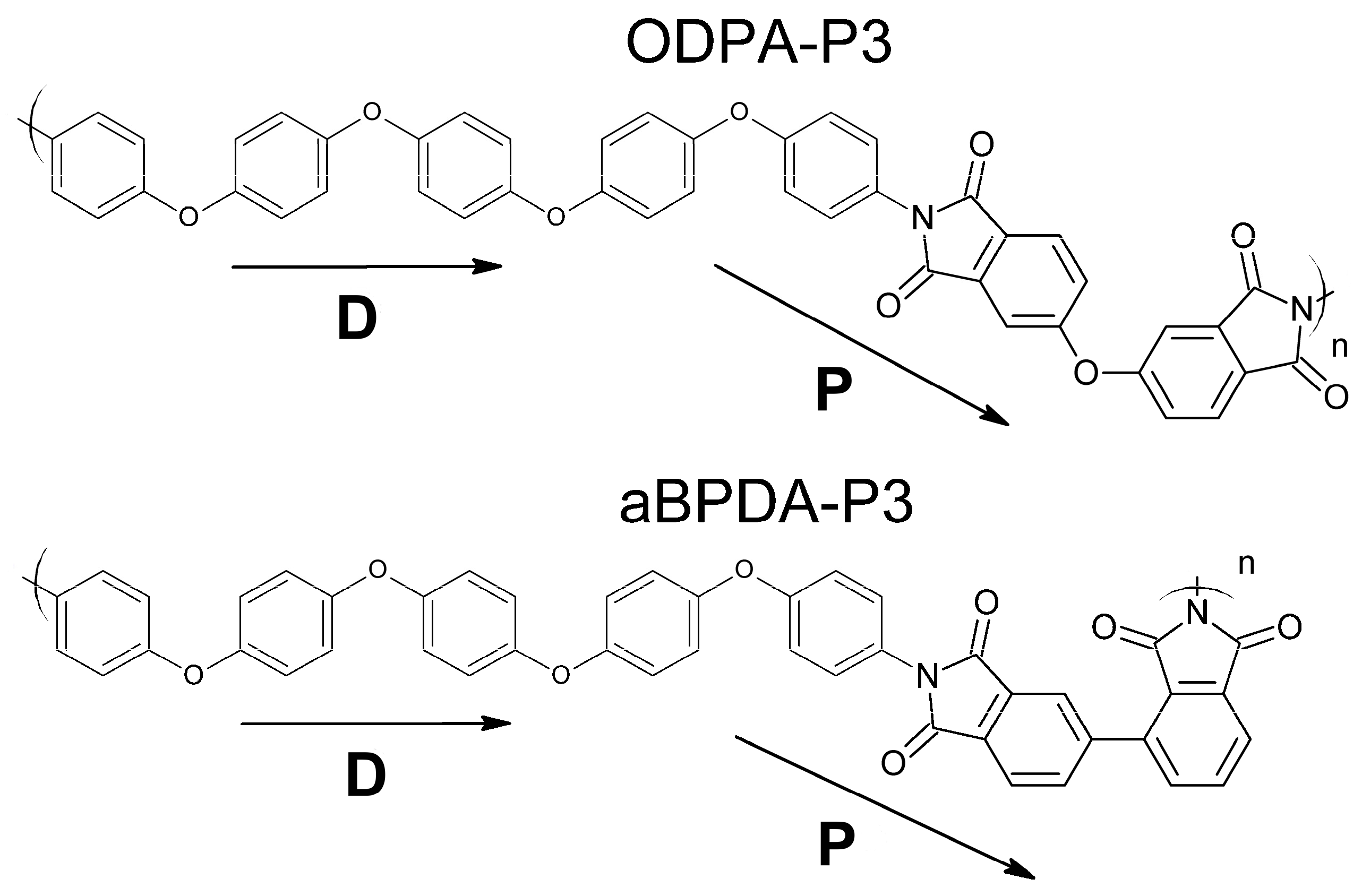
Models and Simulation Method
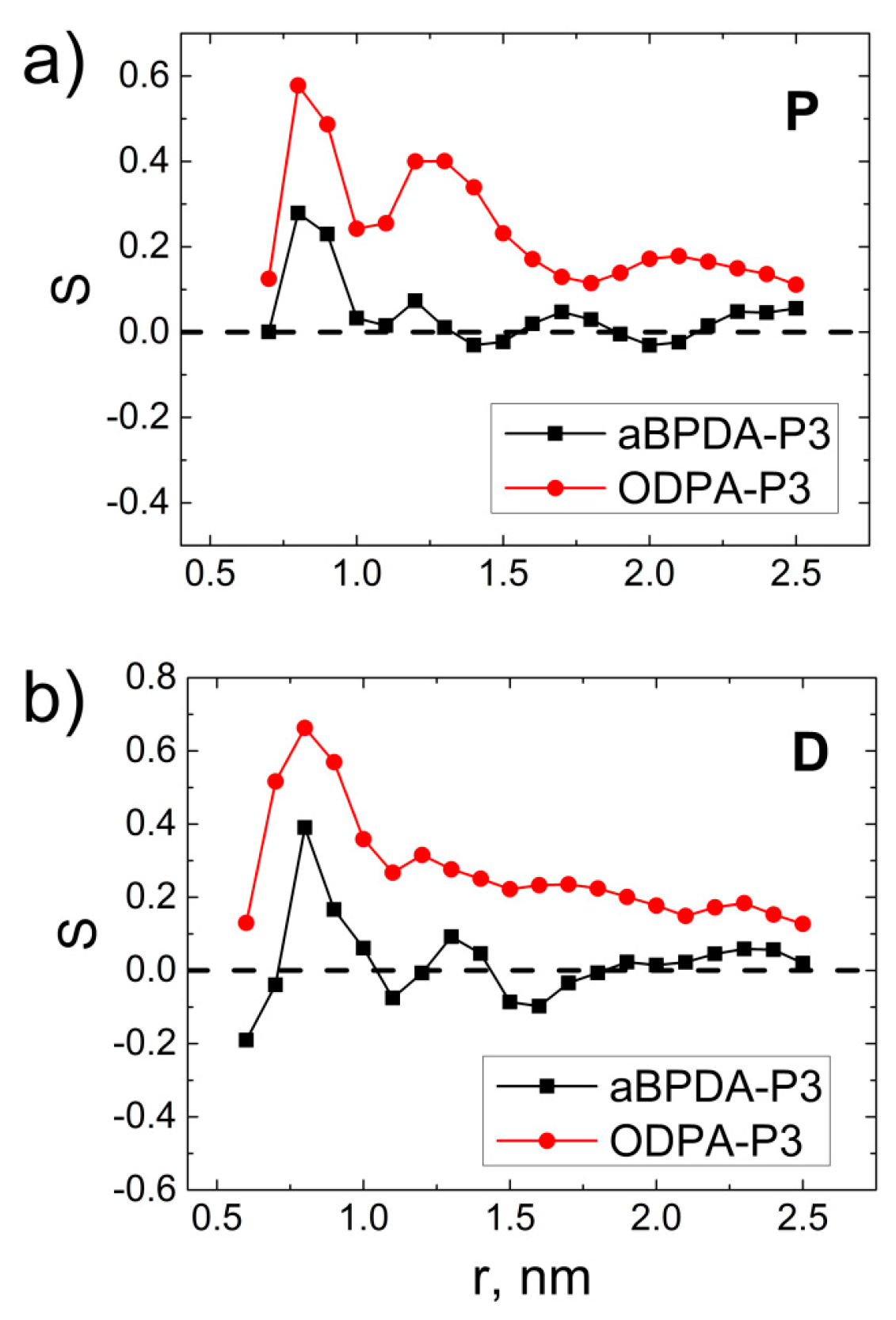
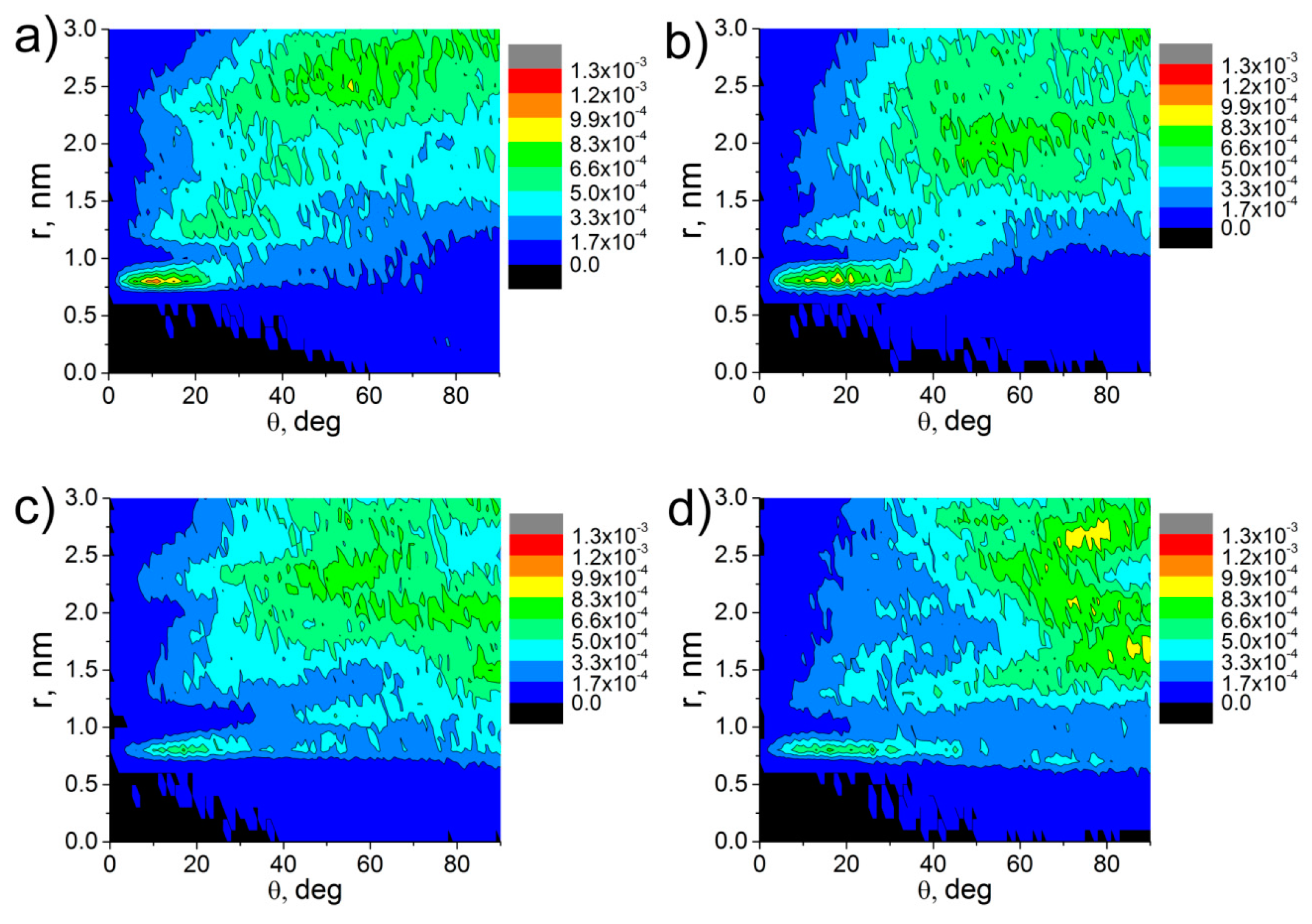
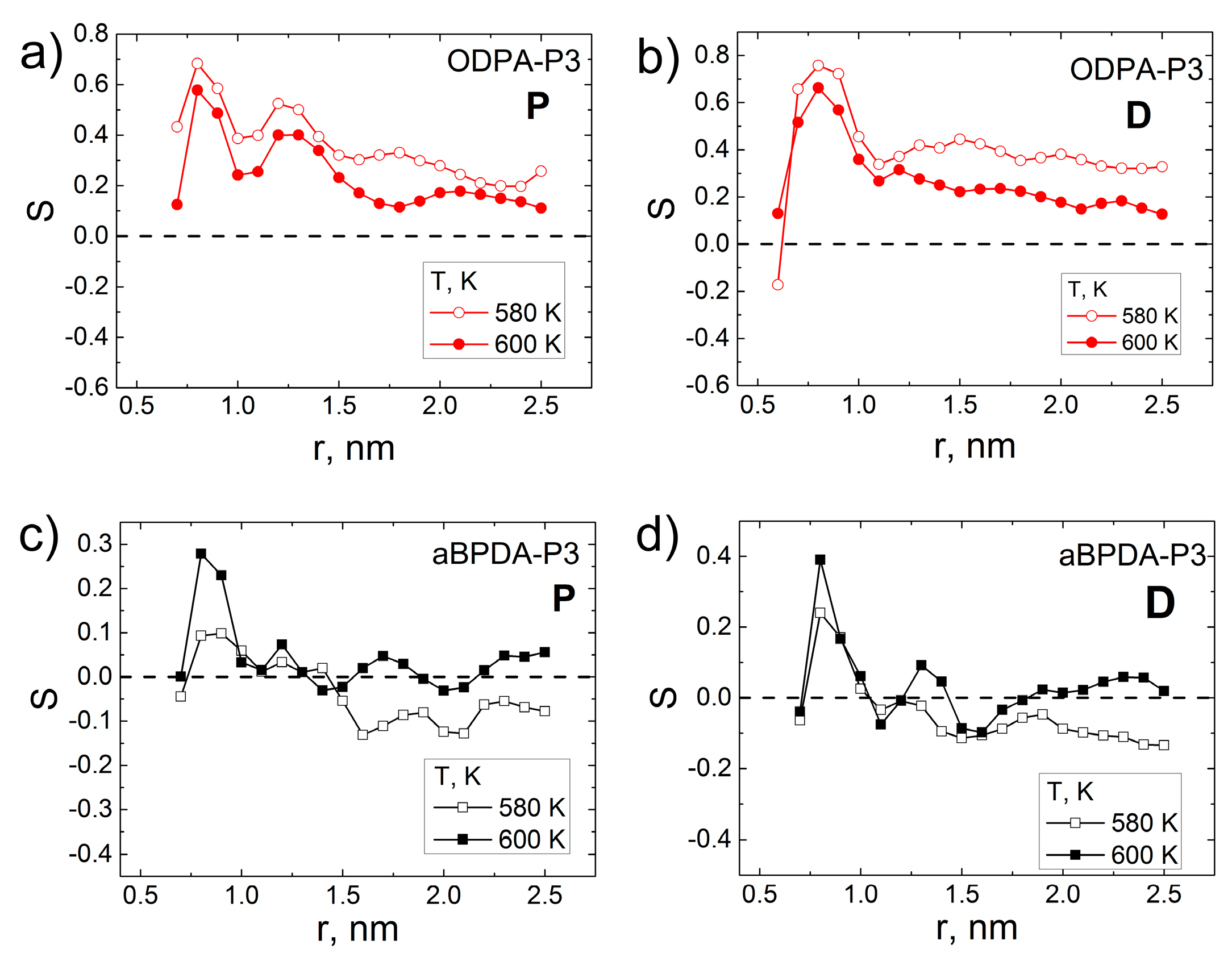
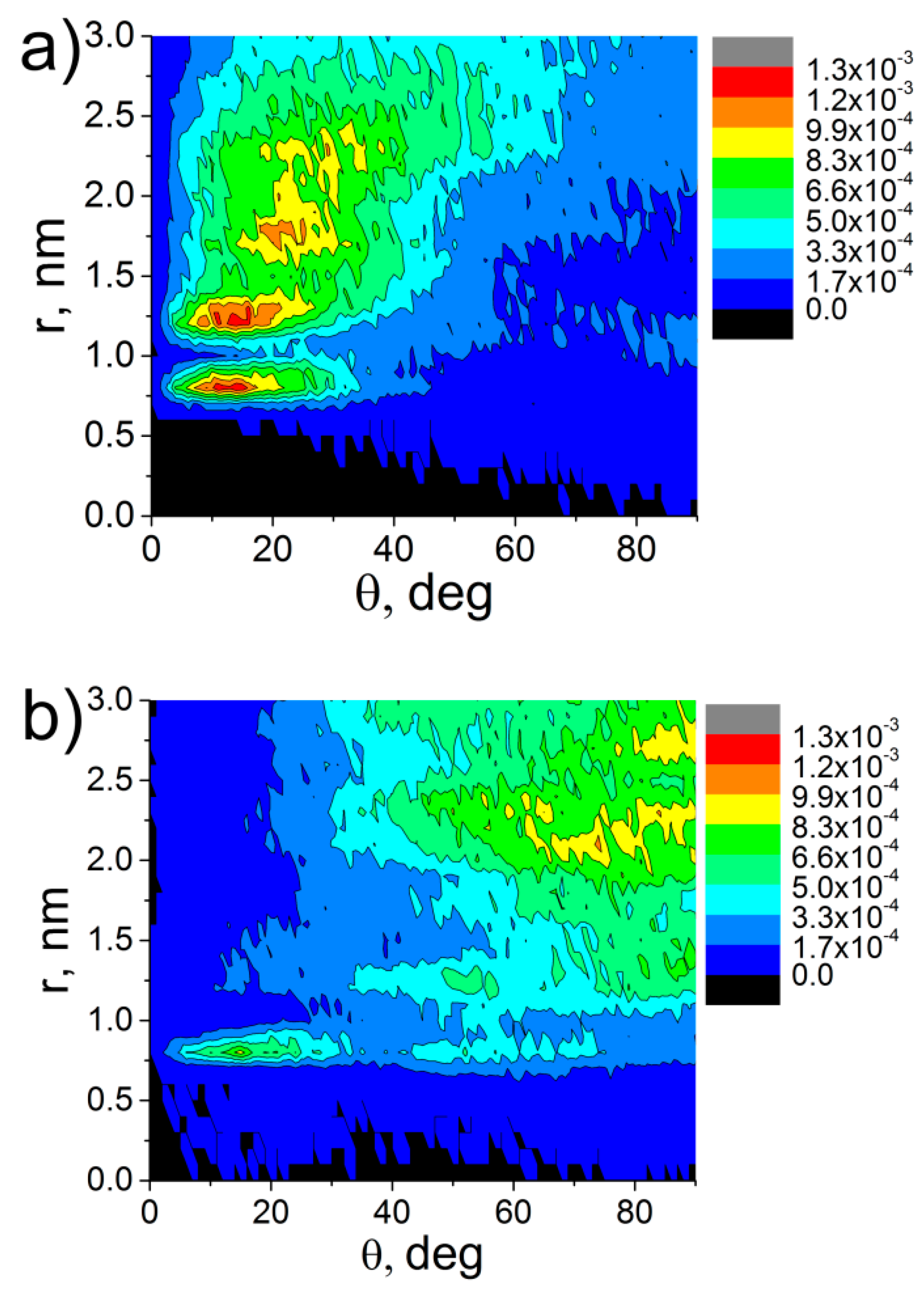
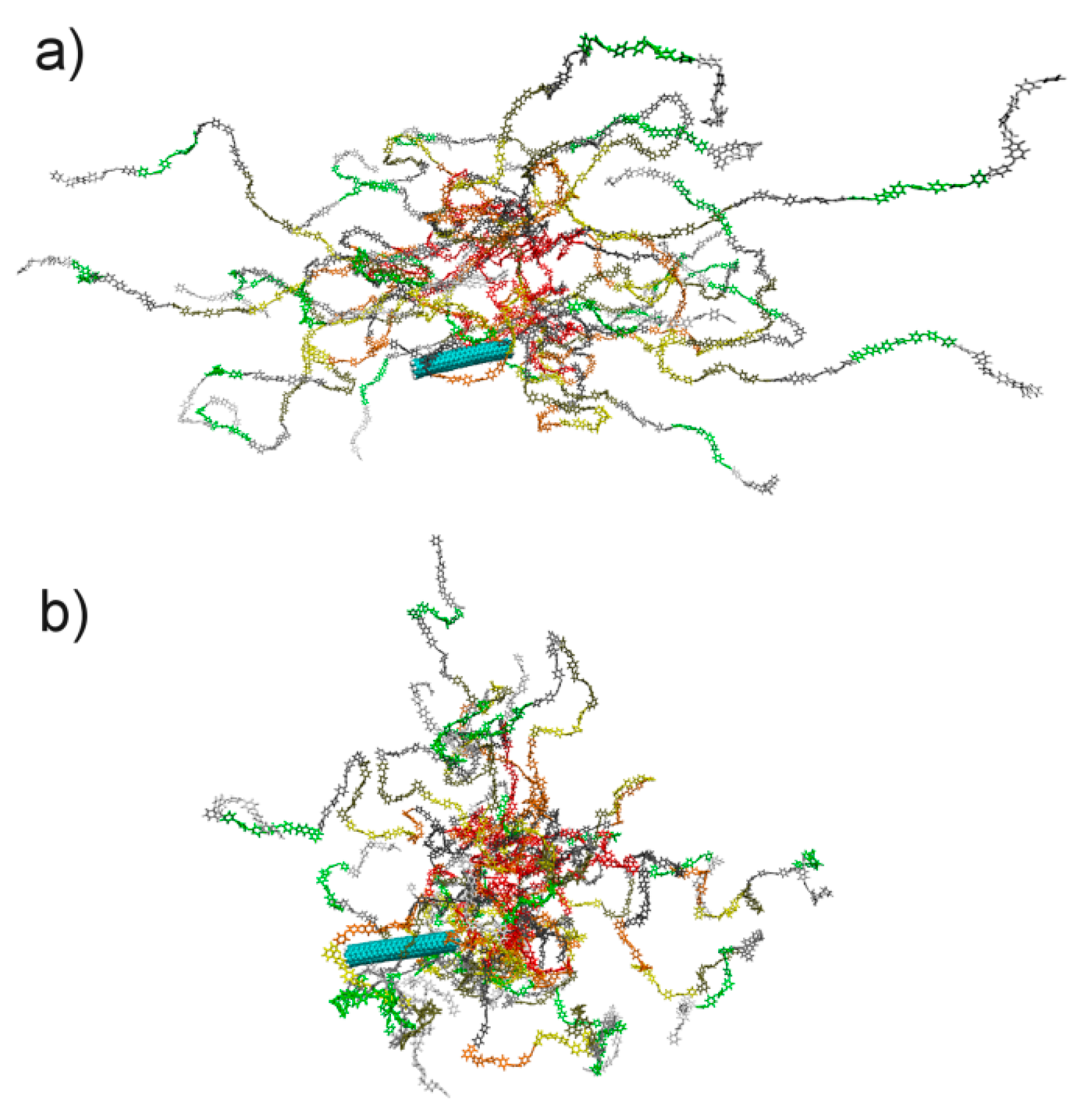
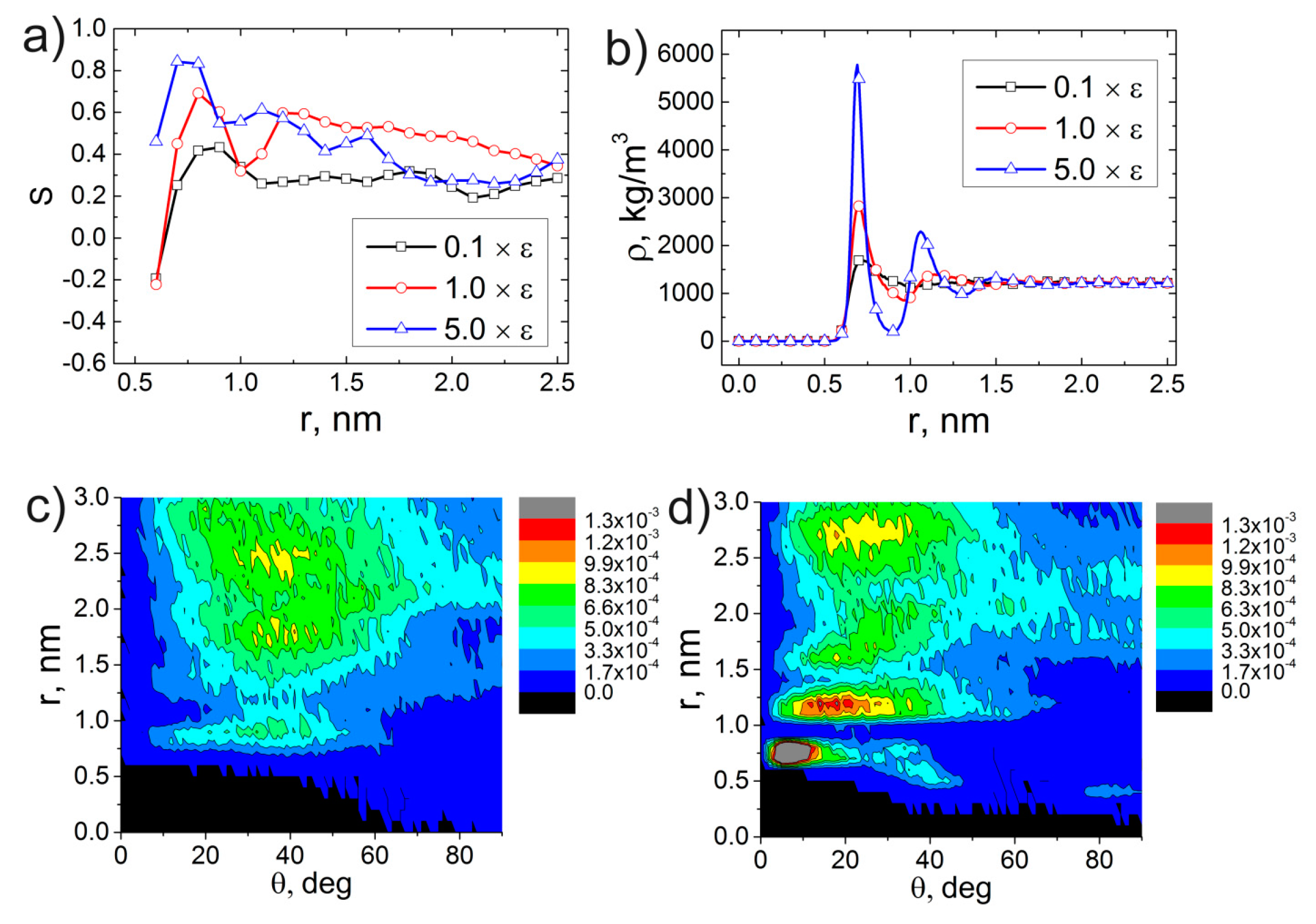
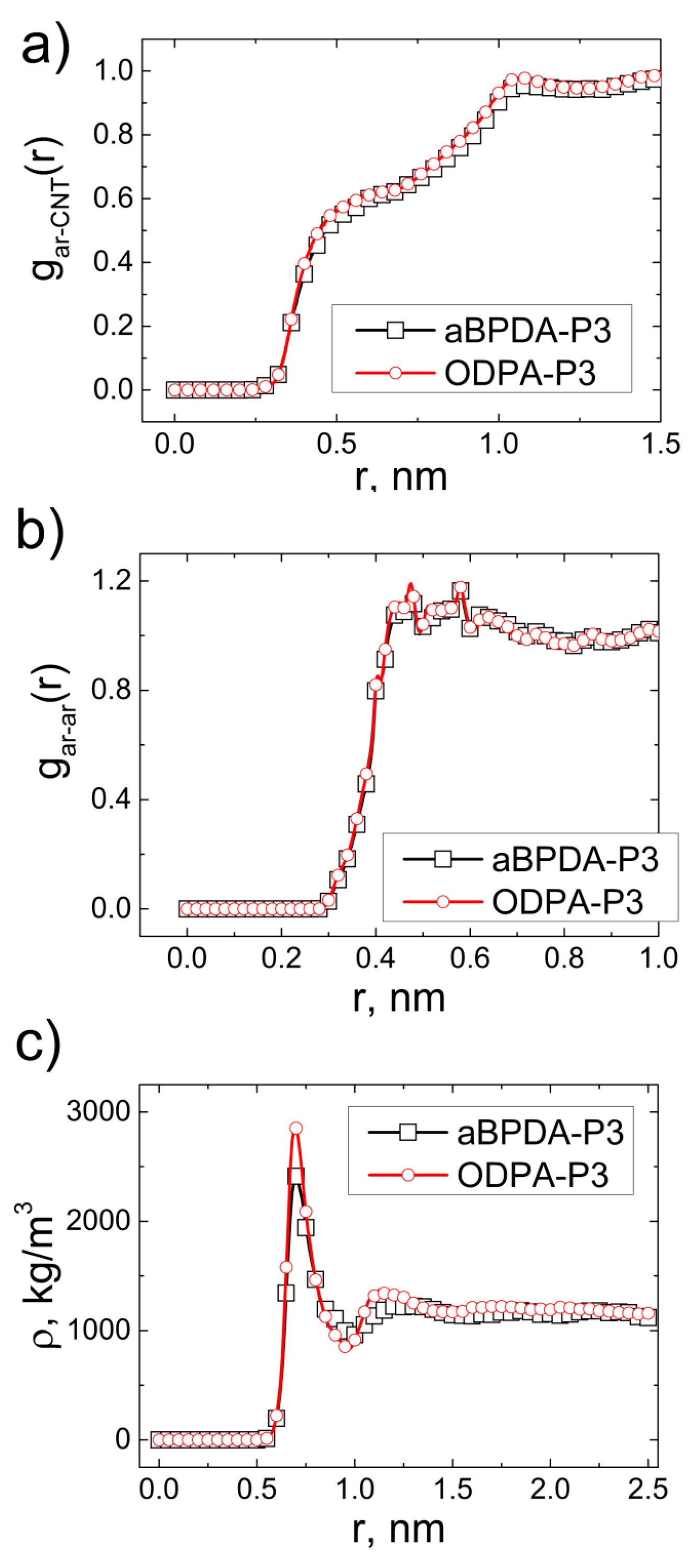
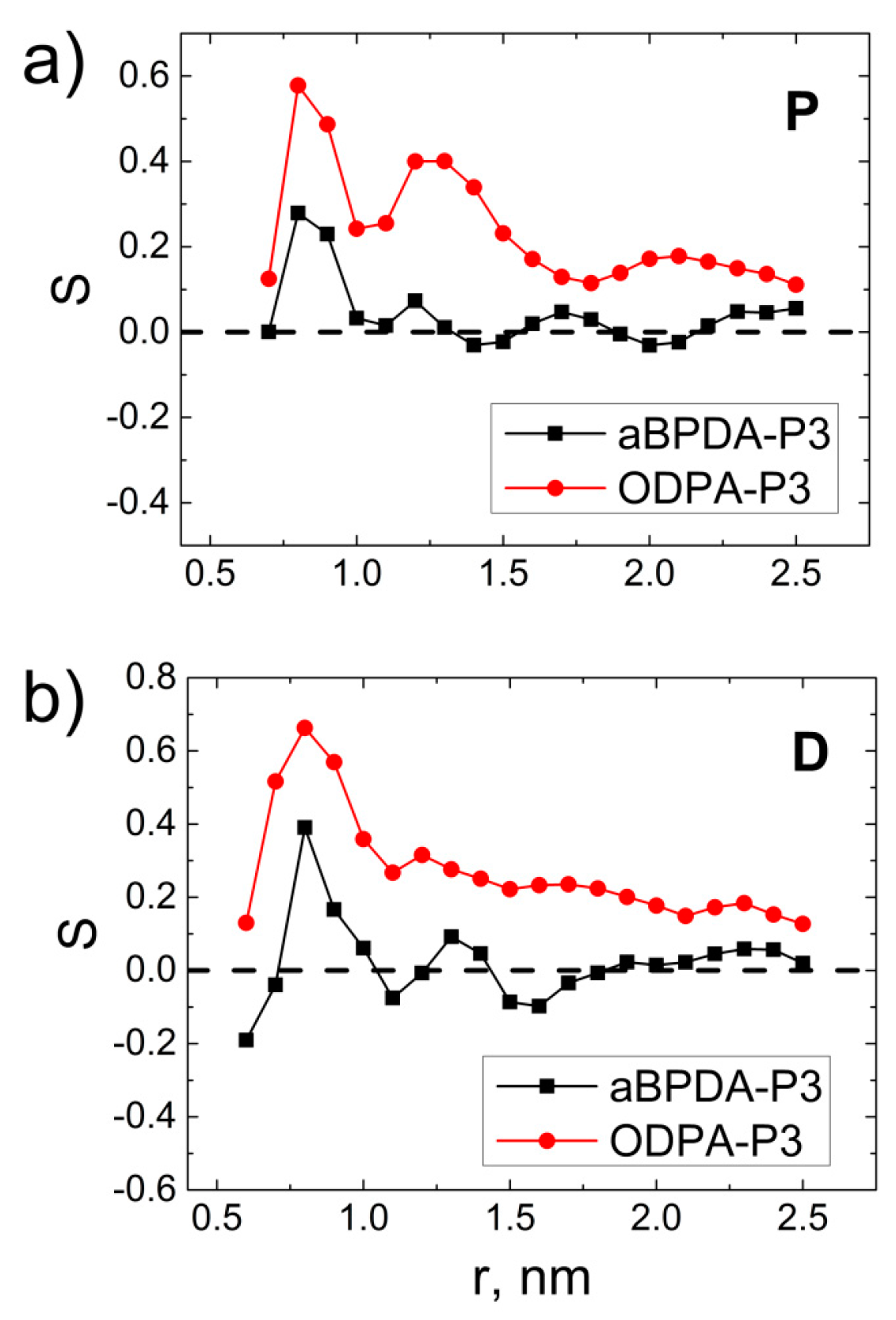
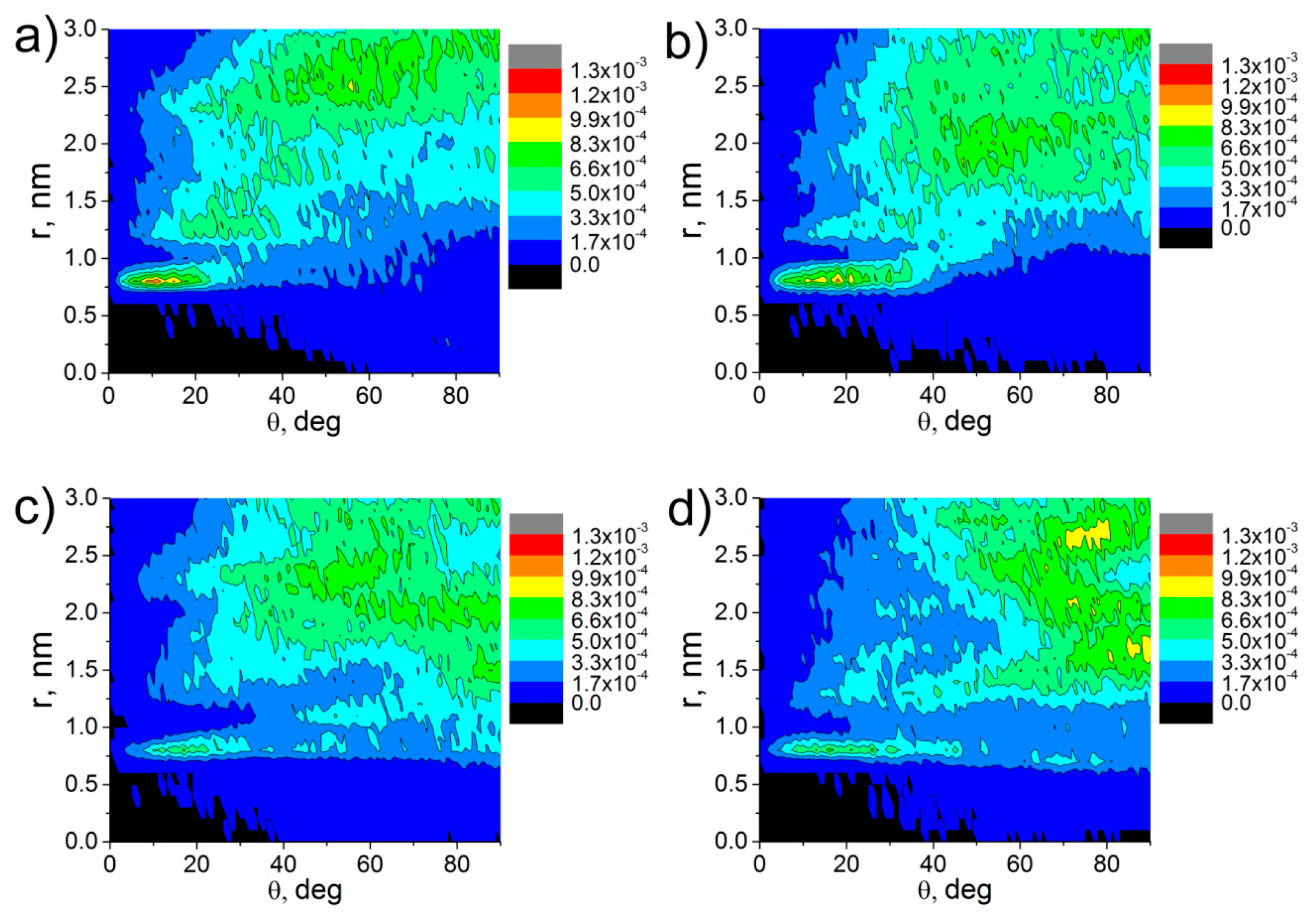
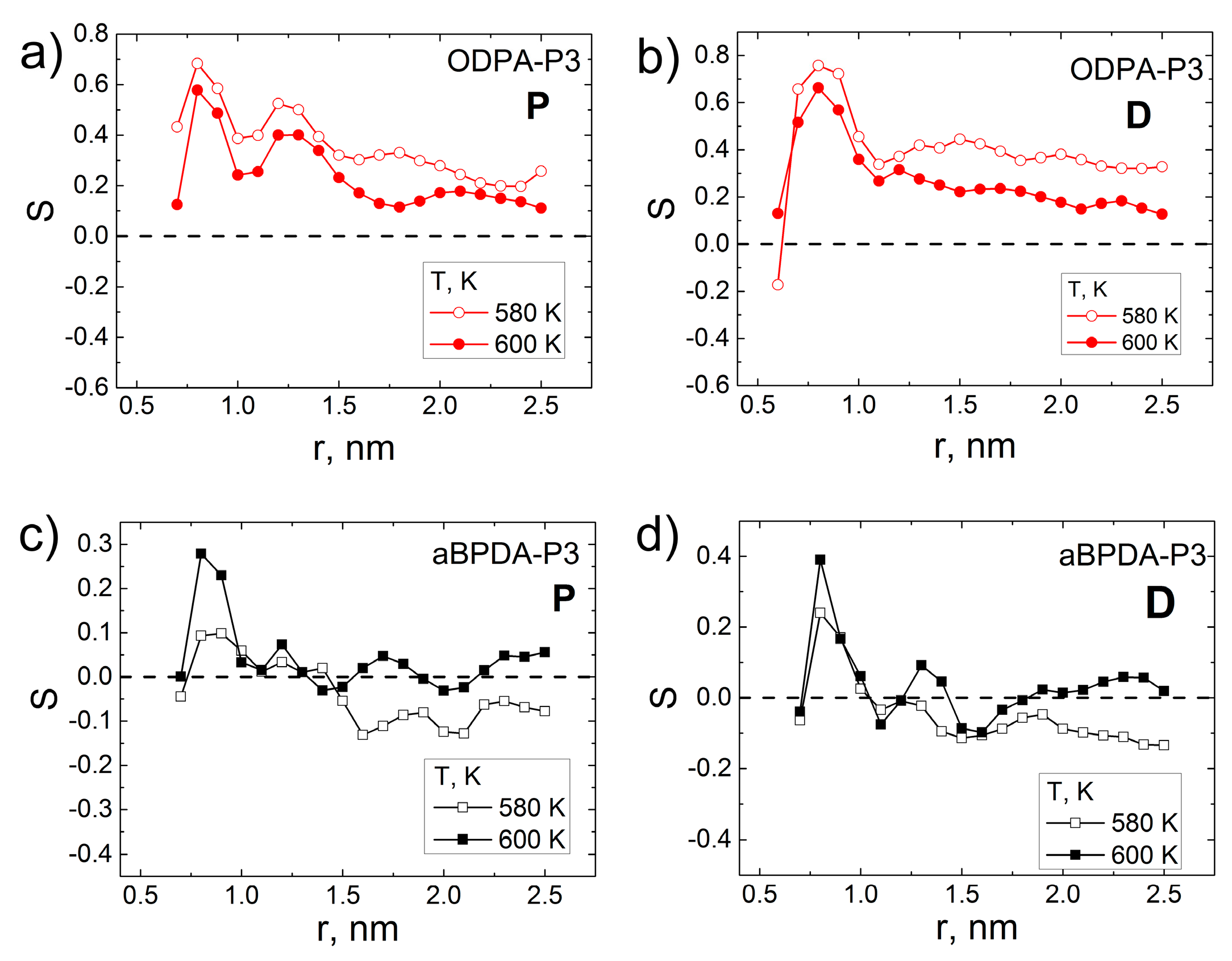
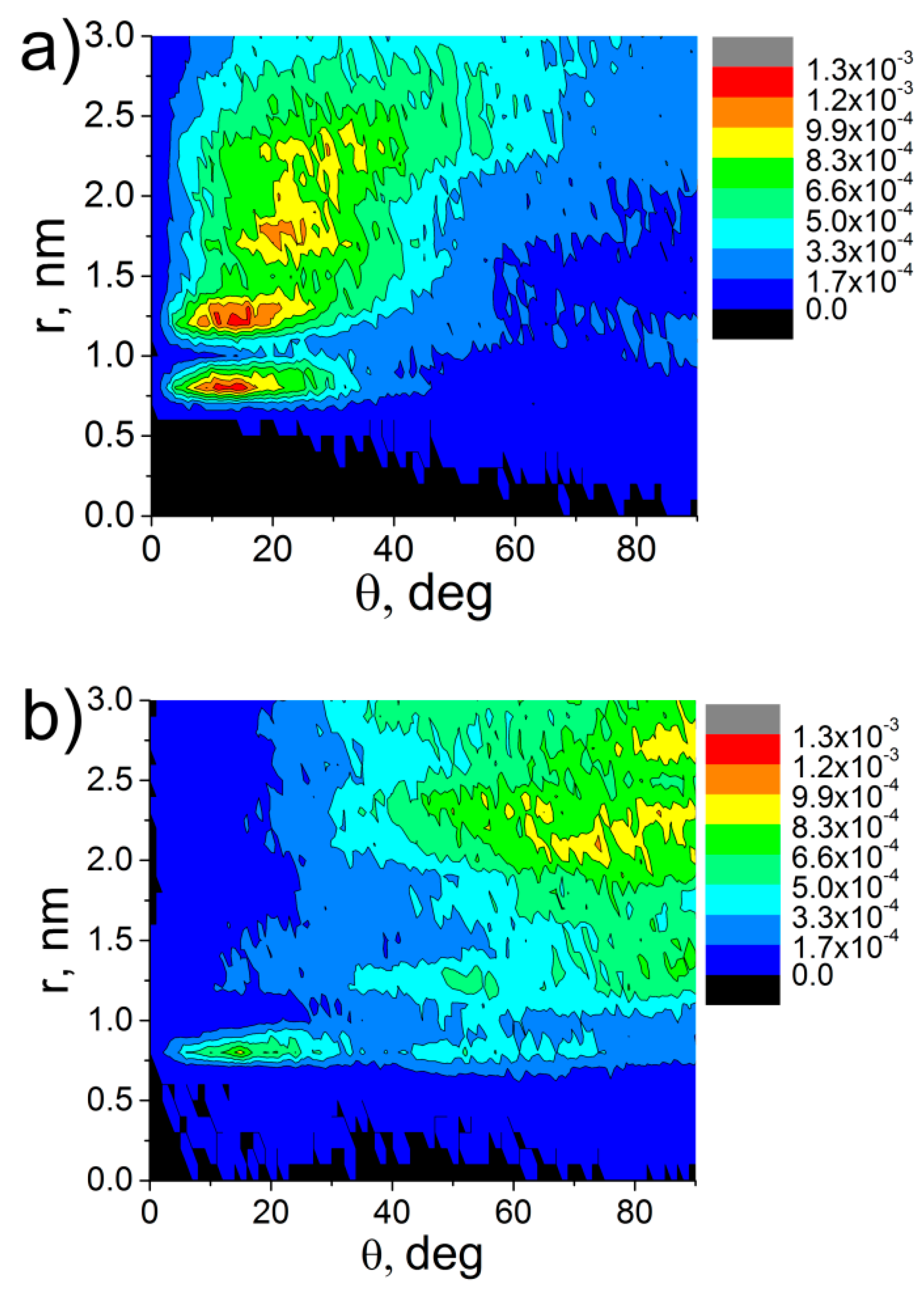
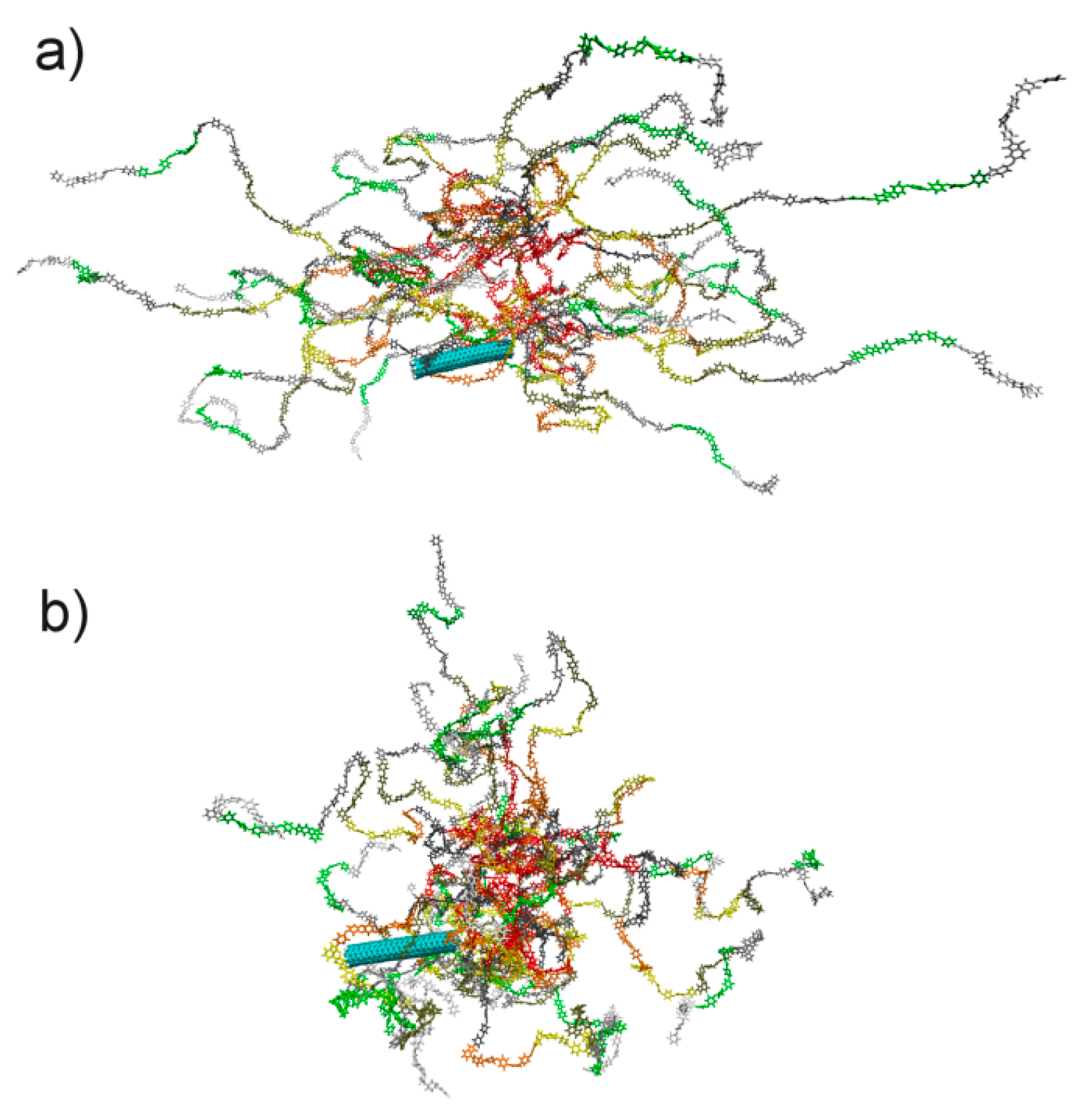
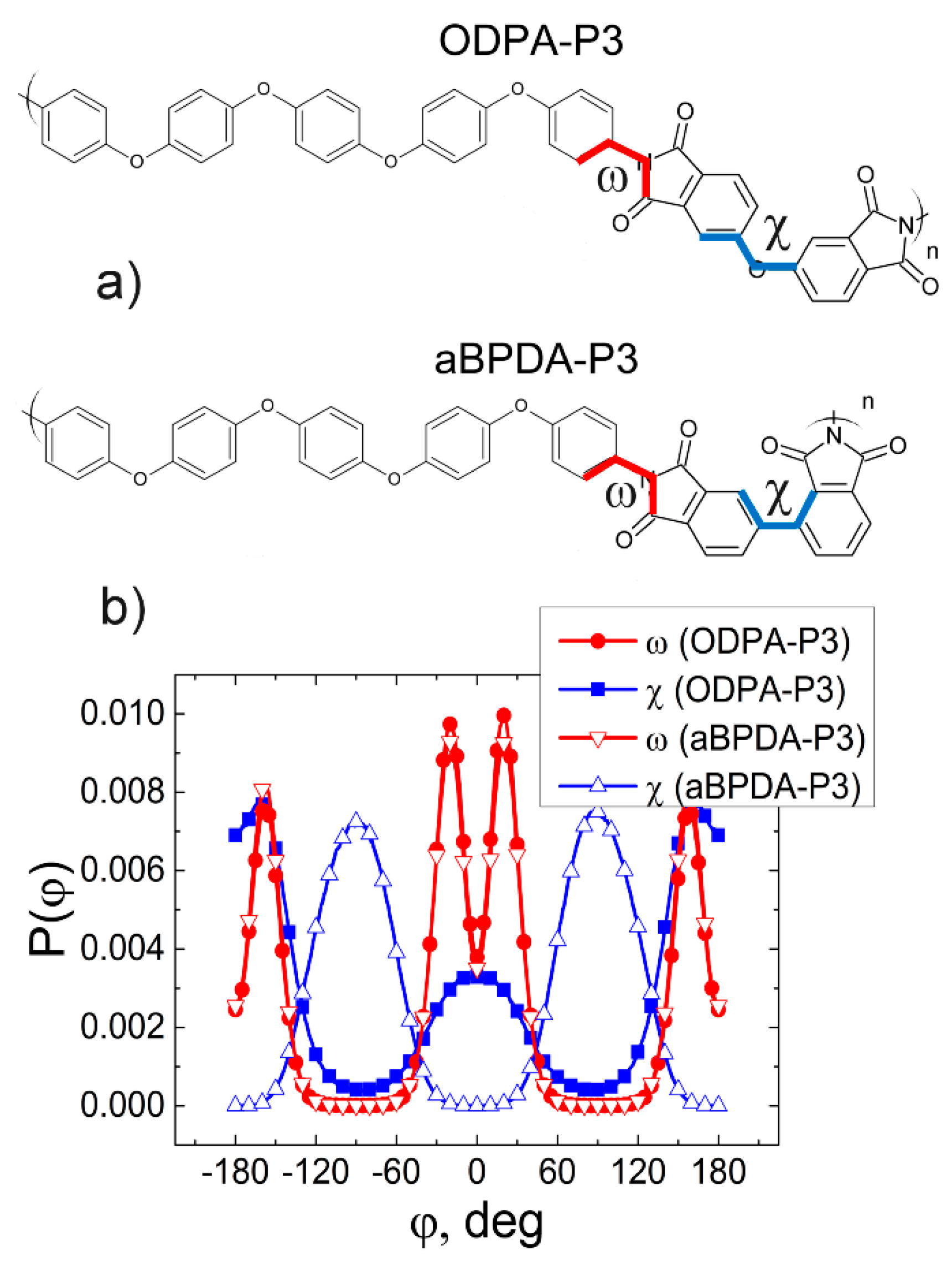
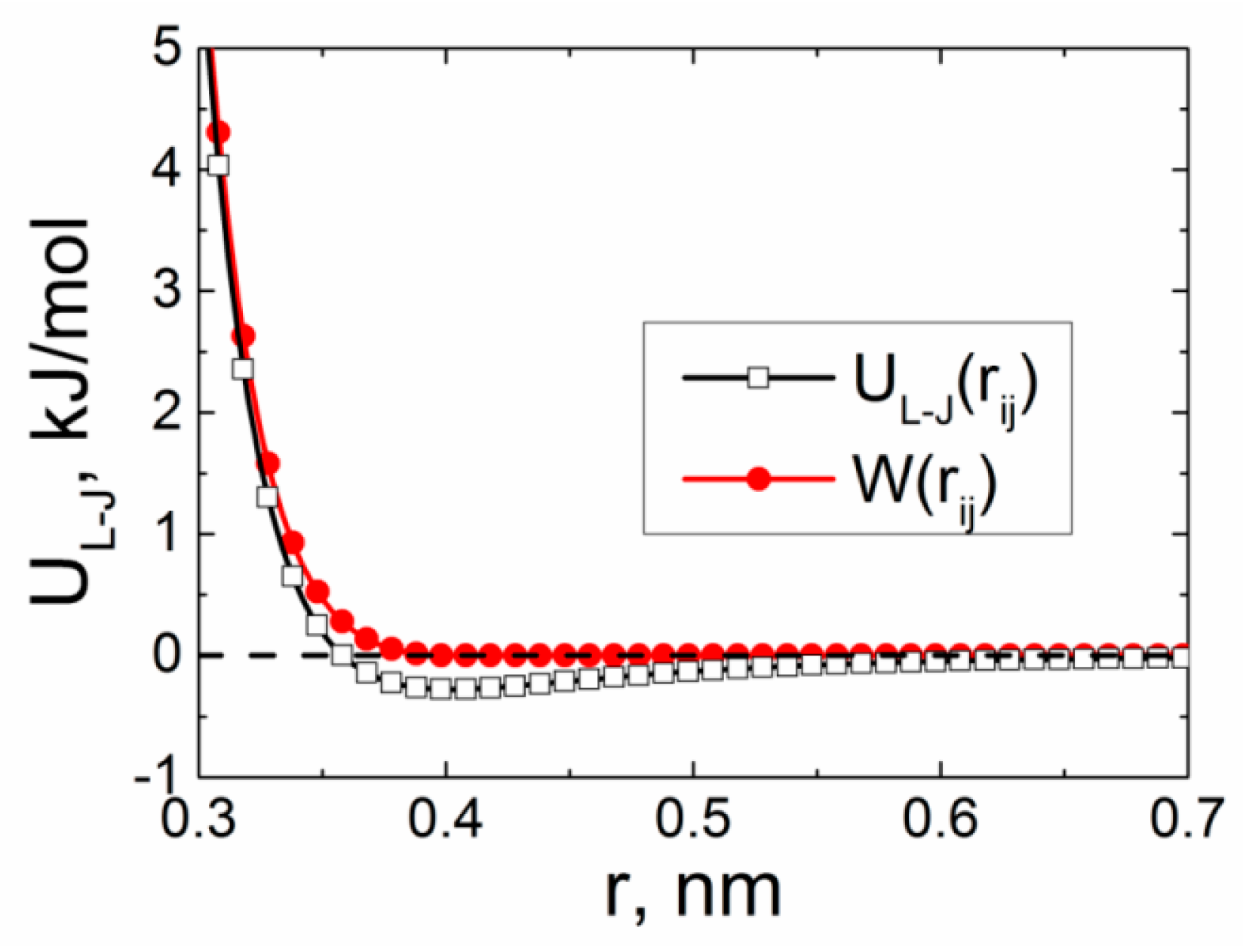
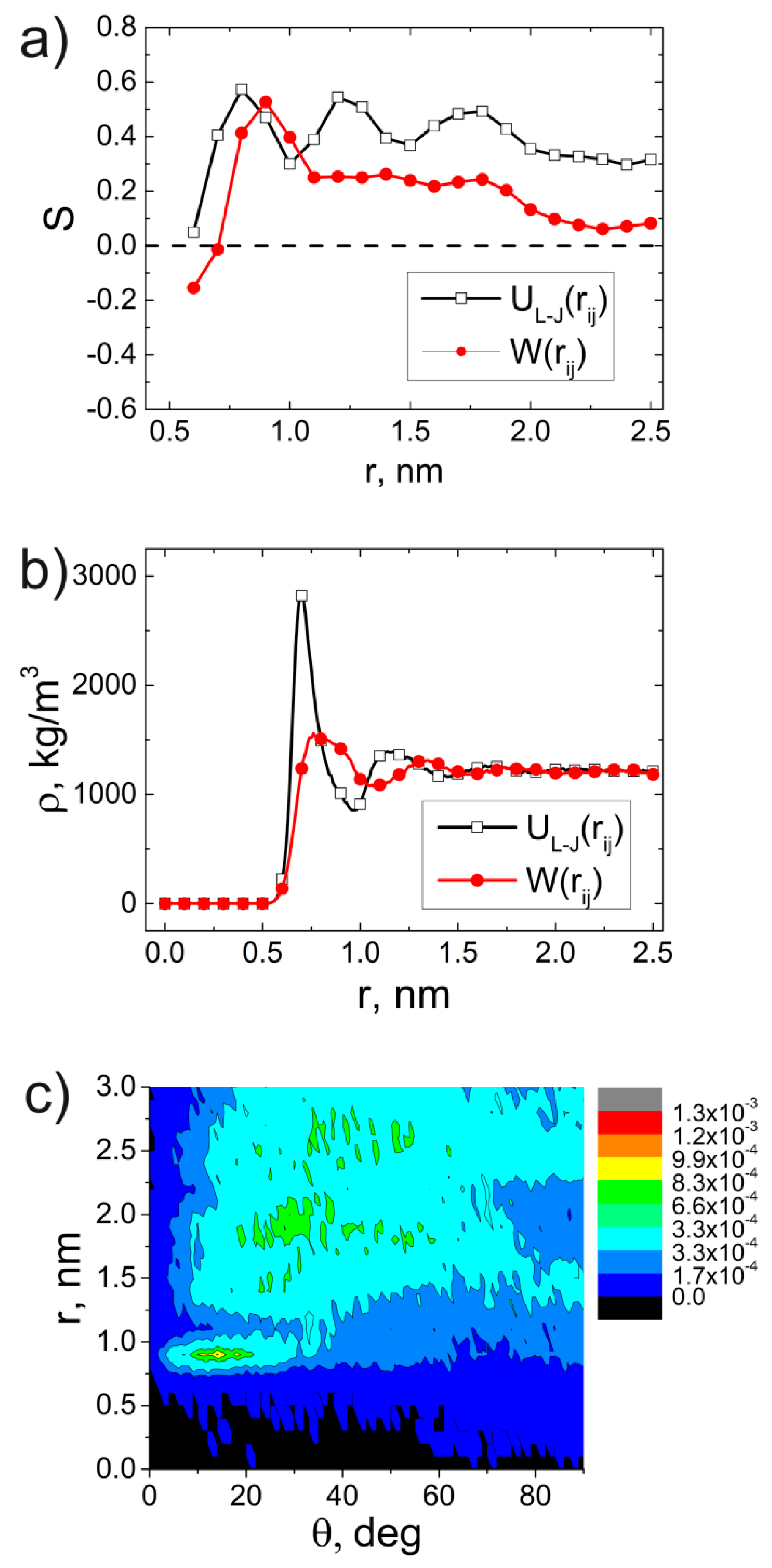
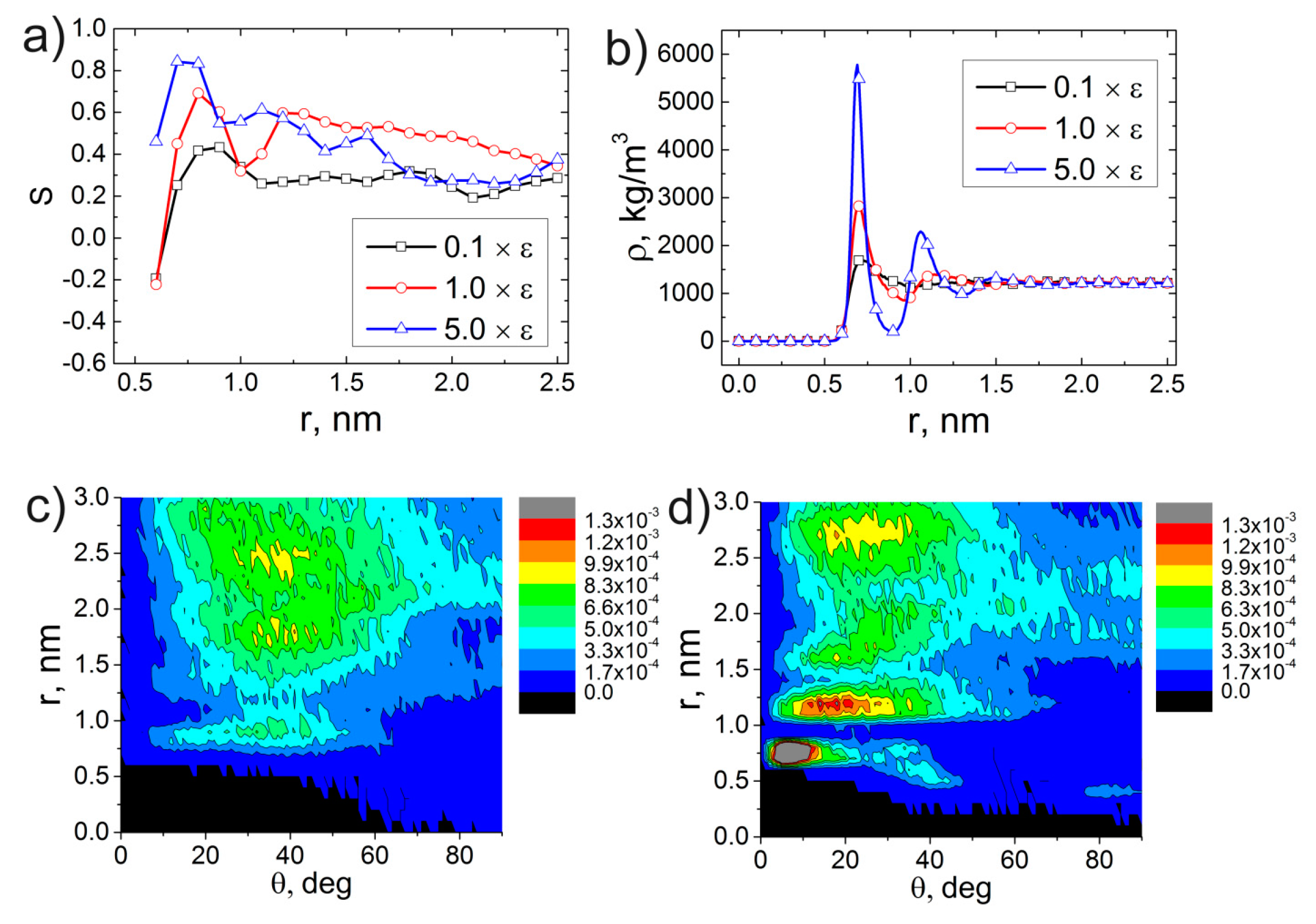
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ishida, H.; Huang, M.T. Molecular level study of the crystallization of a thermoplastic polyimide by infrared spectroscopy. J. Polym. Sci. B 1994, 32, 2271–2282. [Google Scholar] [CrossRef]
- Tamai, S.; Kuroki, T.; Shibuya, A.; Yamaguchi, A. Synthesis and characterization of thermally stable semicrystalline polyimide based on 3,4′-oxydianiline and 3,3′,4,4′-biphenyltetracarboxylic dianhydride. Polymer 2001, 42, 2373–2378. [Google Scholar] [CrossRef]
- Ratta, V.; Ayambem, A.; McGrath, J.E.; Wilkes, G.L. Crystallization and multiple melting behavior of a new semicrystalline polyimide based on 1,3-bis(4-aminophenoxy)benzene (TPER) and 3,3′,4,4′-biphenonetetracarboxylic dianhydride (BTDA). Polymer 2001, 42, 6173–6186. [Google Scholar] [CrossRef]
- Heberer, D.P.; Cheng, S.Z.D.; Barley, J.S.; Lien, S.H.-S.; Bryant, R.G.; Harris, F.W. Crystallization and morphology of semicrystalline polyimides. Macromolecules 1991, 24, 1890–1898. [Google Scholar] [CrossRef]
- Fu, Q.; Livengood, B.P.; Shen, C.C.; Lin, F.; Harris, F.W.; Cheng, S.Z.D.; Hsiao, B.S.; Yeh, F. Crystallization and phase behavior in nylon 6/aromatic polyimide triblock copolymers. Macromol. Chem. Phys. 1998, 199, 1107–1118. [Google Scholar] [CrossRef]
- Srinivas, S.; Caputo, F.E.; Graham, M.; Gardner, S.; Davis, R.M.; McGrath, J.E.; Wilkes, G.L. Semicrystalline polyimides based on controlled molecular weight phthalimide end-capped 1,3-bis(4-aminophenoxy)benzene and 3,3′,4,4′-biphenyltetracarboxylic dianhydride: Synthesis, crystallization, melting, and thermal stability. Macromolecules 1997, 30, 1012–1022. [Google Scholar] [CrossRef]
- Takekoshi, T. Polyimides. In Advances in Polymer Science; Overberger, C.G., Ed.; Springer: Berlin/Heidelberg, Germany, 1990; Volume 94, pp. 1–25. [Google Scholar] [CrossRef]
- Cheng, S.Z.D.; Mittleman, M.L.; Janimak, J.J.; Shen, D.; Chalmers, T.M.; Lien, H.S.; Tso, C.C.; Gabori, P.A.; Harris, F.W. Crystal structure, crystallization kinetics and morphology of a new polyimide. Polym. Int. 1992, 29, 201–208. [Google Scholar] [CrossRef]
- Muellerleile, J.T.; Risch, B.G.; Rodrigues, D.E.; Wilkes, G.L.; Jones, D.M. Crystallization behaviour and morphological features of LARC-CPI. Polymer 1993, 34, 789–806. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Winey, K.I. Nanocomposites Containing Carbon Nanotubes. Macromolecules 2006, 39, 5194–5205. [Google Scholar] [CrossRef]
- Miltner, H.E.; Grossiord, N.; Lu, K.; Loos, J.; Koning, C.E.; Van Mele, B. Isotactic polypropylene/carbon nanotube composites prepared by latex technology. Thermal analysis of carbon nanotube-induced nucleation. Macromolecules 2008, 41, 5753–5762. [Google Scholar] [CrossRef]
- Sandler, J.; Broza, G.; Nolte, M.; Schulte, K.; Lam, Y.-M.; Shaffer, M.S.P. Crystallization of carbon nanotube and nanofiber polypropylene composites. J. Macromol. Sci. B 2003, 42, 479–488. [Google Scholar] [CrossRef]
- Assouline, E.; Lustiger, A.; Barber, A.H.; Cooper, C.A.; Klein, E.; Wachtel, E.; Wagner, H.D. Nucleation ability of multiwall carbon nanotubes in polypropylene composites. J. Polym. Sci. B 2003, 41, 520–527. [Google Scholar] [CrossRef]
- Manchado, M.A.L.; Valentini, L.; Biagiotti, J.; Kenny, J.M. Thermal and mechanical properties of single-walled carbon nanotubes-polypropylene composites prepared by melt processing. Carbon 2005, 43, 1499–1505. [Google Scholar] [CrossRef]
- Li, L.; Yang, Y.; Yang, G.; Chen, X.; Hsiao, B.S.; Chu, B.; Spanier, J.E.; Li, C.Y. Patterning polyethylene oligomers on carbon nanotubes using physical vapor deposition. Nano Lett. 2006, 6, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, C.Y.; Ni, C. Polymer crystallization-driven, periodic patterning on carbon nanotubes. J. Am. Chem. Soc. 2006, 128, 1692–1699. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Zhuang, Y.; Xia, R.; Cheng, J.; Zhang, Y. Carbon nanotubes induced nonisothermal crystallization of ultrahigh molecular weight polyethylene with reduced chain entanglements. Mater. Lett. 2012, 89, 272–275. [Google Scholar] [CrossRef]
- Chae, H.G.; Minus, M.L.; Kumar, S. Oriented and exfoliated single wall carbon nanotubes in polyacrylonitrile. Polymer 2006, 47, 3494–3504. [Google Scholar] [CrossRef]
- Lu, K.; Grossiord, N.; Koning, C.E.; Miltner, H.E.; van Mele, B.; Loos, J. Carbon nanotube/isotactic polypropylene composites prepared by latex technology: Morphology analysis of CNT-induced nucleation. Macromolecules 2008, 41, 8081–8085. [Google Scholar] [CrossRef]
- Phang, I.Y.; Ma, J.; Shen, L.; Liu, T.; De Zhang, W. Crystallization and melting behavior of multi-walled carbon nanotube-reinforced Nylon-6 Composites. Polym. Int. 2006, 55, 71–79. [Google Scholar] [CrossRef]
- Chatterjee, S.; Nüesch, F.A.; Chu, B.T.T. Comparing carbon nanotubes and graphene nanoplatelets as reinforcements in polyamide 12 composites. Nanotechnology 2011, 22, 275714–275721. [Google Scholar] [CrossRef] [PubMed]
- Sandler, J.K.W.; Pegel, S.; Cadek, M.; Gojny, F.; Van Es, M.; Lohmar, J.; Blau, W.J.; Schulte, K.; Windle, A.H.; Shafferf, M.S.P. A comparative study of melt spun polyamide-12 fibres reinforced with carbon nanotubes and nanofibres. Polymer 2004, 45, 2001–2015. [Google Scholar] [CrossRef]
- Yudin, V.E.; Svetlichnyi, V.M.; Shumakov, A.N.; Schechter, R.; Harel, H.; Marom, G. Morphology and mechanical properties of carbon fiber reinforced composites based on semicrystalline polyimides modified by carbon nanofibers. Compos. A 2008, 39, 85–90. [Google Scholar] [CrossRef]
- Yudin, V.E.; Svetlichnyi, V.M.; Shumakov, A.N.; Letenko, D.G.; Feldman, A.Y.; Marom, G. The nucleating effect of carbon nanotubes on crystallinity in R-BAPB-type thermoplastic polyimide. Macromol. Rapid Commun. 2005, 26, 885–888. [Google Scholar] [CrossRef]
- Yudin, V.E.; Feldman, A.Y.; Svetlichnyi, V.M.; Shumakov, A.N.; Marom, G. Crystallization of R-BAPB type polyimide modified by carbon nano-particles. Compos. Sci. Technol. 2007, 67, 789–794. [Google Scholar] [CrossRef]
- Ning, N.; Fu, S.; Zhang, W.; Chen, F.; Wang, K.; Deng, H.; Zhang, Q.; Fu, Q. Realizing the enhancement of interfacial interaction in semicrystalline polymer/filler composites via interfacial crystallization. Prog. Polym. Sci. 2012, 37, 1425–1455. [Google Scholar] [CrossRef]
- Batistakis, C.; Lyulin, A.V.; Michels, M.A.J. Slowing down versus acceleration in the dynamics of confined polymer films. Macromolecules 2012, 45, 7282–7292. [Google Scholar] [CrossRef]
- Solar, M.; Paul, W. Dielectric α-relaxation of 1,4-polybutadiene confined between graphite walls. Eur. Phys. J. E 2015, 38, 37–39. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Santos, C.; Martinez-Hernandez, A.L.; Castano, V.M. Carbon nanotube-polymer nanocomposites: The role of interfaces. Compos. Interfaces 2005, 11, 567–586. [Google Scholar] [CrossRef]
- Rahmat, M.; Hubert, P. Interactions in nanocomposites: A review. Compos. Sci. Technol. 2011, 72, 72–84. [Google Scholar] [CrossRef]
- Andrews, R.; Weisenberger, M.C. Carbon nanotube polymer composites. Curr. Opin. Solid State Mater. Sci. 2004, 8, 31–37. [Google Scholar] [CrossRef]
- Coleman, J.N.; Khan, U.; Blau, W.J.; Gun’ko, Y.K. Small but strong: A review of the mechanical properties of carbon nanotube-polymer composites. Carbon 2006, 44, 1624–1652. [Google Scholar] [CrossRef]
- Mark, J.E. Physical Properties of Polymers Handbook; Springer Science & Business Media: New York, NY, USA, 2007; p. 1096. ISBN 978-0-387-69002-5. [Google Scholar]
- Green, M.J.; Behabtu, N.; Pasquali, M.; Adams, W.W. Nanotubes as polymers. Polymer 2009, 50, 4979–4997. [Google Scholar] [CrossRef]
- Baskaran, D.; Mays, J.W.; Bratcher, M.S. Noncovalent and nonspecific molecular interactions of polymers with multiwalled carbon nanotubes. Chem. Mater. 2005, 17, 3389–3397. [Google Scholar] [CrossRef]
- Tallury, S.S.; Pasquinelli, M.A. Molecular dynamics simulations of flexible polymer chains wrapping single-walled carbon nanotubes. J. Phys. Chem. B 2010, 114, 4122–4129. [Google Scholar] [CrossRef] [PubMed]
- Steuerman, D.W.; Star, A.; Narizzano, R.; Choi, H.; Ries, R.S.; Nicolini, C.; Stoddart, J.F.; Heath, J.R. Interactions between conjugated polymers and single-walled carbon nanotubes. J. Phys. Chem. B 2002, 106, 3124–3130. [Google Scholar] [CrossRef]
- Li, H.; Yan, S. Surface-induced polymer crystallization and the resultant structures and morphologies. Macromolecules 2011, 44, 417–428. [Google Scholar] [CrossRef]
- Azimi, M.; Mirjavadi, S.S.; Hamouda, A.M.S.; Makki, H. Heterogeneities in polymer structural and dynamic properties in graphene and graphene oxide nanocomposites: Molecular dynamics simulations. Macromol. Theory Simul. 2017, 26, 1600086–1600096. [Google Scholar] [CrossRef]
- Yang, M.; Koutsos, V.; Zaiser, M. Interactions between polymers and carbon nanotubes: A molecular dynamics study. J. Phys. Chem. B 2005, 109, 10009–10014. [Google Scholar] [CrossRef] [PubMed]
- Minoia, A.; Chen, L.; Beljonne, D.; Lazzaroni, R. Molecular modeling study of the structure and stability of polymer/carbon nanotube interfaces. Polymer 2012, 53, 5480–5490. [Google Scholar] [CrossRef]
- Jiang, Q.; Tallury, S.S.; Qiu, Y.; Pasquinelli, M.A. Molecular dynamics simulations of the effect of the volume fraction on unidirectional polyimide-carbon nanotube nanocomposites. Carbon 2014, 67, 440–448. [Google Scholar] [CrossRef]
- Liu, W.; Yang, C.-L.; Zhu, Y.-T.; Wang, M. Interactions between single-walled carbon nanotubes and polyethylene/polypropylene/polystyrene/poly(phenylacetylene)/poly(p-phenylenevinylene) considering repeat unit arrangements and conformations: A molecular dynamics simulation study. J. Phys. Chem. C 2008, 112, 1803–1811. [Google Scholar] [CrossRef]
- Asadinezhad, A.; Kelich, P. Effects of carbon nanofiller characteristics on ptt chain conformation and dynamics: A computational study. Appl. Surf. Sci. 2017, 392, 981–990. [Google Scholar] [CrossRef]
- Yang, H.; Chen, Y.; Liu, Y.; Cai, W.S.; Li, Z.S. Molecular dynamics simulation of polyethylene on single wall carbon nanotube. J. Chem. Phys. 2007, 127, 94902–94907. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-S.; Yang, C.-L.; Wang, M.-S.; Chen, B.-D.; Ma, X.-G. Crystallization of alkane melts induced by carbon nanotubes and graphene nanosheets: A molecular dynamics simulation study. Phys. Chem. Chem. Phys. 2011, 13, 15476–15482. [Google Scholar] [CrossRef] [PubMed]
- Liaw, D.-J.; Wang, K.-L.; Huang, Y.-C.; Lee, K.-R.; Lai, J.-Y.; Ha, C.-S. Advanced polyimide materials: Syntheses, physical properties and applications. Prog. Polym. Sci. 2012, 37, 907–974. [Google Scholar] [CrossRef]
- Bessonov, M.I.; Koton, M.M.; Kudryavtsev, V.V.; Laius, L.A. Polyimides: Thermally Stable Polymers; Plenum: New York, NY, USA, 1987; p. 318. ISBN 978-1-4615-7636-5. [Google Scholar]
- Yudin, V.E.; Svetlichnyi, V.M.; Gubanova, G.N.; Didenko, A.L.; Sukhanova, T.E.; Kudryavtsev, V.V.; Ratner, S.; Marom, G. Semicrystalline polyimide matrices for composites: Crystallization and properties. J. Appl. Polym. Sci. 2002, 83, 2873–2882. [Google Scholar] [CrossRef]
- Larin, S.V.; Falkovich, S.G.; Nazarychev, V.M.; Gurtovenko, A.A.; Lyulin, A.V.; Lyulin, S.V. Molecular-dynamics simulation of polyimide matrix pre-crystallization near the surface of a single-walled carbon nanotube. RSC Adv. 2014, 4, 830–844. [Google Scholar] [CrossRef]
- Larin, S.V.; Glova, A.D.; Serebryakov, E.B.; Nazarychev, V.M.; Kenny, J.M.; Lyulin, S.V. Influence of the carbon nanotube surface modification on the microstructure of thermoplastic binders. RSC Adv. 2015, 5, 51621–51630. [Google Scholar] [CrossRef]
- Falkovich, S.G.; Nazarychev, V.M.; Larin, S.V.; Kenny, J.M.; Lyulin, S.V. Mechanical properties of a polymer at the interface structurally ordered by graphene. J. Phys. Chem. C 2016, 120, 6771–6777. [Google Scholar] [CrossRef]
- Falkovich, S.G.; Larin, S.V.; Lyulin, A.V.; Yudin, V.E.; Kenny, J.M.; Lyulin, S.V. Influence of the carbon nanofiller surface curvature on the initiation of crystallization in thermoplastic polymers. RSC Adv. 2014, 4, 48606–48612. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Larin, S.V.; Gurtovenko, A.A.; Lukasheva, N.V.; Yudin, V.E.; Svetlichnyi, V.M.; Lyulin, A.V. Effect of the SO2 group in the diamine fragment of polyimides on their structural, thermophysical, and mechanical properties. Polym. Sci. Ser. A 2012, 54, 631–643. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Gurtovenko, A.A.; Larin, S.V.; Nazarychev, V.M.; Lyulin, A.V. Microsecond atomic-scale molecular dynamics simulations of polyimides. Macromolecules 2013, 46, 6357–6363. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Larin, S.V.; Gurtovenko, A.A.; Nazarychev, V.M.; Falkovich, S.G.; Yudin, V.E.; Svetlichnyi, V.M.; Gofmana, I.V.; Lyulin, A.V. Thermal properties of bulk polyimides: Insights from computer modeling versus experiment. Soft Matter 2014, 10, 1224–1232. [Google Scholar] [CrossRef] [PubMed]
- Falkovich, S.G.; Lyulin, S.V.; Nazarychev, V.M.; Larin, S.V.; Gurtovenko, A.A.; Lukasheva, N.V.; Lyulin, A.V. Influence of the electrostatic interactions on thermophysical properties of polyimides: Molecular-dynamics simulations. J. Polym. Sci. B 2014, 52, 640–646. [Google Scholar] [CrossRef]
- Nazarychev, V.M.; Larin, S.V.; Yakimansky, A.V.; Lukasheva, N.V.; Gurtovenko, A.A.; Gofman, I.V.; Yudin, V.E.; Svetlichnyi, V.M.; Kenny, J.M.; Lyulin, S.V. Parameterization of electrostatic interactions for molecular dynamics simulations of heterocyclic polymers. J. Polym. Sci. B 2015, 53, 912–923. [Google Scholar] [CrossRef]
- Nazarychev, V.M.; Lyulin, A.V.; Larin, S.V.; Gurtovenko, A.A.; Kenny, J.M.; Lyulin, S.V. Molecular Dynamics Simulations of Uniaxial Deformation of Thermoplastic Polyimides. Soft Matter 2016, 12, 3972–3981. [Google Scholar] [CrossRef] [PubMed]
- Nazarychev, V.M.; Lyulin, A.V.; Larin, S.V.; Gofman, I.V.; Kenny, J.M.; Lyulin, S.V. Correlation between the high-temperature local mobility of heterocyclic polyimides and their mechanical properties. Macromolecules 2016, 49, 6700–6710. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Larin, S.V.; Nazarychev, V.M.; Fal’kovich, S.G.; Kenny, J.M. Multiscale computer simulation of polymer nanocomposites based on thermoplastics. Polym. Sci. Ser. C 2016, 58, 2–15. [Google Scholar] [CrossRef]
- Nazarychev, V.M.; Larin, S.V.; Lukasheva, N.V.; Glova, A.D.; Lyulin, S.V. Evaluation of the characteristic equilibration times of bulk polyimides via full-atomic computer simulation. Polym. Sci. Ser. A 2013, 55, 570–576. [Google Scholar] [CrossRef]
- Hegde, M.; Lafont, U.; Norder, B.; Picken, S.J.; Samulski, E.T.; Rubinstein, M.; Dingemans, T. SWCNT Induced crystallization in an amorphous all-aromatic poly(ether imide). Macromolecules 2013, 46, 1492–1503. [Google Scholar] [CrossRef]
- Hegde, M.; Lafont, U.; Norder, B.; Samulski, E.T.; Rubinstein, M.; Dingemans, T.J. SWCNT Induced crystallization in amorphous and semi-crystalline poly(etherimide)s: Morphology and thermo-mechanical properties. Polymer 2014, 55, 3746–3757. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Karatrantos, A.; Composto, R.J.; Winey, K.I.; Clarke, N. Structure and conformations of Polymer/SWCNT nanocomposites. Macromolecules 2011, 44, 9830–9838. [Google Scholar] [CrossRef]
- Chakraborty, S.; Roy, S. Structural, dynamical, and thermodynamical properties of carbon nanotube polycarbonate composites: A molecular dynamics study. J. Phys. Chem. B 2012, 116, 3083–3091. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.; Turci, F.; Schilling, T. Crystallization mechanism in melts of short n-alkane chains. J. Chem. Phys. 2012, 139, 214904–214909. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.; Berryman, J.T.; Schilling, T. Crystal nucleation mechanism in melts of short polymer chains under quiescent conditions and under shear flow. J. Chem. Phys. 2014, 141, 124910–124919. [Google Scholar] [CrossRef] [PubMed]
- Šponer, J.; Jurečka, P.; Marchan, I.; Luque, F.J.; Orozco, M.; Hobza, P. Nature of base stacking: Reference quantum-chemical stacking energiesin ten unique B-DNA base-pair steps. Chem. Eur. J. 2006, 12, 2854–2865. [Google Scholar] [CrossRef] [PubMed]
- Oostenbrink, C.; Soares, T.A.; Van Der Vegt, N.F.A.; Van Gunsteren, W.F. Validation of the 53A6 GROMOS force field. Eur. Biophys. J. 2005, 34, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Weeks, J.D.; Chandler, D.; Andersen, H.C. Role of Repulsive forces in determining the equilibrium structure of simple liquids. J. Chem. Phys. 1971, 54, 5237–5247. [Google Scholar] [CrossRef]
- Chen, J.; Liu, H.; Weimer, W.A.; Halls, M.D.; Waldeck, D.H.; Walker, G.C. Noncovalent engineering of carbon nanotube surfaces by rigid, functional conjugated polymers. J. Am. Chem. Soc. 2002, 124, 9034–9035. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Zhang, Y.; Wang, D.; Dai, H. Noncovalent sidewall functionalization of single-walled carbon nanotubes for protein immobilization. J. Am. Chem. Soc. 2001, 123, 3838–3839. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazarychev, V.M.; Larin, S.V.; Lyulin, A.V.; Dingemans, T.; Kenny, J.M.; Lyulin, S.V. Atomistic Molecular Dynamics Simulations of the Initial Crystallization Stage in an SWCNT-Polyetherimide Nanocomposite. Polymers 2017, 9, 548. https://doi.org/10.3390/polym9100548
Nazarychev VM, Larin SV, Lyulin AV, Dingemans T, Kenny JM, Lyulin SV. Atomistic Molecular Dynamics Simulations of the Initial Crystallization Stage in an SWCNT-Polyetherimide Nanocomposite. Polymers. 2017; 9(10):548. https://doi.org/10.3390/polym9100548
Chicago/Turabian StyleNazarychev, Victor M., Sergey V. Larin, Alexey V. Lyulin, Theo Dingemans, Jose M. Kenny, and Sergey V. Lyulin. 2017. "Atomistic Molecular Dynamics Simulations of the Initial Crystallization Stage in an SWCNT-Polyetherimide Nanocomposite" Polymers 9, no. 10: 548. https://doi.org/10.3390/polym9100548