
Anti-Proliferation and DNA Cleavage Activities of Copper(II) Complexes of N3O Tripodal Polyamine Ligands
 , and
, and
Abstract
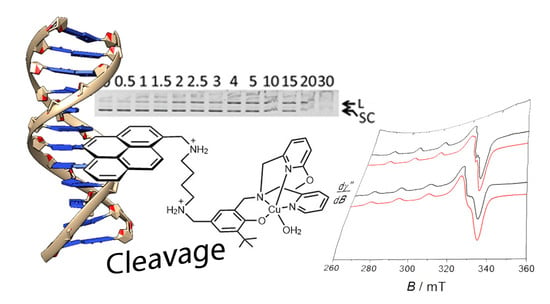
:
1. Introduction
2. Results and Discussion
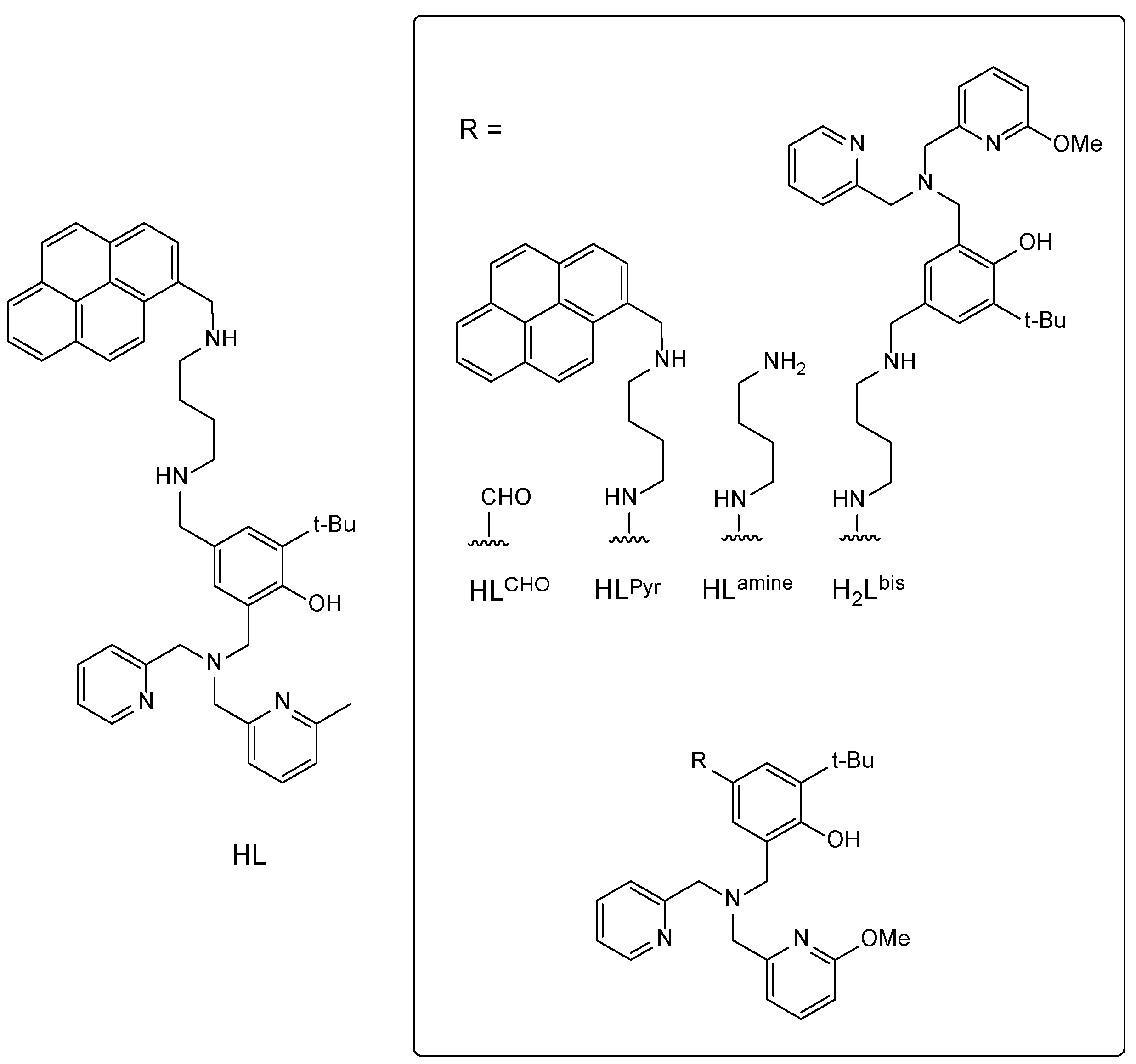
2.1. Synthesis and Structure of the Complexes
2.2. Spectroscopic Characterization
2.3. Electrochemistry
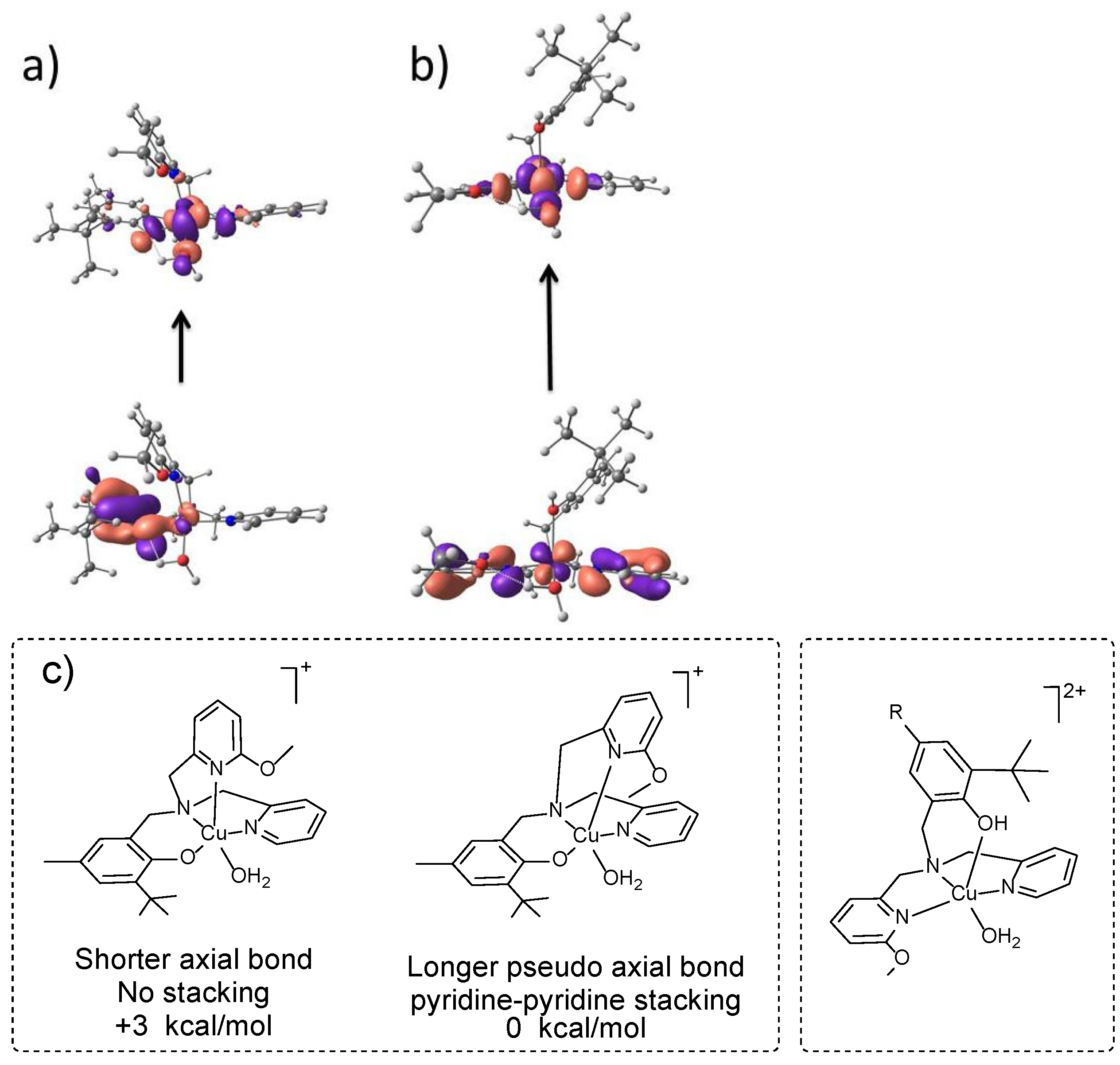
2.4. DFT and TD-DFT Calculations
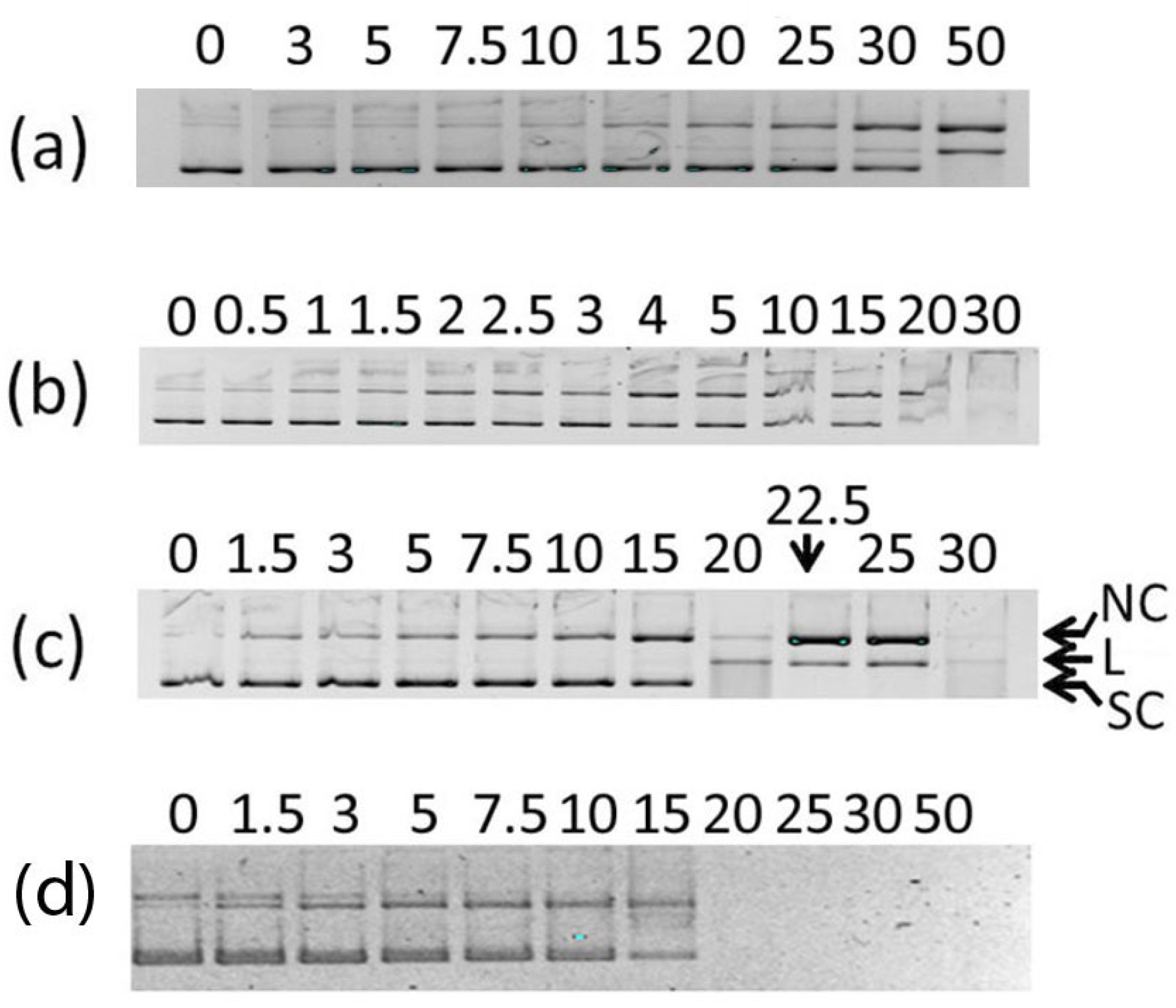
2.5. DNA Cleavage
2.6. Mode of Binding of the Best Nuclease 2
2.7. Anti-Proliferative Activity
3. Materials and Methods
3.1. Materials and Instruments
3.2. Synthesis
3.3. Crystal Structure Analysis
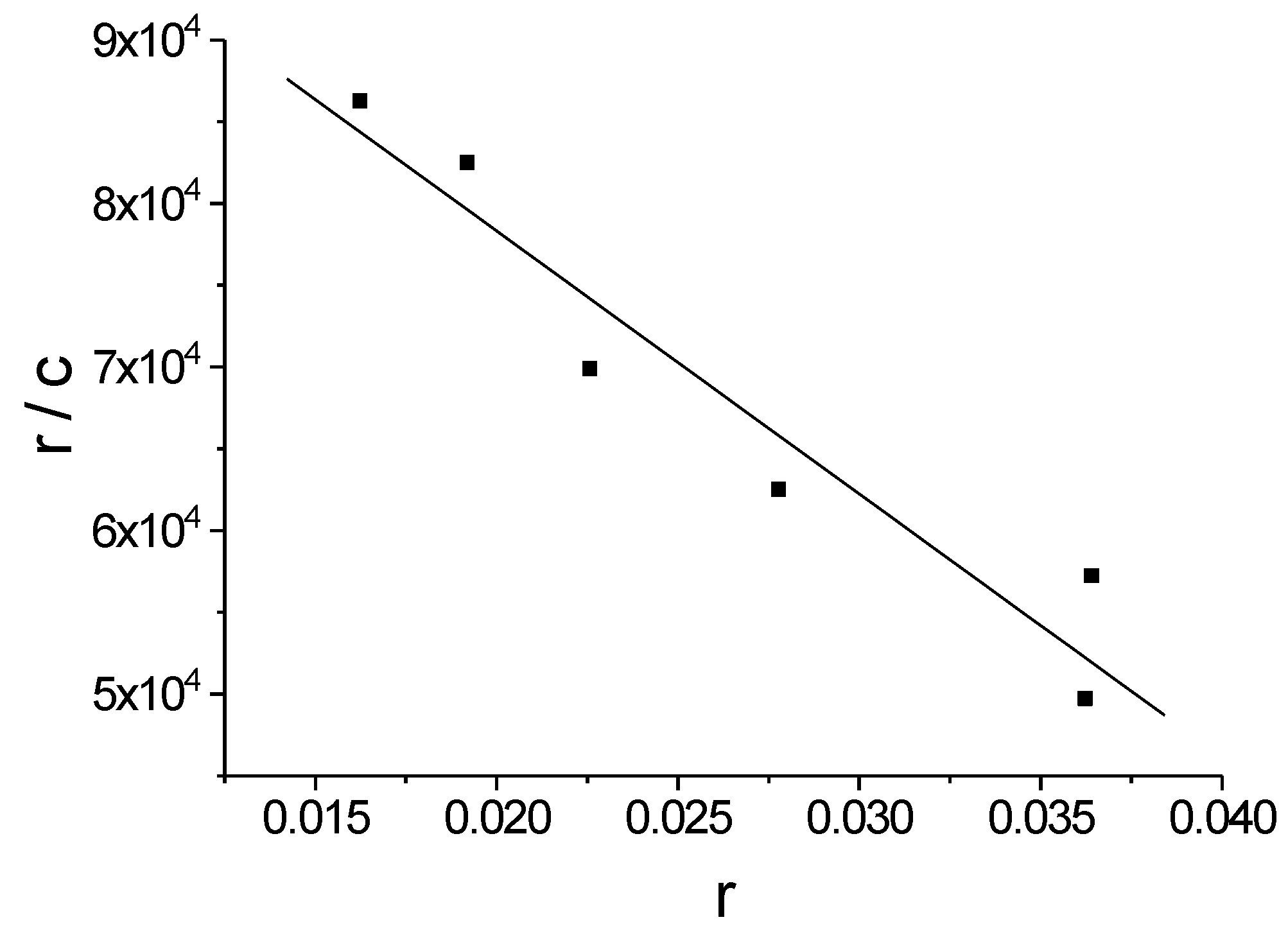
3.4. Determination of the DNA Binding Constant
3.5. Procedure for DNA Cleavage Experiments
3.6. Cell Culture
3.7. Cell Proliferation Assay
3.8. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fantoni, N.Z.; Molphy, Z.; Slator, C.; Menounou, G.; Toniolo, G.; Mitrikas, G.; McKee, V.; Chatgilialoglu, C.; Kellett, A. Polypyridyl-Based Copper Phenanthrene Complexes: A New Type of Stabilized Artificial Chemical Nuclease. Chem. Eur. J. 2019, 25, 221–237. [Google Scholar] [CrossRef]
- Desbouis, D.; Troitsky, I.P.; Belousoff, M.J.; Spiccia, L.; Graham, B. Copper(II), zinc(II) and nickel(II) complexes as nuclease mimetics. Coord. Chem. Rev. 2012, 256, 897–937. [Google Scholar] [CrossRef]
- Mancin, F.; Scrimin, P.; Tecilla, P. Progress in artificial metallonucleases. Chem. Commun. 2012, 48, 5545–5559. [Google Scholar] [CrossRef]
- Wende, C.; Lüdtke, C.; Kulak, N. Copper Complexes of N-Donor Ligands as Artificial Nucleases. Eur. J. Inorg. Chem. 2014, 2014, 2597–2612. [Google Scholar] [CrossRef]
- Cristina, M.; Maura, P.; Francesco, T.; Carlo, S. Copper Complexes as Anticancer Agents. Anti Cancer Agents Med. Chem. 2009, 9, 185–211. [Google Scholar] [CrossRef]
- Saverio, T.; Luciano, M. Copper Compounds in Anticancer Strategies. Curr. Med. Chem. 2009, 16, 1325–1348. [Google Scholar] [CrossRef]
- Tisato, F.; Marzano, C.; Porchia, M.; Pellei, M.; Santini, C. Copper in diseases and treatments, and copper-based anticancer strategies. Med. Res. Rev. 2010, 30, 708–749. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.; White, A.R. Copper complexes as therapeutic agents. Metallomics 2012, 4, 127–138. [Google Scholar] [CrossRef]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in Copper Complexes as Anticancer Agents. Chem. Rev. 2014, 114, 815–862. [Google Scholar] [CrossRef]
- Denoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M.A. Targeting copper in cancer therapy: ‘Copper That Cancer’. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef]
- Huiqi, X.; Kang, Y.J. Role of Copper in Angiogenesis and Its Medicinal Implications. Curr. Med. Chem. 2009, 16, 1304–1314. [Google Scholar] [CrossRef]
- Mazdak, H.; Yazdekhasti, F.; Movahedian, A.; Mirkheshti, N.; Shafieian, M. The comparative study of serum iron, copper, and zinc levels between bladder cancer patients and a control group. Int. Urol. Nephrol. 2010, 42, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Lelièvre, P.; Sancey, L.; Coll, J.-L.; Deniaud, A.; Busser, B. The Multifaceted Roles of Copper in Cancer: A Trace Metal Element with Dysregulated Metabolism, but Also a Target or a Bullet for Therapy. Cancers 2020, 12, 3594. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Chatterjee, S.; Pal, S.; Biswas, J.; Efferth, T.; Choudhuri, S.K. The role of copper in drug-resistant murine and human tumors. BioMetals 2008, 22, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Pitié, M.; Pratviel, G. Activation of DNA Carbon−Hydrogen Bonds by Metal Complexes. Chem. Rev. 2010, 110, 1018–1059. [Google Scholar] [CrossRef]
- Villarreal, W.; Colina-Vegas, L.; Visbal, G.; Corona, O.; Corrêa, R.S.; Ellena, J.; Cominetti, M.R.; Batista, A.A.; Navarro, M. Copper(I)–Phosphine Polypyridyl Complexes: Synthesis, Characterization, DNA/HSA Binding Study, and Antiproliferative Activity. Inorg. Chem. 2017, 56, 3781–3793. [Google Scholar] [CrossRef]
- Banasiak, A.; Zuin Fantoni, N.; Kellett, A.; Colleran, J. Mapping the DNA Damaging Effects of Polypyridyl Copper Complexes with DNA Electrochemical Biosensors. Molecules 2022, 27, 645. [Google Scholar] [CrossRef]
- Jaividhya, P.; Dhivya, R.; Akbarsha, M.A.; Palaniandavar, M. Efficient DNA cleavage mediated by mononuclear mixed ligand copper(II) phenolate complexes: The role of co-ligand planarity on DNA binding and cleavage and anticancer activity. J. Inorg. Biochem. 2012, 114, 94–105. [Google Scholar] [CrossRef]
- Loganathan, R.; Ramakrishnan, S.; Suresh, E.; Riyasdeen, A.; Akbarsha, M.A.; Palaniandavar, M. Mixed Ligand Copper(II) Complexes of N,N-Bis(benzimidazol-2-ylmethyl)amine (BBA) with Diimine Co-Ligands: Efficient Chemical Nuclease and Protease Activities and Cytotoxicity. Inorg. Chem. 2012, 51, 5512–5532. [Google Scholar] [CrossRef]
- Silva, P.P.; Guerra, W.; dos Santos, G.C.; Fernandes, N.G.; Silveira, J.N.; da Costa Ferreira, A.M.; Bortolotto, T.; Terenzi, H.; Bortoluzzi, A.J.; Neves, A.; et al. Correlation between DNA interactions and cytotoxic activity of four new ternary compounds of copper(II) with N-donor heterocyclic ligands. J. Inorg. Biochem. 2014, 132, 67–76. [Google Scholar] [CrossRef]
- Bhat, S.S.; Kumbhar, A.A.; Heptullah, H.; Khan, A.A.; Gobre, V.V.; Gejji, S.P.; Puranik, V.G. Synthesis, Electronic Structure, DNA and Protein Binding, DNA Cleavage, and Anticancer Activity of Fluorophore-Labeled Copper(II) Complexes. Inorg. Chem. 2011, 50, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.P.; Guerra, W.; Silveira, J.N.; Ferreira, A.M.d.C.; Bortolotto, T.; Fischer, F.L.; Terenzi, H.; Neves, A.; Pereira-Maia, E.C. Two New Ternary Complexes of Copper(II) with Tetracycline or Doxycycline and 1,10-Phenanthroline and Their Potential as Antitumoral: Cytotoxicity and DNA Cleavage. Inorg. Chem. 2011, 50, 6414–6424. [Google Scholar] [CrossRef] [PubMed]
- Li, M.-J.; Lan, T.-Y.; Cao, X.-H.; Yang, H.-H.; Shi, Y.; Yi, C.; Chen, G.-N. Synthesis, characterization, DNA binding, cleavage activity and cytotoxicity of copper(ii) complexes. Dalton Trans. 2014, 43, 2789–2798. [Google Scholar] [CrossRef] [PubMed]
- Leite, S.M.G.; Lima, L.M.P.; Gama, S.; Mendes, F.; Orio, M.; Bento, I.; Paulo, A.; Delgado, R.; Iranzo, O. Copper(II) Complexes of Phenanthroline and Histidine Containing Ligands: Synthesis, Characterization and Evaluation of their DNA Cleavage and Cytotoxic Activity. Inorg. Chem. 2016, 55, 11801–11814. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, Q.; Huang, Y.; Ma, X.; Xiong, X.; Li, H. Synthesis, structure, and biological evaluation of a copper(ii) complex with fleroxacin and 1,10-phenanthroline. Dalton Trans. 2016, 45, 10928–10935. [Google Scholar] [CrossRef]
- Bortolotto, T.; Silva, P.P.; Neves, A.; Pereira-Maia, E.C.; Terenzi, H. Photoinduced DNA Cleavage Promoted by Two Copper(II) Complexes of Tetracyclines and 1,10-Phenanthroline. Inorg. Chem. 2011, 50, 10519–10521. [Google Scholar] [CrossRef]
- Lüdtke, C.; Sobottka, S.; Heinrich, J.; Liebing, P.; Wedepohl, S.; Sarkar, B.; Kulak, N. Forty Years after the Discovery of Its Nucleolytic Activity: [Cu(phen)2]2+ Shows Unattended DNA Cleavage Activity upon Fluorination. Chem. Eur. J. 2021, 27, 3273–3277. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, X.; Hu, M.; Guo, Z. Improving nuclease activity of copper(II)–terpyridine complex through solubilizing and charge effects of glycine. J. Inorg. Biochem. 2013, 121, 114–120. [Google Scholar] [CrossRef]
- Molphy, Z.; Prisecaru, A.; Slator, C.; Barron, N.; McCann, M.; Colleran, J.; Chandran, D.; Gathergood, N.; Kellett, A. Copper Phenanthrene Oxidative Chemical Nucleases. Inorg. Chem. 2014, 53, 5392–5404. [Google Scholar] [CrossRef]
- Berthet, N.; Martel-Frachet, V.; Michel, F.; Philouze, C.; Hamman, S.; Ronot, X.; Thomas, F. Nuclease and anti-proliferative activities of copper(ii) complexes of N3O tripodal ligands involving a sterically hindered phenolate. Dalton Trans. 2013, 42, 8468–8483. [Google Scholar] [CrossRef]
- Xu, W.; Craft, J.; Fontenot, P.; Barens, M.; Knierim, K.; Albering, J.; Mautner, F.; Massoud, S. Effect of the central metal ion on the cleavage of DNA by [M(TPA)Cl]ClO4 complexes (M = CoII, CuII and ZnII, TPA = tris(2-pyridylmethyl)amine): An efficient artificial nuclease for DNA cleavage. Inorg. Chim. Acta 2011, 373, 159–166. [Google Scholar] [CrossRef]
- Doniz Kettenmann, S.; Nossol, Y.; Louka, F.R.; Legrande, J.R.; Marine, E.; Fischer, R.C.; Mautner, F.A.; Hergl, V.; Kulak, N.; Massoud, S.S. Copper(II) Complexes with Tetradentate Piperazine-Based Ligands: DNA Cleavage and Cytotoxicity. Inorganics 2021, 9, 12. [Google Scholar] [CrossRef]
- Castilho, N.; Gabriel, P.; Camargo, T.P.; Neves, A.; Terenzi, H. Targeting an Artificial Metal Nuclease to DNA by a Simple Chemical Modification and Its Drastic Effect on Catalysis. ACS Med. Chem. Lett. 2020, 11, 286–291. [Google Scholar] [CrossRef]
- Levín, P.; Balsa, L.M.; Silva, C.P.; Herzog, A.E.; Vega, A.; Pavez, J.; León, I.E.; Lemus, L. Artificial Chemical Nuclease and Cytotoxic Activity of a Mononuclear Copper(I) Complex and a Related Binuclear Double-Stranded Helicate. Eur. J. Inorg. Chem. 2021, 2021, 4103–4112. [Google Scholar] [CrossRef]
- McGorman, B.; Fantoni, N.Z.; O’Carroll, S.; Ziemele, A.; El-Sagheer, A.H.; Brown, T.; Kellett, A. Enzymatic Synthesis of Chemical Nuclease Triplex-Forming Oligonucleotides with Gene-Silencing Applications. Nucleic Acids Res. 2022, 50, 5467–5481. [Google Scholar] [CrossRef] [PubMed]
- Philibert, A.; Thomas, F.; Philouze, C.; Hamman, S.; Saint-Aman, E.; Pierre, J.-L. Galactose Oxidase Models: Tuning the Properties of CuII–Phenoxyl Radicals. Chem. Eur. J. 2003, 9, 3803–3812. [Google Scholar] [CrossRef]
- Thomas, F. Ten Years of a Biomimetic Approach to the Copper(II) Radical Site of Galactose Oxidase. Eur. J. Inorg. Chem. 2007, 2007, 2379–2404. [Google Scholar] [CrossRef]
- Kunert, R.; Philouze, C.; Berthiol, F.; Jarjayes, O.; Storr, T.; Thomas, F. Distorted copper(ii) radicals with sterically hindered salens: Electronic structure and aerobic oxidation of alcohols. Dalton Trans. 2020, 49, 12990–13002. [Google Scholar] [CrossRef]
- Maheswari, P.U.; Roy, S.; den Dulk, H.; Barends, S.; van Wezel, G.; Kozlevčar, B.; Gamez, P.; Reedijk, J. The Square-Planar Cytotoxic [CuII(pyrimol)Cl] Complex Acts as an Efficient DNA Cleaver without Reductant. J. Am. Chem. Soc. 2006, 128, 710–711. [Google Scholar] [CrossRef]
- Ghosh, K.; Kumar, P.; Tyagi, N.; Singh, U.P.; Aggarwal, V.; Baratto, M.C. Synthesis and reactivity studies on new copper(II) complexes: DNA binding, generation of phenoxyl radical, SOD and nuclease activities. Eur. J. Med. Chem. 2010, 45, 3770–3779. [Google Scholar] [CrossRef]
- Ghosh, K.; Kumar, P.; Mohan, V.; Singh, U.P.; Kasiri, S.; Mandal, S.S. Nuclease Activity via Self-Activation and Anticancer Activity of a Mononuclear Copper(II) Complex: Novel Role of the Tertiary Butyl Group in the Ligand Frame. Inorg. Chem. 2012, 51, 3343–3345. [Google Scholar] [CrossRef] [PubMed]
- Cuzan, O.; Kochem, A.; Simaan, A.J.; Bertaina, S.; Faure, B.; Robert, V.; Shova, S.; Giorgi, M.; Maffei, M.; Réglier, M.; et al. Oxidative DNA Cleavage Promoted by a Phenoxyl-Radical Copper(II) Complex. Eur. J. Inorg. Chem. 2016, 2016, 5575–5584. [Google Scholar] [CrossRef]
- Gentil, S.; Serre, D.; Philouze, C.; Holzinger, M.; Thomas, F.; Le Goff, A. Electrocatalytic O2 Reduction at a Bio-inspired Mononuclear Copper Phenolato Complex Immobilized on a Carbon Nanotube Electrode. Angew. Chem. Int. Ed. 2016, 55, 2517–2520. [Google Scholar] [CrossRef]
- Serre, D.; Erbek, S.; Berthet, N.; Ronot, X.; Martel-Frachet, V.; Thomas, F. Copper(II) complexes of N3O tripodal ligands appended with pyrene and polyamine groups: Anti-proliferative and nuclease activities. J. Inorg. Biochem. 2018, 179, 121–134. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2[prime or minute]-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Vaidyanathan, M.; Viswanathan, R.; Palaniandavar, M.; Balasubramanian, T.; Prabhaharan, P.; Muthiah, P. Copper(II) Complexes with Unusual Axial Phenolate Coordination as Structural Models for the Active Site in Galactose Oxidase: X-ray Crystal Structures and Spectral and Redox Properties of [Cu(bpnp)X] Complexes. Inorg. Chem. 1998, 37, 6418–6427. [Google Scholar] [CrossRef]
- Michel, F.; Thomas, F.; Hamman, S.; Philouze, C.; Saint-Aman, E.; Pierre, J.-L. Galactose Oxidase Models: Creation and Modification of Proton Transfer Coupled to Copper(II) Coordination Processes in Pro-Phenoxyl Ligands. Eur. J. Inorg. Chem. 2006, 2006, 3684–3696. [Google Scholar] [CrossRef]
- Colomban, C.; Philouze, C.; Molton, F.; Leconte, N.; Thomas, F. Copper(II) Complexes of N3O Ligands as Models for Galactose Oxidase: Effect of Variation of Steric Bulk of Coordinated Phenoxyl Moiety on the Radical Stability and Spectroscopy. Inorg. Chim. Acta 2017, 481, 129–142. [Google Scholar] [CrossRef]
- Michel, F.; Thomas, F.; Hamman, S.; Saint-Aman, E.; Bucher, C.; Pierre, J.-L. Galactose Oxidase Models: Solution Chemistry, and Phenoxyl Radical Generation Mediated by the Copper Status. Chem. Eur. J. 2004, 10, 4115–4125. [Google Scholar] [CrossRef]
- Michel, F.; Hamman, S.; Thomas, F.; Philouze, C.; Gautier-Luneau, I.; Pierre, J.-L. Galactose Oxidase models: 19F NMR as a powerful tool to study the solution chemistry of tripodal ligands in the presence of copper(ii). Chem. Commun. 2006, 4122–4124. [Google Scholar] [CrossRef]
- Michel, F.; Hamman, S.; Philouze, C.; Del Valle, C.P.; Saint-Aman, E.; Thomas, F. Galactose oxidase models: Insights from 19F NMR spectroscopy. Dalton Trans. 2009, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Iacomino, G.; Picariello, G.; D’Agostino, L. DNA and nuclear aggregates of polyamines. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Li, J.; Huang, G.; Yan, L.; Ma, J. Interacting mechanism of benzo(a)pyrene with free DNA in vitro. Int. J. Biol. Macromol. 2021, 167, 854–861. [Google Scholar] [CrossRef] [PubMed]
- McGhee, J.D.; von Hippel, P.H. Theoretical aspects of DNA-protein interactions: Co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974, 86, 469–489. [Google Scholar] [CrossRef]
- Hinton, D.M.; Bode, V.C. Ethidium binding affinity of circular lambda deoxyribonucleic acid determined fluorometrically. J. Biol. Chem. 1975, 250, 1061–1067. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS, Bruker–Siemens Area Detector Absorption and Other Correction; Version 2008/1; University of Göttingen: Göttingen, Germany, 2006. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–340. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Petersson, G.A.; Al-Laham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Casida, M.E. Recent Advances in Density Functional Methods; Chong, D.P., Ed.; World Scientific: Singapore, 1995. [Google Scholar]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilizaion of Ab Initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Kozlowski, A.; Charles, S.A.; Harris, J.M. Development of Pegylated Interferons for the Treatment of Chronic Hepatitis C. BioDrugs 2001, 15, 419–429. [Google Scholar] [CrossRef] [PubMed]
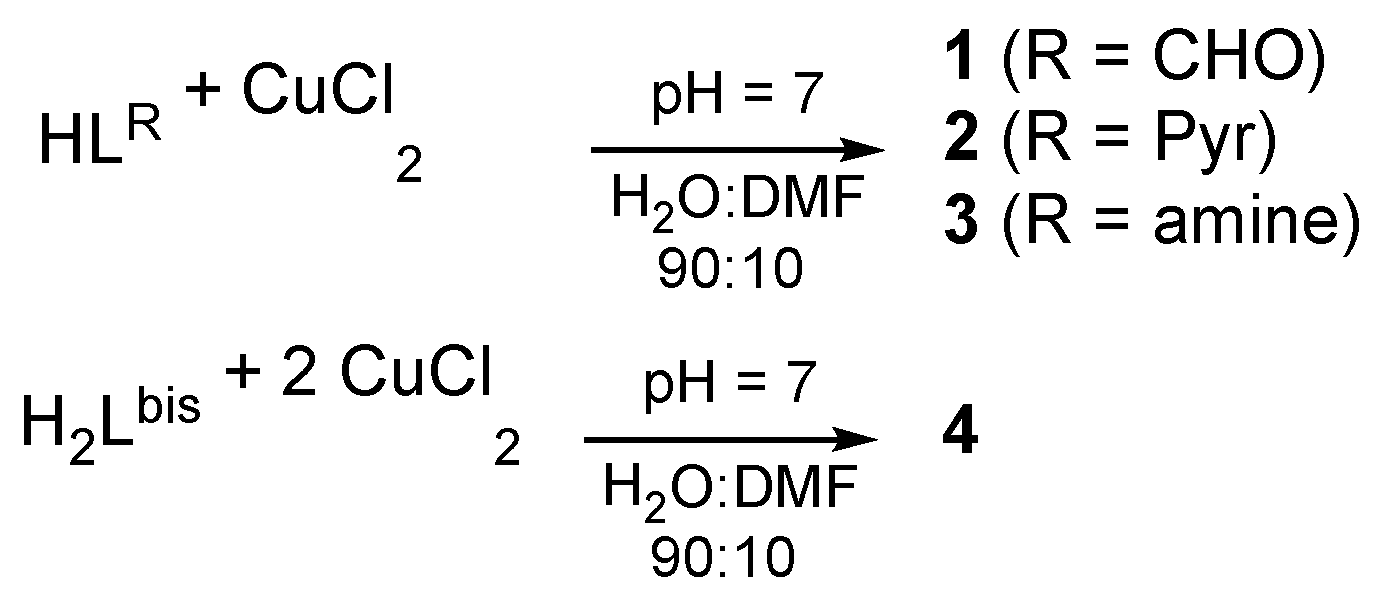







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | pH | g_|_ | g// | A_|_ | A// | g/A c |
|---|---|---|---|---|---|---|
| 1 | 1.80 a | 2.056 | 2.266 | 1.7 | 16.9 | 128 |
| 2 | 3.11 a | 2.056 | 2.266 | 1.7 | 16.9 | 128 |
| 3 | 3.25 a | 2.055 | 2.266 | 1.4 | 16.9 | 128 |
| 4 | 3.34 a | 2.055 | 2.266 | 1.4 | 16.9 | 128 |
| 1 | 3.80 a | 2.053 | 2.264 | 1.4 | 16.4 | 132 |
| 2 | 5.15 a | 2.053 | 2.261 | 1.4 | 15.9 | 136 |
| 3 | 5.26 a | 2.053 | 2.261 | 1.4 | 15.9 | 136 |
| 4 | 5.35 a | 2.054 | 2.262 | 1.4 | 16.0 | 135 |
| 1 | 7 b | 2.056 | 2.257 | 1.4 | 16.3 | 132 |
| 2 | 7 b | 2.054 | 2.254 | 1.5 | 16.6 | 130 |
| 3 | 7 b | 2.054 | 2.254 | 1.5 | 16.6 | 130 |
| 4 | 7 b | 2.054 | 2.254 | 1.5 | 16.6 | 130 |
| Phenolate Form | Phenol Form c | ||||||
|---|---|---|---|---|---|---|---|
| Charge Transfer | d-d Band | d-d Band | |||||
| Complex | pKa | λmax | ε | λmax | ε | λmax | ε |
| 1 | 2.82 ± 0.02 | 482 | 662 b | 680 | 170 | 680 c | <90 c |
| 2 | 4.15 ± 0.01 | 462 | 1040 | 670 | 101 | 680 | 66 |
| 3 | 4.25 ± 0.01 | 461 | 780 | 670 | 232 | 687 | 147 |
| 4 | 8.77 ± 0.03 d | 457 | 2005 | 670 | 328 | 687 | 147 |
 2H+ + [(Lbis)Cu2]2+.
2H+ + [(Lbis)Cu2]2+.| Complex | Ep c,red | Ep a,red | Ep a,ox |
|---|---|---|---|
| 1 | −0.45 | −0.14 | 0.96 |
| 2 | −0.51 | +0.16 | 0.73, 0.88 |
| 3 | −0.45 | - b | 0.69, 0.9 b |
| 4 | −0.47 | +0.07 | 0.56, 0.73, 0.88 |
| PyrPt c | - | - | 0.86, 1.0 b |
| Protonation State | Exogenous Ligand | Main Contribution to the Electronic Excitation | λcald [nm] (f) |
|---|---|---|---|
| Phenolate | chloride | βHOMO → βLUMO (LMCT) | 510 (0.0376) |
| Phenolate | water | βHOMO → βLUMO (LMCT) | 563 (0.0646) |
| Phenol | chloride | βHOMO-7 → βLUMO (LMCT) | 855 (0.0031) |
| Phenol | water | βHOMO-5 → βLUMO (dd) | 661 (0.0012) |
| Complex | No Exogenous Agent | Mercaptan | Ascorbic Acid |
|---|---|---|---|
| 1 | >50 | 40 | 30 |
| 2 | - b | 13 | 7 |
| 3 | >40 | 22.5 | 16 |
| 4 | - b | - b | >18 |
| Complex | IC50 (µM) | RF | |
|---|---|---|---|
| RT112 | RT112-CP | ||
| Cisplatin | 9.1 ± 1.2 | 23.8 ± 1.2 | 2.6 |
| 1 | 10.9 ± 0.2 | 13.2 ± 0.2 | 1.2 |
| 2 | 2.0 ± 0.2 | 1.2 ± 0.1 | 0.6 |
| 3 | 10.8 ± 0.2 | 10.5 ± 0.9 | 1.0 |
| 4 | 1.6 ± 0.2 | 1.0 ± 0.1 | 0.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serre, D.; Erbek, S.; Berthet, N.; Philouze, C.; Ronot, X.; Martel-Frachet, V.; Thomas, F. Anti-Proliferation and DNA Cleavage Activities of Copper(II) Complexes of N3O Tripodal Polyamine Ligands. Inorganics 2023, 11, 396. https://doi.org/10.3390/inorganics11100396
Serre D, Erbek S, Berthet N, Philouze C, Ronot X, Martel-Frachet V, Thomas F. Anti-Proliferation and DNA Cleavage Activities of Copper(II) Complexes of N3O Tripodal Polyamine Ligands. Inorganics. 2023; 11(10):396. https://doi.org/10.3390/inorganics11100396
Chicago/Turabian StyleSerre, Doti, Sule Erbek, Nathalie Berthet, Christian Philouze, Xavier Ronot, Véronique Martel-Frachet, and Fabrice Thomas. 2023. "Anti-Proliferation and DNA Cleavage Activities of Copper(II) Complexes of N3O Tripodal Polyamine Ligands" Inorganics 11, no. 10: 396. https://doi.org/10.3390/inorganics11100396