
The Cryptic Nature of Fe-S Clusters: A Case Study of the Hepatitis B HBx Oncoprotein




Abstract
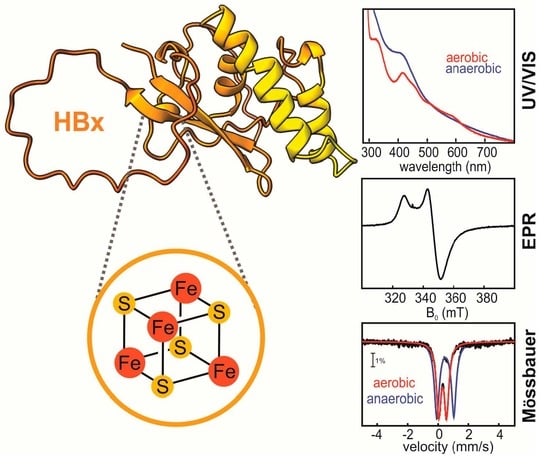
:
1. Introduction
2. Methods for Fingerprinting Fe-S Clusters
2.1. UV/VIS Spectroscopy
2.1.1. [2Fe-2S] Clusters
2.1.2. [3Fe-4S] Clusters
2.1.3. [4Fe-4S] Clusters
2.2. Electron Paramagnetic Resonance (EPR) Spectroscopy
2.2.1. [2Fe-2S] Clusters
2.2.2. [3Fe-4S] Clusters
2.2.3. [4Fe-4S] Clusters
2.3. Mössbauer Spectroscopy
2.3.1. [2Fe-2S] Clusters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Formal Valence | δ (mm/s) | |ΔEQav| (mm/s) |
|---|---|---|---|
| [2Fe-2S]2+ | |||
| Rieske (CCHH) [38] | Fe3+ Fe3+ | 0.24 0.32 | 0.52 0.91 |
| mitoNEET (CCCH) | Fe3+ Fe3+ | 0.26 0.30 | 0.47 0.96 |
| IscR (CCCH) [35] | Fe3+ Fe3+ | 0.27 0.30 | 0.48 0.72 |
| Grx3 (CC(GSH)2) [52] | Fe3+ Fe2+ | 0.29 0.29 | 0.55 0.76 |
| Grx-Fra2 (CHXGSH) [52] | Fe3+ Fe2+ | 0.30 0.32 | 0.50 0.82 |
| IscU (CCCD) [90] | Fe3+ Fe3+ | 0.27 0.32 | 0.66 0.94 |
| Apd1/Aim32 (CCHH) [36] | Fe3+ Fe3+ | 0.24 0.35 | 0.54 1.06 |
| RsrR (CCHE) [60,61] | Fe3+ Fe3+ | 0.28 0.29 | 0.54 0.76 |
| Biotin synthase (CCCR) [57,58] | Fe3+ Fe3+ | 0.29 0.29 | 0.51 0.51 |
| [2Fe-2S]1+ | |||
| Rieske (CCHH) [38] | Fe3+ Fe2+ | 0.31 0.74 | 0.63 3.05 |
| mitoNEET (CCCH) [34] | Fe3+ Fe2+ | 0.32 0.68 | 1.07 3.15 |
| IscR CCCH [35] | Fe3+ Fe2+ | 0.33 0.70 | 1.09 3.40 |
| Apd1/Aim32 (CCHH) [36] | Fe3+ Fe2+ | 0.32 0.75 | 0.81 3.16 |
2.3.2. [3Fe-4S] Clusters
2.3.3. [4Fe-4S] Clusters
2.4. Supplementary or Alternative Approaches
2.4.1. Chemoproteomics
2.4.2. Pulse EPR
2.4.3. NMR Spectroscopy
3. Identification of a Fe-S Cluster in the Hepatitis Virus HBx Oncoprotein
3.1. The Hepatitis Virus HBx
3.2. Spectroscopic Identification of the Cofactor
3.3. Whole-Cell Mössbauer Studies to Establish the Physiological Fe-S Cluster Form in HBx
3.4. Redox Transformations of the HBx Fe-S Cluster
3.5. HBx Preferentially Binds a Fe-S Cluster and Not Zn
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beinert, H.; Holm, R.H.; Münck, E. Iron-sulfur clusters: Nature’s modular, multipurpose structures. Science 1997, 277, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 2005, 74, 247–281. [Google Scholar] [CrossRef] [PubMed]
- Fontecave, M. Iron-sulfur clusters: Ever-expanding roles. Nat. Chem. Biol. 2006, 2, 171–174. [Google Scholar] [CrossRef]
- Vernis, L.; El Banna, N.; Baïlle, D.; Hatem, E.; Heneman, A.; Huang, M.E. Fe-S Clusters Emerging as Targets of Therapeutic Drugs. Oxid. Med. Cell. Longev. 2017, 2017, 3647657. [Google Scholar] [CrossRef] [PubMed]
- Honarmand Ebrahimi, K.; Ciofi-Baffoni, S.; Hagedoorn, P.L.; Nicolet, Y.; Le Brun, N.E.; Hagen, W.R.; Armstrong, F.A. Iron–sulfur clusters as inhibitors and catalysts of viral replication. Nat. Chem. 2022, 14, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, E.; Bes, M.T.; González, A.; Peleato, M.L.; Fillat, M.F. Redox-Based Transcriptional Regulation in Prokaryotes: Revisiting Model Mechanisms. Antioxid. Redox Signal. 2019, 30, 1651–1696. [Google Scholar] [CrossRef]
- Fuss, J.O.; Tsai, C.L.; Ishida, J.P.; Tainer, J.A. Emerging critical roles of Fe-S clusters in DNA replication and repair. Biochim. Biophys. Acta 2015, 1853, 1253–1271. [Google Scholar] [CrossRef]
- Crack, J.C.; Green, J.; Thomson, A.J.; Brun, N.E.L. Iron–Sulfur Clusters as Biological Sensors: The Chemistry of Reactions with Molecular Oxygen and Nitric Oxide. Acc. Chem. Res. 2014, 47, 3196–3205. [Google Scholar] [CrossRef]
- Schwartz, C.J.; Giel, J.L.; Patschkowski, T.; Luther, C.; Ruzicka, F.J.; Beinert, H.; Kiley, P.J. IscR, an Fe-S cluster-containing transcription factor, represses expression of Escherichia coli genes encoding Fe-S cluster assembly proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 14895–14900. [Google Scholar] [CrossRef] [PubMed]
- Tse, E.C.M.; Zwang, T.J.; Barton, J.K. The Oxidation State of [4Fe-4S] Clusters Modulates the DNA-Binding Affinity of DNA Repair Proteins. J. Am. Chem. Soc. 2017, 139, 12784–12792. [Google Scholar] [CrossRef]
- Bak, D.W.; Elliott, S.J. Alternative FeS cluster ligands: Tuning redox potentials and chemistry. Curr. Opin. Chem. Biol. 2014, 19, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Beinert, H. Iron-sulfur proteins: Ancient structures, still full of surprises. J. Biol. Inorg. Chem. 2000, 5, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A. Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol. 2006, 59, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Raulfs, E.C.; O’Carroll, I.P.; Dos Santos, P.C.; Unciuleac, M.-C.; Dean, D.R. In vivo iron-sulfur cluster formation. Proc. Natl. Acad. Sci. USA 2008, 105, 8591–8596. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Cash, V.L.; Flint, D.H.; Dean, D.R. Assembly of Iron-Sulfur Clusters: Identification of an iscsua-hscba-fdx gene cluster from Azotobacter vinelandii. J. Biol. Chem. 1998, 273, 13264–13272. [Google Scholar] [CrossRef] [PubMed]
- Outten, F.W.; Wood, M.J.; Munoz, F.M.; Storz, G. The SufE protein and the SufBCD complex enhance SufS cysteine desulfurase activity as part of a sulfur transfer pathway for Fe-S cluster assembly in Escherichia coli. J. Biol. Chem. 2003, 278, 45713–45719. [Google Scholar] [CrossRef] [PubMed]
- Wehrspan, Z.J.; McDonnell, R.T.; Elcock, A.H. Identification of Iron-Sulfur (Fe-S) Cluster and Zinc (Zn) Binding Sites within Proteomes Predicted by DeepMind’s AlphaFold2 Program Dramatically Expands the Metalloproteome. J. Mol. Biol. 2022, 434, 167377. [Google Scholar] [CrossRef]
- Pritts, J.D.; Michel, S.L.J. Fe-S clusters masquerading as zinc finger proteins. J. Inorg. Biochem. 2022, 230, 111756. [Google Scholar] [CrossRef]
- Lewis, B.E.; Mason, Z.; Rodrigues, A.V.; Nuth, M.; Dizin, E.; Cowan, J.A.; Stemmler, T.L. Unique roles of iron and zinc binding to the yeast Fe–S cluster scaffold assembly protein “Isu1”. Metallomics 2019, 11, 1820–1835. [Google Scholar] [CrossRef]
- Chen, J.; Calderone, L.A.; Pan, L.; Quist, T.; Pandelia, M.-E. The Fe and Zn cofactor dilemma. Biochim. Biophys. Acta—Proteins Proteom. 2023, 1871, 140931. [Google Scholar] [CrossRef]
- Maio, N.; Raza, M.K.; Li, Y.; Zhang, D.L.; Bollinger, J.M., Jr.; Krebs, C.; Rouault, T.A. An iron-sulfur cluster in the zinc-binding domain of the SARS-CoV-2 helicase modulates its RNA-binding and -unwinding activities. Proc. Natl. Acad. Sci. USA 2023, 120, e2303860120. [Google Scholar] [CrossRef]
- Maio, N.; Lafont, B.A.P.; Sil, D.; Li, Y.; Bollinger, J.M.; Krebs, C.; Pierson, T.C.; Linehan, W.M.; Rouault, T.A. Fe-S cofactors in the SARS-CoV-2 RNA-dependent RNA polymerase are potential antiviral targets. Science 2021, 373, 236–241. [Google Scholar] [CrossRef]
- Ueda, C.; Langton, M.; Chen, J.; Pandelia, M.E. The HBx protein from hepatitis B virus coordinates a redox-active Fe-S cluster. J. Biol. Chem. 2022, 298, 101698. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Wang, R.; Nakamaru-Ogiso, E.; Knight, S.A.; Buck, C.B.; You, J. The Oncogenic Small Tumor Antigen of Merkel Cell Polyomavirus Is an Iron-Sulfur Cluster Protein That Enhances Viral DNA Replication. J. Virol. 2016, 90, 1544–1556. [Google Scholar] [CrossRef] [PubMed]
- Villalta, A.; Srour, B.; Lartigue, A.; Clémancey, M.; Byrne, D.; Chaspoul, F.; Loquet, A.; Guigliarelli, B.; Blondin, G.; Abergel, C. Evidence for [2Fe-2S]2+ and Linear [3Fe-4S]1+ Clusters in a Unique Family of Glycine/Cysteine-Rich Fe-S Proteins from Megavirinae Giant Viruses. J. Am. Chem. Soc. 2023, 145, 2733–2738. [Google Scholar] [CrossRef]
- Maio, N.; Singh, A.; Uhrigshardt, H.; Saxena, N.; Tong, W.H.; Rouault, T.A. Cochaperone binding to LYR motifs confers specificity of iron sulfur cluster delivery. Cell Metab. 2014, 19, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Lippard, S.J.; Berg, J.M. Principles of Bioinorganic Chemistry; University Science Books: Herndon, VA, USA, 1994. [Google Scholar]
- Betinol, I.O.; Nader, S.; Mansy, S.S. Spectral decomposition of iron-sulfur clusters. Anal. Biochem. 2021, 629, 114269. [Google Scholar] [CrossRef]
- Nasta, V.; Giachetti, A.; Ciofi-Baffoni, S.; Banci, L. Structural insights into the molecular function of human [2Fe-2S] BOLA1-GRX5 and [2Fe-2S] BOLA3-GRX5 complexes. Biochim. Biophys. Acta 2017, 1861, 2119–2131. [Google Scholar] [CrossRef]
- Gibney, B.R.; Mulholland, S.E.; Rabanal, F.; Dutton, P.L. Ferredoxin and ferredoxin–heme maquettes. Proc. Natl. Acad. Sci. USA 1996, 93, 15041–15046. [Google Scholar] [CrossRef]
- Dailey, H.A.; Finnegan, M.G.; Johnson, M.K. Human ferrochelatase is an iron-sulfur protein. Biochemistry 1994, 33, 403–407. [Google Scholar] [CrossRef]
- Liu, J.; Chakraborty, S.; Hosseinzadeh, P.; Yu, Y.; Tian, S.; Petrik, I.; Bhagi, A.; Lu, Y. Metalloproteins Containing Cytochrome, Iron–Sulfur, or Copper Redox Centers. Chem. Rev. 2014, 114, 4366–4469. [Google Scholar] [CrossRef] [PubMed]
- Valer, L.; Rossetto, D.; Scintilla, S.; Hu, Y.J.; Tomar, A.; Nader, S.; Betinol, I.O.; Mansy, S.S. Methods to identify and characterize iron–sulfur oligopeptides in water. Can. J. Chem. 2022, 100, 475–483. [Google Scholar] [CrossRef]
- Camponeschi, F.; Piccioli, M.; Banci, L. The Intriguing mitoNEET: Functional and Spectroscopic Properties of a Unique [2Fe-2S] Cluster Coordination Geometry. Molecules 2022, 27, 8218. [Google Scholar] [CrossRef] [PubMed]
- Fleischhacker, A.S.; Stubna, A.; Hsueh, K.-L.; Guo, Y.; Teter, S.J.; Rose, J.C.; Brunold, T.C.; Markley, J.L.; Münck, E.; Kiley, P.J. Characterization of the [2Fe-2S] Cluster of Escherichia coli Transcription Factor IscR. Biochemistry 2012, 51, 4453–4462. [Google Scholar] [CrossRef] [PubMed]
- Stegmaier, K.; Blinn, C.M.; Bechtel, D.F.; Greth, C.; Auerbach, H.; Müller, C.S.; Jakob, V.; Reijerse, E.J.; Netz, D.J.A.; Schünemann, V.; et al. Apd1 and Aim32 Are Prototypes of Bishistidinyl-Coordinated Non-Rieske [2Fe–2S] Proteins. J. Am. Chem. Soc. 2019, 141, 5753–5765. [Google Scholar] [CrossRef] [PubMed]
- Cline, J.F.; Hoffman, B.M.; Mims, W.; LaHaie, E.; Ballou, D.; Fee, J. Evidence for N coordination to Fe in the [2Fe-2S] clusters of Thermus Rieske protein and phthalate dioxygenase from Pseudomonas. J. Biol. Chem. 1985, 260, 3251–3254. [Google Scholar] [CrossRef] [PubMed]
- Fee, J.A.; Findling, K.L.; Yoshida, T.; Hille, R.; Tarr, G.E.; Hearshen, D.O.; Dunham, W.R.; Day, E.P.; Kent, T.A.; Münck, E. Purification and characterization of the Rieske iron-sulfur protein from Thermus thermophilus. Evidence for a [2Fe-2S] cluster having non-cysteine ligands. J. Biol. Chem. 1984, 259, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Emptage, M.H.; Dreyers, J.L.; Kennedy, M.C.; Beinert, H. Optical and EPR characterization of different species of active and inactive aconitase. J. Biol. Chem. 1983, 258, 11106–11111. [Google Scholar] [CrossRef]
- Broderick, J.B.; Duffus, B.R.; Duschene, K.S.; Shepard, E.M. Radical S-Adenosylmethionine Enzymes. Chem. Rev. 2014, 114, 4229–4317. [Google Scholar] [CrossRef]
- Sweeney, W.; Rabinowitz, J.; Yoch, D. High and low reduction potential 4Fe-4S clusters in Azotobacter vinelandii (4Fe-4S) 2ferredoxin I. Influence of the polypeptide on the reduction potentials. J. Biol. Chem. 1975, 250, 7842–7847. [Google Scholar] [CrossRef]
- Guigliarelli, B.; Bertrand, P. Application of EPR Spectroscopy to the Structural and Functional Study of Iron-Sulfur Proteins. In Advances in Inorganic Chemistry; Sykes, A.G., Ed.; Academic Press: Cambridge, MA, USA, 1999; pp. 421–497. [Google Scholar]
- Hagen, W.R. EPR spectroscopy of complex biological iron-sulfur systems. J. Biol. Inorg. Chem. 2018, 23, 623–634. [Google Scholar] [CrossRef]
- Beinert, H.; Sands, R.H. Studies on succinic and DPNH dehydrogenase preparations by paramagnetic resonance (EPR) spectroscopy. Biochem. Biophys. Res. Commun. 1960, 3, 41–46. [Google Scholar] [CrossRef]
- Gibson, J.; Hall, D.; Thornley, J.; Whatley, F. The iron complex in spinach ferredoxin. Proc. Natl. Acad. Sci. USA 1966, 56, 987–990. [Google Scholar] [CrossRef]
- Rupp, H.; Rao, K.K.; Hall, D.O.; Cammack, R. Electron spin relaxation of iron-sulphur proteins studied by microwave power saturation. Biochim. Biophys. Acta Protein Struct. 1978, 537, 255–269. [Google Scholar] [CrossRef]
- Cavazza, C.; Guigliarelli, B.; Bertrand, P.; Bruschi, M. Biochemical and EPR characterization of a high potential iron-sulfur protein in Thiobacillus ferrooxidans. FEMS Microbiol. Lett. 1995, 130, 193–199. [Google Scholar] [CrossRef]
- Ta, D.T.; Vickery, L.E. Cloning, sequencing, and overexpression of a [2Fe-2S] ferredoxin gene from Escherichia coli. J. Biol. Chem. 1992, 267, 11120–11125. [Google Scholar] [CrossRef]
- Meyer, J.; Clay, M.D.; Johnson, M.K.; Stubna, A.; Münck, E.; Higgins, C.; Wittung-Stafshede, P. A hyperthermophilic plant-type [2Fe-2S] ferredoxin from Aquifex aeolicus is stabilized by a disulfide bond. Biochemistry 2002, 41, 3096–3108. [Google Scholar] [CrossRef] [PubMed]
- Birrell, J.A.; Laurich, C.; Reijerse, E.J.; Ogata, H.; Lubitz, W. Importance of Hydrogen Bonding in Fine Tuning the [2Fe-2S] Cluster Redox Potential of HydC from Thermotoga maritima. Biochemistry 2016, 55, 4344–4355. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Mapolelo, D.T.; Randeniya, S.; Johnson, M.K.; Outten, C.E. Human glutaredoxin 3 forms [2Fe-2S]-bridged complexes with human BolA2. Biochemistry 2012, 51, 1687–1696. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Mapolelo, D.T.; Dingra, N.N.; Naik, S.G.; Lees, N.S.; Hoffman, B.M.; Riggs-Gelasco, P.J.; Huynh, B.H.; Johnson, M.K.; Outten, C.E. The Yeast Iron Regulatory Proteins Grx3/4 and Fra2 Form Heterodimeric Complexes Containing a [2Fe-2S] Cluster with Cysteinyl and Histidyl Ligation. Biochemistry 2009, 48, 9569–9581. [Google Scholar] [CrossRef]
- Dlouhy, A.C.; Li, H.; Albetel, A.-N.; Zhang, B.; Mapolelo, D.T.; Randeniya, S.; Holland, A.A.; Johnson, M.K.; Outten, C.E. The Escherichia coli BolA Protein IbaG Forms a Histidine-Ligated [2Fe-2S]-Bridged Complex with Grx4. Biochemistry 2016, 55, 6869–6879. [Google Scholar] [CrossRef]
- Dicus, M.M.; Conlan, A.; Nechushtai, R.; Jennings, P.A.; Paddock, M.L.; Britt, R.D.; Stoll, S. Binding of histidine in the (Cys) 3 (His) 1-coordinated [2Fe−2S] cluster of human mitoNEET. J. Am. Chem. Soc. 2010, 132, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Landry, A.P.; Wang, Y.; Ding, H. Binding of Nitric Oxide in CDGSH-type [2Fe-2S] Clusters of the Human Mitochondrial Protein Miner2. J. Biol. Chem. 2017, 292, 3146–3153. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Mapolelo, D.T.; Dingra, N.N.; Keller, G.; Riggs-Gelasco, P.J.; Winge, D.R.; Johnson, M.K.; Outten, C.E. Histidine 103 in Fra2 Is an Iron-Sulfur Cluster Ligand in the [2Fe-2S] Fra2-Grx3 Complex and Is Required for In Vivo Iron Signaling in Yeast*. J. Biol. Chem. 2011, 286, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Stoll, S.; Britt, R.D.; Jarrett, J.T. Reduction of the [2Fe-2S] cluster accompanies formation of the intermediate 9-mercaptodethiobiotin in Escherichia coli biotin synthase. Biochemistry 2011, 50, 7953–7963. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, I.; Cohen, G.; Flint, D.H. Biotin Synthase: Purification, Characterization as a [2Fe-2S] Cluster Protein, and in vitro Activity of the Escherichia coli bioB Gene Product. Biochemistry 1994, 33, 3625–3631. [Google Scholar] [CrossRef] [PubMed]
- Roret, T.; Tsan, P.; Couturier, J.; Zhang, B.; Johnson, M.K.; Rouhier, N.; Didierjean, C. Structural and spectroscopic insights into BolA-glutaredoxin complexes. J. Biol. Chem. 2014, 289, 24588–24598. [Google Scholar] [CrossRef]
- Volbeda, A.; Martinez, M.T.P.; Crack, J.C.; Amara, P.; Gigarel, O.; Munnoch, J.T.; Hutchings, M.I.; Darnault, C.; Le Brun, N.E.; Fontecilla-Camps, J.C. Crystal Structure of the Transcription Regulator RsrR Reveals a [2Fe–2S] Cluster Coordinated by Cys, Glu, and His Residues. J. Am. Chem. Soc. 2019, 141, 2367–2375. [Google Scholar] [CrossRef]
- Munnoch, J.T.; Martinez, M.T.; Svistunenko, D.A.; Crack, J.C.; Le Brun, N.E.; Hutchings, M.I. Characterization of a putative NsrR homologue in Streptomyces venezuelae reveals a new member of the Rrf2 superfamily. Sci. Rep. 2016, 6, 31597. [Google Scholar] [CrossRef]
- Rousset, M.; Montet, Y.; Guigliarelli, B.; Forget, N.; Asso, M.; Bertrand, P.; Fontecilla-Camps, J.C.; Hatchikian, E.C. [3Fe-4S] to [4Fe-4S] cluster conversion in Desulfovibrio fructosovorans [NiFe] hydrogenase by site-directed mutagenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 11625–11630. [Google Scholar] [CrossRef]
- Priem, A.H.; Klaassen, A.A.K.; Reijerse, E.J.; Meyer, T.E.; Luchinat, C.; Capozzi, F.; Dunham, W.R.; Hagen, W.R. EPR analysis of multiple forms of [4Fe–4S]3+ clusters in HiPIPs. J. Biol. Inorg. Chem. 2005, 10, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Heering, H.A.; Bulsink, Y.B.; Hagen, W.R.; Meyer, T.E. Reversible Super-Reduction of the Cubane [4Fe-4S](3+; 2+; 1+) in the High-Potential Iron-Sulfur Protein Under Non-Denaturing Conditions: EPR Spectroscopic and Electrochemical Studies. Eur. J. Biochem. 1995, 232, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Nakamaru-Ogiso, E.; Yano, T.; Yagi, T.; Ohnishi, T. Characterization of the iron-sulfur cluster N7 (N1c) in the subunit NuoG of the proton-translocating NADH-quinone oxidoreductase from Escherichia coli. J. Biol. Chem. 2005, 280, 301–307. [Google Scholar] [CrossRef]
- Rothery, R.A.; Weiner, J.H. Alteration of the iron-sulfur cluster composition of Escherichia coli dimethyl sulfoxide reductase by site-directed mutagenesis. Biochemistry 1991, 30, 8296–8305. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Heinnickel, M.; Krebs, C.; Shen, G.; Golbeck, J.; Bryant, D. Biogenesis of Iron-Sulfur Clusters in Photosystem I. J. Biol. Chem. 2008, 283, 28426–28435. [Google Scholar] [CrossRef]
- Lubner, C.E.; Artz, J.H.; Mulder, D.W.; Oza, A.; Ward, R.J.; Williams, S.G.; Jones, A.K.; Peters, J.W.; Smalyukh, I.I.; Bharadwaj, V.S. A site-differentiated [4Fe–4S] cluster controls electron transfer reactivity of Clostridium acetobutylicum [FeFe]-hydrogenase I. Chem. Sci. 2022, 13, 4581–4588. [Google Scholar] [CrossRef]
- Berto, P.; Di Valentin, M.; Cendron, L.; Vallese, F.; Albertini, M.; Salvadori, E.; Giacometti, G.M.; Carbonera, D.; Costantini, P. The [4Fe-4S]-cluster coordination of [FeFe]-hydrogenase maturation protein HydF as revealed by EPR and HYSCORE spectroscopies. Biochim. Biophys. Acta 2012, 1817, 2149–2157. [Google Scholar] [CrossRef]
- Reents, H.; Gruner, I.; Harmening, U.; Bottger, L.H.; Layer, G.; Heathcote, P.; Trautwein, A.X.; Jahn, D.; Hartig, E. Bacillus subtilis Fnr senses oxygen via a [4Fe-4S] cluster coordinated by three cysteine residues without change in the oligomeric state. Mol. Microbiol. 2006, 60, 1432–1445. [Google Scholar] [CrossRef]
- Gruner, I.; Fradrich, C.; Bottger, L.H.; Trautwein, A.X.; Jahn, D.; Hartig, E. Aspartate 141 is the fourth ligand of the oxygen-sensing [4Fe-4S]2+ cluster of Bacillus subtilis transcriptional regulator Fnr. J. Biol. Chem. 2011, 286, 2017–2021. [Google Scholar] [CrossRef]
- Yukl, E.T.; Elbaz, M.A.; Nakano, M.M.; Moënne-Loccoz, P. Transcription Factor NsrR from Bacillus subtilis Senses Nitric Oxide with a 4Fe-4S Cluster (†). Biochemistry 2008, 47, 13084–13092. [Google Scholar] [CrossRef]
- Conover, R.C.; Kowal, A.T.; Fu, W.G.; Park, J.B.; Aono, S.; Adams, M.W.; Johnson, M.K. Spectroscopic characterization of the novel iron-sulfur cluster in Pyrococcus furiosus ferredoxin. J. Biol. Chem. 1990, 265, 8533–8541. [Google Scholar] [CrossRef]
- Cicchillo, R.M.; Lee, K.-H.; Baleanu-Gogonea, C.; Nesbitt, N.M.; Krebs, C.; Booker, S.J. Escherichia coli Lipoyl Synthase Binds Two Distinct [4Fe−4S] Clusters per Polypeptide. Biochemistry 2004, 43, 11770–11781. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Saleh, L.; Anton, B.P.; Madinger, C.L.; Benner, J.S.; Iwig, D.F.; Roberts, R.J.; Krebs, C.; Booker, S.J. Characterization of RimO, a new member of the methylthiotransferase subclass of the radical SAM superfamily. Biochemistry 2009, 48, 10162–10174. [Google Scholar] [CrossRef] [PubMed]
- Pandelia, M.E.; Lanz, N.D.; Booker, S.J.; Krebs, C. Mössbauer spectroscopy of Fe/S proteins. Biochim. Biophys. Acta 2015, 1853, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, P.; Guigliarelli, B.; Gayda, J.-P.; Peter, B.; Gibson, J.F. A ligand-field model to describe a new class of 2Fe-2S clusters in proteins and their synthetic analogues. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1985, 831, 261–266. [Google Scholar] [CrossRef]
- Conlan, A.R.; Axelrod, H.L.; Cohen, A.E.; Abresch, E.C.; Zuris, J.; Yee, D.; Nechushtai, R.; Jennings, P.A.; Paddock, M.L. Crystal Structure of Miner1: The Redox-active 2Fe-2S Protein Causative in Wolfram Syndrome 2. J. Mol. Biol. 2009, 392, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Vinyard, D.J.; Reesbeck, M.E.; Suzuki, T.; Manakongtreecheep, K.; Holland, P.L.; Brudvig, G.W.; Söll, D. A [3Fe-4S] cluster is required for tRNA thiolation in archaea and eukaryotes. Proc. Natl. Acad. Sci. USA 2016, 113, 12703–12708. [Google Scholar] [CrossRef]
- Kent, T.A.; Emptage, M.H.; Merkle, H.; Kennedy, M.C.; Beinert, H.; Münck, E. Mössbauer studies of aconitase. Substrate and inhibitor binding, reaction intermediates, and hyperfine interactions of reduced 3Fe and 4Fe clusters. J. Biol. Chem. 1985, 260, 6871–6881. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.; Gomes, C.M.; Huber, H.; Teixeira, M.; Wittung-Stafshede, P. Formation of a linear [3Fe-4S] cluster in a seven-iron ferredoxin triggered by polypeptide unfolding. JBIC J. Biol. Inorg. Chem. 2002, 7, 357–362. [Google Scholar] [CrossRef]
- Kennedy, M.C.; Kent, T.; Emptage, M.; Merkle, H.; Beinert, H.; Münck, E. Evidence for the formation of a linear [3Fe-4S] cluster in partially unfolded aconitase. J. Biol. Chem. 1984, 259, 14463–14471. [Google Scholar] [CrossRef]
- Flint, D.; Emptage, M.; Finnegan, M.; Fu, W.; Johnson, M. The role and properties of the iron-sulfur cluster in Escherichia coli dihydroxy-acid dehydratase. J. Biol. Chem. 1993, 268, 14732–14742. [Google Scholar] [CrossRef] [PubMed]
- Brereton, P.S.; Duderstadt, R.E.; Staples, C.R.; Johnson, M.K.; Adams, M.W.W. Effect of Serinate Ligation at Each of the Iron Sites of the [Fe4S4] Cluster of Pyrococcus furiosus Ferredoxin on the Redox, Spectroscopic, and Biological Properties. Biochemistry 1999, 38, 10594–10605. [Google Scholar] [CrossRef] [PubMed]
- Rothery, R.A.; Bertero, M.G.; Cammack, R.; Palak, M.; Blasco, F.; Strynadka, N.C.J.; Weiner, J.H. The Catalytic Subunit of Escherichia coli Nitrate Reductase A Contains a Novel [4Fe-4S] Cluster with a High-Spin Ground State. Biochemistry 2004, 43, 5324–5333. [Google Scholar] [CrossRef] [PubMed]
- Moss, T.H.; Bearden, A.J.; Bartsch, R.G.; Cusanovich, M.A.; San Pietro, A. Mössbauer spectroscopy of nonheme iron proteins. Biochemistry 1968, 7, 1591–1596. [Google Scholar] [CrossRef]
- Bill, E. Iron-sulfur clusters—New features in enzymes and synthetic models. Hyperfine Interact. 2012, 205, 139–147. [Google Scholar] [CrossRef]
- Münck, E.; Debrunner, P.G.; Tsibris, J.C.M.; Gunsalus, I.C. Mössbauer parameters of putidaredoxin and its selenium analog. Biochemistry 1972, 11, 855–863. [Google Scholar] [CrossRef]
- Banci, L.; Ciofi-Baffoni, S.; Mikolajczyk, M.; Winkelmann, J.; Bill, E.; Pandelia, M.-E. Human anamorsin binds [2Fe–2S] clusters with unique electronic properties. J. Biol. Inorg. Chem. 2013, 18, 883–893. [Google Scholar] [CrossRef]
- Agar, J.N.; Krebs, C.; Frazzon, J.; Huynh, B.H.; Dean, D.R.; Johnson, M.K. IscU as a scaffold for iron-sulfur cluster biosynthesis: Sequential assembly of [2Fe-2S] and [4Fe-4S] clusters in IscU. Biochemistry 2000, 39, 7856–7862. [Google Scholar] [CrossRef]
- Huynh, B.H.; Moura, J.J.; Moura, I.; Kent, T.A.; LeGall, J.; Xavier, A.V.; Münck, E. Evidence for a three-iron center in a ferredoxin from Desulfovibrio gigas. Mössbauer and EPR studies. J. Biol. Chem. 1980, 255, 3242–3244. [Google Scholar] [CrossRef]
- Hans, M.; Buckel, W.; Bill, E. Spectroscopic evidence for an all-ferrous [4Fe-4S]0 cluster in the superreduced activator of 2-hydroxyglutaryl-CoA dehydratase from Acidaminococcus fermentans. J. Biol. Inorg. Chem. 2008, 13, 563–574. [Google Scholar] [CrossRef]
- Crack, J.C.; Le Brun, N.E. Native Mass Spectrometry of Iron-Sulfur Proteins. Methods Mol. Biol. 2021, 2353, 231–258. [Google Scholar] [PubMed]
- Crack, J.C.; Gray, E.; Le Brun, N.E. Sensing mechanisms of iron–sulfur cluster regulatory proteins elucidated using native mass spectrometry. Dalton Trans. 2021, 50, 7887–7897. [Google Scholar] [CrossRef]
- Bak, D.W.; Weerapana, E. Monitoring Fe-S cluster occupancy across the E. coli proteome using chemoproteomics. Nat. Chem. Biol. 2023, 19, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Camponeschi, F.; Ciofi-Baffoni, S.; Piccioli, M. The NMR contribution to protein-protein networking in Fe-S protein maturation. J. Biol. Inorg. Chem. 2018, 23, 665–685. [Google Scholar] [CrossRef] [PubMed]
- Camponeschi, F.; Gallo, A.; Piccioli, M.; Banci, L. The long-standing relationship between paramagnetic NMR and iron–sulfur proteins: The mitoNEET example. An old method for new stories or the other way around? Magn. Reson. 2021, 2, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Trindade, I.B.; Coelho, A.; Cantini, F.; Piccioli, M.; Louro, R.O. NMR of paramagnetic metalloproteins in solution. J. Inorg. Biochem. 2022, 234, 111871. [Google Scholar] [CrossRef] [PubMed]
- Pochapsky, T.C.; Kostic, M.; Jain, N.; Pejchal, R. Redox-Dependent Conformational Selection in a Cys4Fe2S2 Ferredoxin. Biochemistry 2001, 40, 5602–5614. [Google Scholar] [CrossRef]
- Jain, N.U.; Pochapsky, T.C. Redox Dependence of Hyperfine-Shifted 13C and 15N Resonances in Putidaredoxin. J. Am. Chem. Soc. 1998, 120, 12984–12985. [Google Scholar] [CrossRef]
- Xu, X.; Kim, S.K.; Schürmann, P.; Hirasawa, M.; Tripathy, J.N.; Smith, J.; Knaff, D.B.; Ubbink, M. Ferredoxin/ferredoxin-thioredoxin reductase complex: Complete NMR mapping of the interaction site on ferredoxin by gallium substitution. FEBS Lett. 2006, 580, 6714–6720. [Google Scholar] [CrossRef]
- Ramelot, T.A.; Cort, J.R.; Goldsmith-Fischman, S.; Kornhaber, G.J.; Xiao, R.; Shastry, R.; Acton, T.B.; Honig, B.; Montelione, G.T.; Kennedy, M.A. Solution NMR structure of the iron-sulfur cluster assembly protein U (IscU) with zinc bound at the active site. J. Mol. Biol. 2004, 344, 567–583. [Google Scholar] [CrossRef]
- Tiollais, P.; Pourcel, C.; Dejean, A. The hepatitis B virus. Nature 1985, 317, 489–495. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Duan, L.X. Hepatitis B virus X antigen in the pathogenesis of chronic infections and the development of hepatocellular carcinoma. Am. J. Pathol. 1997, 150, 1141–1157. [Google Scholar] [PubMed]
- Nguyen, V.T.; Law, M.G.; Dore, G.J. Hepatitis B-related hepatocellular carcinoma: Epidemiological characteristics and disease burden. J. Viral Hepat. 2009, 16, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Arbuthnot, P.; Kew, M. Hepatitis B virus and hepatocellular carcinoma. Int. J. Exp. Pathol. 2001, 82, 77–100. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.J.; Schneider, R.J. The enigmatic X gene of hepatitis B virus. J. Virol. 2004, 78, 12725–12734. [Google Scholar] [CrossRef]
- Benhenda, S.; Cougot, D.; Buendia, M.A.; Neuveut, C. Hepatitis B virus X protein molecular functions and its role in virus life cycle and pathogenesis. Adv. Cancer Res. 2009, 103, 75–109. [Google Scholar]
- Rossner, M.T. Review: Hepatitis B virus X-gene product: A promiscuous transcriptional activator. J. Med. Virol. 1992, 36, 101–117. [Google Scholar] [CrossRef]
- Ueda, C. The Biophysical and Spectroscopic Characterization of Redox-Active Metallocofactors in a Viral Fe-S Protein and a Diiron HD-Domain Phosphohydrolase. Doctoral Dissertation, Brandeis University, Waltham, MA, USA, 2022. [Google Scholar]
- Matteucci, S.; Camponeschi, F.; Clémancey, M.; Ciofi-Baffoni, S.; Blondin, G.; Banci, L. In Cellulo Mössbauer and EPR Studies Bring New Evidence to the Long-Standing Debate on Iron-Sulfur Cluster Binding in Human Anamorsin. Angew. Chem. Int. Ed. Engl. 2021, 60, 14841–14845. [Google Scholar] [CrossRef]
- Beilschmidt, L.K.; Ollagnier de Choudens, S.; Fournier, M.; Sanakis, I.; Hograindleur, M.-A.; Clémancey, M.; Blondin, G.; Schmucker, S.; Eisenmann, A.; Weiss, A.; et al. ISCA1 is essential for mitochondrial Fe4S4 biogenesis in vivo. Nat. Commun. 2017, 8, 15124. [Google Scholar] [CrossRef]
- Yang, J.; Naik, S.G.; Ortillo, D.O.; García-Serres, R.; Li, M.; Broderick, W.E.; Huynh, B.H.; Broderick, J.B. The iron-sulfur cluster of pyruvate formate-lyase activating enzyme in whole cells: Cluster interconversion and a valence-localized [4Fe-4S]2+ state. Biochemistry 2009, 48, 9234–9241. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.P.; Crack, J.C.; Puglisi, R.; Pastore, A.; Le Brun, N.E. Native mass spectrometric studies of IscSU reveal a concerted, sulfur-initiated mechanism of iron–sulfur cluster assembly. Chem. Sci. 2023, 14, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Chandramouli, K.; Unciuleac, M.-C.; Naik, S.; Dean, D.R.; Huynh, B.H.; Johnson, M.K. Formation and Properties of [4Fe-4S] Clusters on the IscU Scaffold Protein. Biochemistry 2007, 46, 6804–6811. [Google Scholar] [CrossRef]
- Mapolelo, D.T.; Zhang, B.; Naik, S.G.; Huynh, B.H.; Johnson, M.K. Spectroscopic and functional characterization of iron-sulfur cluster-bound forms of Azotobacter vinelandii (Nif)IscA. Biochemistry 2012, 51, 8071–8084. [Google Scholar] [CrossRef]
- Ramakrishnan, D.; Xing, W.; Beran, R.K.; Chemuru, S.; Rohrs, H.; Niedziela-Majka, A.; Marchand, B.; Mehra, U.; Zábranský, A.; Doležal, M.; et al. Hepatitis B Virus X Protein Function Requires Zinc Binding. J. Virol. 2019, 93, e00250-19. [Google Scholar] [CrossRef] [PubMed]





| Coordination | Protein | Stot | (g1, g2, g3) a | gav | Temperature (K) a,b |
|---|---|---|---|---|---|
| [2Fe-2S]1+ | |||||
| CCCC | Mitochondrial FXD1 [48] | 1/2 | 2.03, 1.94, 1.94 | 1.97 | 4–100 |
| Vegetative Fd [49] | 1/2 | 2.05, 1.95, 1.88 | 1.96 | 4–100 | |
| HydC thioredoxin-like [50] | 1/2 | 2.01, 1.95, 1.92 | 1.96 | 40 | |
| CC(GSH)2 | Glrx3 [51] | 1/2 | 2.01, 1.97, 1.92 | 1.97 | 26 |
| Grx3 [52] | 1/2 | 2.03, 1.94, 1.94 | 1.97 | 26 | |
| Grx4 [53] | 1/2 | 2.03, 1.94, 1.94 | 1.97 | 20 | |
| CCCH | mitoNEET [54,55] | 1/2 | 2.01, 1.94, 1.90 | 1.95 | 5–60 |
| Miner1 [55] | 1/2 | 2.01, 1.94, 1.90 | 1.95 | 20 | |
| Miner2 [55] Miner2 [55] | 1/2 1/2 | 2.00, 1.92, 1.90 2.01, 1.94, 1.89 | 1.94 1.95 | 20 20 | |
| IscR [9,35] | 1/2 | 1.99, 1.93, 1.88 | 1.93 | 20 | |
| CC(GSH)H | Glrx3−BolA2 [51] | 1/2 | 2.01, 1.91, 1.88 | 1.93 | 26 |
| Grx3-Fra2 C [52,56] | 1/2 | 2.01, 1.92, 1.87 | 1.93 | 4–70 | |
| Glrx4-IbaG C [53] | 1/2 | 2.01, 1.92, 1.87 | 1.93 | 20 | |
| CCCR | Biotin synthase [57,58] | 1/2 | 2.03, 1.95, 1.90 | 1.96 | 6–80 |
| CCHH | Rieske [38] | 1/2 | 2.02, 1.90, 1.80 | 1.91 | 4–100 |
| Apd1 [36] | 1/2 | 2.01, 1.91, 1.86 | 1.93 | 10 | |
| C(GSH)HH | GrxS14-BolA1 [59] | 1/2 | 2.02, 1.96, 1.65 | 1.88 | 10 |
| CCHE | RsrR [60,61] | 1/2 | 2.00, 1.92, 1.87 | 1.93 | 10 |
| [3Fe-4S]1+ | |||||
| CCC cube | [NiFe] hydrogenase [62] | 1/2 | 2.02, 2.00, 1.97 | 2.00 | 2–12 |
| CCC linear | GciS [25] | 5/2 | 9.10, 4.30, 4.15 | 5.85 | 4–40 |
| [4Fe-4S]3+ | |||||
| CCCC | HiPIP [41,63,64] | 1/2 | 2.12, 2.03, 2.03 | 2.06 | 4–20 |
| [4Fe-4S]1+ | |||||
| CCCC | Super-reduced HiPIP [64] | 1/2 | 2.04, 1.92, 1.92 | 1.96 | 4–40 |
| NuoG [65] | 1/2 | 2.06, 1.94, 1.89 | 1.96 | 4–60 | |
| DMSO reductase [66] | 1/2 | 2.03, 1.94, 1.94 | 1.97 | 4–30 | |
| NfuA [67] | 1/2 | 2.04, 1.95, 1.90 | 1.96 | 5–35 | |
| CCCH | [FeFe]-hydrogenase I [68] [FeFe]-hydrogenase I [68] [FeFe]-hydrogenase I [68] | 1/2 | 2.07, 1.93, 1.87 | 1.96 | 4–20 |
| 3/2 | 5.60, 1.71, 1.31 | 2.87 | 4–30 | ||
| 7/2 | 5.21, 5.13, 4.91 | 5.08 | 4–80 | ||
| HydF [69] | 1/2 | 2.04, 1.90, 1.85 | 1.93 | 10 | |
| CCCD | FNR [70,71] | 1/2 | 2.05, 1.94, 1.89 | 1.96 | 15 |
| NsrR [72] | 1/2 | 2.04, 1.93, 1.93 | 1.97 | 10 | |
| Fd [73] | 1/2 | 2.10, 1.87, 1.80 | 1.92 | 4–15 | |
| CCCS | LipA Aux [74] | 1/2 | 2.05, 1.91, 1.91 | 1.96 | 13 |
| CCC(H2O) | Aconitase [39] | 1/2 | 2.06, 1.93, 1.86 | 1.95 | 13 |
| CCC(citrate) | Aconitase + citrate [39] | 1/2 | 2.04, 1.85, 1.78 | 1.89 | 13 |
| CCC(H2O)-RS | PFL-AE [40] | 1/2 | 2.02, 1.94, 1.88 | 1.95 | 12 |
| RimO [75] | 1/2 | 2.04, 1.93, 1.93 | 1.97 | 14 | |
| CCC(SAM)-RS | PFL-AE + SAM [40] | 1/2 | 2.01, 1.89, 1.88 | 1.93 | 4–40 |
| RimO + SAM [75] | 1/2 | 2.04, 1.93, 1.93 | 1.97 | 14 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quist, T.; Chen, J.; MacNeil, A.; Pandelia, M.-E. The Cryptic Nature of Fe-S Clusters: A Case Study of the Hepatitis B HBx Oncoprotein. Inorganics 2023, 11, 475. https://doi.org/10.3390/inorganics11120475
Quist T, Chen J, MacNeil A, Pandelia M-E. The Cryptic Nature of Fe-S Clusters: A Case Study of the Hepatitis B HBx Oncoprotein. Inorganics. 2023; 11(12):475. https://doi.org/10.3390/inorganics11120475
Chicago/Turabian StyleQuist, Trent, Jiahua Chen, Alex MacNeil, and Maria-Eirini Pandelia. 2023. "The Cryptic Nature of Fe-S Clusters: A Case Study of the Hepatitis B HBx Oncoprotein" Inorganics 11, no. 12: 475. https://doi.org/10.3390/inorganics11120475