Preparation and Utility of N-Alkynyl Azoles in Synthesis

Abstract
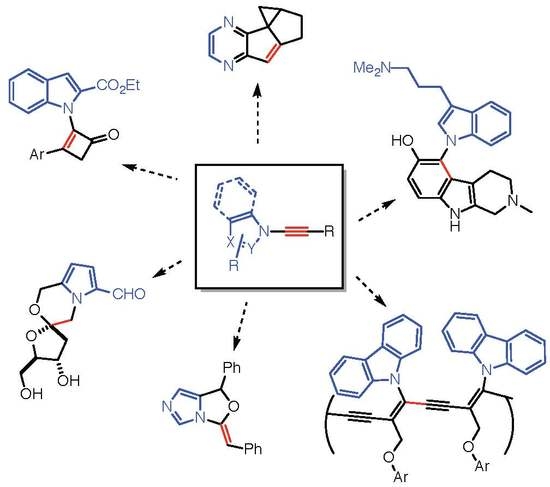
:
1. Introduction
2. Synthesis of N-Alkynyl Azoles
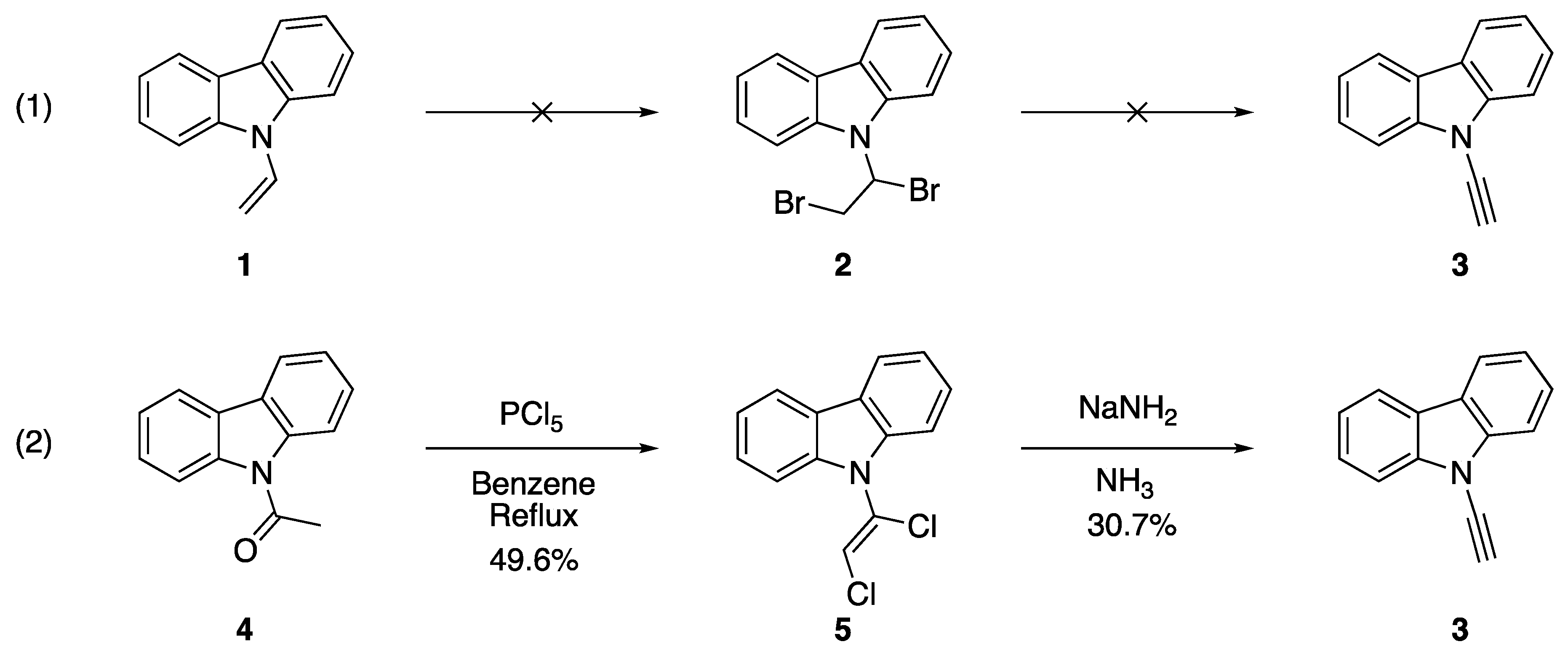
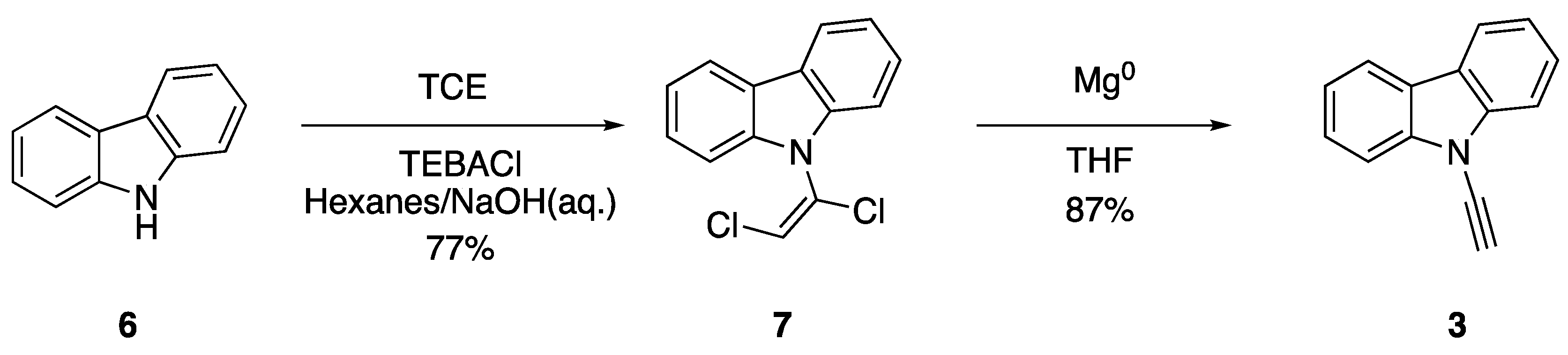
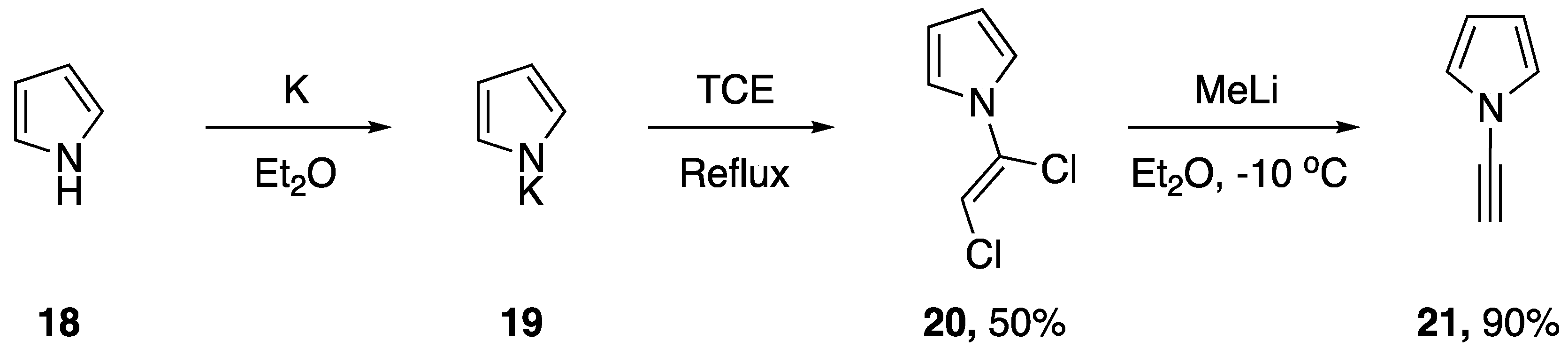
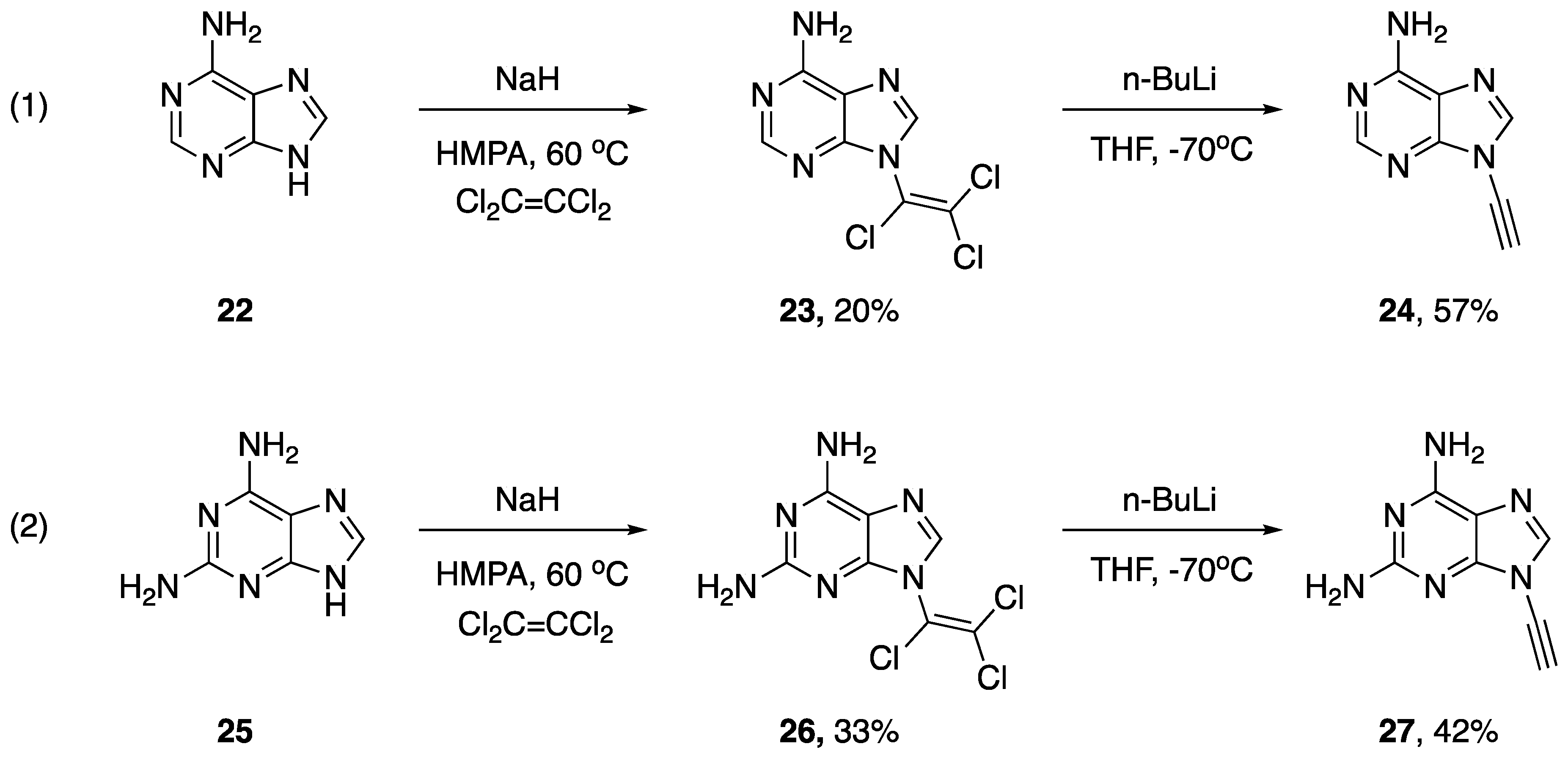
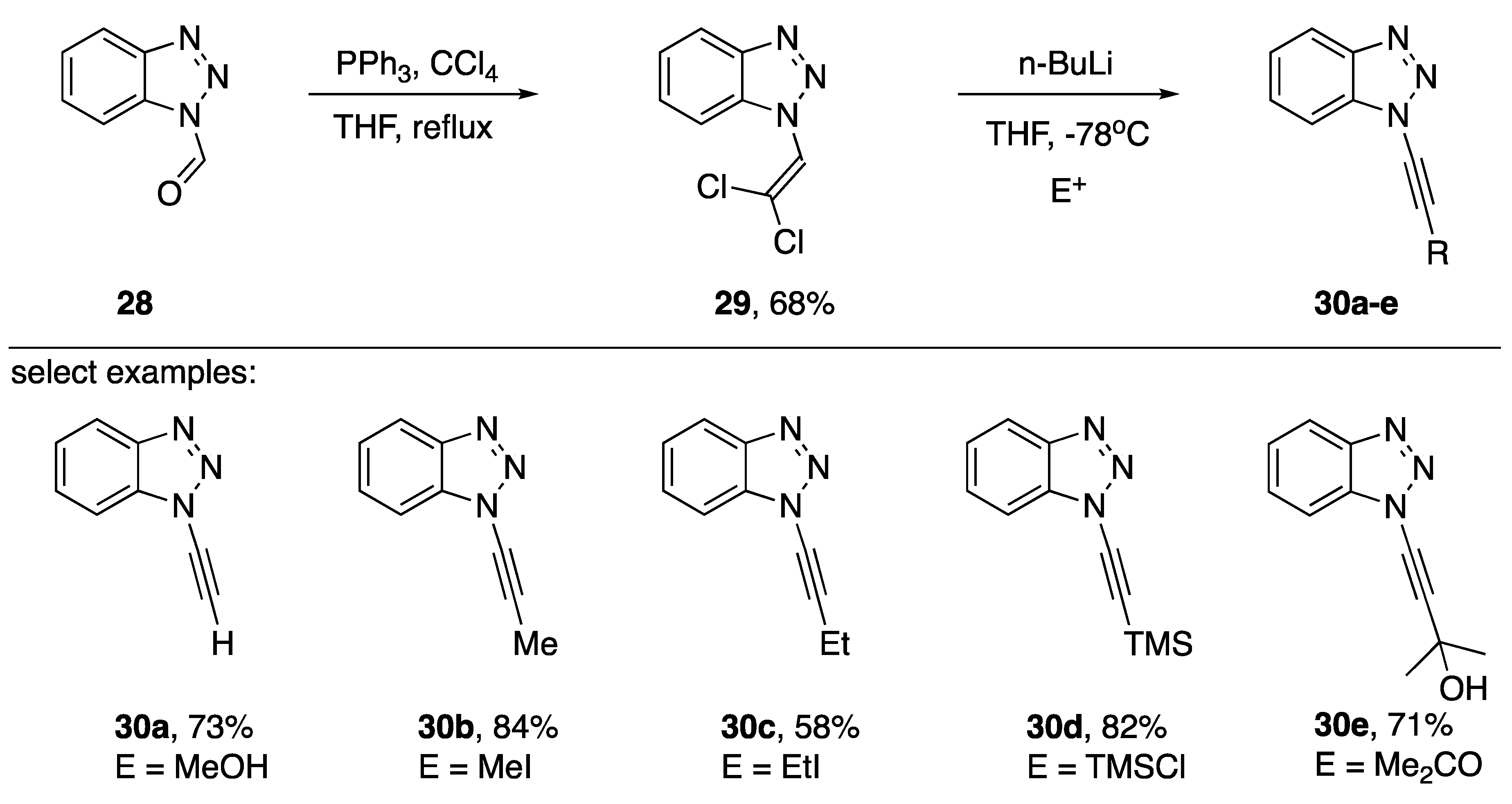
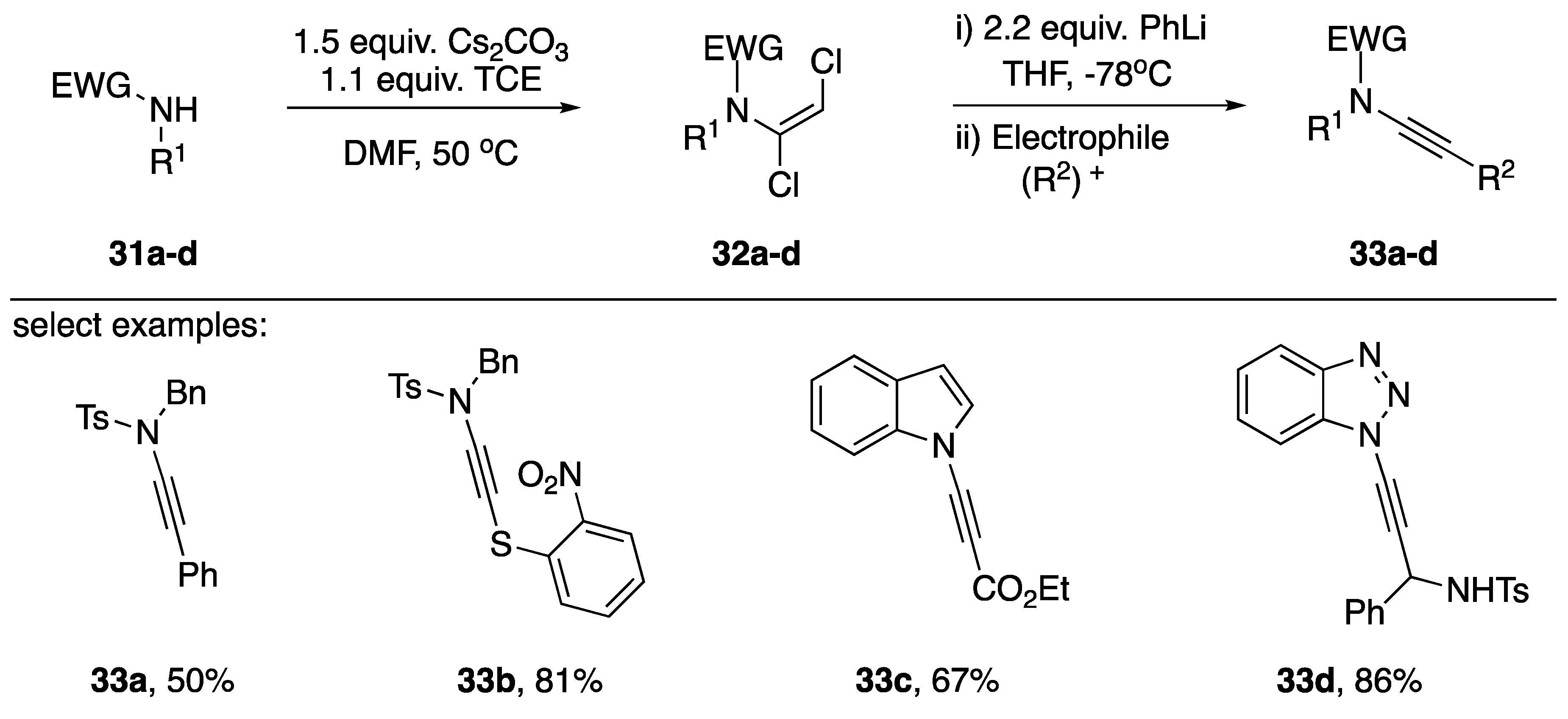
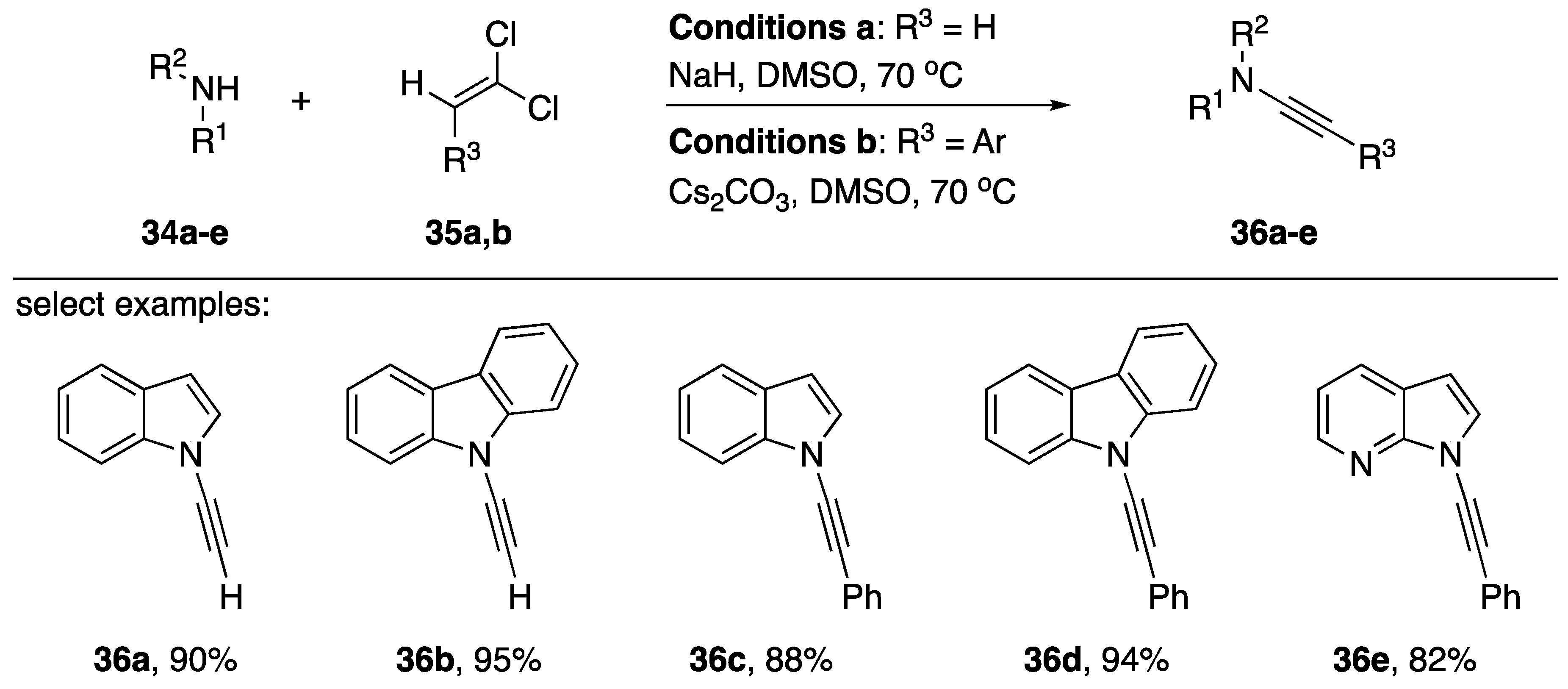
2.1. Elimination
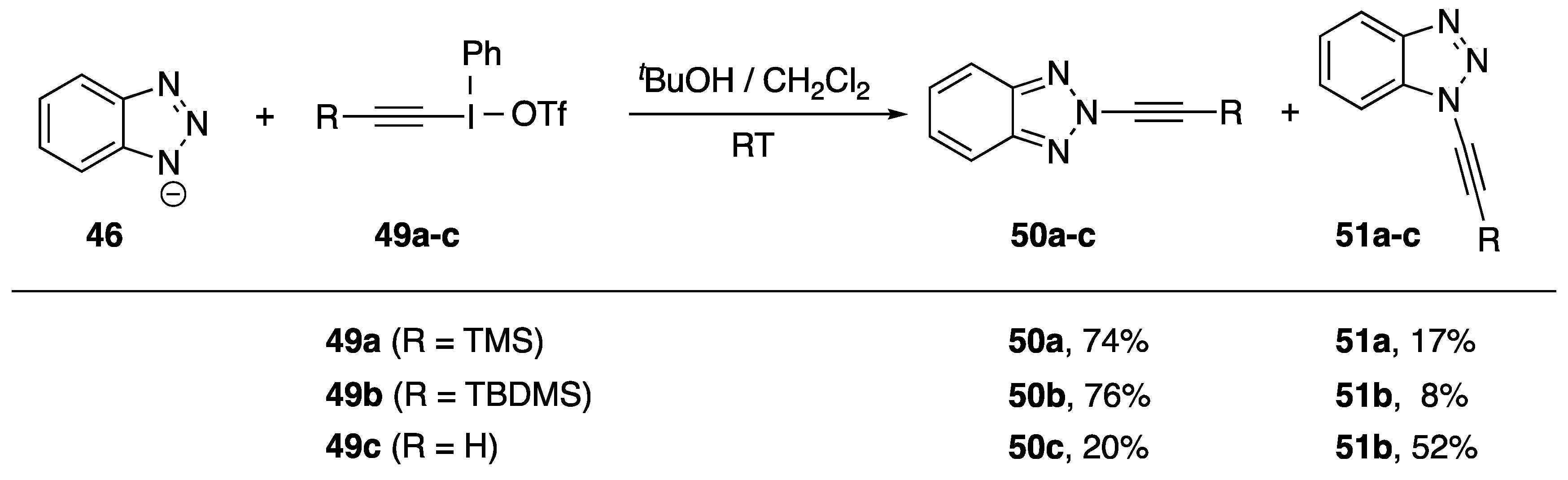
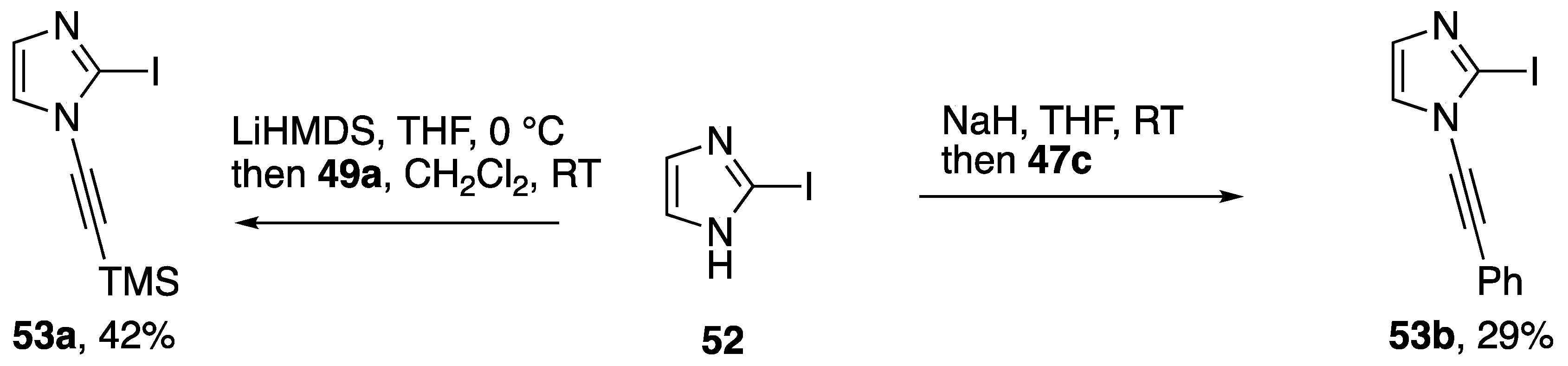
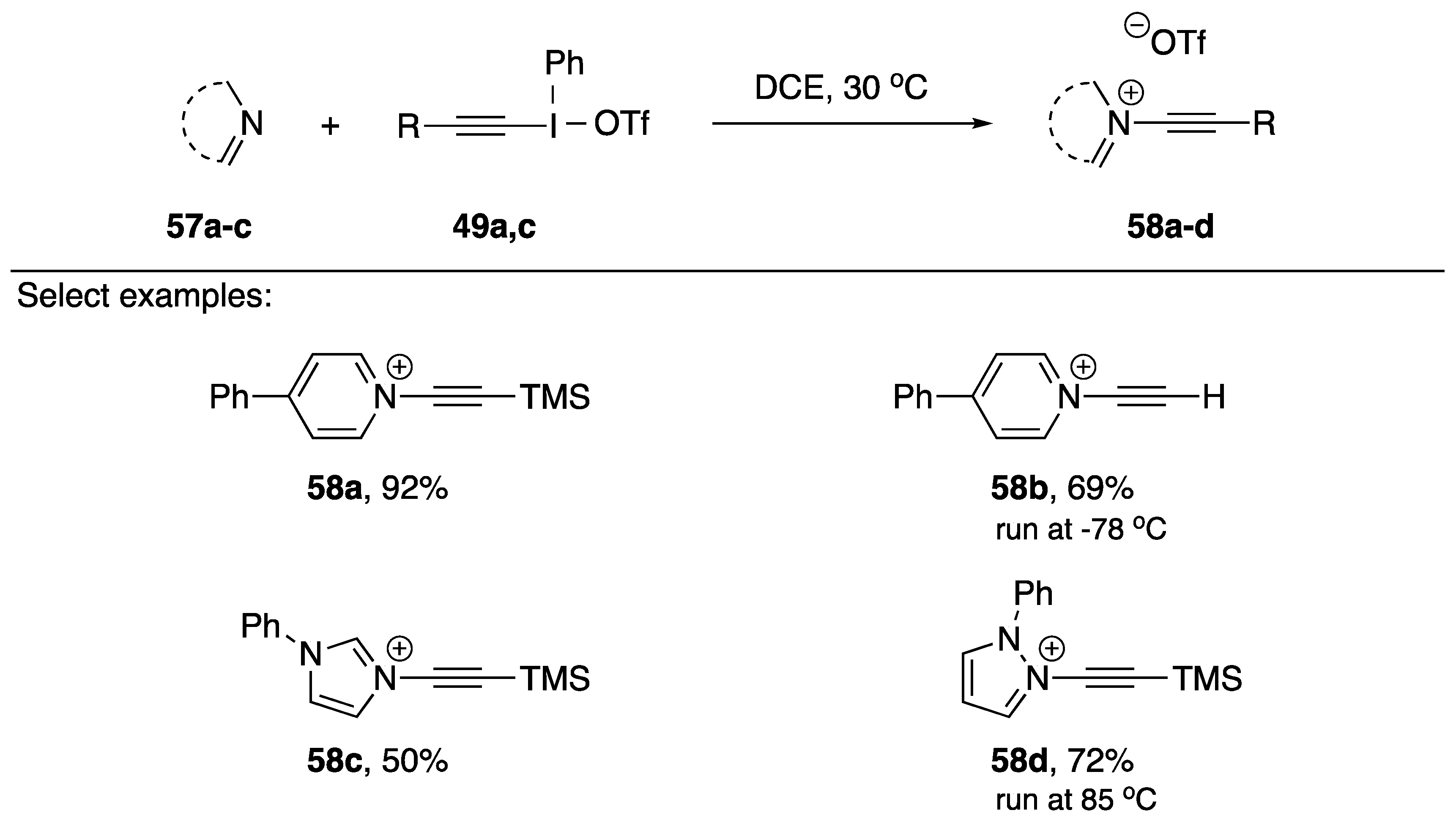
2.2. Alkynyliodonium Salts
2.3. Cross-Coupling
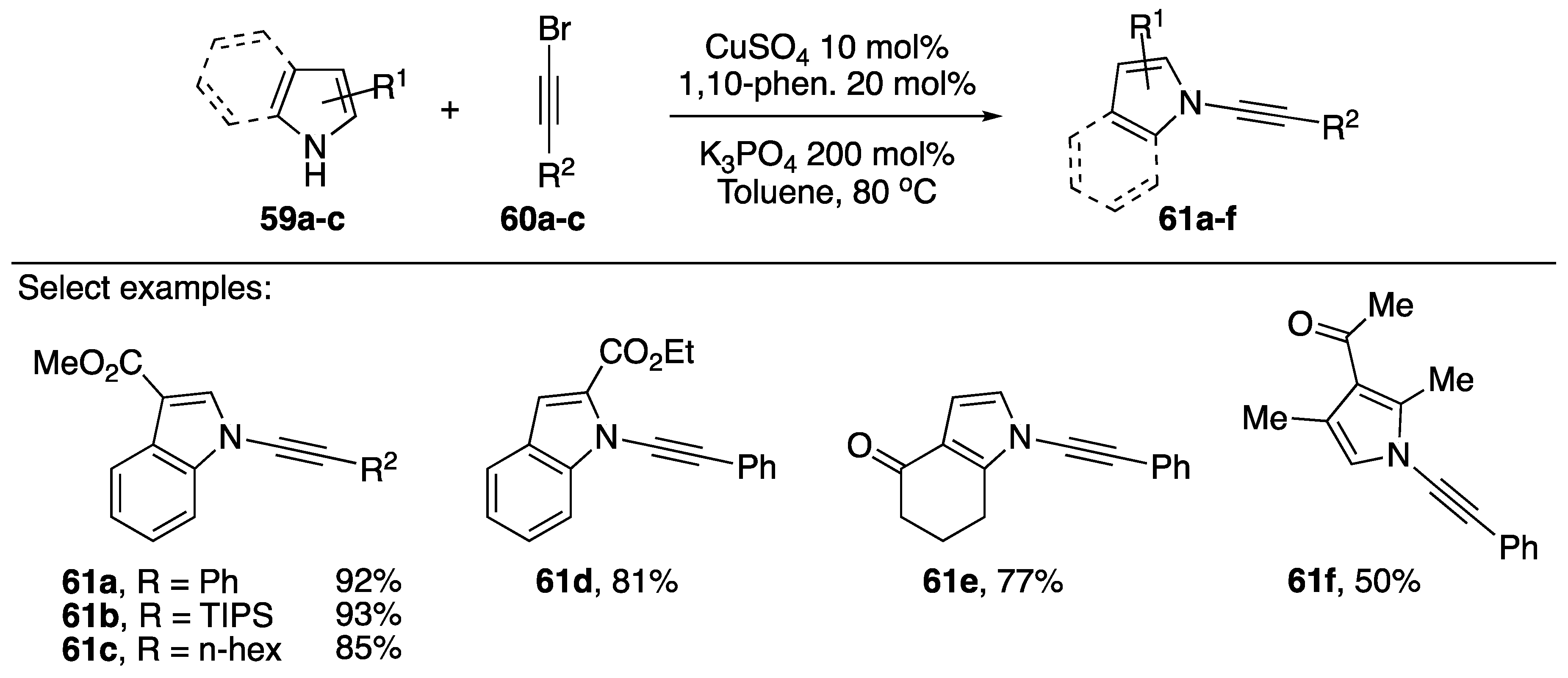
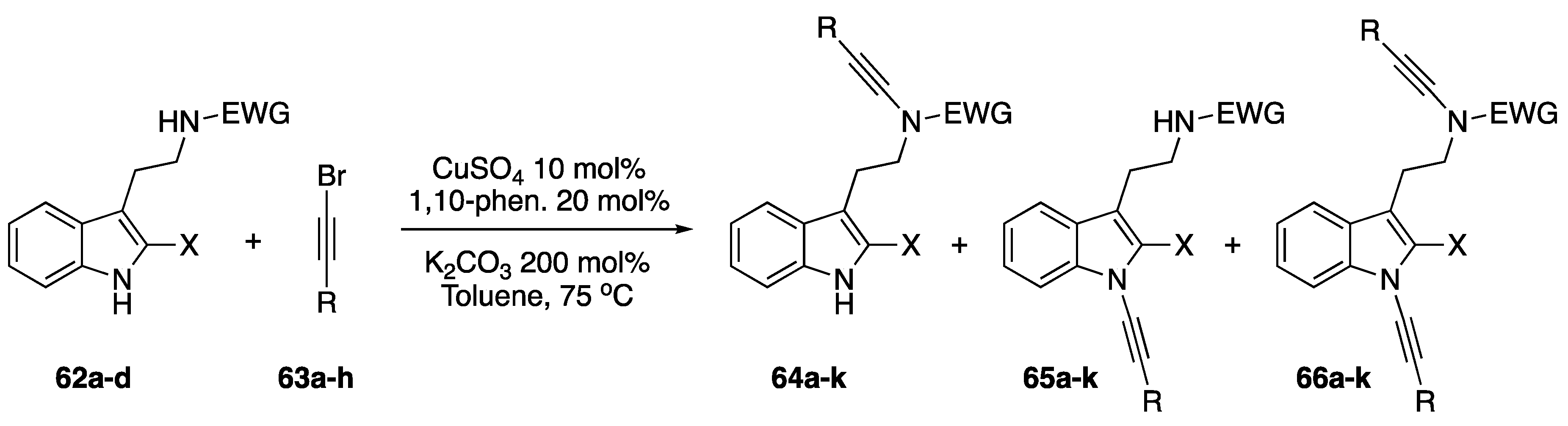
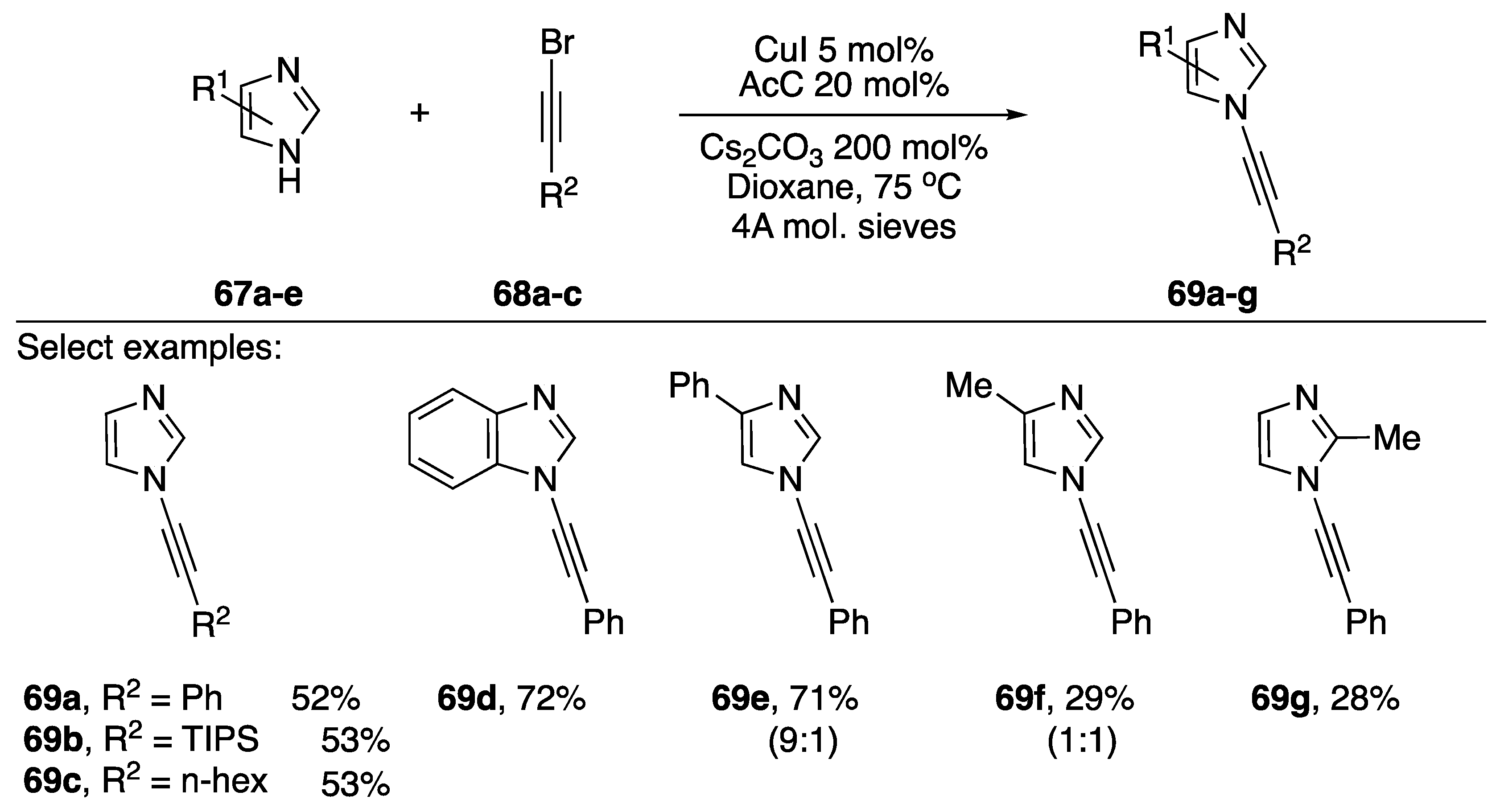
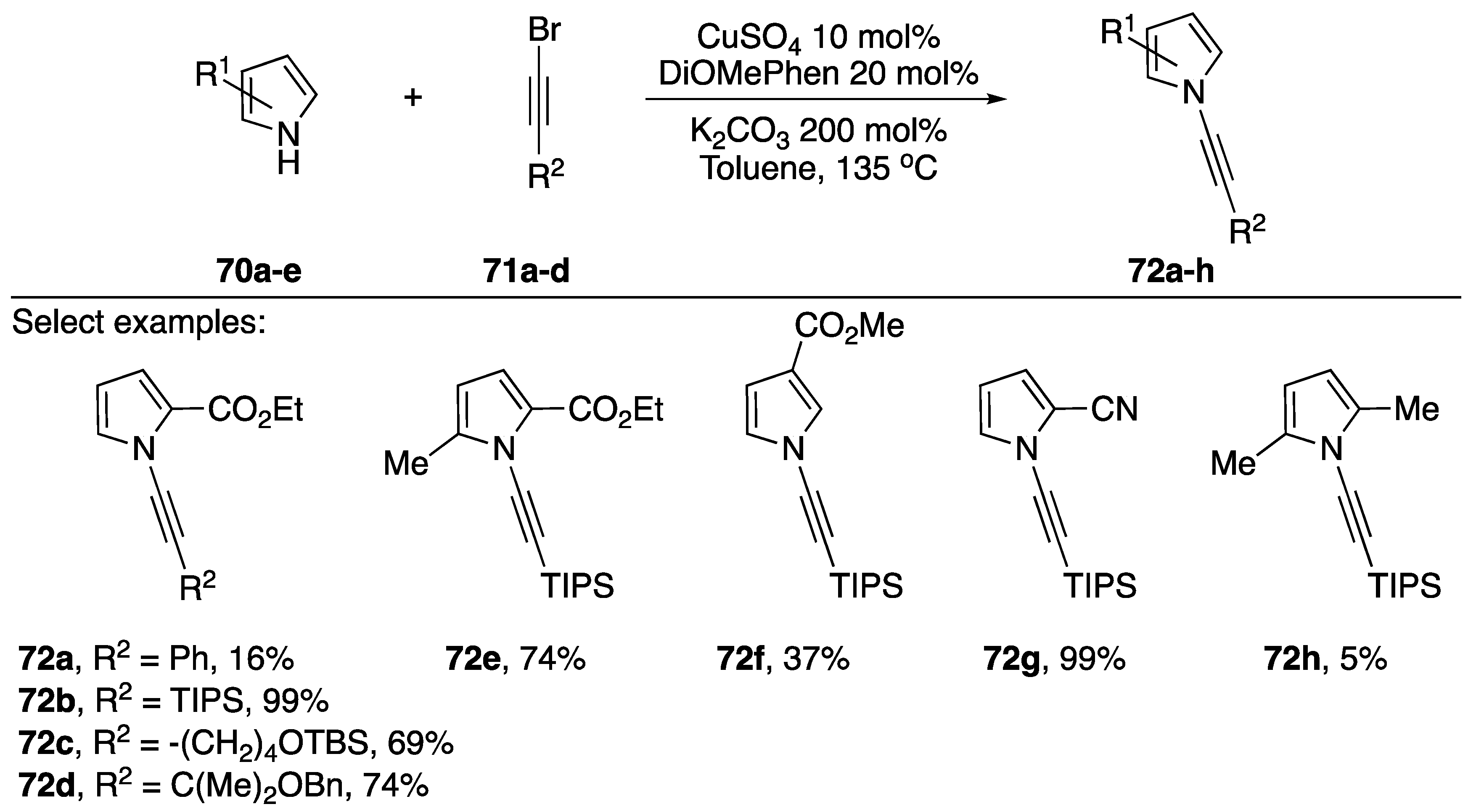
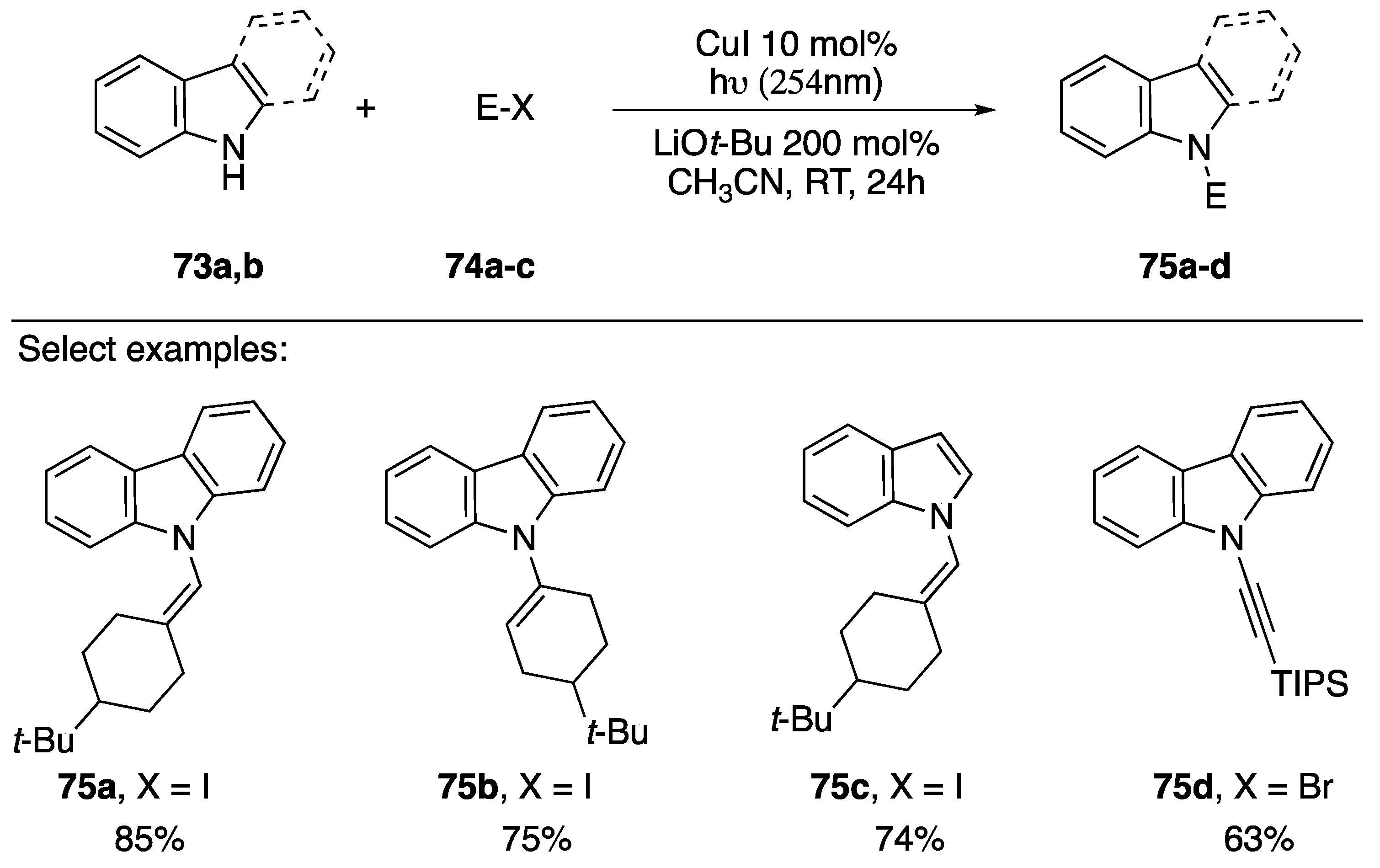
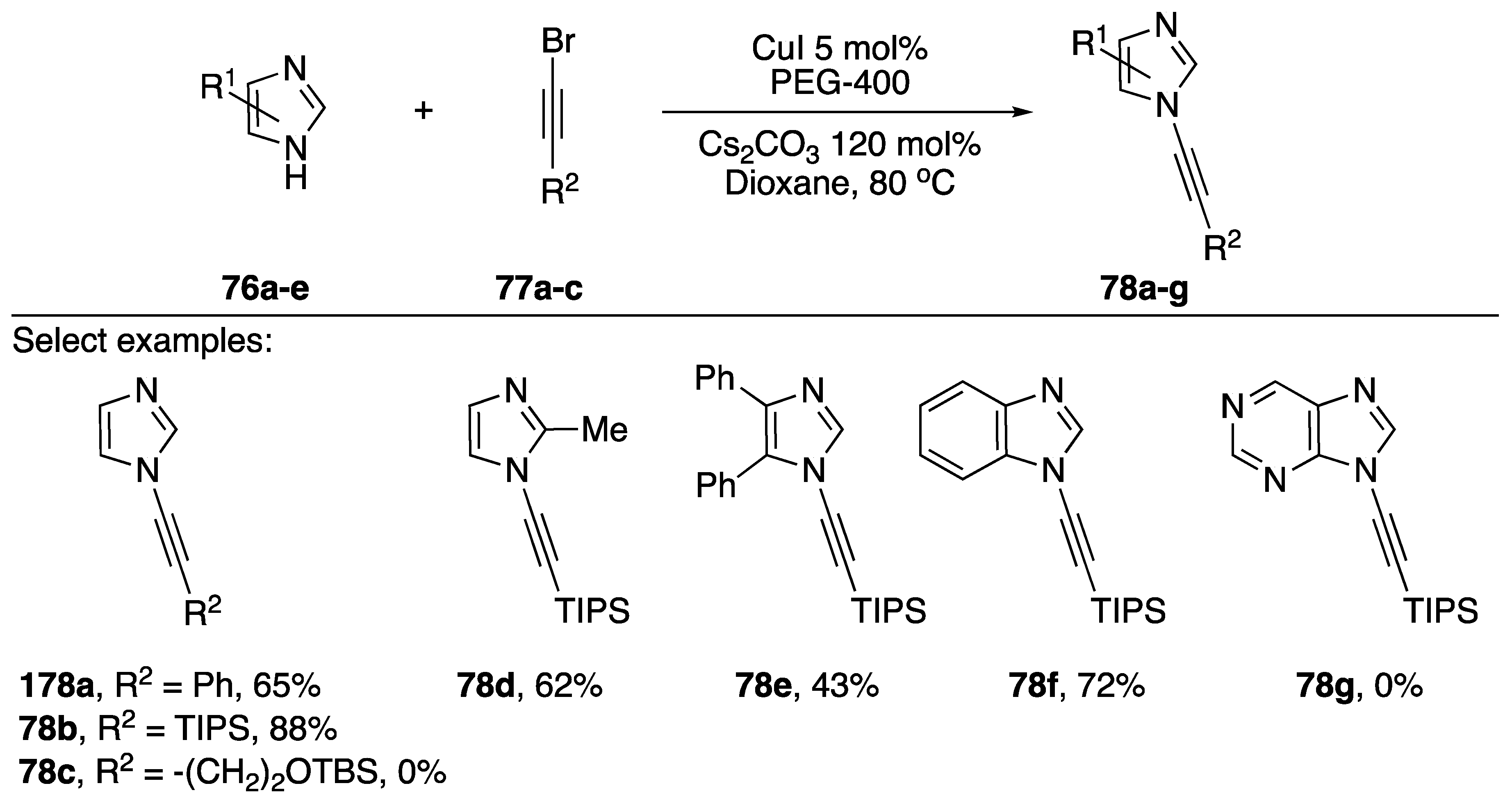
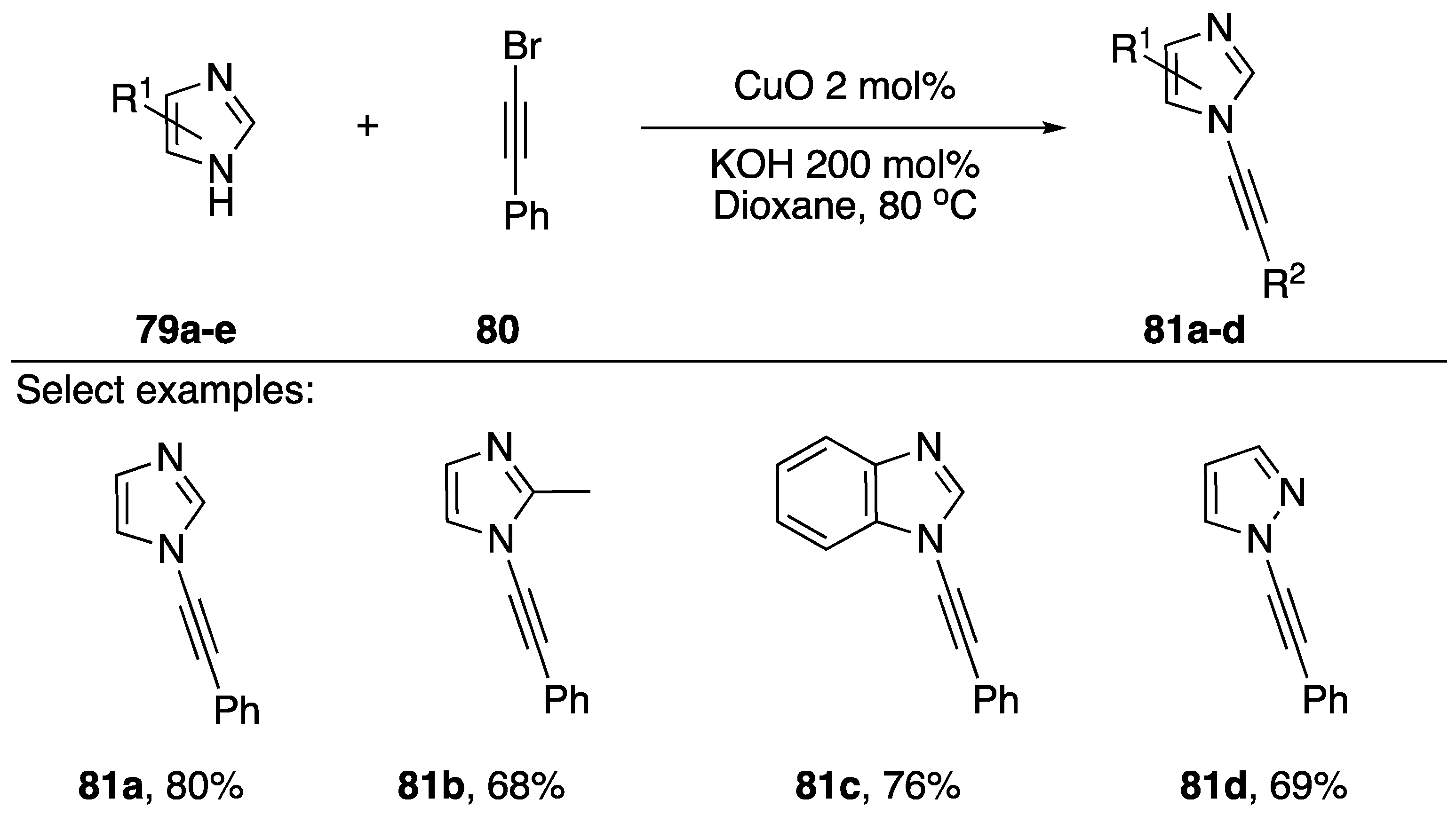
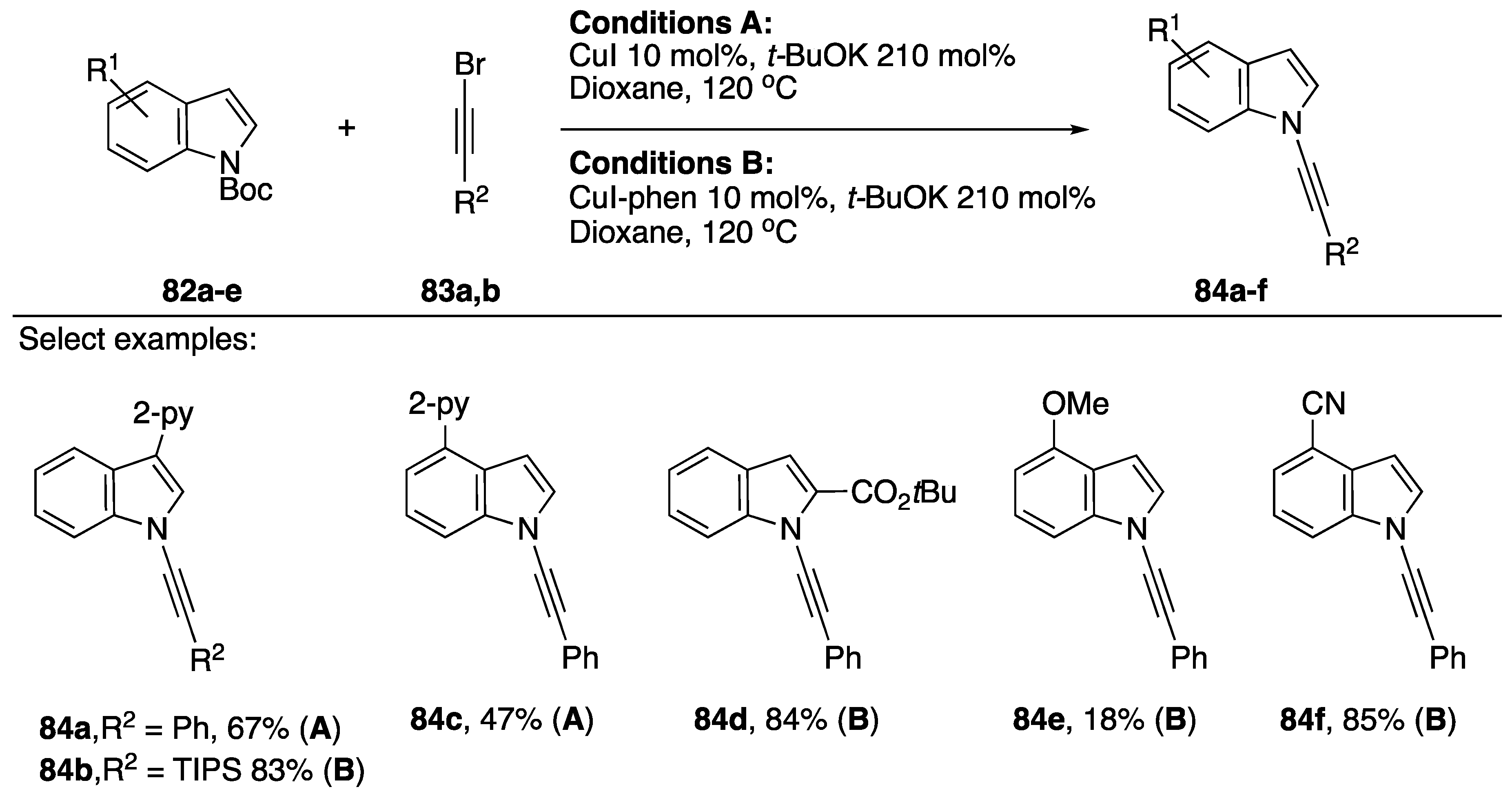
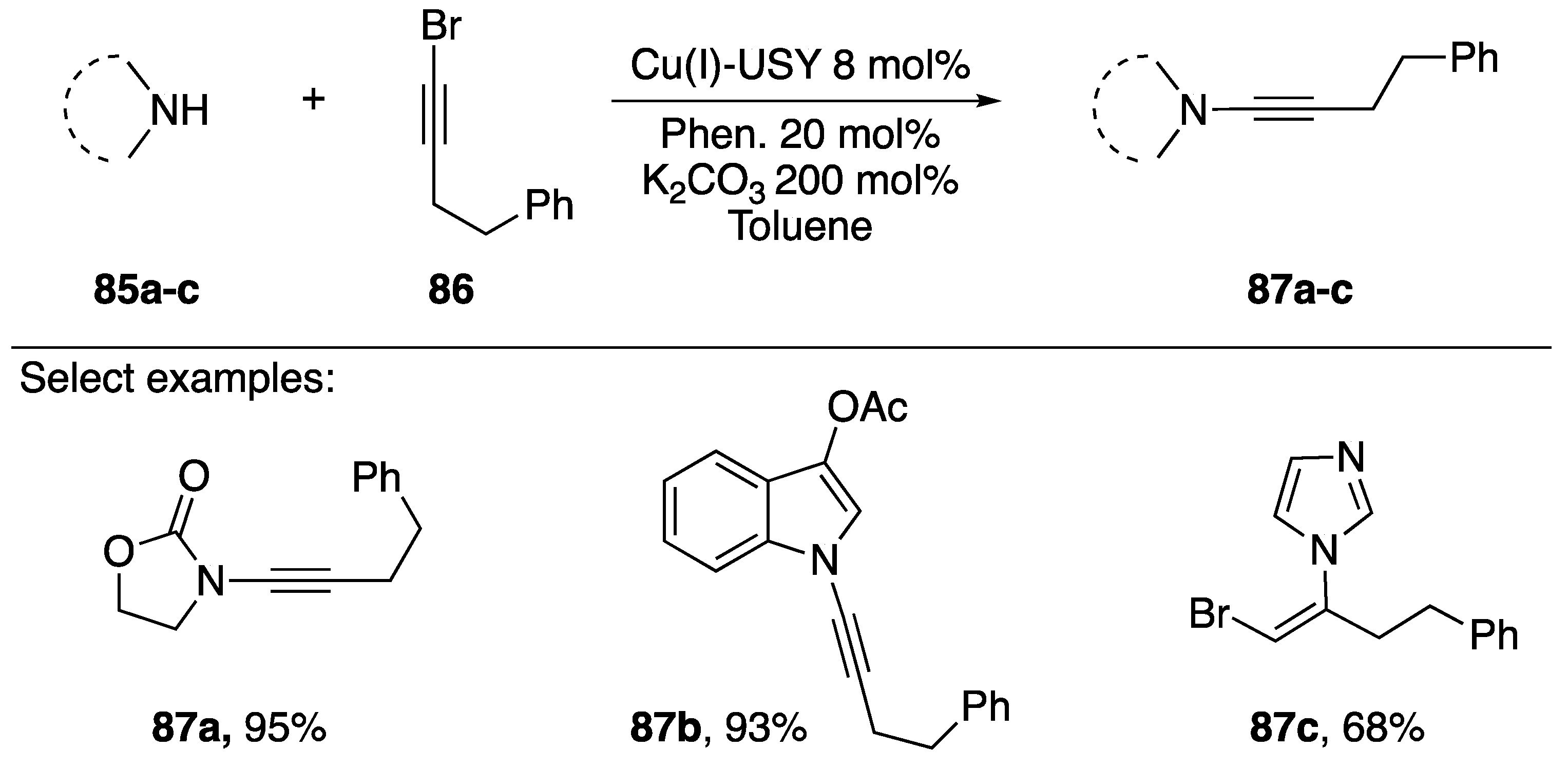
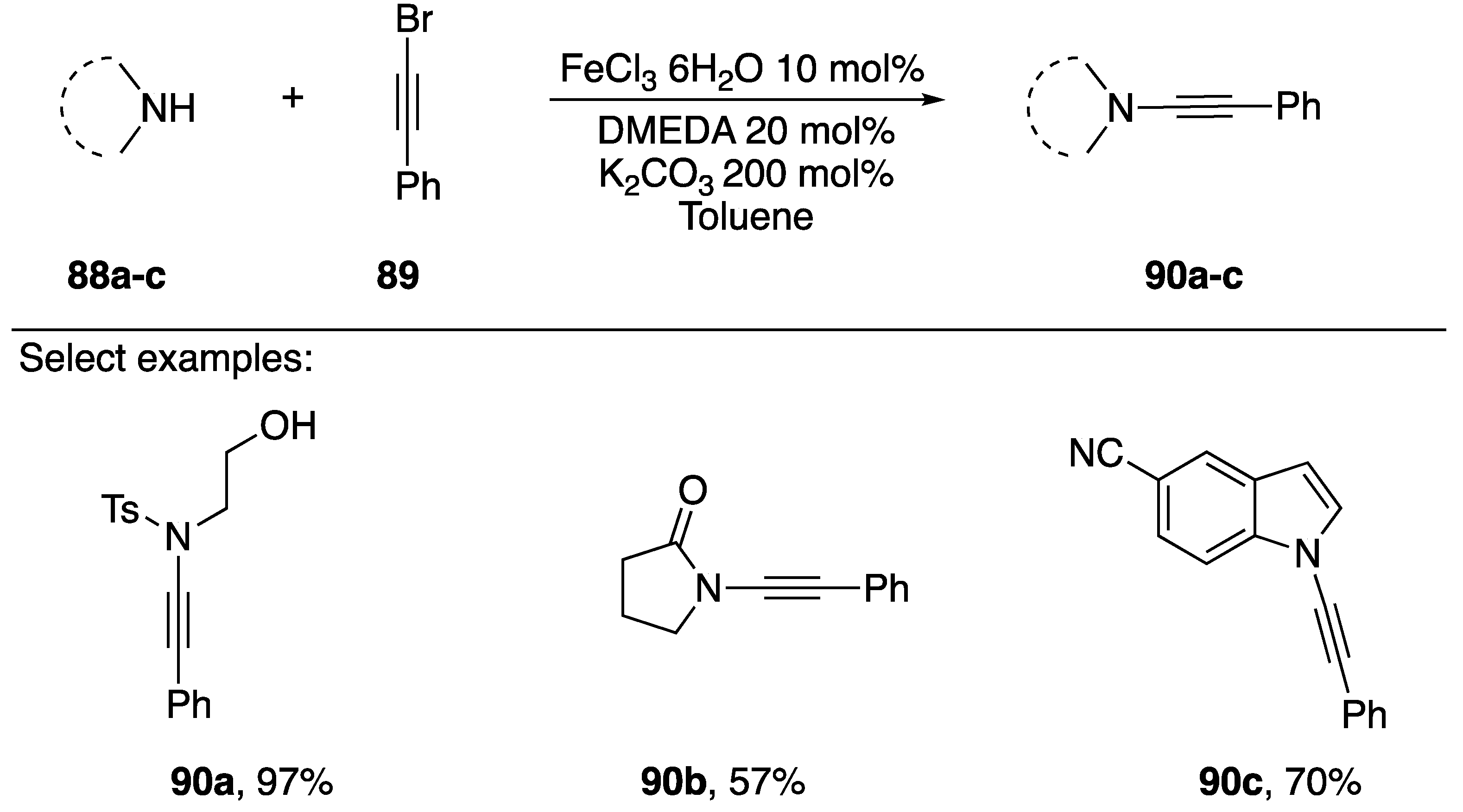
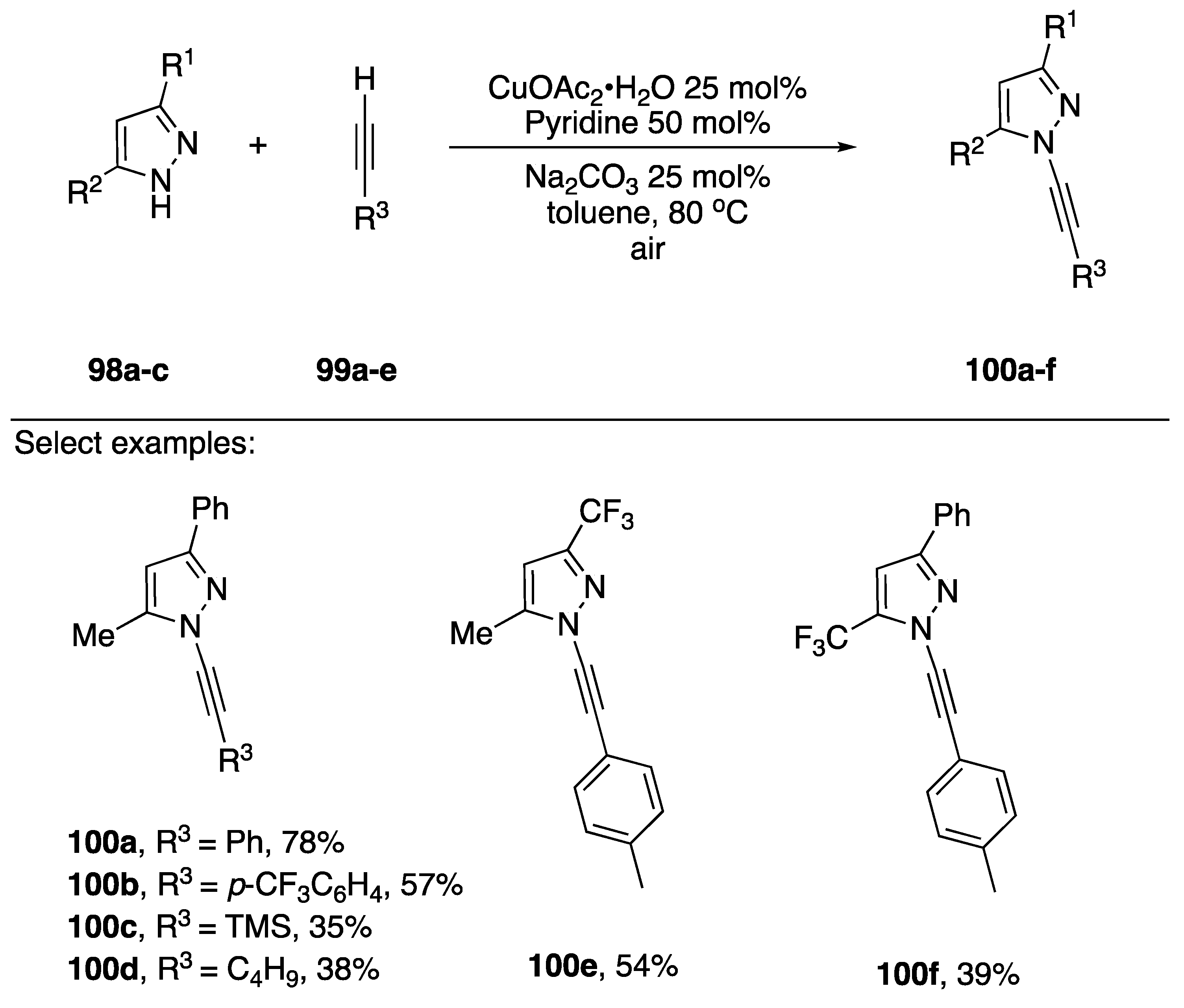
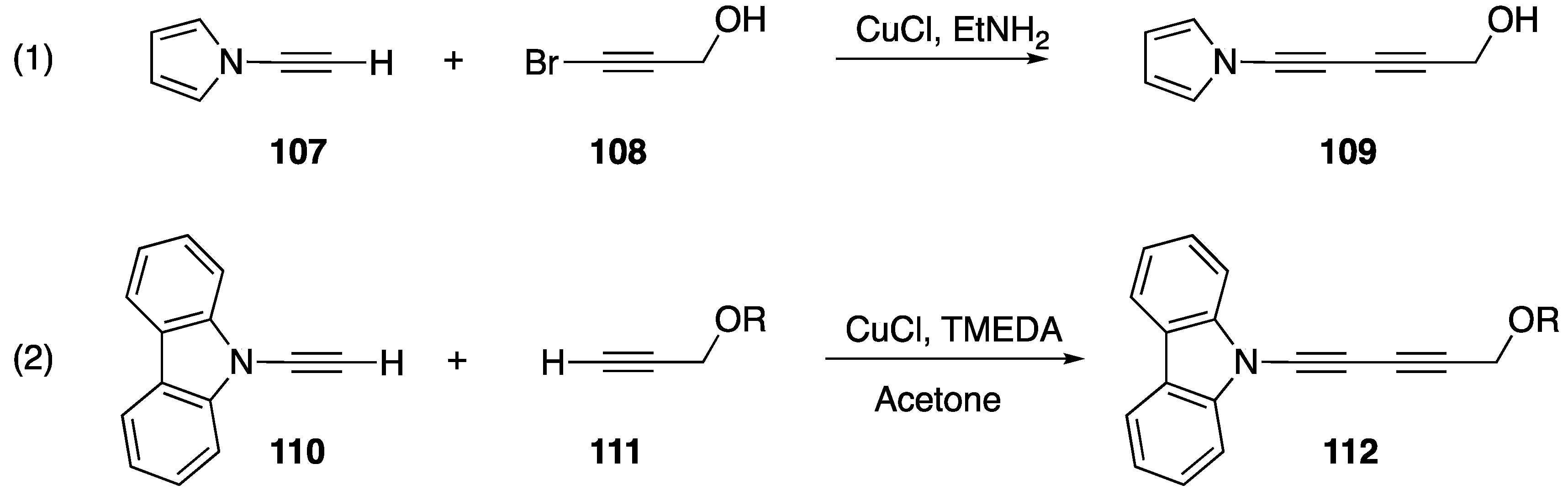
2.3.1. 1-Bromoalkynes
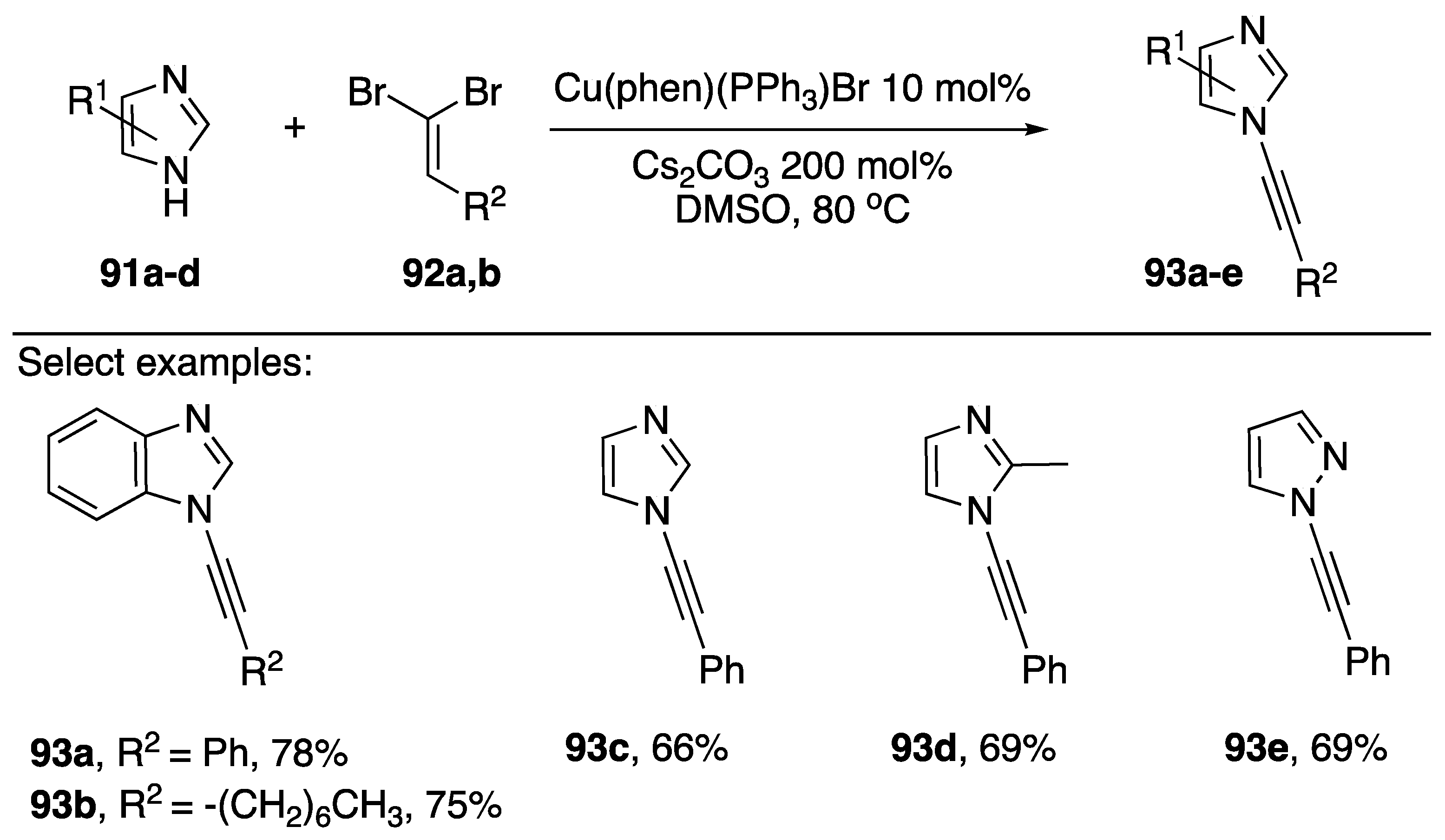
2.3.2. 1,1-Dibromoalkenes
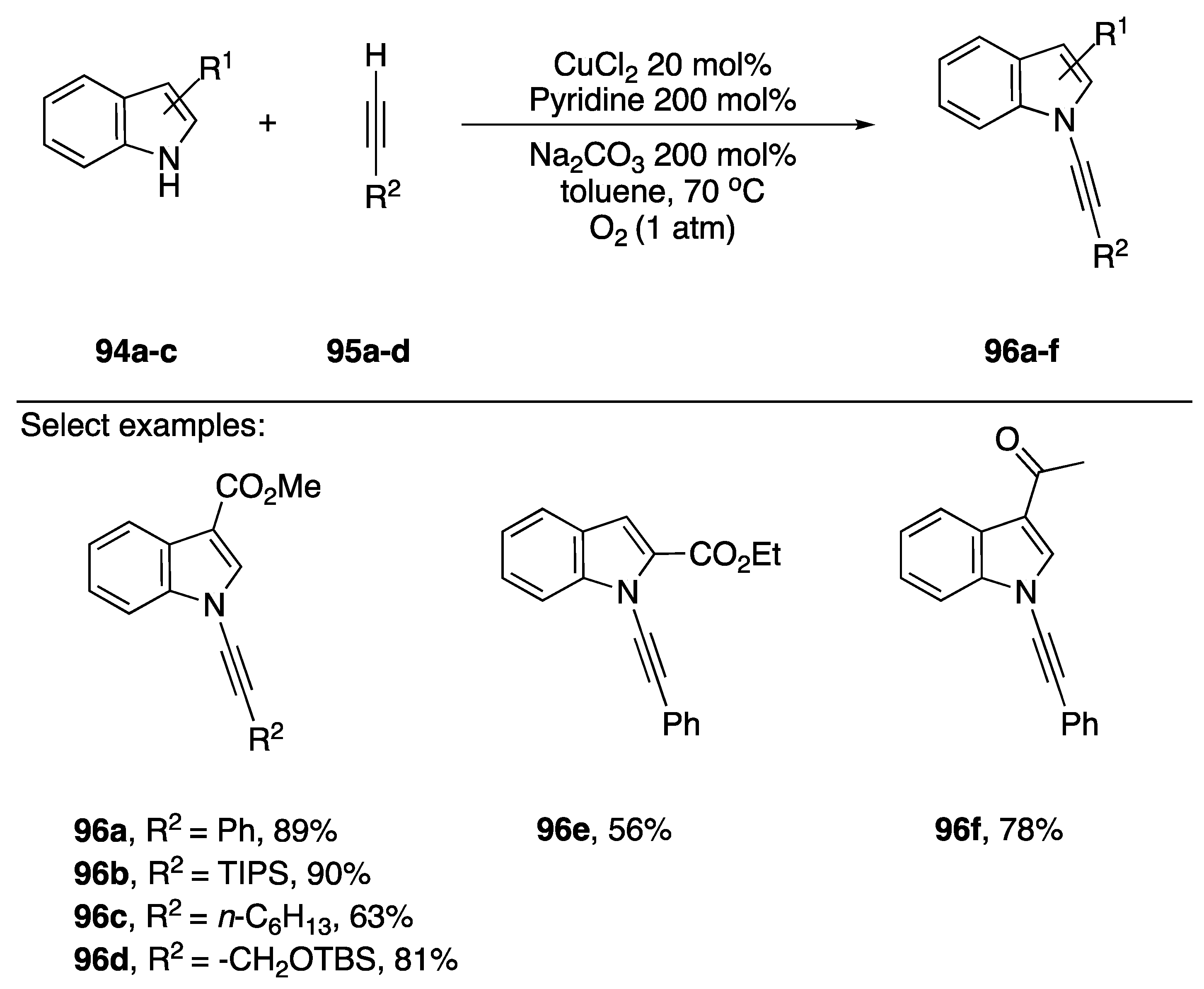
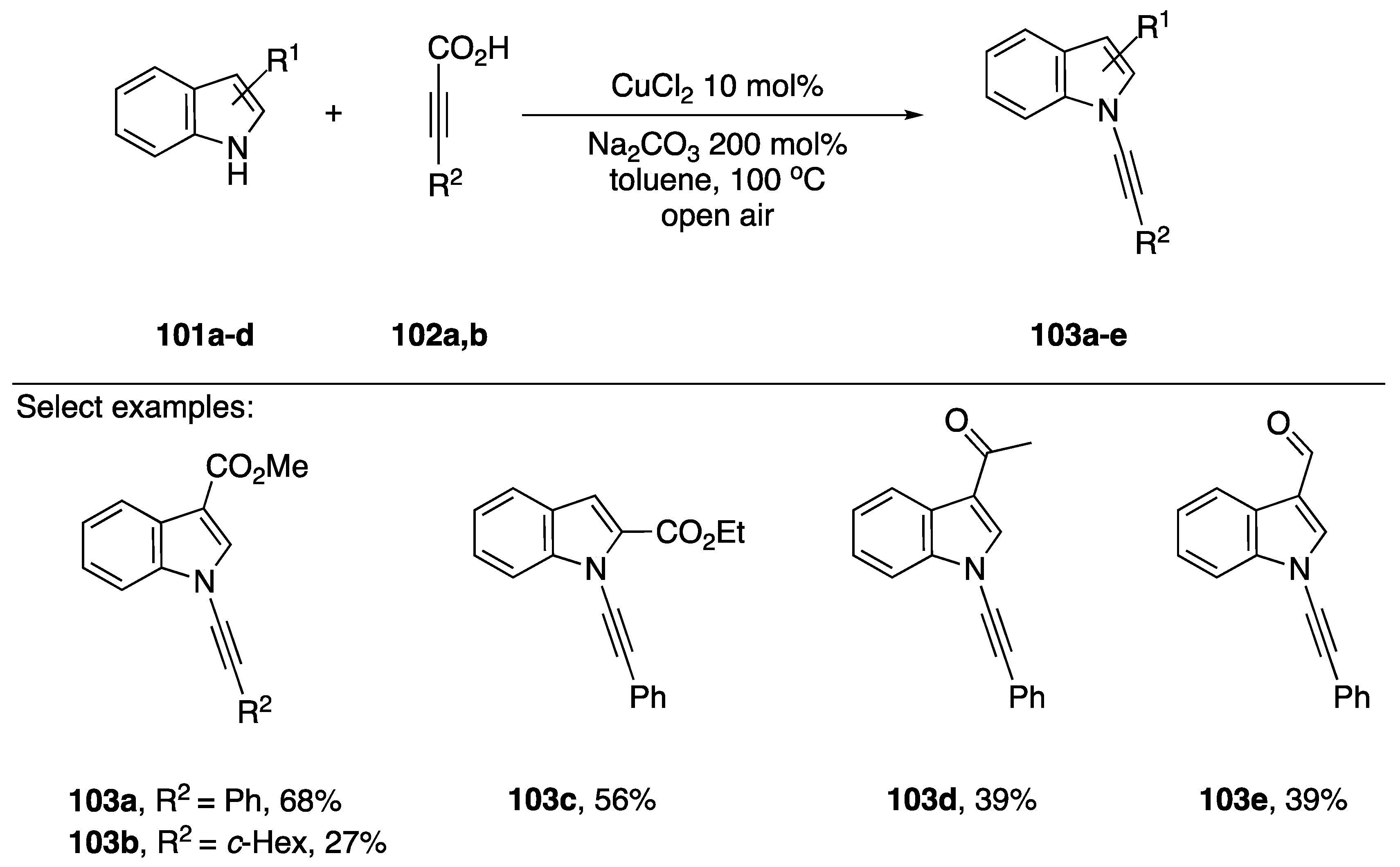
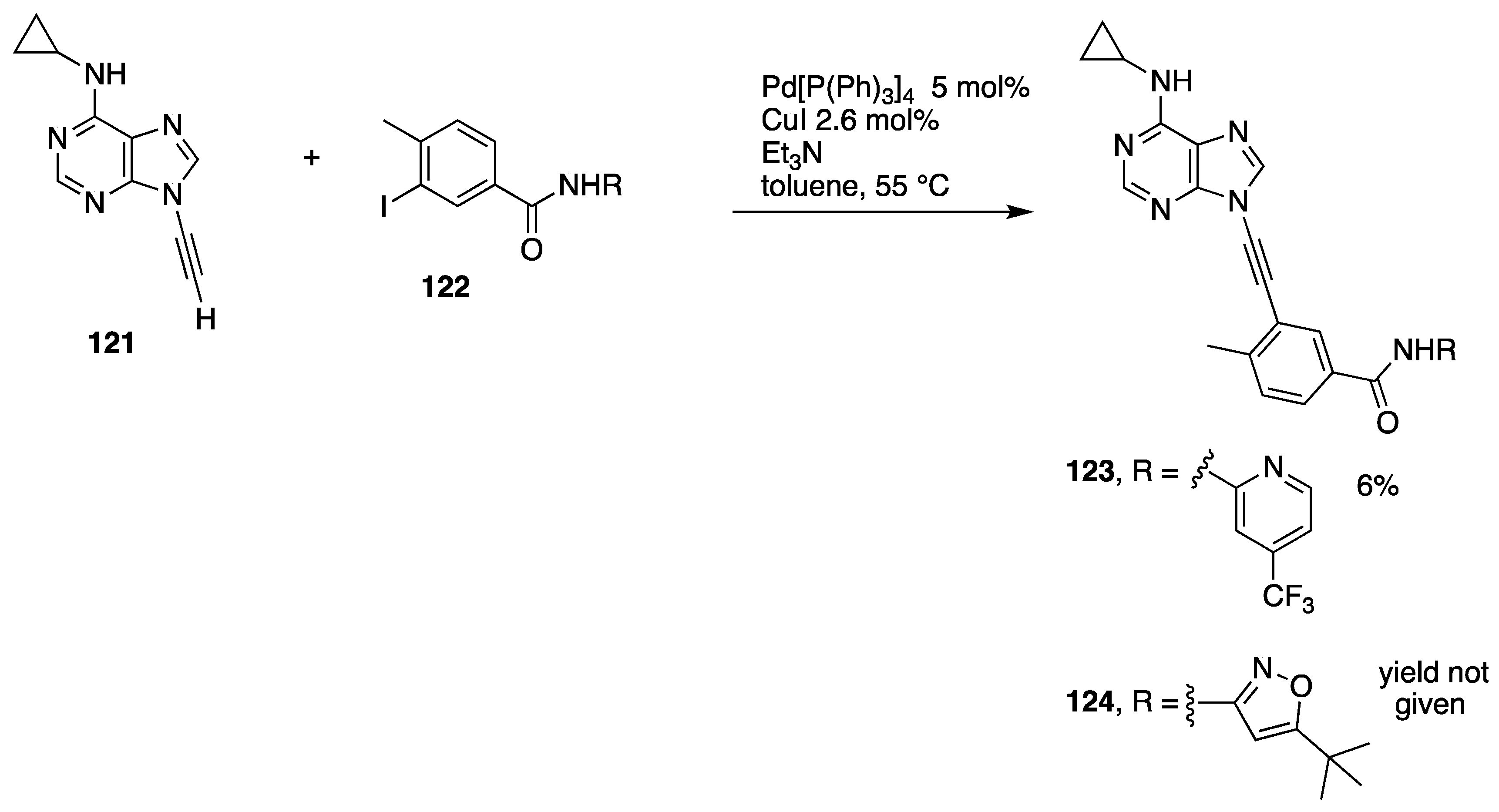
2.3.3. Alternative Coupling Partners
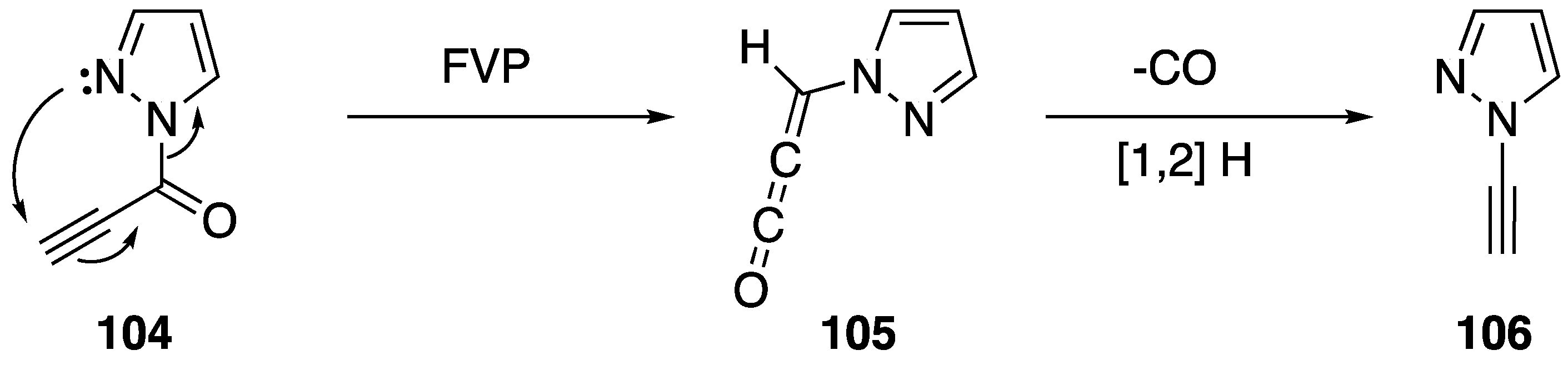
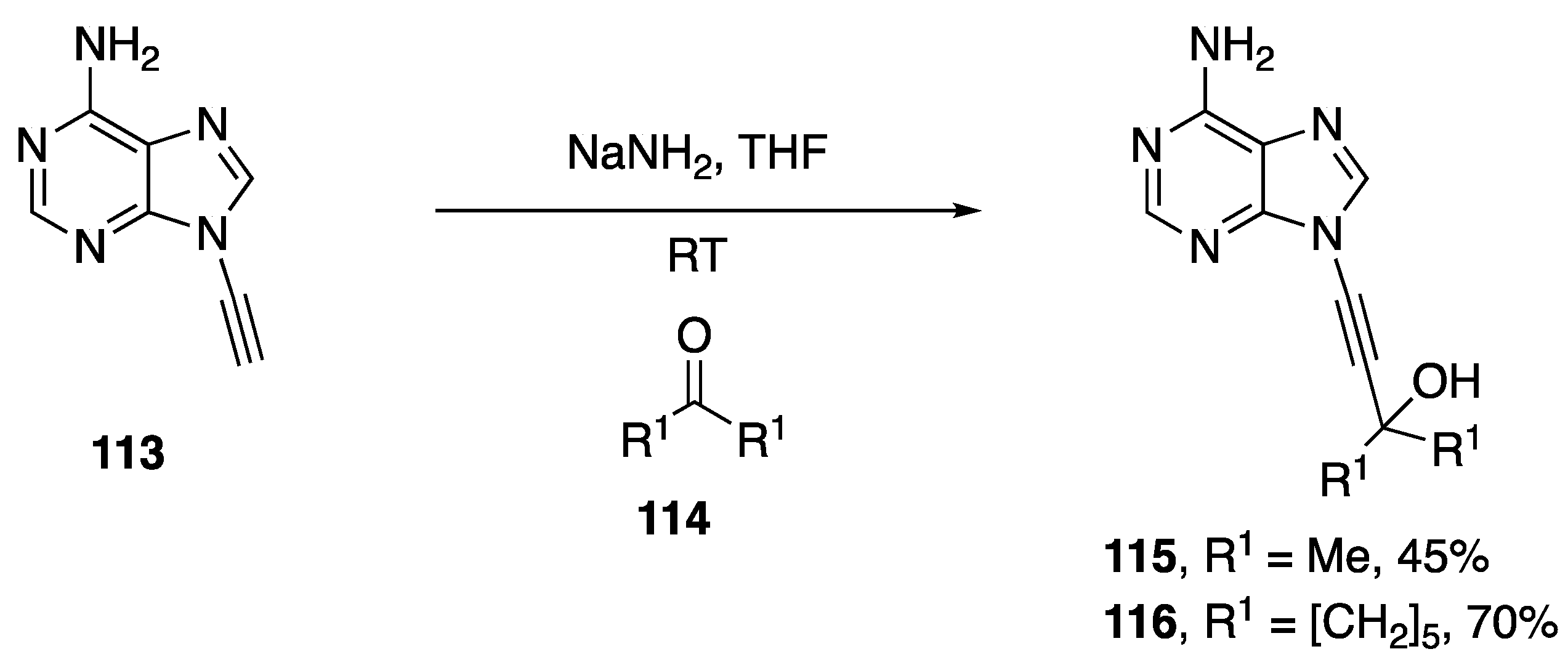
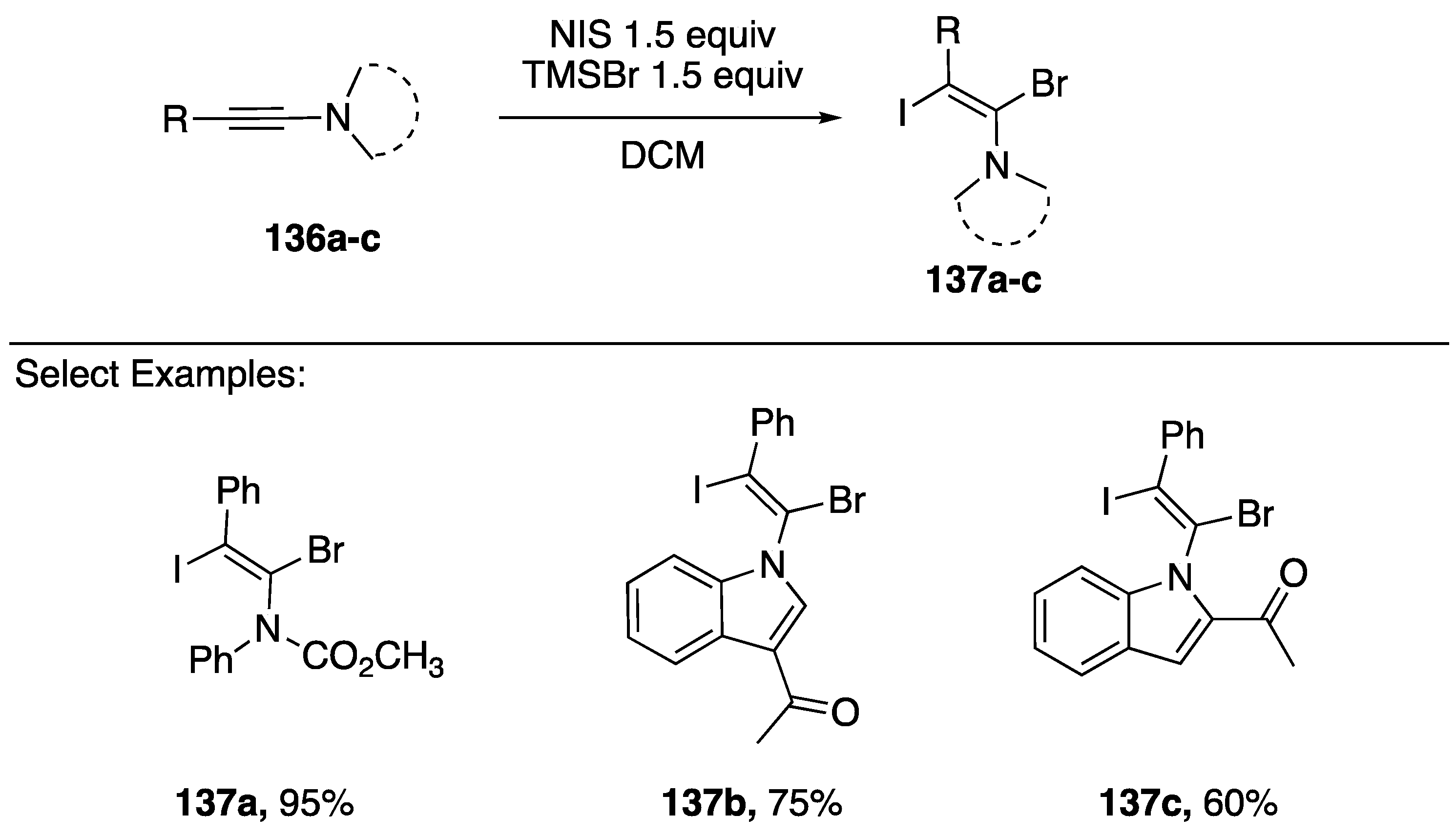
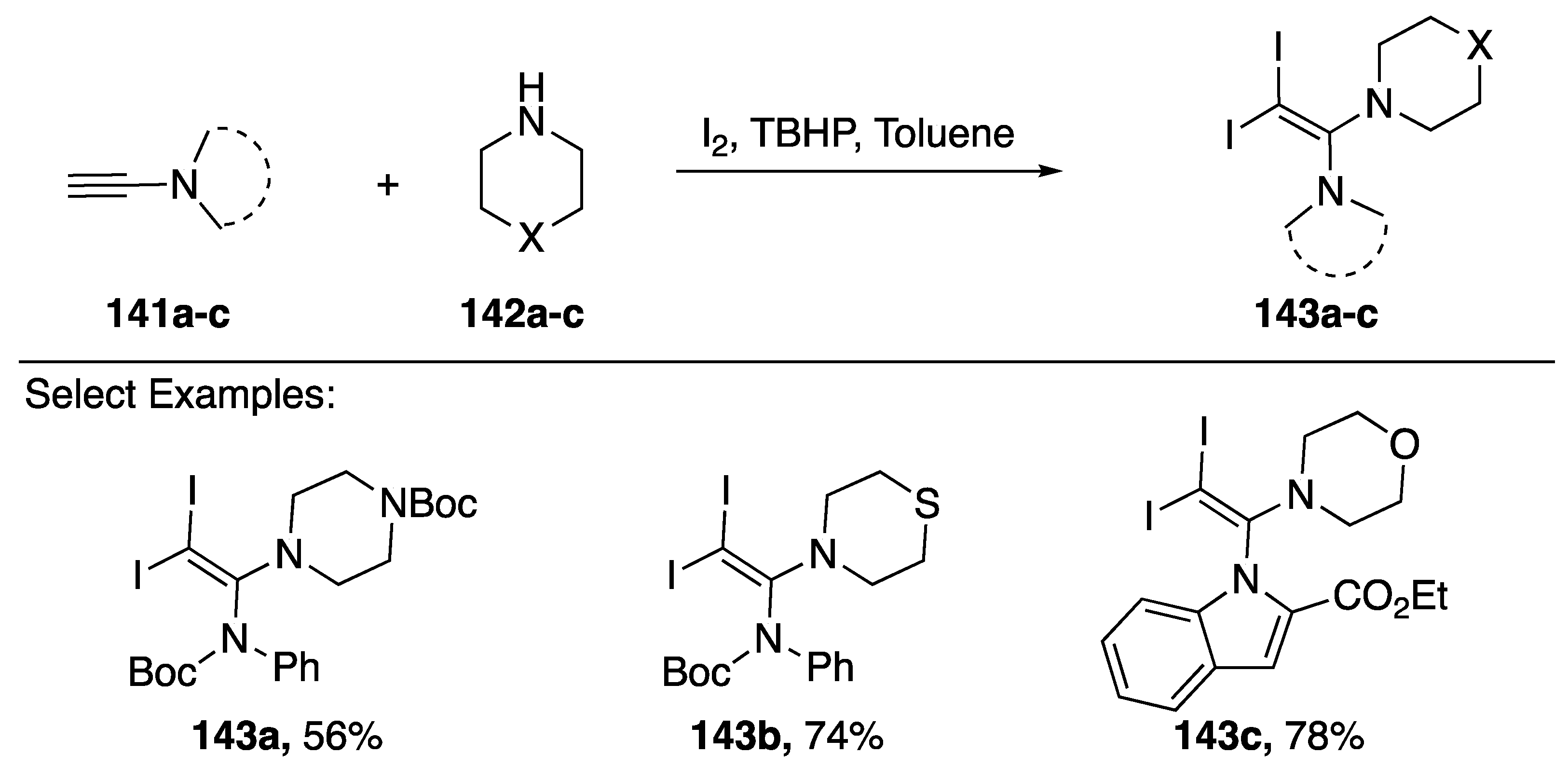
2.4. Other Methods
3. Reactions of N-Alkynyl Azoles
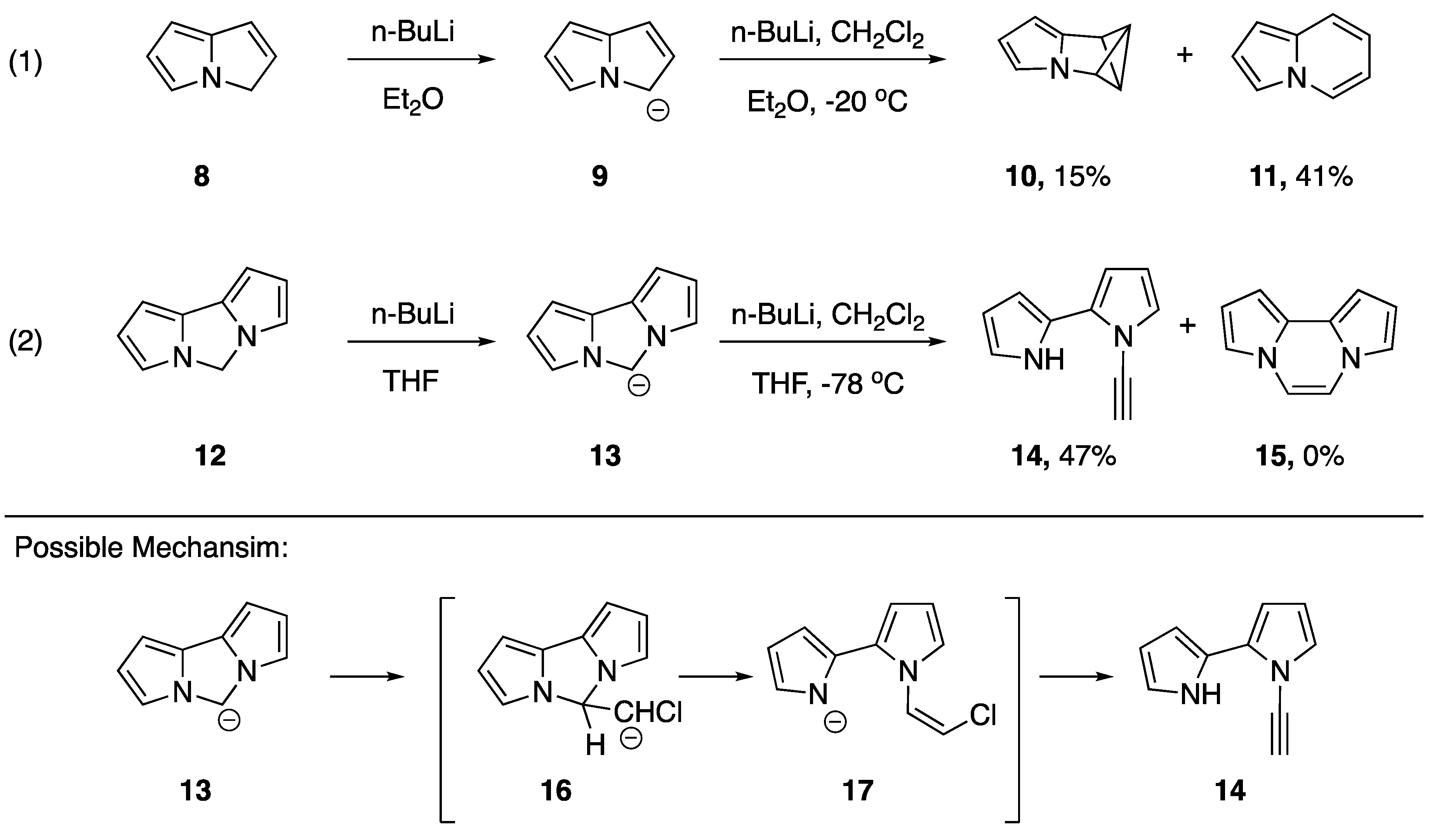
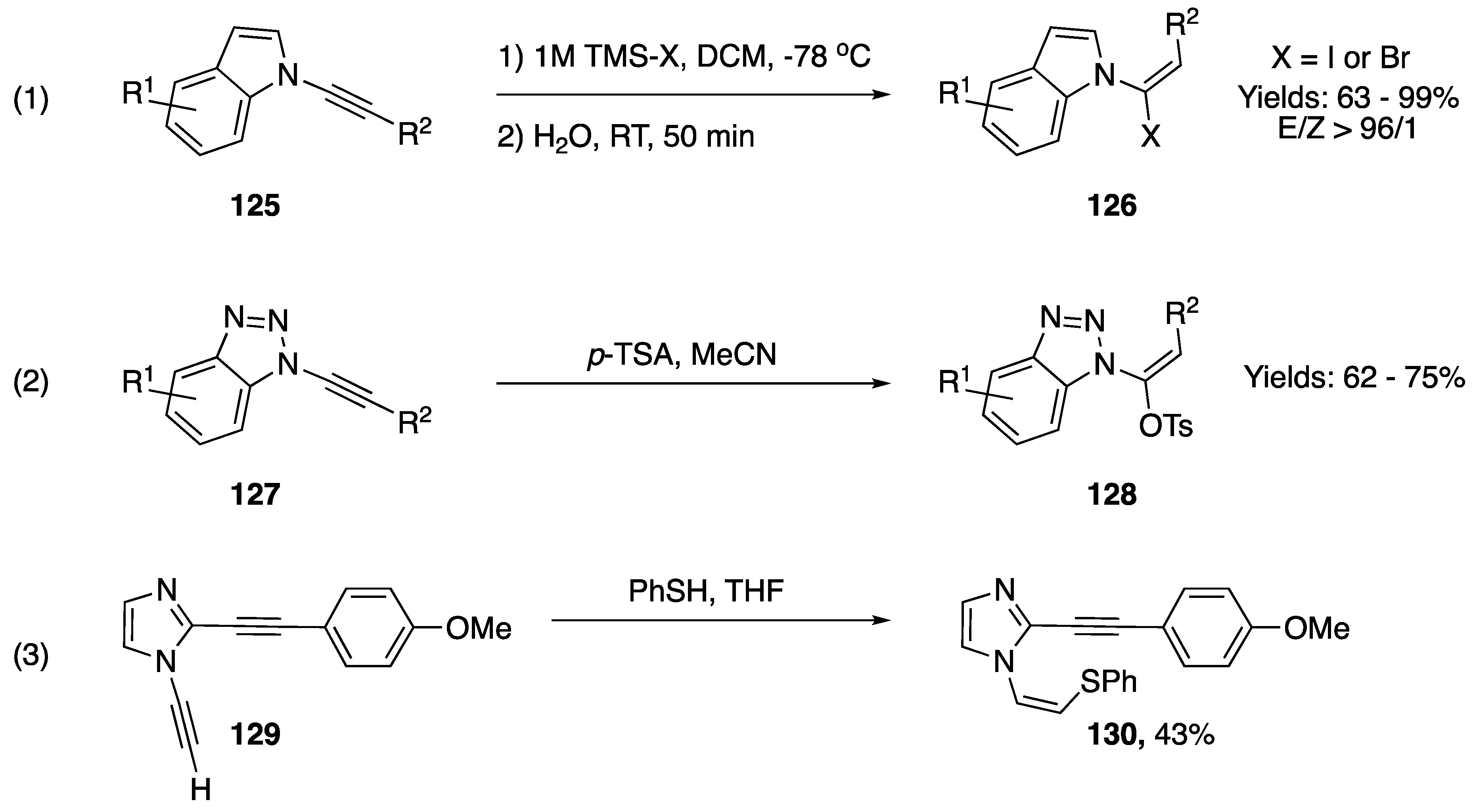
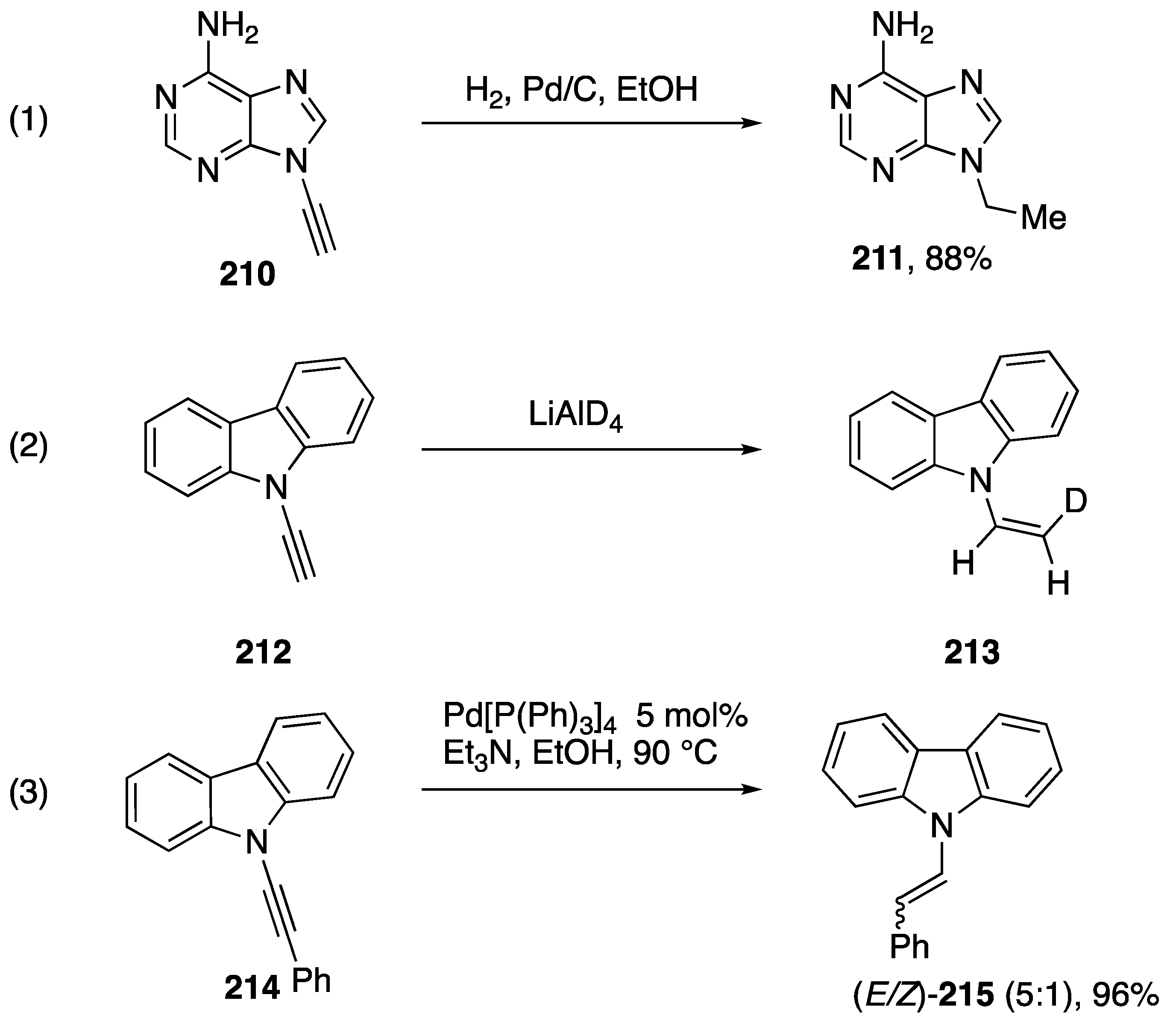
3.1. Reactions of Alkyne C-H
3.2. Addition Reactions
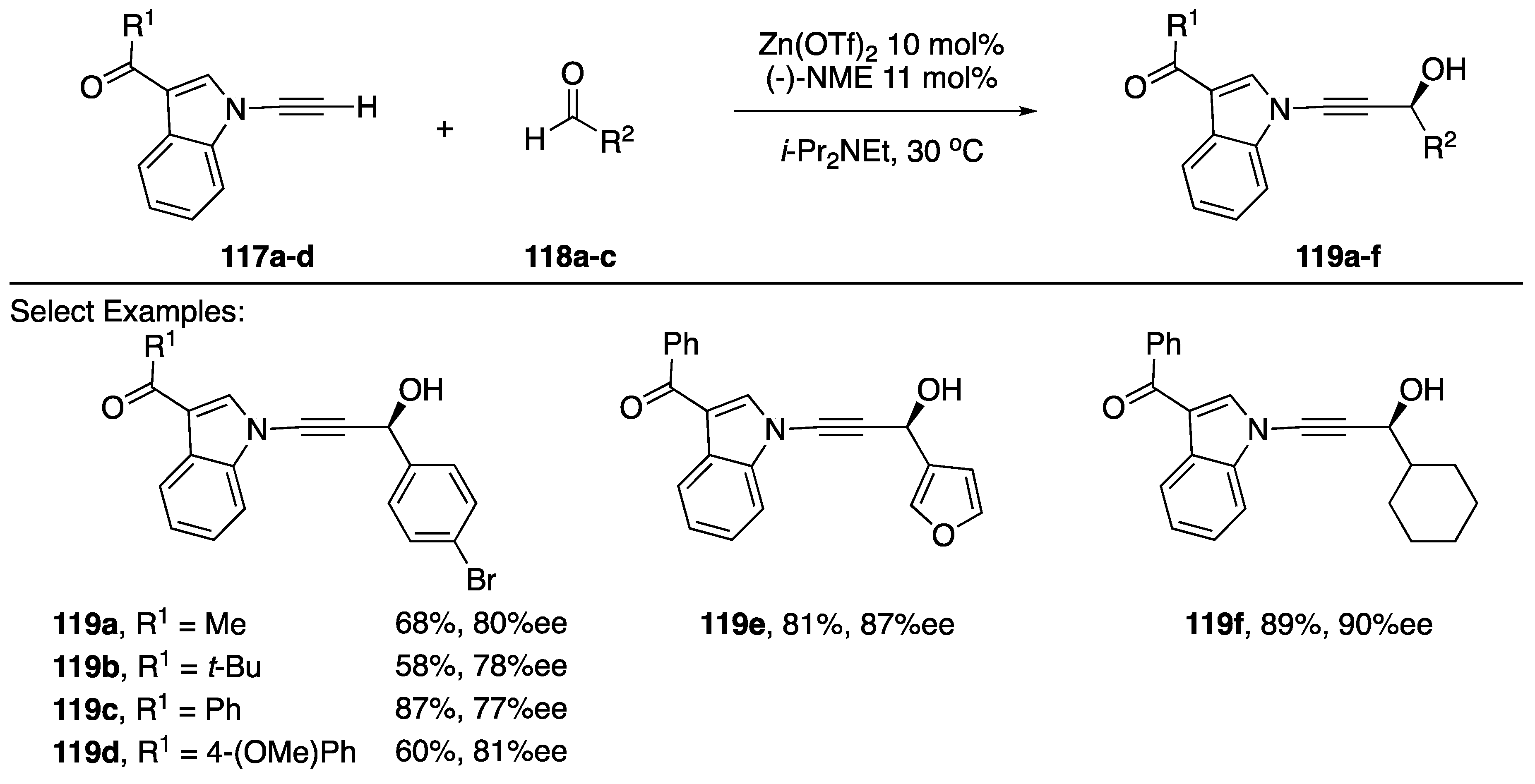
3.2.1. Carbon-Heteroatom Bond Formation
3.2.2. Carbon-Carbon Bond Formation
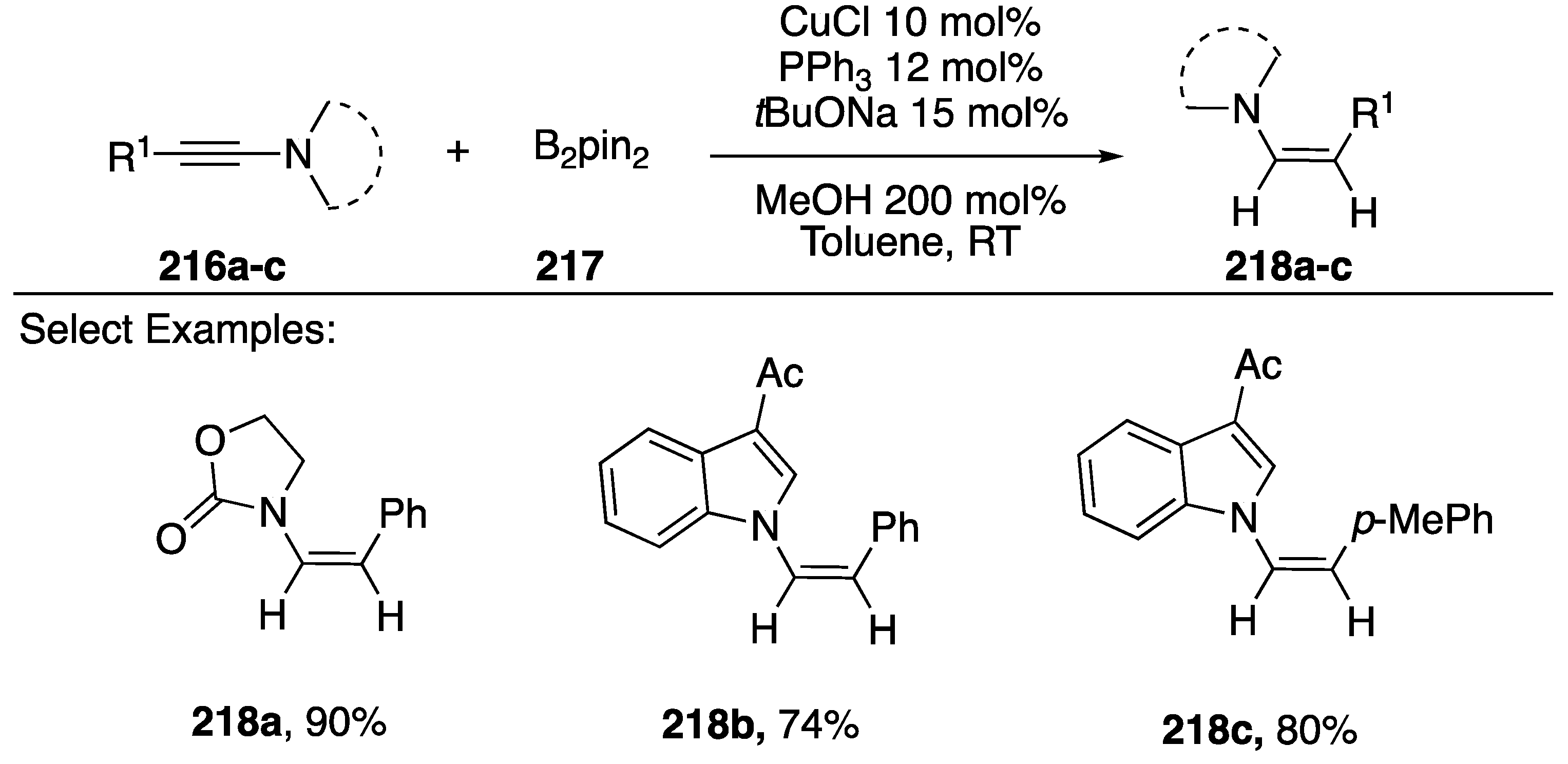
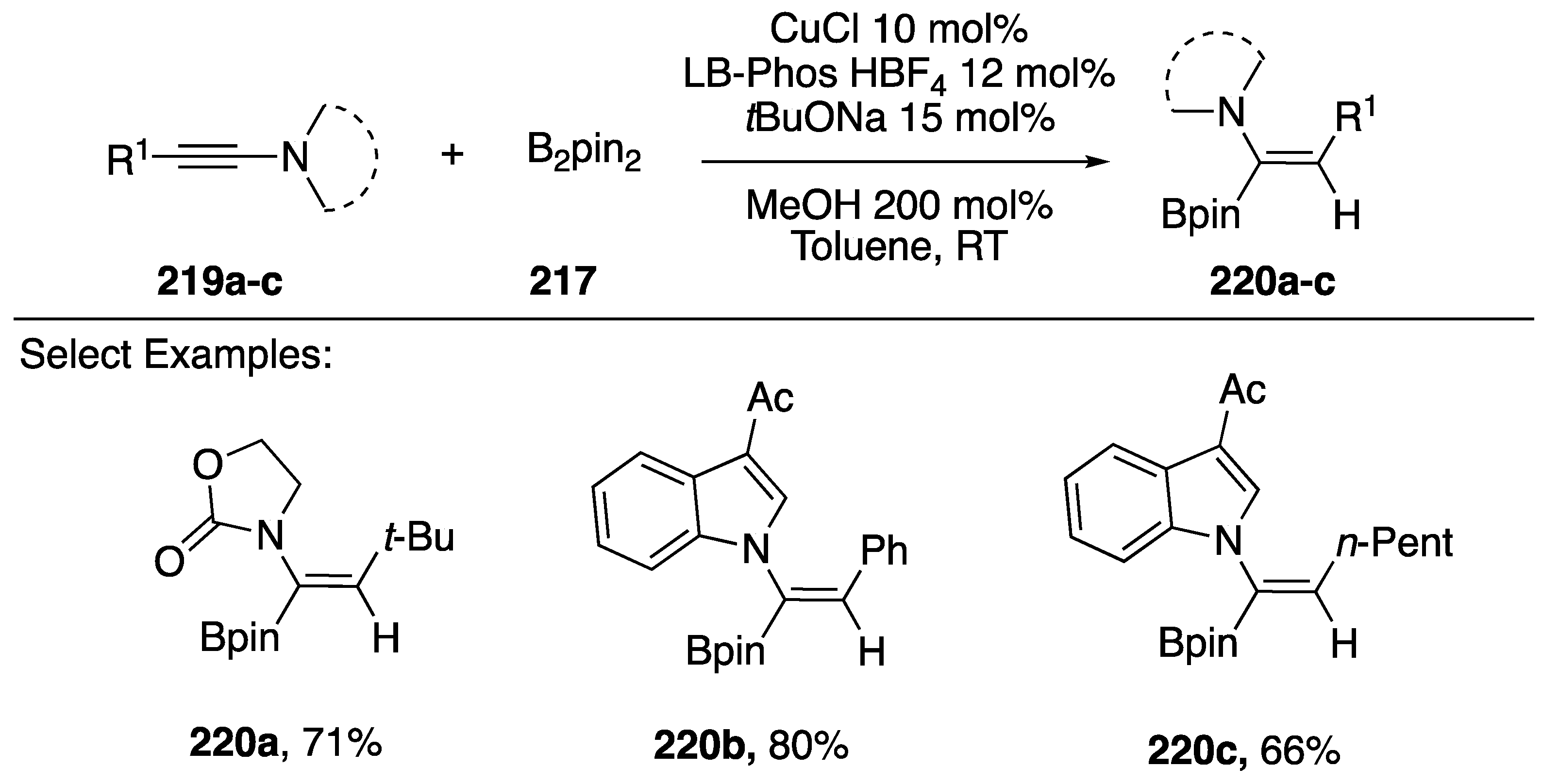
3.2.3. Carbon-Boron and Carbon-Hydrogen Bond Formation
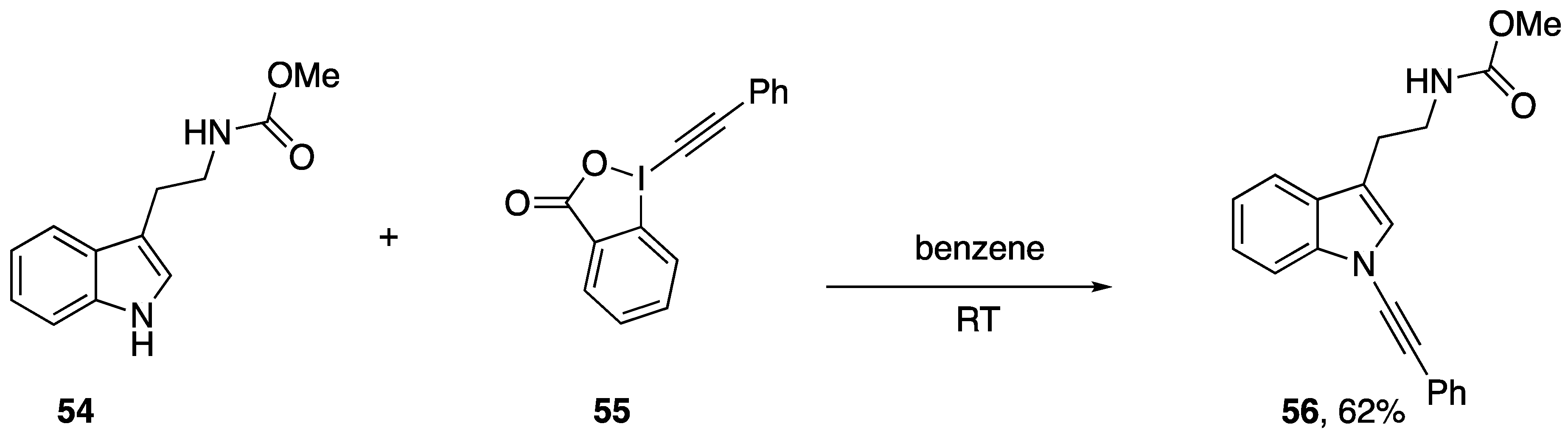
3.3. Cycloadditions and Annulations
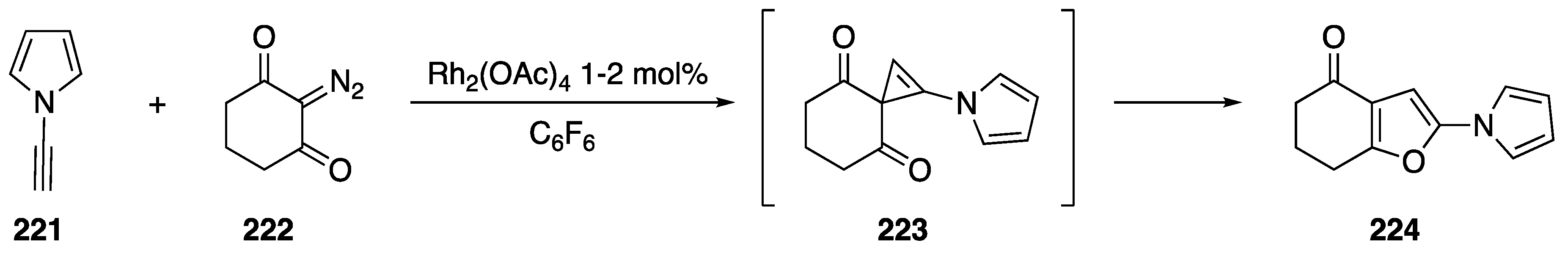
3.3.1. [2 + 1] Cycloaddition
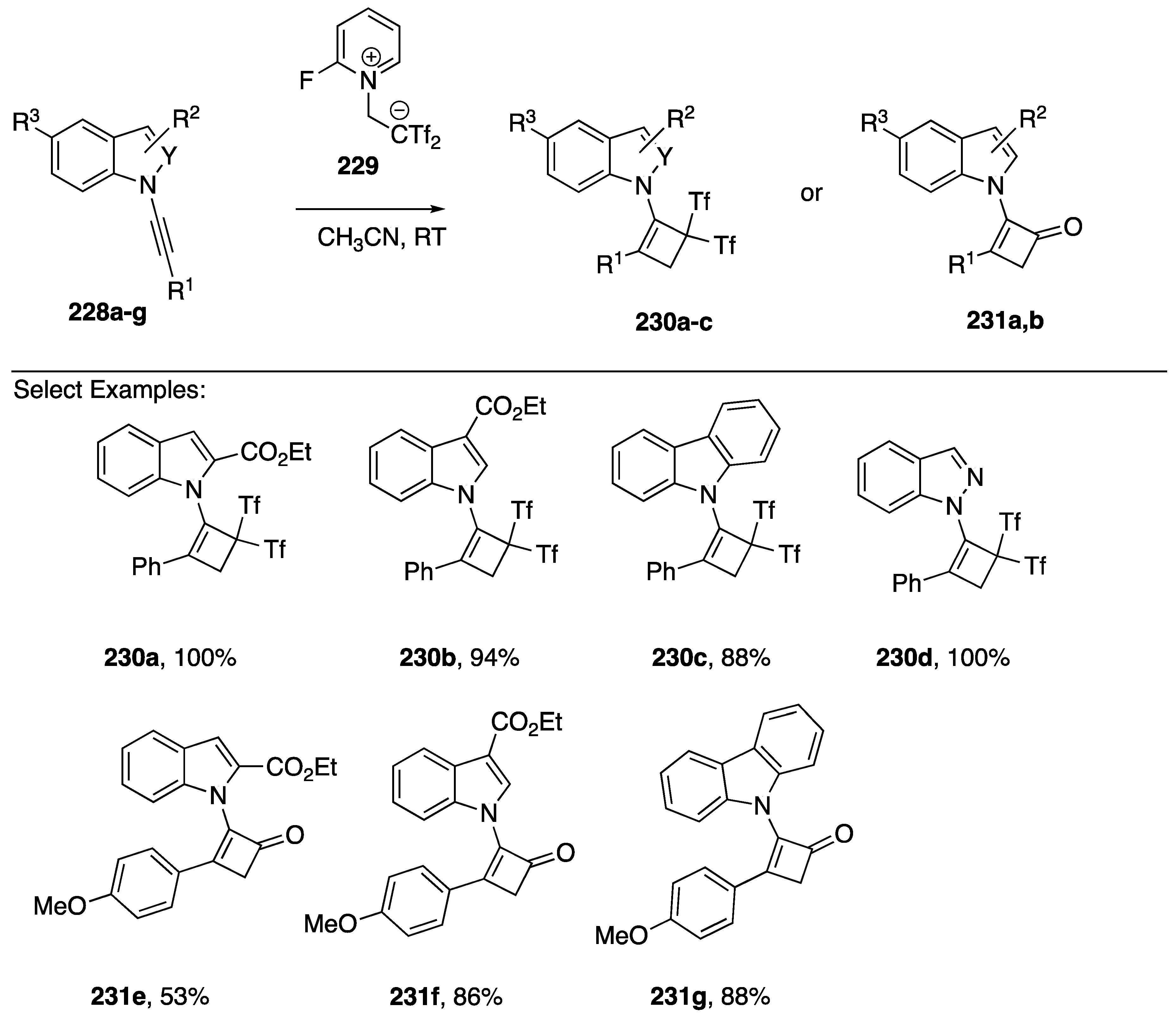
3.3.2. [2 + 2] Cycloaddition
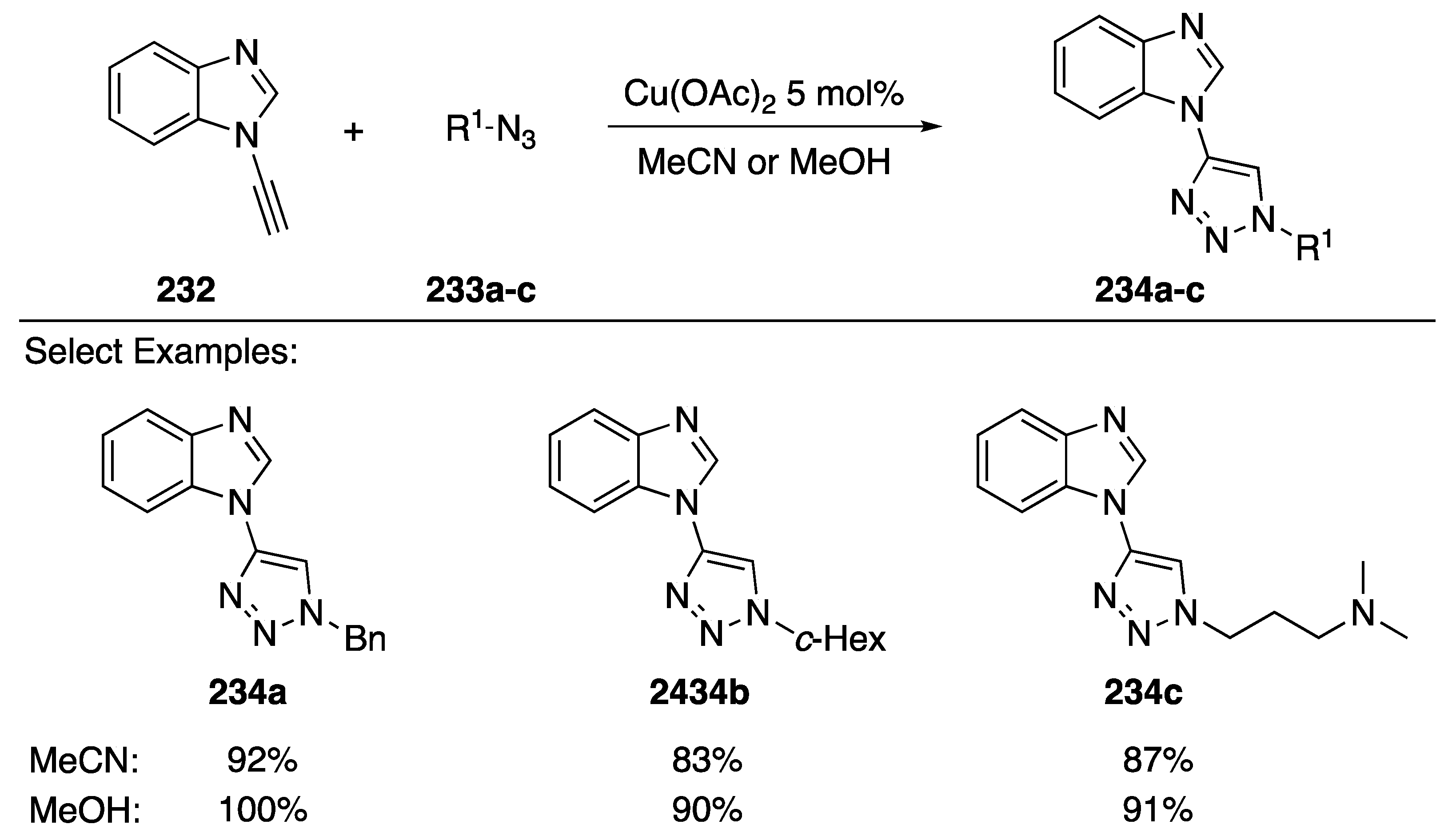
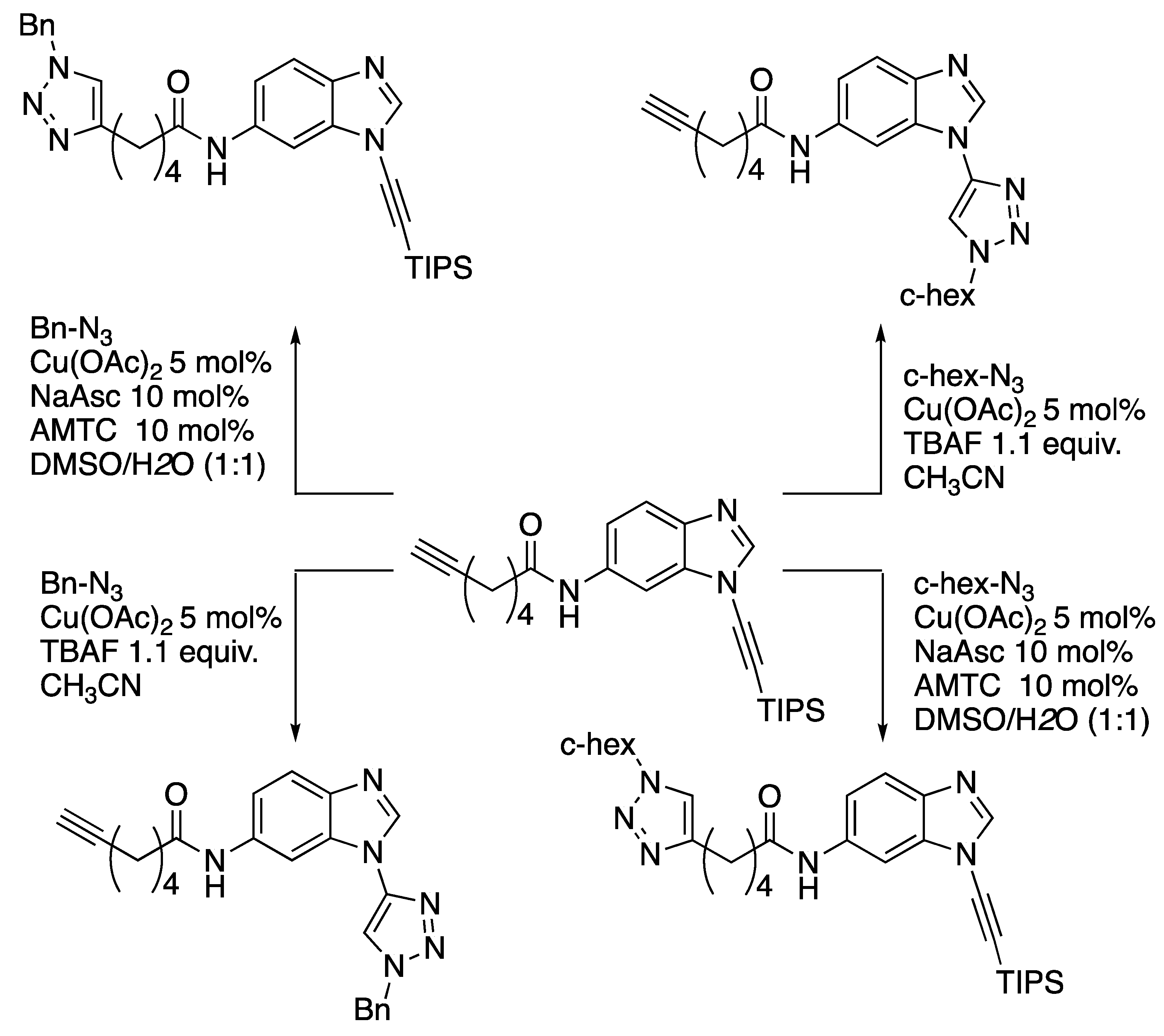
3.3.3. [3 + 2] Cycloaddition
3.3.4. [4 + 2] Cycloaddition
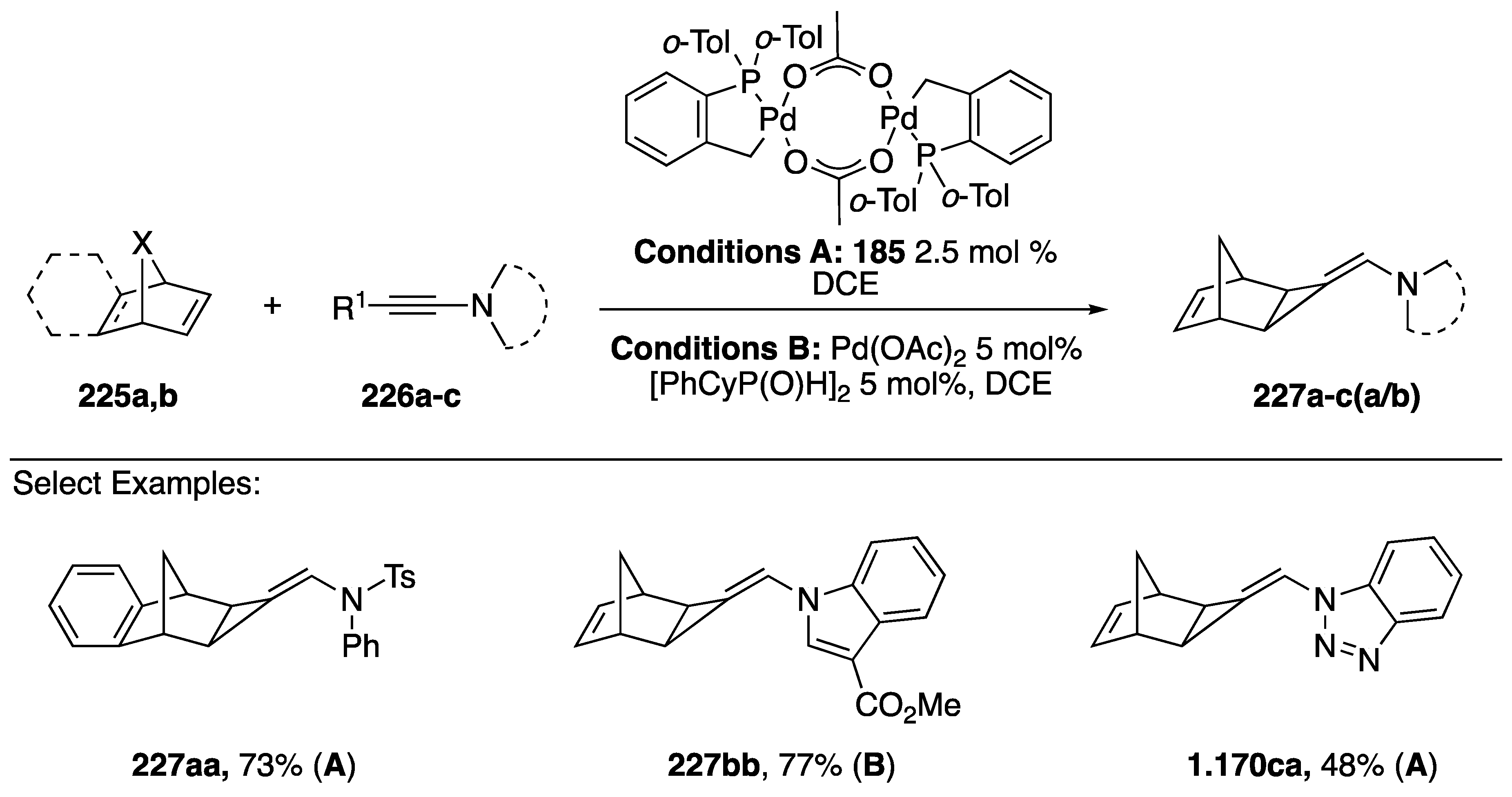
3.3.5. [2 + 2 + 2] Cycloaddition
3.3.6. [3 + 2 + 2] Cycloaddition
3.3.7. [4 + 3 + 2] Cycloaddition
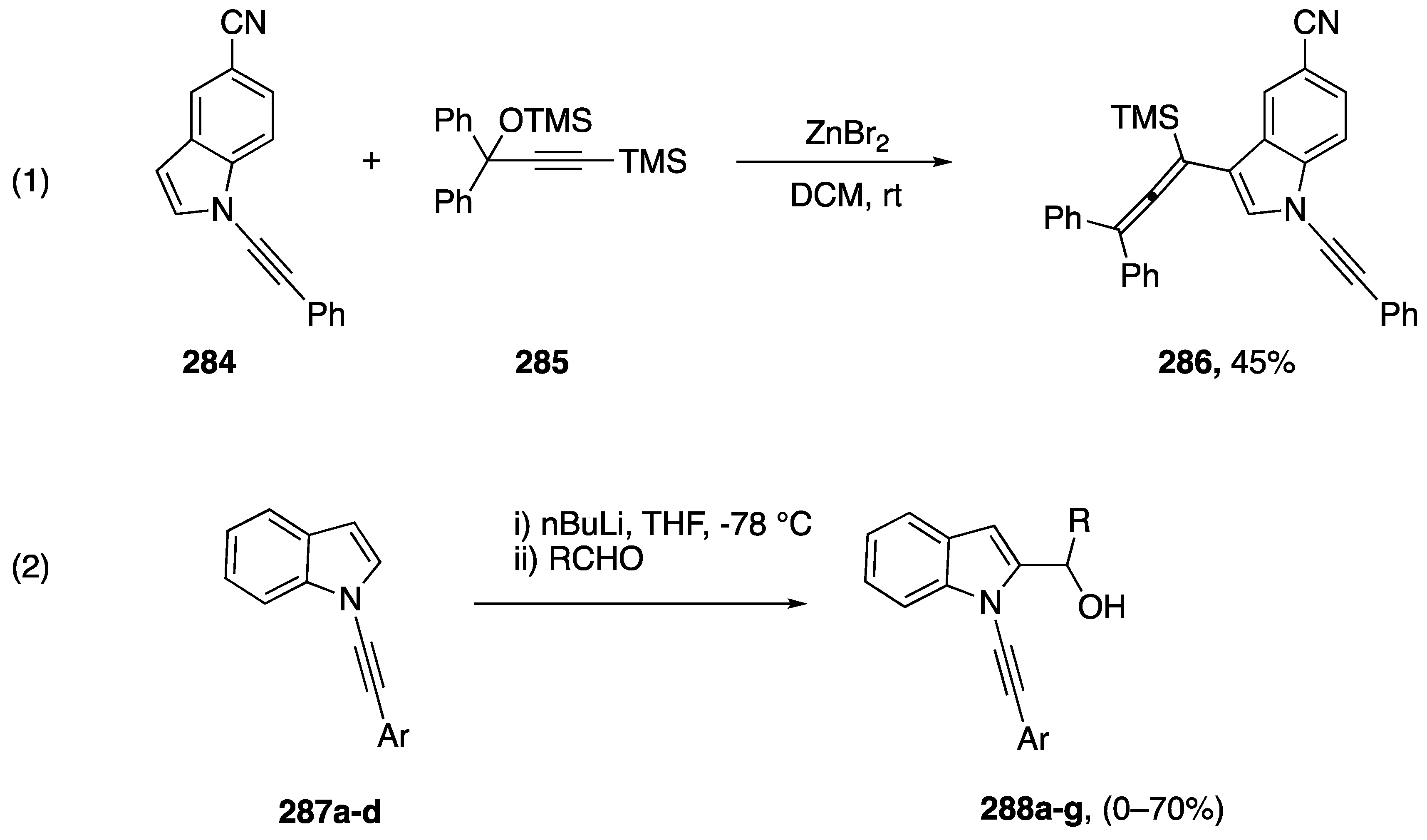
3.3.8. Other Annulations
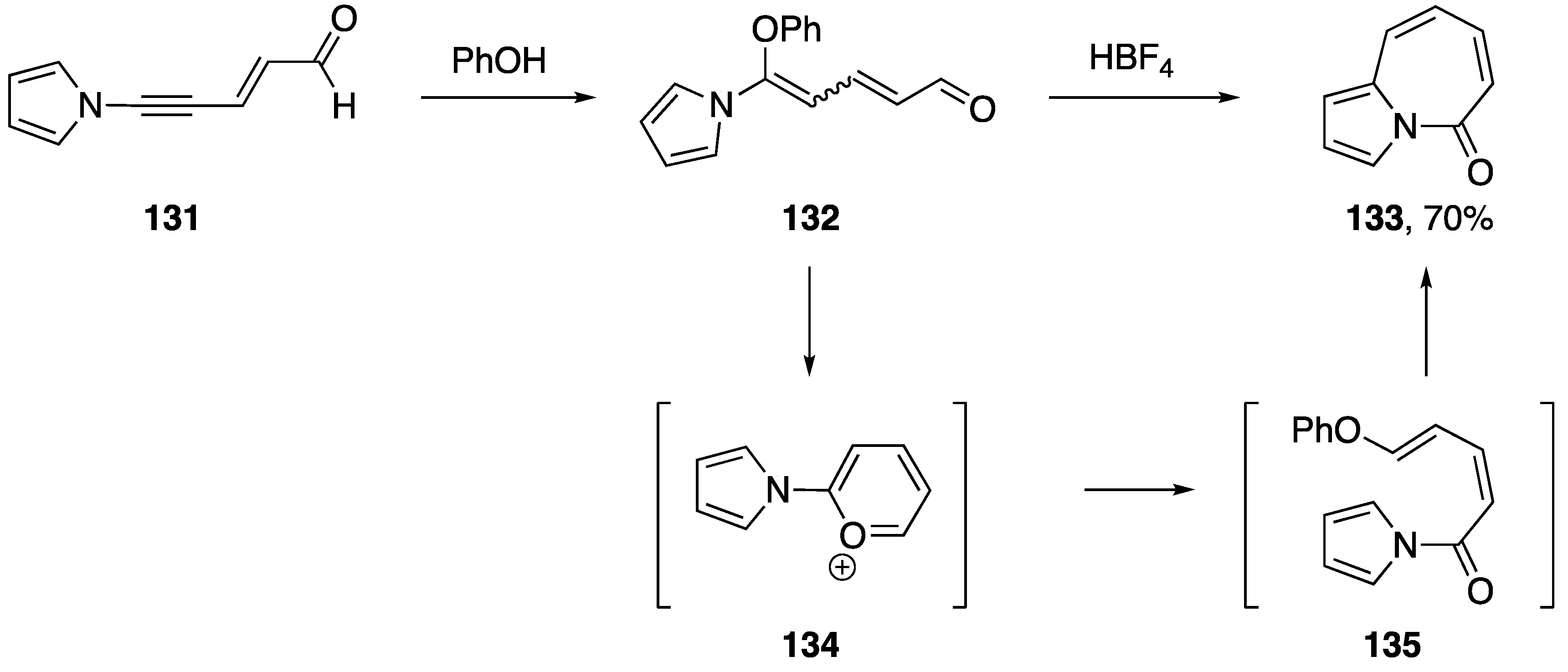
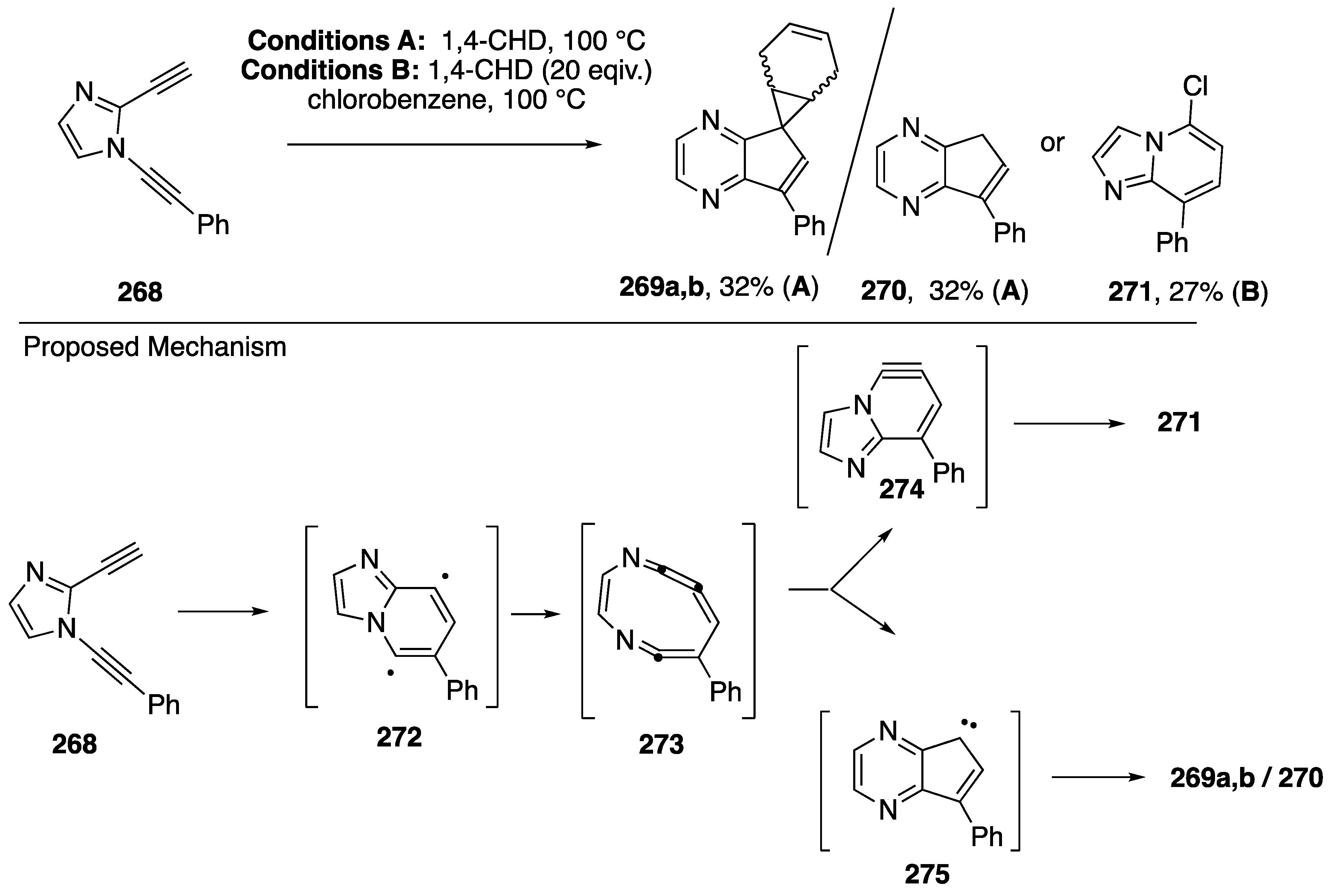
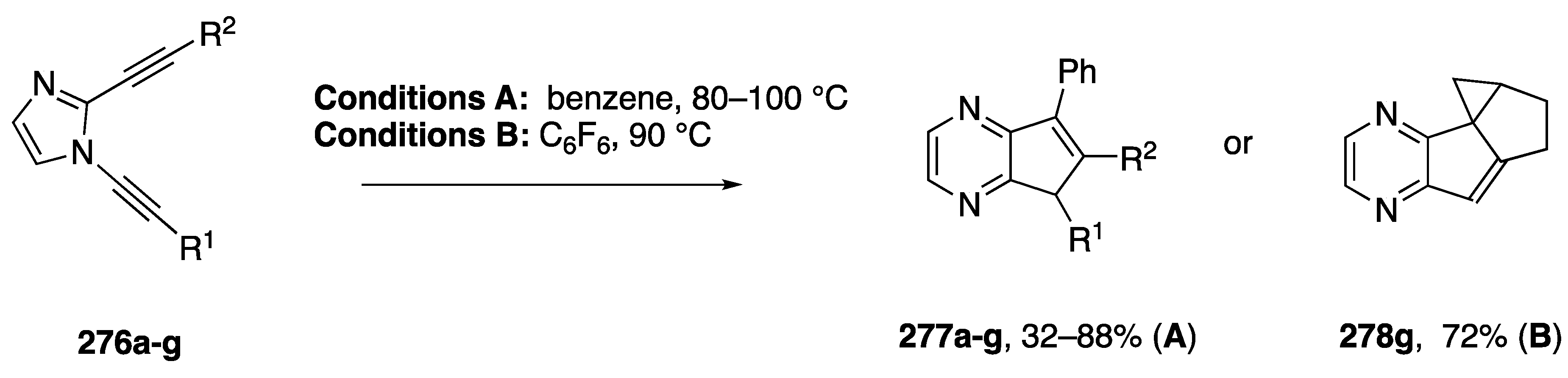
3.4. Bergman Cyclizations
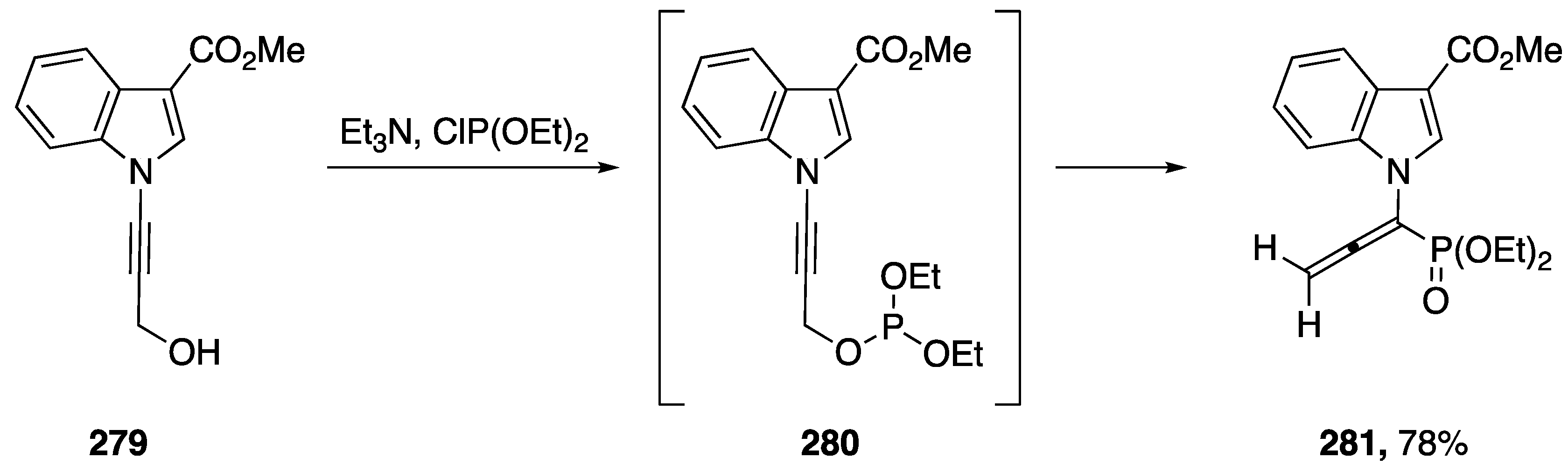
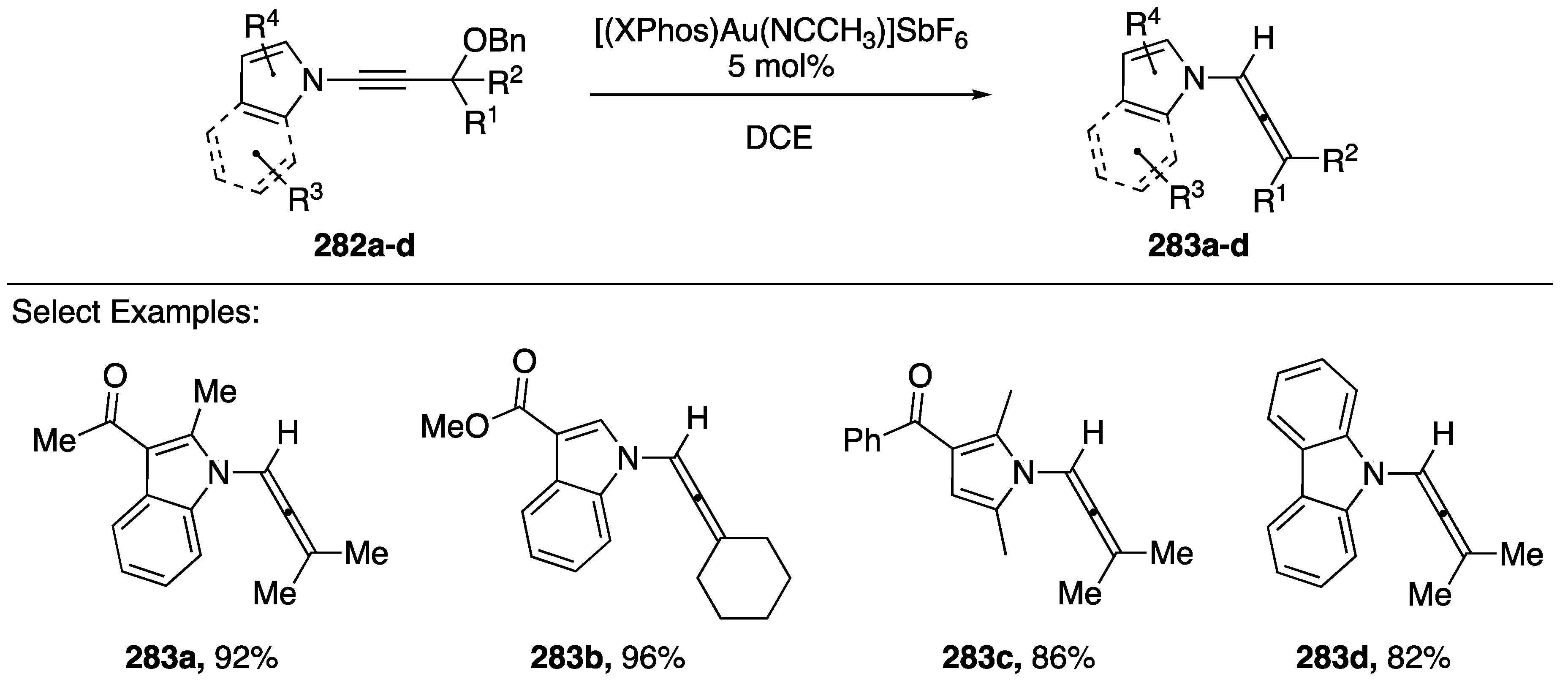
3.5. Sigmatropic Rearrangements
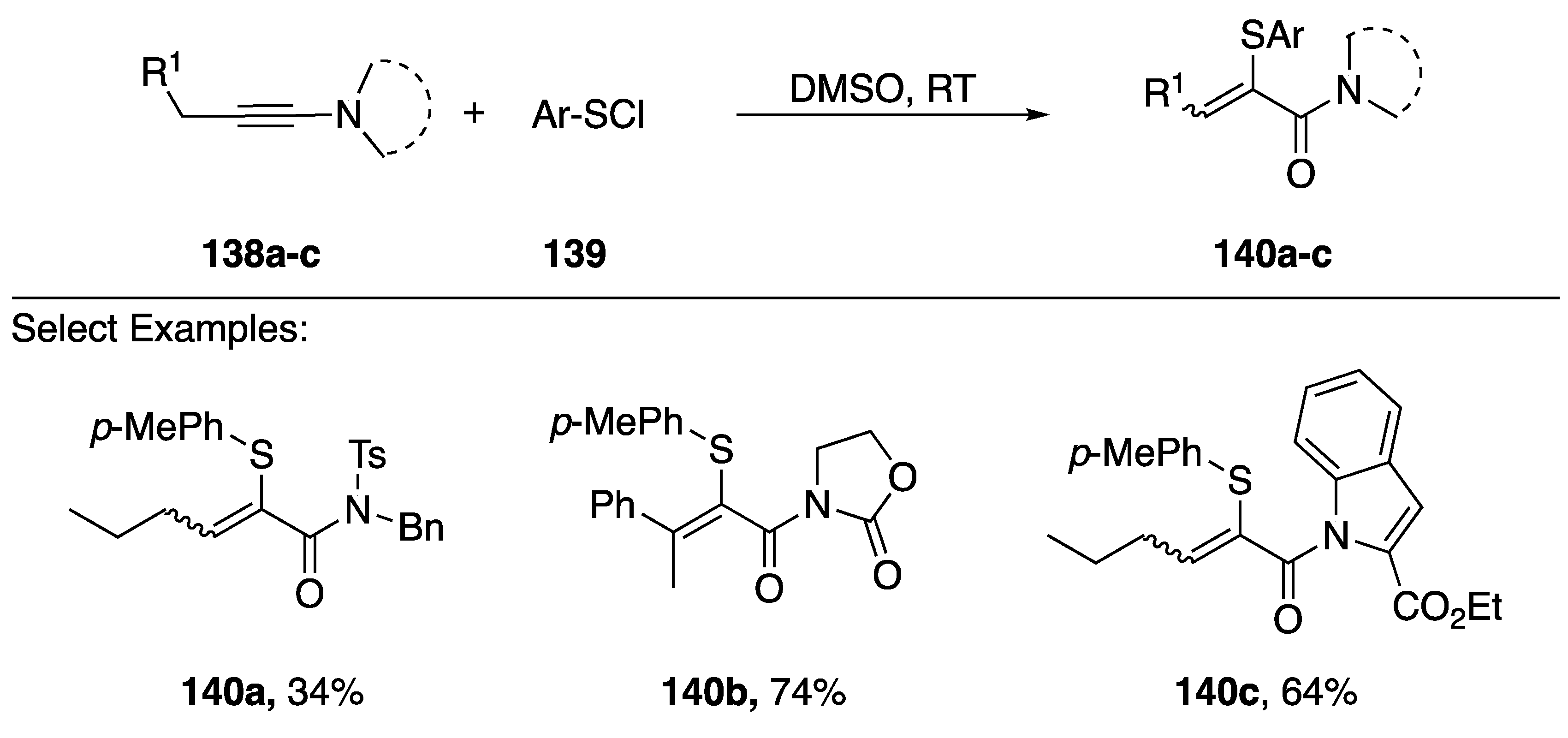
3.6. Other Reactions
4. Applications of N-Alkynyl Azoles
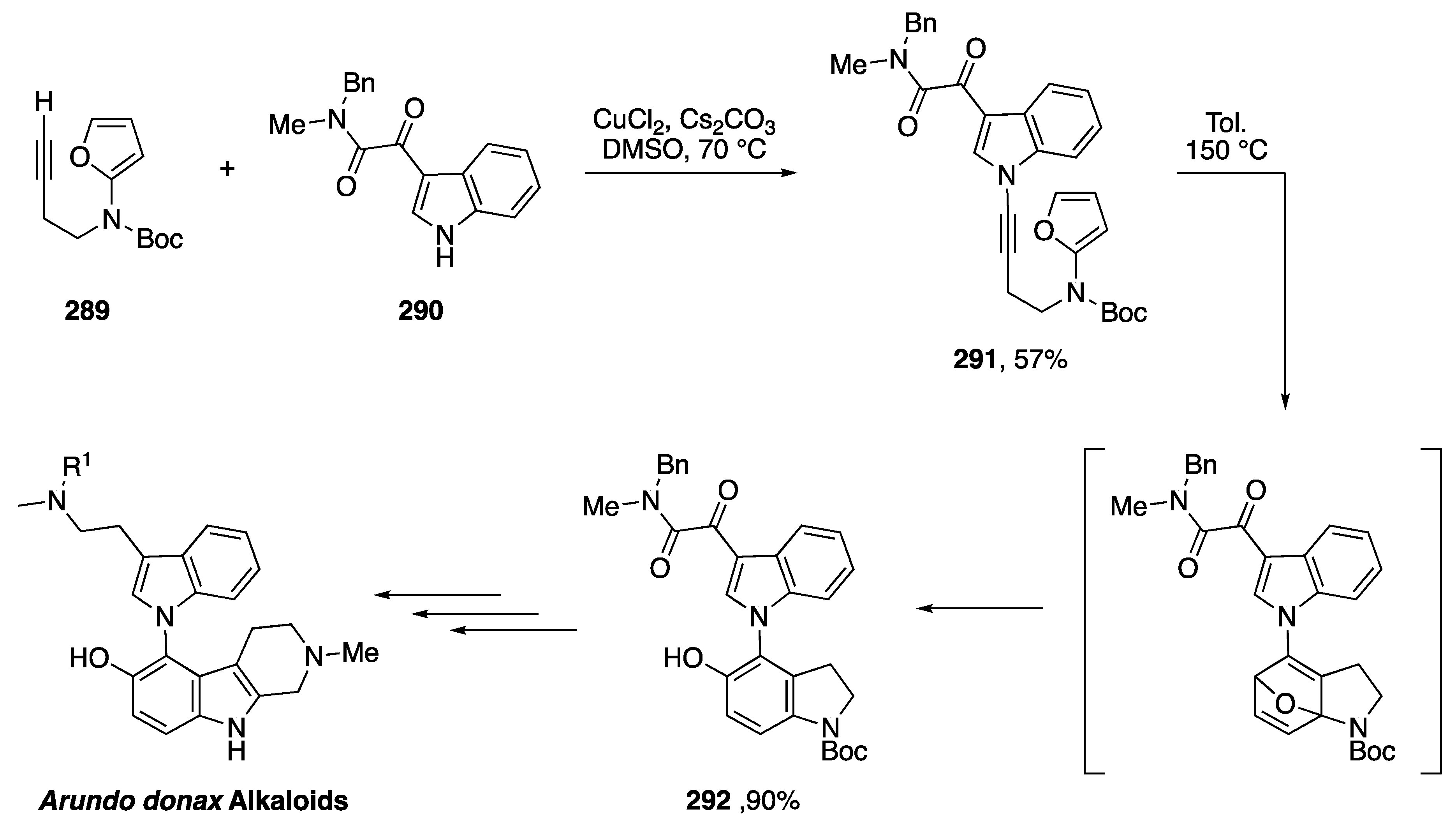
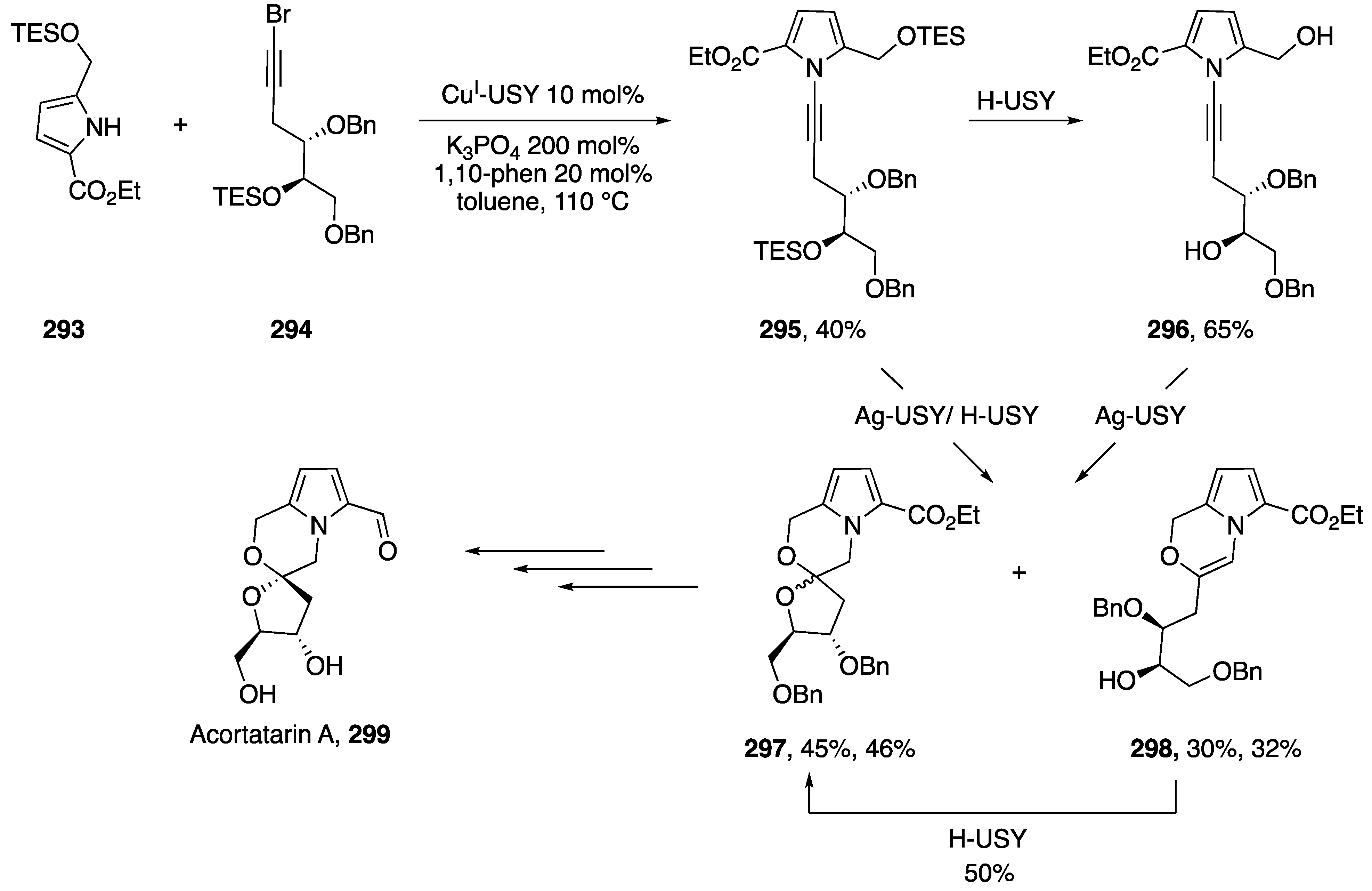
4.1. Total Syntheses
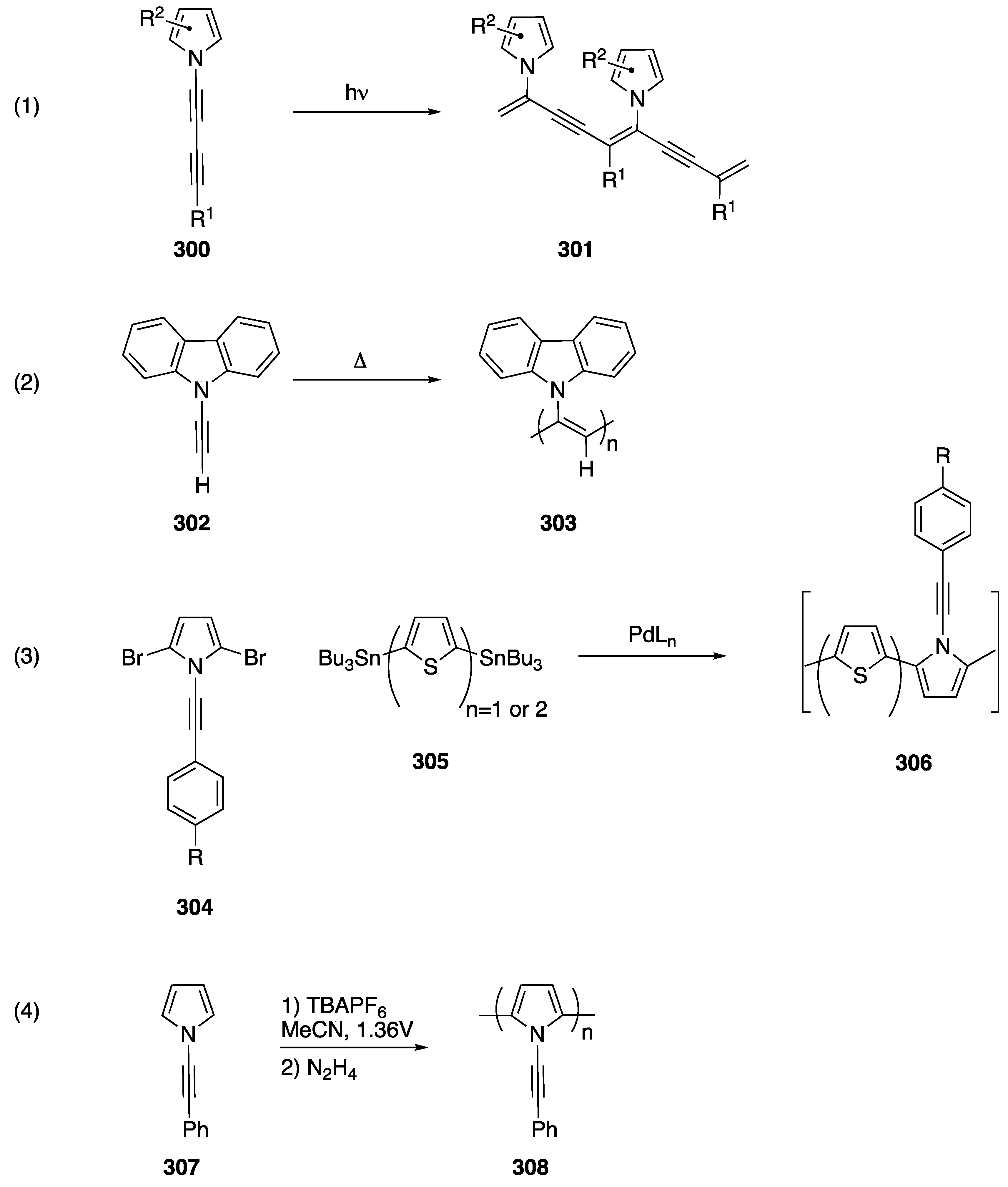
4.2. Polymer Synthesis
4.3. Other Applications
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ficini, J. Ynamine—Versatile Tool in Organic-Synthesis. Tetrahedron 1976, 32, 1449–1486. [Google Scholar] [CrossRef]
- Chiang, Y.; Kresge, A.J.; Paine, S.W.; Popik, V.V. Reactive Species: Ynols and Ynamines. J. Phys. Org. Chem. 1996, 9, 361–370. [Google Scholar] [CrossRef]
- Zificsak, C.A.; Mulder, J.A.; Hsung, R.P.; Rameshkumar, C.; Wei, L.L. Recent advances in the chemistry of ynamines and ynamides. Tetrahedron 2001, 57, 7575–7606. [Google Scholar] [CrossRef]
- Pan, F.; Shu, C.; Ye, L.W. Recent progress towards gold-catalyzed synthesis of N-containing tricyclic compounds based on ynamides. Org. Biomol. Chem. 2016, 14, 9456–9465. [Google Scholar] [CrossRef] [PubMed]
- DeKorver, K.A.; Li, H.; Lohse, A.G.; Hayashi, R.; Lu, Z.; Zhang, Y.; Hsung, R.P. Ynamides: A Modern Functional Group for the New Millennium. Chem. Rev. 2010, 110, 5064–5106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evano, G.; Blanchard, N.; Compain, G.; Coste, A.; Demmer, C.S.; Gati, W.; Guissart, C.; Heimburger, J.; Henry, N.; Jouvin, K.; et al. A Journey in the Chemistry of Ynamides: From Synthesis to Applications. Chem. Lett. 2016, 45, 574–585. [Google Scholar] [CrossRef] [Green Version]
- Katritzky, A.R.; Jiang, R.; Singh, S.K. Synthesis and reactions of N-ethynyl-heterocycles. Heterocycles 2004, 63, 1455–1475. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kundu, S.K. Synthesis and Properties of N-Ethynylcarbazoleand Poly-N-ethynylcarbazole. J. Org. Chem. 1970, 35, 4250–4252. [Google Scholar] [CrossRef]
- Viehe, H.G. Synthesis and Reactions of Alkynylamines. Angew. Chem. Int. Ed. 1967, 6, 767–778. [Google Scholar] [CrossRef]
- Pielichowski, J.; Chrzaszcz, R. A New Method of Synthesis of 9-Ethynylcarbazole and Its Derivatives. Bull. Soc. Chim. Belg. 1995, 104, 117–118. [Google Scholar] [CrossRef]
- Burger, U.; Dreier, F. Reactions of Nitrogen Containing Aromatic Anions with Chlorocarbene. Tetrahedron 1983, 39, 2065–2071. [Google Scholar] [CrossRef]
- Deangelis, F.; Gambacorta, A.; Nicoletti, R. Improved Route for Ring Expansion of 5-Membered Heterocyclic-Compounds by Use of Phase Transfer Catalysts. Synthesis 1976, 798–800. [Google Scholar] [CrossRef]
- Paley, M.S.; Frazier, D.O.; Abeledeyem, H.; McManus, S.P.; Zutaut, S.E. Synthesis, Vapor Growth, Polymerization, and Characterization of Thin-Films of Novel Diacetylene Derivatives of Pyrrole—The Use of Computer Modeling to Predict Chemical and Optical-Properties of these Diacetylenes and Poly(Diacetylenes). J. Am. Chem. Soc. 1992, 114, 3247–3251. [Google Scholar] [CrossRef]
- Brandsma, L.; Mal’kina, A.G.; Trofimov, B.A. An Improved Procedure for N-Ethynylpyrrole. Synth. Commun. 1994, 24, 2721–2724. [Google Scholar] [CrossRef]
- Joshi, R.V.; Xu, Z.-Q.; Ksebati, M.B.; Kessel, D.; Corbett, T.H.; Drach, J.C.; Zemlicka, J. Synthesis, Transformations and Biological Activity of Chloro Enamines and Ynamines Derived from Chloroalkenyl- and Alkynyl-N-substituted Purine and Pyrimidine Bases of Nucleic Acids. J. Chem. Soc. Perkin Trans. 1 1994, 1089–1098. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Zhang, S.M.; Hussein, A.H.M.; Fang, Y.F.; Steel, P.J. One-carbon homologation of carboxylic acids via BtCH(2)TMS: A safe alternative to the Arndt-Eistert reaction. J. Org. Chem. 2001, 66, 5606–5612. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Abdel-Fattah, A.A.A.; Wang, M. A Novel Access to Disubstituted Acetylenes. J. Org. Chem. 2002, 67, 7526–7529. [Google Scholar] [CrossRef] [PubMed]
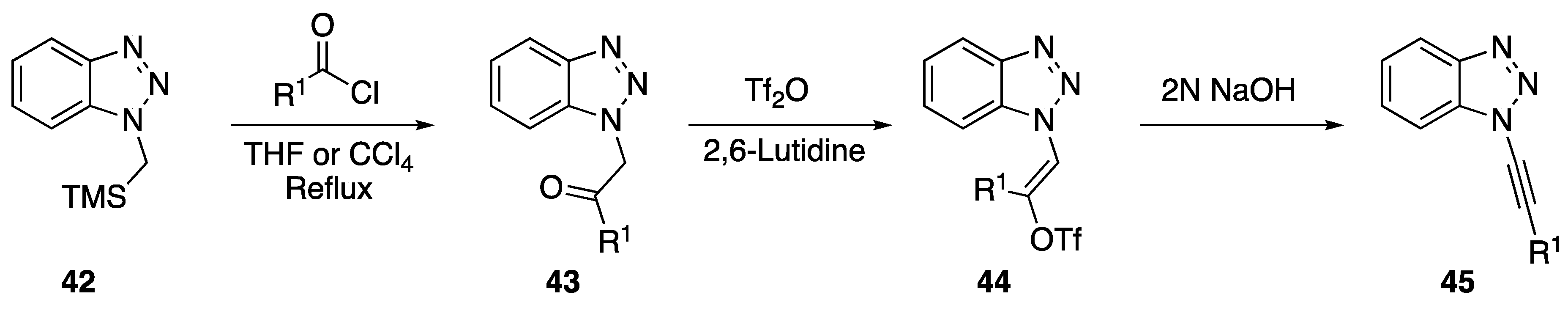
- Katritzky, A.R.; Singh, S.K.; Jiang, R. A convenient synthesis of functionalized N-(ethynyl)benzotriazoles. Tetrahedron 2006, 62, 3794–3797. [Google Scholar] [CrossRef]
- Mansfield, S.J.; Campbell, C.D.; Jones, M.W.; Anderson, E.A. A robust and modular synthesis of ynamides. Chem. Commun. 2015, 51, 3316–3319. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.L.; Zeng, X.Z.; Wang, H.; Zhao, J.F. A Robust One-Step Approach to Ynamides. Org. Lett. 2018, 20, 280–283. [Google Scholar] [CrossRef]
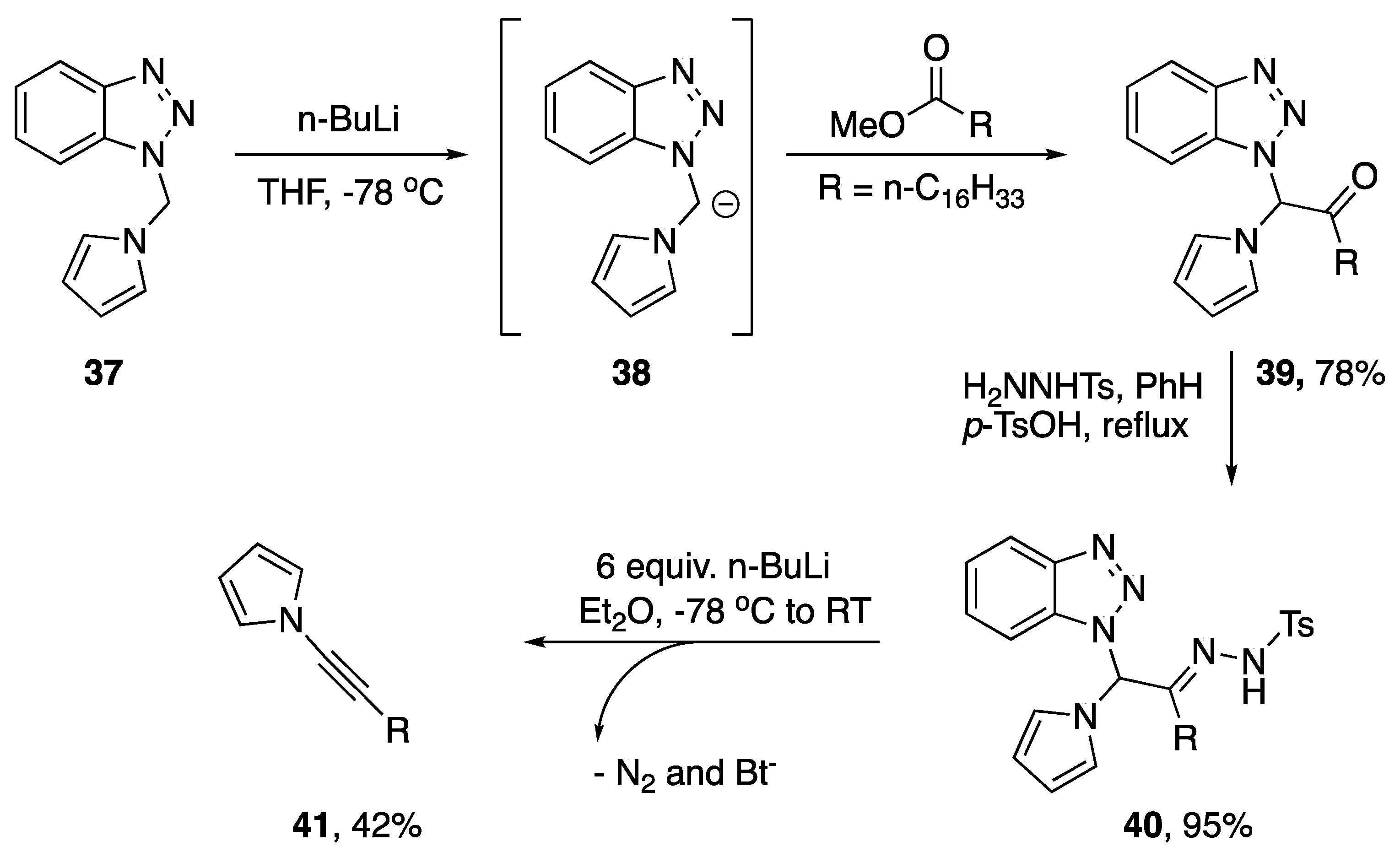
- Katritzky, A.R.; Wang, J.; Karodia, N.; Li, J. A Novel Transformation of Esters to Alkynes with 1-Substituted Benzotriazoles. J. Org. Chem. 1997, 62, 4142–4147. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Xiao, J.; Peng, Z.; Dong, W.; An, D. Base-Induced One-Pot Preparation of N- or P-Substituted Alkynes. Eur. J. Org. Chem. 2015, 2015, 7806–7815. [Google Scholar] [CrossRef]
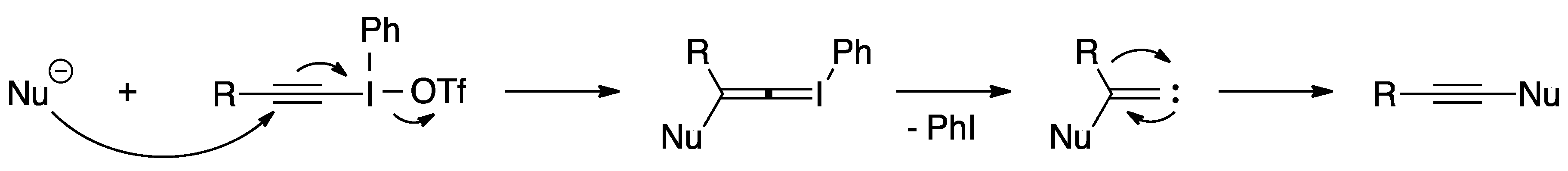
- Zhdankin, V.V.; Stang, P.J. Alkynyliodonium salts in organic synthesis. Tetrahedron 1998, 54, 10927–10966. [Google Scholar] [CrossRef]
- Brand, J.P.; Waser, J. Electrophilic alkynylation: The dark side of acetylene chemistry. Chem. Soc. Rev. 2012, 41, 4165–4179. [Google Scholar] [CrossRef] [PubMed]
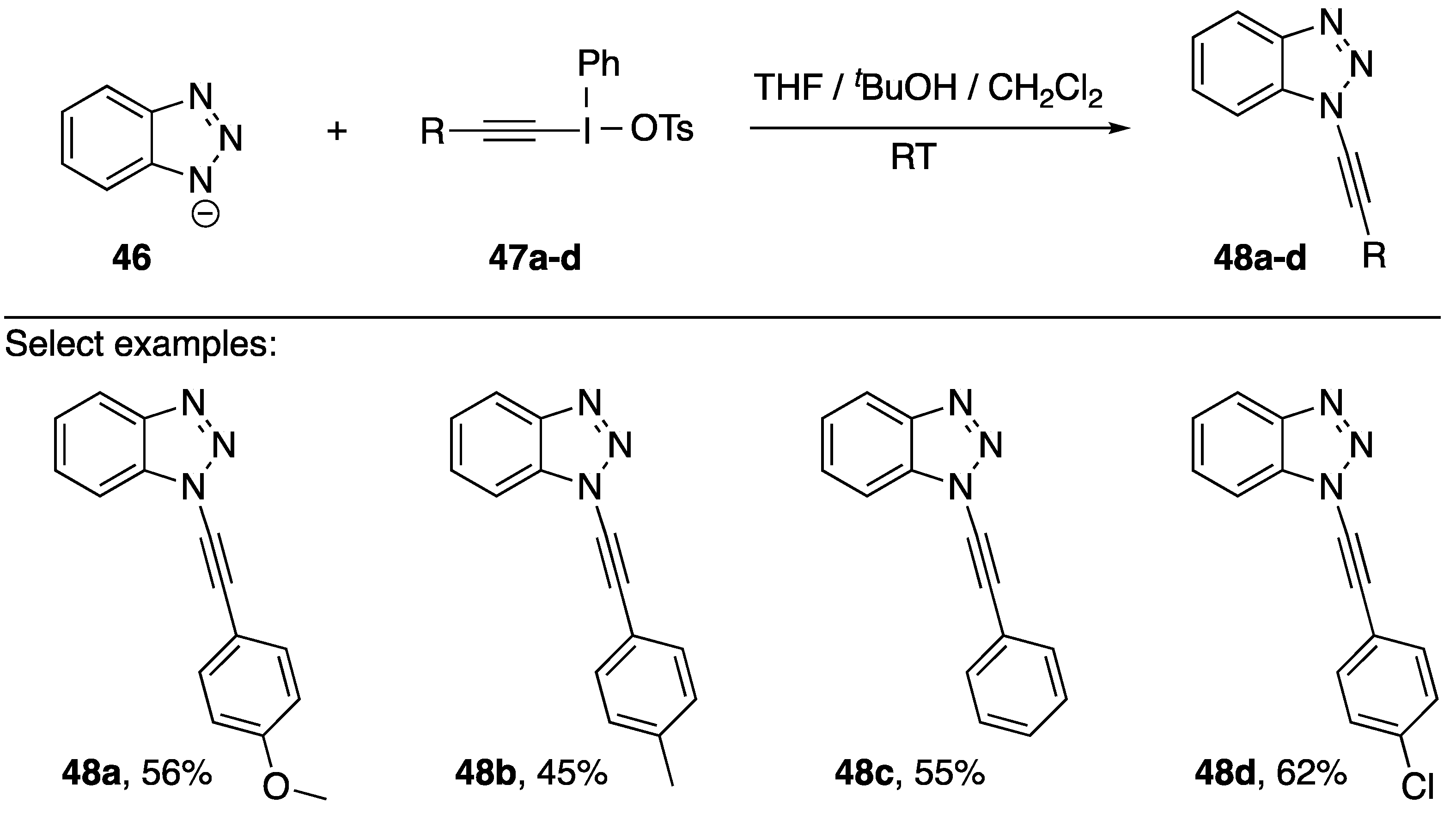
- Kitamura, T.; Tashi, N.; Tsuda, K.; Chen, H.Y.; Fujiwara, Y. A convenient synthesis of new 1-alkynyl-1H-benzotriazoles by reaction of alkynyl(phenyl)iodonium salts with benzotriazole ion. Heterocycles 2000, 52, 303–312. [Google Scholar] [CrossRef]
- Kitamura, T.; Tashi, N.; Tsuda, K.; Fujiwara, Y. Alkynylbenzotriazoles by direct alkynylation of benzotriazole using alkynyliodonium salts. Tetrahedron Lett. 1998, 39, 3787–3790. [Google Scholar] [CrossRef]
- Kitamura, T.; Morshed, M.H.; Tsukada, S.; Miyazaki, Y.; Iguchi, N.; Inoue, D. Alkynylation of benzotriazole with silylethynyliodonium triflates. Regioselective synthesis of 2-ethynyl-2H-benzotriazole derivatives. J. Org. Chem. 2011, 76, 8117–8120. [Google Scholar] [CrossRef] [PubMed]
- Davydov, D.Y.; Oprunenko, Y.F.; Beletskaya, I.P. Pd/Al2O3-catalysed regioselective N-1-modification of benzotriazoles using iodonium salts. Tetrahedron Lett. 2017, 58, 4465–4467. [Google Scholar] [CrossRef]
- Huang, X.; Zhu, Q. Preparation of resin-bound alkynyl iodonium salts and their application in organic synthesis as alkynyl transfer reagents. Tetrahedron Lett. 2001, 42, 6373–6375. [Google Scholar] [CrossRef]
- Nadipuram, A.N.; David, W.M.; Kumar, D.; Kerwin, S.M. Synthesis and Thermolysis of Heterocyclic 3-Aza-3-ene-1,5-diynes. Org. Lett. 2002, 4, 4543–4546. [Google Scholar] [CrossRef]
- Kerwin, S.M.; Nadipuram, A. 5H-Cyclopentapyrazines from 1,2-Dialkynylimidazoles. Synlett 2004, 1404–1408. [Google Scholar] [CrossRef]
- Roy, A.; Das, M.K.; Chaudhuri, S.; Bisai, A. Transition-Metal Free Oxidative Alkynylation of 2-Oxindoles with Ethynylbenziodoxolone (EBX) Reagents. J. Org. Chem. 2018, 83, 403–421. [Google Scholar] [CrossRef] [PubMed]
- Toriumi, N.; Asano, N.; Miyamoto, K.; Muranaka, A.; Uchiyama, M. N-Alkynylpyridinium Salts: Highly Electrophilic Alkyne-Pyridine Conjugates as Precursors of Cationic Nitrogen-Embedded Polycyclic Aromatic Hydrocarbons. J. Am. Chem. Soc. 2018, 140, 3858–3862. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Review on Recent Progress in Nitrogen-Doped Graphene: Synthesis, Characterization, and Its Potential Applications. ACS Catal. 2012, 2, 781–794. [Google Scholar] [CrossRef] [Green Version]
- Frederick, M.O.; Mulder, J.A.; Tracey, M.R.; Hsung, R.P.; Huang, J.; Kurtz, K.C.M.; Shen, L.C.; Douglas, C.J. A copper-catalyzed C-N bond formation involving sp-hybridized carbons. A direct entry to chiral ynamides via N-alkynylation of amides. J. Am. Chem. Soc. 2003, 125, 2368–2369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.S.; Hsung, R.P.; Tracey, M.R.; Kurtz, K.C.M.; Vera, E.L. Copper sulfate-pentahydrate-1,10-phenanthroline catalyzed amidations of alkynyl bromides. Synthesis of heteroaromatic amine substituted ynamides. Org. Lett. 2004, 6, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Zhang, Y.S.; Huang, J.; Hsung, R.P.; Kurtz, K.C.M.; Oppenheimer, J.; Petersen, M.E.; Sagamanova, I.K.; Shen, L.C.; Tracey, M.R. Copper(II)-catalyzed amidations of alkynyl bromides as a general synthesis of ynamides and Z-enamides. An intramolecular amidation for the synthesis of macrocyclic ynamides. J. Org. Chem. 2006, 71, 4170–4177. [Google Scholar] [CrossRef] [PubMed]
- Laroche, C.; Li, J.; Freyer, M.W.; Kerwin, S.M. Coupling reactions of bromoalkynes with imidazoles mediated by copper salts: Synthesis of novel N-alkynylimidazoles. J. Org. Chem. 2008, 73, 6462–6465. [Google Scholar] [CrossRef]
- Hirano, S.; Tanaka, R.; Urabe, H.; Sato, F. Practical preparation of N-(1-alkynyl)sulfonamides and their remote diastereoselective addition to aldehydes via titanation. Org. Lett. 2004, 6, 727–729. [Google Scholar] [CrossRef]
- Dunetz, J.R.; Danheiser, R.L. Copper-mediated N-alkynylation of carbamates, ureas, and sulfonamides. A general method for the synthesis of ynamides. Org. Lett. 2003, 5, 4011–4014. [Google Scholar] [CrossRef]
- Reinus, B.J.; Kerwin, S.M. A Copper-Catalyzed N-Alkynylation Route to 2-Substituted N-Alkynyl Pyrroles and Their Cyclization into Pyrrolo 2,1-c oxazin-1-ones: A Formal Total Synthesis of Peramine. Synthesis 2017, 49, 2544–2554. [Google Scholar]
- Ziegler, D.T.; Choi, J.; Munoz-Molina, J.M.; Bissember, A.C.; Peters, J.C.; Fu, G.C. A Versatile Approach to Ullmann C-N Couplings at Room Temperature: New Families of Nucleophiles and Electrophiles for Photoinduced, Copper-Catalyzed Processes. J. Am. Chem. Soc. 2013, 135, 13107–13112. [Google Scholar] [CrossRef] [PubMed]
- Burley, G.A.; Davies, D.L.; Griffith, G.A.; Lee, M.; Singh, K. Cu-Catalyzed N-Alkynylation of Imidazoles, Benzimidazoles, Indazoles, and Pyrazoles Using PEG as Solvent Medium. J. Org. Chem. 2010, 75, 980–983. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Reddy, G.C.; Balasubramanyam, P.; Salvanna, N. Copper(II) Oxide Catalyzed Ligand-Free Coupling Reaction of Heteroarenes with Bromoalkynes. Synthesis 2011, 816–820. [Google Scholar] [CrossRef]
- Song, J.H.; Wang, Q.B.; Fu, H.X.; Fu, S.L.; Li, S.B.; Shi, F.; Wu, C.R. Copper-Catalyzed N-Alkynylation of N-tert-Butyloxycarbonyl (Boc)-Protected Indoles. Asian J. Org. Chem. 2013, 2, 877–881. [Google Scholar] [CrossRef]
- Harkat, H.; Borghèse, S.; Nigris, M.D.; Kiselev, S.; Bénéteau, V.; Pale, P. Zeo-Click Synthesis: Copper-Zeolite-Catalyzed Synthesis of Ynamides. Adv. Synth. Catal. 2014, 356, 3842–3848. [Google Scholar] [CrossRef]
- Yao, B.B.; Liang, Z.J.; Niu, T.M.; Zhang, Y. Iron-Catalyzed Amidation of Alkynyl Bromides: A Facile Route for the Preparation of Ynamides. J. Org. Chem. 2009, 74, 4630–4633. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Salvanna, N.; Reddy, G.C.; Balasubramanyam, P. Copper-catalyzed coupling of imidazoles and s with 1,1-dibromo-1-alkenes: A distinct approach for direct N-alkynylation of heteroarenes. Tetrahedron Lett. 2011, 52, 6497–6500. [Google Scholar] [CrossRef]
- Wang, M.G.; Wu, J.; Shang, Z.C. A Simple and Efficient Copper-Catalyzed Synthesis of N-Alkynylimidazoles. Synlett 2012, 23, 589–594. [Google Scholar]
- Jouvin, K.; Coste, A.; Bayle, A.; Legrand, F.; Karthikeyan, G.; Tadiparthi, K.; Evano, G. Copper-Mediated Selective Cross-Coupling of 1,1-Dibromo-1-alkenes and Heteronucleophiles: Development of General Routes to Heterosubstituted Alkynes and Alkenes. Organometallics 2012, 31, 7933–7947. [Google Scholar] [CrossRef]
- Hamada, T.; Ye, X.; Stahl, S.S. Copper-catalyzed aerobic oxidative amidation of terminal alkynes: Efficient synthesis of ynamides. J. Am. Chem. Soc. 2008, 130, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Siemsen, P.; Livingston, R.C.; Diederich, F. Acetylenic coupling: A powerful tool in molecular construction. Angew. Chem. Int. Ed. 2000, 39, 2633–2657. [Google Scholar] [CrossRef]
- Chan, D.M.T.; Lam, P.Y.S. Boronic Acids in Organic Synthesis and Chemical Biology; Wiley-VCH: New York, NY, USA, 2005; pp. 205–240. [Google Scholar]
- Le, H.T.N.; Tran, T.V.; Phan, N.T.S.; Truong, T. Efficient and recyclable Cu-2(BDC)(2)(BPY)-catalyzed oxidative amidation of terminal alkynes: Role of bipyridine ligand. Catal. Sci. Technol. 2015, 5, 851–859. [Google Scholar] [CrossRef]
- Sau, M.C.; Rajesh, Y.; Mandal, M.; Bhattacharjee, M. Copper Catalyzed Regioselective N-Alkynylation of Pyrazoles and Evaluation of the Anticancer Activity of Ethynyl-Pyrazoles. ChemistrySelect 2018, 3, 3511–3515. [Google Scholar] [CrossRef]
- Jia, W.; Jiao, N. Cu-Catalyzed Oxidative Amidation of Propiolic Acids Under Air via Decarboxylative Coupling. Org. Lett. 2010, 12, 2000–2003. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.F.C.; Eastwood, F.W.; Fallon, G.D.; Lee, S.C.; McGeary, R.P. The Pyrolytic Rearrangement of 1-Alkynoyl-3-methylpyrazoles: Synthesis of Pyrazolo[1,5-a]pyridin-5-ols and Related Compounds. Aust. J. Chem. 1994, 47, 991–1007. [Google Scholar] [CrossRef]
- Cook, A.M.; Wolf, C. Terminal ynamides: Synthesis, coupling reactions, and additions to common electrophiles. Tetrahedron Lett. 2015, 56, 2377–2392. [Google Scholar] [CrossRef]
- Tabata, H.; Kuwamoto, K.; Okuno, T. Conformational polymorphs and solid-state polymerization of 9-(1,3-butadiynyl)carbazole derivatives. J. Mol. Struct. 2016, 1106, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Cook, A.M.; Wolf, C. Catalytic enantioselective nucleophilic addition of ynamides to aldehydes. Chem. Commun. 2014, 50, 3151–3154. [Google Scholar] [CrossRef]
- Huang, W.-S.; Metcalf, C.A.; Sundaramoorthi, R.; Wang, Y.; Zou, D.; Thomas, R.M.; Zhu, X.; Cai, L.; Wen, D.; Liu, S.; et al. Discovery of 3-[2-(Imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-[4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl]benzamide (AP24534), a Potent, Orally Active Pan-Inhibitor of Breakpoint Cluster Region-Abelson (BCR-ABL) Kinase Including the T315I Gatekeeper Mutant. J. Med. Chem. 2010, 53, 4701–4719. [Google Scholar]
- Sato, A.H.; Ohashi, K.; Ito, K.; Iwasawa, T. Regio- and stereoselective synthesis of 1-(1-halovinyl)-1H-indoles from 1-ethynyl-1H-indoles with in situ generated HX. Tetrahedron Lett. 2013, 54, 2878–2881. [Google Scholar] [CrossRef]
- Laroche, C.; Li, J.; Kerwin, S.M. Cytotoxic 1,2-Dialkynylimidazole-Based Aza-Enediynes: Aza-Bergman Rearrangement Rates Do Not Predict Cytotoxicity. J. Med. Chem. 2011, 54, 5059–5069. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Berger, D.; Neuenschwander, M. Rearrangement of 5-Substituted 5-Aminopentadienals. Angew. Chem. Int. Ed. 1998, 37, 2138–2140. [Google Scholar] [CrossRef]
- Ide, M.; Yauchi, Y.; Iwasawa, T. Regio-, and stereoselective iodobromination of ynamides for synthesis of (E)-1-bromo-2-iodoenamides. Eur. J. Org. Chem. 2014, 2014, 3262–3267. [Google Scholar] [CrossRef]
- Huang, H.; Tang, L.; Liu, Q.; Xi, Y.; He, G.; Zhu, H. Formation of α-chalcogenyl acrylamides through unprecedented chalcogen-mediated metal-free oxyfunctionalization of ynamides with DMSO as an oxidant. Chem. Commun. (Camb. UK) 2016, 52, 5605–5608. [Google Scholar] [CrossRef] [PubMed]
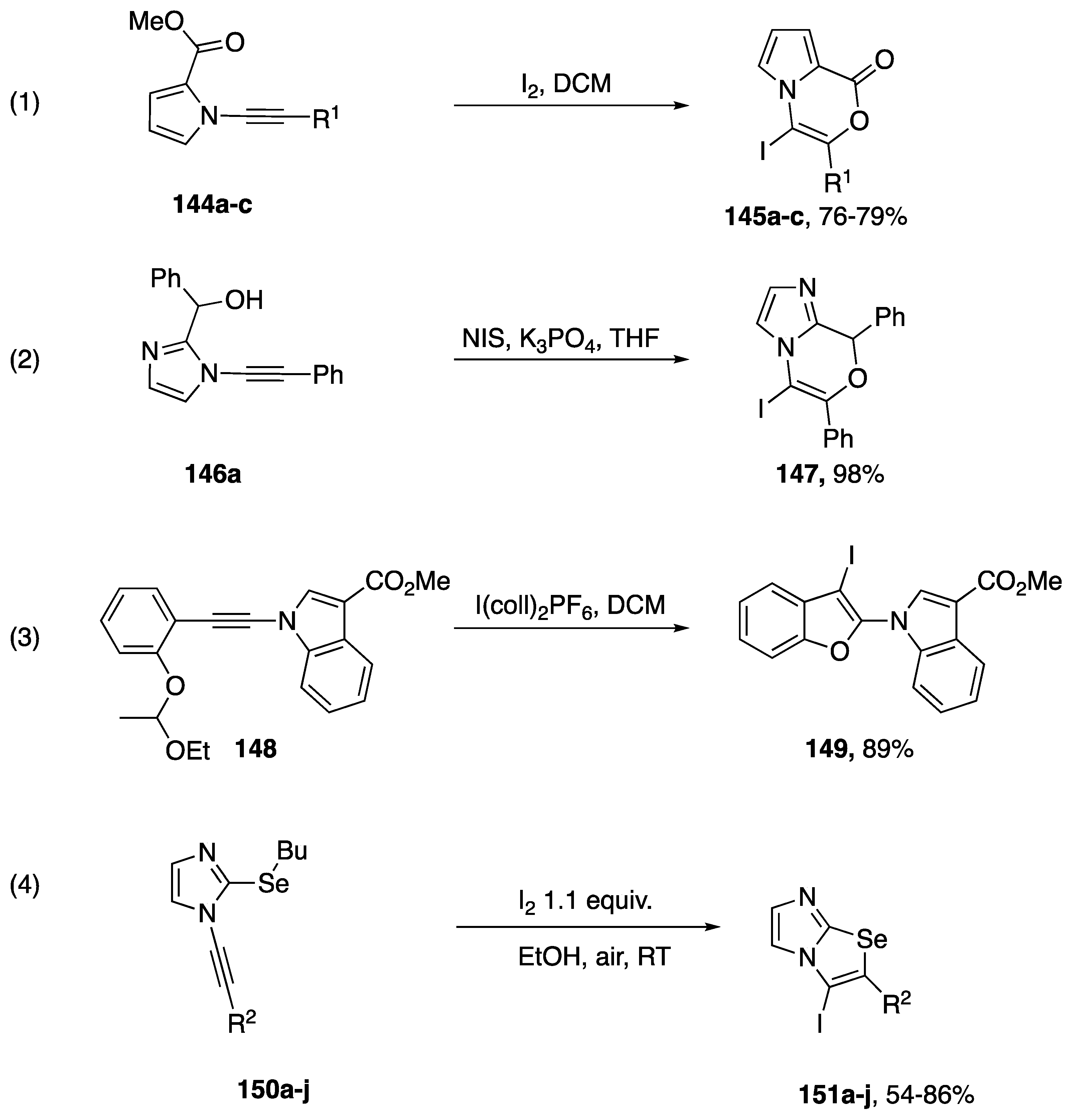
- Mphahlele, M.J. Molecular Iodine-Mediated Cyclization of Tethered Heteroatom-Containing Alkenyl or Alkynyl Systems. Molecules 2009, 14, 4814–4837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Tang, L.; Han, X.; He, G.; Xi, Y.; Zhu, H. Regioselective iodoamination of terminal ynamides for the synthesis of α-amino-β,β-diiodo-enamides. Chem. Commun. 2016, 52, 4321–4324. [Google Scholar] [CrossRef]
- Yenice, I.; Basceken, S.; Balci, M. Nucleophilic and electrophilic cyclization of N-alkyne-substituted pyrrole derivatives: Synthesis of pyrrolopyrazinone, pyrrolotriazinone, and pyrrolooxazinone moieties. Beilstein J. Org. Chem. 2017, 13, 825–834. [Google Scholar] [CrossRef]
- Laroche, C.; Kerwin, S.M. Efficient, Regioselective Access to Bicyclic Imidazo 1,2-x-Heterocycles via Gold- and Base-Promoted Cyclization of 1-Alkynylimidazoles. J. Org. Chem. 2009, 74, 9229–9232. [Google Scholar] [CrossRef]
- Okitsu, T.; Nakata, K.; Nishigaki, K.; Michioka, N.; Karatani, M.; Wada, A. Iodocyclization of Ethoxyethyl Ethers to Ynamides: An Immediate Construction to Benzo b furans. J. Org. Chem. 2014, 79, 5914–5920. [Google Scholar] [CrossRef]
- Grimaldi, T.B.; Godoi, B.; Roehrs, J.A.; Speranca, A.; Zeni, G. Electrophilic Cyclization of N-Alkynyl-2-(organochalcogen)imidazoles: An Alternative Access to Imidazo[2,1-b]chalcogenazoles. Eur. J. Org. Chem. 2013, 2013, 2646–2652. [Google Scholar] [CrossRef]
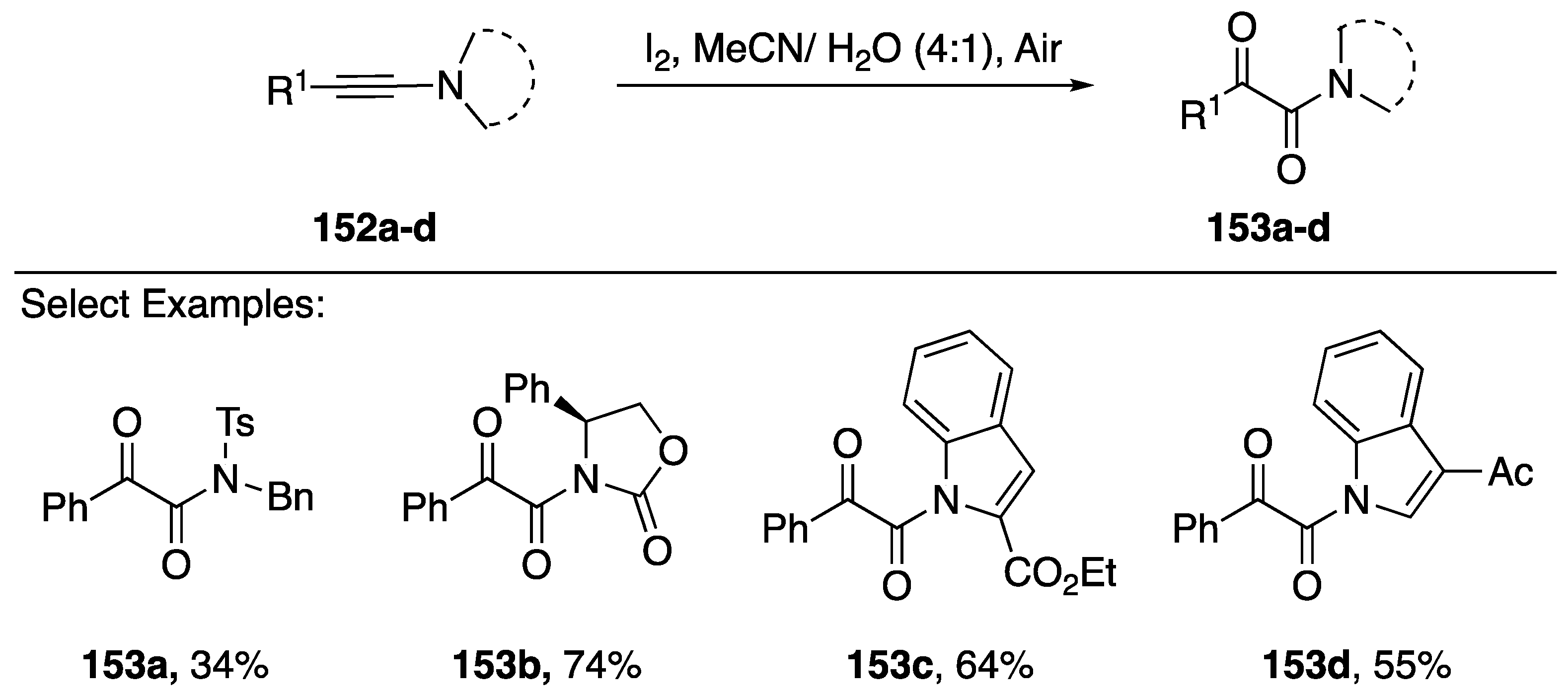
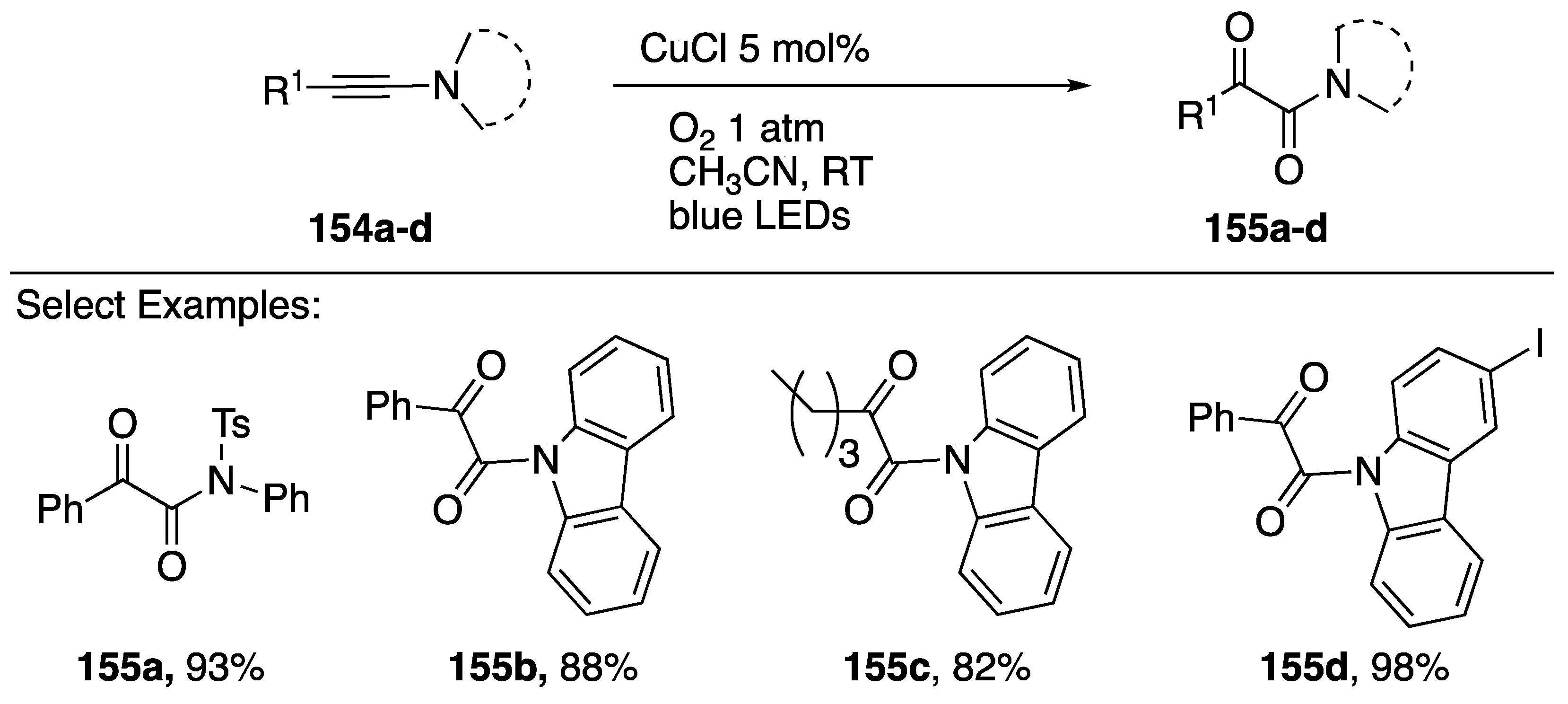
- Huang, H.; He, G.; Zhu, X.; Jin, X.; Qiu, S.; Zhu, H. Iodine-Mediated Oxidation of Ynamides: A Facile Access to N-Monosubstituted α-Ketoamides and α-Ketoimides. Eur. J. Org. Chem. 2014, 2014, 7174–7183. [Google Scholar] [CrossRef]
- Chikugo, T.; Yauchi, Y.; Ide, M.; Iwasawa, T. Transition metal-free oxidation of ynamides for synthesis of α-keto-imides. Tetrahedron 2014, 70, 3988–3993. [Google Scholar] [CrossRef]
- Ragupathi, A.; Charpe, V.P.; Sagadevan, A.; Hwang, K.C. Visible Light-Mediated Copper(I)-catalyzed aerobic oxidation of ynamides/ynamines at room temperature: A sustainable approach to the synthesis of α-ketoimides/α-ketoamides. Adv. Synth. Catal. 2017, 359, 1138–1143. [Google Scholar] [CrossRef]
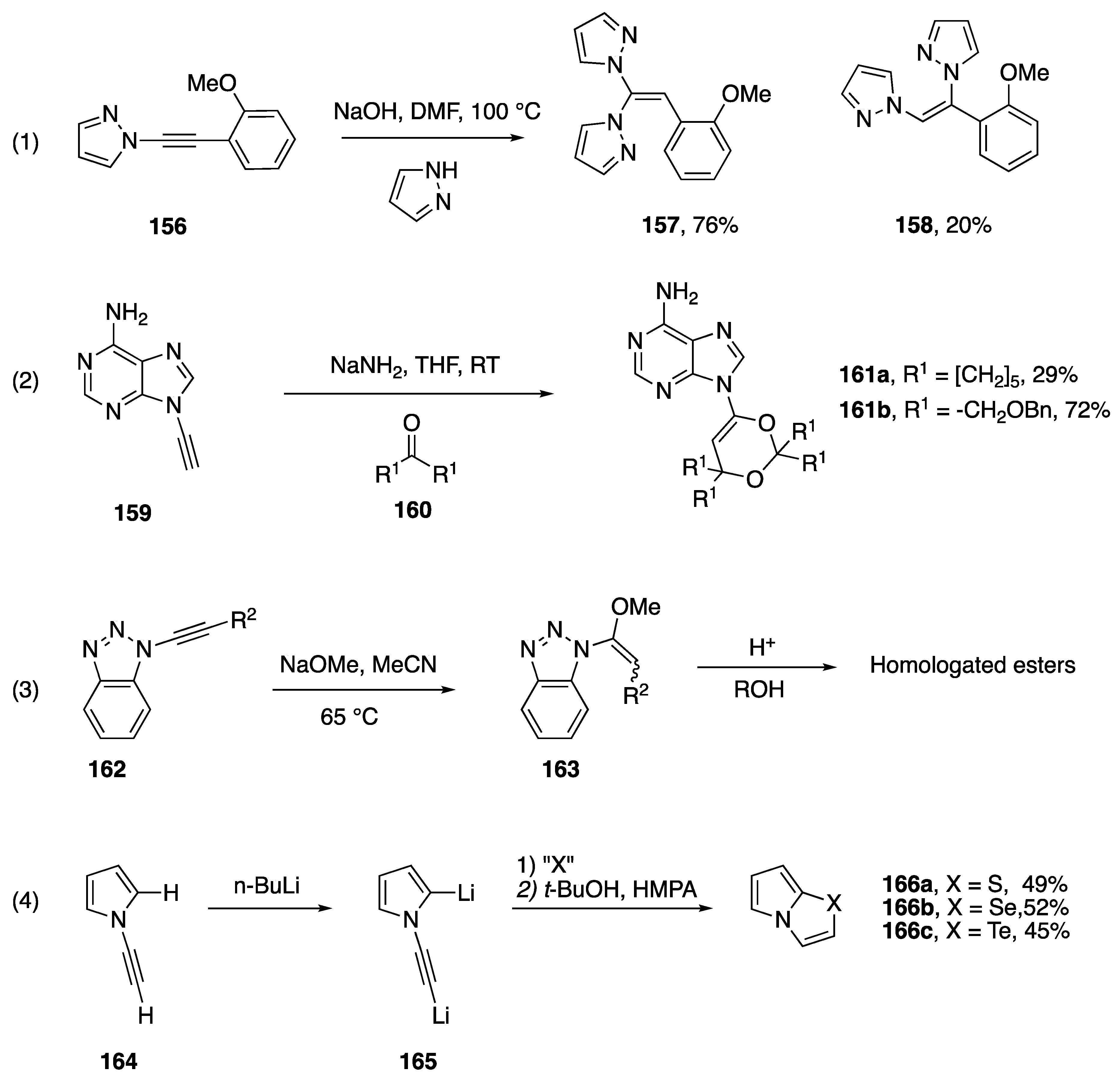
- Zhang, C.; Shi, Y.-L.; Zhang, L.-Y.; Yuan, D.-P.; Ban, M.-T.; Zheng, J.-Y.; Liu, D.-H.; Guo, S.-N.; Cui, D.-M. NaOH-promoted reaction of 1,1-dihaloalkenes and 1H-azoles: Synthesis of dihetaryl substituted alkenes. New J. Chem. 2018, 42, 17732–17739. [Google Scholar] [CrossRef]
- Mal’kina, A.G.; den Besten, R.; van der Kerk, A.C.H.T.M.; Brandsma, L.; Trofimov, B.A. Dimetallation of N-ethynylpyrrole and subsequent regiospecific derivatization. J. Organomet. Chem. 1995, 493, 271–273. [Google Scholar] [CrossRef]
- Roehrs, J.A.; Pistoia, R.P.; Back, D.F.; Zeni, G. Three-Step One-Pot Synthesis of Imidazo[2,1-b]chalcogenazoles via Intramolecular Cyclization of N-Alkynylimidazoles. Adv. Synth. Catal. 2012, 354, 1791–1796. [Google Scholar] [CrossRef]
- Laroche, C.; Gilbreath, B.; Kerwin, S.M. Exploring the synthetic utility of 1-alkynylimidazoles: Regiocontrolled cyclization to diverse imidazoazines and imidazoazoles. Tetrahedron 2014, 70, 4534–4539. [Google Scholar] [CrossRef]
- Habert, L.; Retailleau, P.; Gillaizeau, I. Rapid synthesis of 3-aminoisocoumarin derivatives from ynamides. Org. Biomol. Chem. 2018, 16, 7351–7355. [Google Scholar] [CrossRef]
- Graux, L.V.; Clavier, H.; Buono, G. Palladium-Catalyzed Addition of 1,3-Diones to Ynamides: An Entry to Alkoxy-Substituted Enamides. ChemCatChem 2014, 6, 2544–2548. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, V.; Hari Babu, M.; Kant, R.; Sridhar Reddy, M. N-Substitution dependent stereoselectivity switch in palladium catalyzed hydroalkynylation of ynamides: A regio and stereoselective synthesis of ynenamides. Chem. Commun. 2015, 51, 14996–14999. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.; Hong, S.W.; Oh, K.H.; Park, J.K. Divergent C-H Annulation for Multifused N-Heterocycles: Regio- and Stereospecific Cyclizations of N-Alkynylindoles. Angew. Chem. Int. Ed. 2017, 56, 13387–13391. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, L.; Zhang, F.; Zhu, G. Preparation of (Z)-α,β-Disubstituted Enamides via Palladium-Catalyzed Addition of Boronic Acids to Ynamides. J. Org. Chem. 2014, 79, 9319–9324. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.V.; Kim, S.W.; Nguyen, Q.H.; Kim, H.; Wang, S.; Hoang, T.; Shin, S. Bronsted Acid-Catalyzed Oxygenative Bimolecular Friedel-Crafts-type Coupling of Ynamides. Angew. Chem. Int. Ed. 2017, 56, 3670–3674. [Google Scholar] [CrossRef] [PubMed]
- Bauld, N.L.; Gao, D.X. The mechanism of the prototype cation radical cycloaddition reaction: The cyclodimerization of N-vinylcarbazole. J. Chem. Soc. Perkin Trans. 2 2000, 191–192. [Google Scholar] [CrossRef]
- Siva Reddy, A.; Kumara Swamy, K.C. Ethanol as a Hydrogenating Agent: Palladium-Catalyzed Stereoselective Hydrogenation of Ynamides to Give Enamides. Angew. Chem. Int. Ed. 2017, 56, 6984–6988. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Zhang, Q.; Huang, H.; Chen, S.; Wang, Q.; Zhang, D.; Zhang, R.; Zhu, H. Copper(I)-Catalyzed Highly Regio- and Stereoselective Boron Addition-Protonolysis of Alkynamides to give Alkenamides. Eur. J. Org. Chem. 2013, 2013, 6979–6989. [Google Scholar] [CrossRef]
- Itami, K.; Yoshida, J. Multisubstituted olefins: Platform synthesis and applications to materials science and pharmaceutical chemistry. B Chem Soc Jpn. 2006, 79, 811–824. [Google Scholar] [CrossRef]
- He, G.; Chen, S.; Wang, Q.; Huang, H.; Zhang, Q.; Zhang, D.; Zhang, R.; Zhu, H. Studies on copper(I)-catalyzed highly regio- and stereo-selective hydroboration of alkynamides. Org. Biomol. Chem. 2014, 12, 5945–5953. [Google Scholar] [CrossRef] [PubMed]
- Pirrung, M.C.; Zhang, J.C.; Morehead, A.T. Dipolar Cycloaddition of Cyclic Rhodium Carbenoids to Digonal Carbon—Synthesis of Isoeuparin. Tetrahedron Lett. 1994, 35, 6229–6230. [Google Scholar] [CrossRef]
- Clavier, H.; Lepronier, A.; Bengobesse-Mintsa, N.; Gatineau, D.; Pellissier, H.; Giordano, L.; Tenaglia, A.; Buono, G. Palladium-Mediated [2+1] Cycloaddition of Norbornene Derivatives with Ynamides. Adv. Synth. Catal. 2013, 355, 403–408. [Google Scholar] [CrossRef]
- Audran, G.; Pellissier, H. Synthesis of Methylene- and Alkylidenecyclopropane Derivatives. Adv. Synth. Catal. 2010, 352, 575–608. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Lazaro-Milla, C. Direct Metal-Free Entry to Aminocyclobutenes or Aminocyclobutenols from Ynamides: Synthetic Applications. Chem. Eur. J. 2016, 22, 8998–9005. [Google Scholar] [CrossRef] [Green Version]
- Alcaide, B.; Almendros, P.; Lazaro-Milla, C. Regioselective Synthesis of Heteroatom-Functionalized Cyclobutene-triflones and Cyclobutenones. Adv. Synth. Catal. 2017, 359, 2630–2639. [Google Scholar] [CrossRef]
- Hatit, M.Z.C.; Sadler, J.C.; McLean, L.A.; Whitehurst, B.C.; Seath, C.P.; Humphreys, L.D.; Young, R.J.; Watson, A.J.B.; Burley, G.A. Chemoselective Sequential Click Ligations Directed by Enhanced Reactivity of an Aromatic Ynamine. Org. Lett. 2016, 18, 1694–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seath, C.P.; Burley, G.A.; Watson, A.J.B. Determining the Origin of Rate-Independent Chemoselectivity in CuAAC Reactions: An Alkyne-Specific Shift in Rate-Determining Step. Angew. Chem. Int. Ed. 2017, 56, 3314–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatit, M.Z.C.; Seath, C.P.; Watson, A.J.B.; Burley, G.A. Strategy for Conditional Orthogonal Sequential CuAAC Reactions Using a Protected Aromatic Ynamine. J. Org. Chem. 2017, 82, 5461–5468. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, V.; Kumar, R.; Sharma, K.; Sridhar, B.; Reddy, M.S. Copper-Promoted Regioselective Intermolecular Diamination of Ynamides: Synthesis of Imidazo[1,2-a]pyridines. ACS Omega 2017, 2, 2770–2777. [Google Scholar] [CrossRef]
- Davies, P.W.; Cremonesi, A.; Dumitrescu, L. Intermolecular and Selective Synthesis of 2,4,5-Trisubstituted Oxazoles by a Gold-Catalyzed Formal [3+2] Cycloaddition. Angew. Chem. Int. Ed. 2011, 50, 8931–8935. [Google Scholar] [CrossRef]
- Gillie, A.D.; Jannapu Reddy, R.; Davies, P.W. Efficient and Flexible Synthesis of Highly Functionalised 4-Aminooxazoles by a Gold-Catalysed Intermolecular Formal [3+2] Dipolar Cycloaddition. Adv. Synth. Catal. 2016, 358, 226–239. [Google Scholar] [CrossRef] [Green Version]
- Garzon, M.; Arce, E.M.; Reddy, R.J.; Davies, P.W. General Entry into o-,o’-Heteroatom-Linked N-(Hetero)aryl-Imidazole Motifs by Gold-Catalysed Formal [3+2]-Dipolar Cycloaddition. Adv. Synth. Catal. 2017, 359, 1837–1843. [Google Scholar] [CrossRef]
- Han, X.-L.; Zhou, C.-J.; Liu, X.-G.; Zhang, S.-S.; Wang, H.; Li, Q. Regioselective Synthesis of 5-Aminooxazoles via Cp*Co(III)-Catalyzed Formal [3 + 2] Cycloaddition of N-(Pivaloyloxy)amides with Ynamides. Org. Lett. 2017, 19, 6108–6111. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Zhang, H.; Zhao, X. Selenium-π-Acid Catalyzed Oxidative Functionalization of Alkynes: Facile Access to Ynones and Multisubstituted Oxazoles. ACS Catal. 2018, 8, 6745–6750. [Google Scholar] [CrossRef]
- Duret, G.; Quinlan, R.; Martin, R.E.; Bisseret, P.; Neuburger, M.; Gandon, V.; Blanchard, N. Inverse Electron-Demand 4+2 -Cycloadditions of Ynamides: Access to Novel Pyridine Scaffolds. Org. Lett. 2016, 18, 1610–1613. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.N.; Yeom, H.S.; Fang, L.C.; He, S.H.; Ma, Z.X.; Kedrowski, B.L.; Hsung, R.P. Ynamides in Ring Forming Transformations. Acc. Chem. Res. 2014, 47, 560–578. [Google Scholar] [CrossRef]
- Foster, R.A.A.; Willis, M.C. Tandem inverse-electron-demand hetero-/retro-Diels-Alder reactions for aromatic nitrogen heterocycle synthesis. Chem. Soc. Rev. 2013, 42, 63–76. [Google Scholar] [CrossRef]
- Duret, G.; Quinlan, R.; Yin, B.; Martin, R.E.; Bisseret, P.; Neuburger, M.; Gandon, V.; Blanchard, N. Intramolecular Inverse Electron-Demand [4 + 2] Cycloadditions of Ynamides with Pyrimidines: Scope and Density Functional Theory Insights. J. Org. Chem. 2017, 82, 1726–1742. [Google Scholar] [CrossRef]
- Chowdhury, H.; Chatterjee, N.; Goswami, A. An Eco-Friendly Route to N-Arylindoles by Iron-Catalyzed [2+2+2] Cycloaddition of Diynes with (Indol-1-yl)alkynes. Eur. J. Org. Chem. 2015, 2015, 7735–7742. [Google Scholar] [CrossRef]
- Amasaki, R.; Terashima, N.; Sotome, I.; Komagawa, S.; Saito, S. Nickel-Catalyzed [3+2+2] Cycloaddition of Ethyl Cyclopropylideneacetate and Heteroatom-Substituted Alkynes: Application to Selective Three-Component Reaction with 1,3-Diynes. J. Org. Chem. 2010, 75, 480–483. [Google Scholar] [CrossRef]
- Yamasaki, R.; Ohashi, M.; Maeda, K.; Kitamura, T.; Nakagawa, M.; Kato, K.; Fujita, T.; Kamura, R.; Kinoshita, K.; Masu, H.; et al. Ni-Catalyzed [4+3+2] Cycloaddition of Ethyl Cyclopropylideneacetate and Dienynes: Scope and Mechanistic Insights. Chem. Eur. J. 2013, 19, 3415–3425. [Google Scholar] [CrossRef]
- Jin, H.; Tian, B.; Song, X.; Xie, J.; Rudolph, M.; Rominger, F.; Hashmi, A.S.K. Gold-Catalyzed Synthesis of Quinolines from Propargyl Silyl Ethers and Anthranils through the Umpolung of a Gold Carbene Carbon. Angew. Chem. Int. Ed. 2016, 55, 12688–12692. [Google Scholar] [CrossRef] [PubMed]
- Rode, N.D.; Arcadi, A.; Di Nicola, A.; Marinelli, F.; Michelet, V. Gold-Catalyzed Cascade Reaction of β-(2-Aminophenyl)-α,β-ynones with Ynamides: A Sequential Route to Polysubstituted 2-Aminoquinolines. Org. Lett. 2018, 20, 5103–5106. [Google Scholar] [CrossRef] [PubMed]
- Han, X.-L.; Liu, X.-G.; Lin, E.; Chen, Y.; Chen, Z.; Wang, H.; Li, Q. Cp*Co(III)-Catalyzed oxidative [5+2] annulation: Regioselective synthesis of 2-aminobenzoxepines via C-H/O-H functionalization of 2-vinylphenols with ynamides. Chem. Commun. (Camb. UK) 2018, 54, 11562–11565. [Google Scholar] [CrossRef] [PubMed]
- Nadipuram, A.K.; Kerwin, S.M. Intra- and intermolecular trapping of cyclopentapyrazine carbenes derived from 1,2-dialkynylimidazoles. Tetrahedron Lett. 2006, 47, 353–356. [Google Scholar] [CrossRef]
- Nadipuram, A.K.; Kerwin, S.M. Thermal cyclization of 1,2-dialkynylimidazoles to imidazo 1,2-a pyridines. Tetrahedron 2006, 62, 3798–3808. [Google Scholar] [CrossRef]
- Gomes, F.; Fadel, A.; Rabasso, N. 2,3-Sigmatropic Rearrangement of Ynamides: Preparation of alpha-Amino Allenephosphonates. J. Org. Chem. 2012, 77, 5439–5444. [Google Scholar] [CrossRef]
- Adler, P.; Gomes, F.; Fadel, A.; Rabasso, N. Selective Reduction of Amino Allenephosphonates: Preparation of α-Amino Vinylphosphonates. Eur. J. Org. Chem. 2013, 2013, 7546–7555. [Google Scholar] [CrossRef]
- Zhao, Q.; Gagosz, F. Synthesis of Allenamides and Structurally Related Compounds by a Gold-Catalyzed Hydride Shift Process. Adv. Synth. Catal. 2017, 359, 3108–3113. [Google Scholar] [CrossRef]
- Chen, L.; Yu, L.; Deng, Y.; Zheng, Z.-J.; Xu, Z.; Cao, J.; Xu, L.-W. C-H Functionalization/C-O Bond Cleavage of Benzyl Silyl Ethers with Ynamides for the Chemoselective Synthesis of Skeletally Diverse Compounds. Adv. Synth. Catal. 2016, 358, 480–485. [Google Scholar] [CrossRef]
- Prochnow, T.; Maroneze, A.; Back, D.F.; Jardim, N.S.; Nogueira, C.W.; Zeni, G. Synthesis and anticholinesterase activity of 2-substituted-N-alkynylindoles. Org. Biomol. Chem. 2018, 16, 7926–7934. [Google Scholar] [CrossRef]
- Chen, J.J.; Ferreira, A.J.; Beaudry, C.M. Synthesis of Bis(indole) Alkaloids from Arundo donax: The Ynindole Diels-Alder Reaction, Conformational Chirality, and Absolute Stereochemistry. Angew. Chem. Int. Ed. 2014, 53, 11931–11934. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, E.; Borghese, S.; Blanc, A.; Beneteau, V.; Pale, P. Zeolite-Based Organic Synthesis (ZeoBOS) of Acortatarin A: First Total Synthesis Based on Native and Metal-Doped Zeolite-Catalyzed Steps. Chem. Eur. J. 2017, 23, 1484–1489. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Nakanishi, H.; Hosomi, T.; Kato, M. Synthesis and Solid-State Polymerization of a New Diacetylene—1-(N-carbazolyl)penta-1,3-diyn-5-ol. Macromolecules 1988, 21, 1238–1240. [Google Scholar] [CrossRef]
- Dumitrescu, S.; Percec, V.; Simionescu, C.I. Polymerization of Acetylenic Derivatives. 27. Synthesis and Properties of Isomeric Poly-N-Ethynylcarbazole. J. Polym. Sci. Part A Polym. Chem. 1977, 15, 2893–2907. [Google Scholar] [CrossRef]
- Sata, T.; Nomura, R.; Wada, T.; Sasabe, H.; Masuda, T. Polymerization of N-carbazolylacetylene by various transition metal catalysts and polymer properties. J. Polym. Sci. Part A Polym. Chem. 1998, 36, 2489–2492. [Google Scholar] [CrossRef]
- Yamamoto, T.; Mahmut, A.; Abe, M.; Kuroda, S.I.; Imase, T.; Sasaki, S. Alternating copolymer of thiophene and N-(phenylethynyl)pyrrole. New pi-conjugated alternating five-membered ring copolymer and its packing structure. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 2219–2224. [Google Scholar] [CrossRef]
- Yamashita, R.; Koizumi, T.; Sasaki, S.; Yamamoto, T. Preparation of Soluble Polypyrrole with -C C-p-C6H4-hexyl Side Chains at the N-Position and Its Self-assembling Behavior. Polym. J. 2007, 39, 1202–1206. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yamashita, R. Preparation of New pi-Conjugated Thiophene-Pyrrole Copolymers Having Ethynyl Substituents at the N-Position of Pyrrole. Polym. J. 2008, 40, 775–778. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yamagata, Y.; Yamashita, R.; Abla, M.; Fukumoto, H.; Koizumi, T.-A. Copolymers of pyrrole with N-alkynylpyrroles. Synth. Met. 2012, 162, 2406–2413. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kundu, S.K. Photoconductive Properties of Arylethynylcopper Polymers—Effects of Structure and Oxygen. J. Phys. Chem. 1973, 77, 2677–2680. [Google Scholar] [CrossRef]
- Moggio, I.; Alloisio, M.; Cravino, A.; Comoretto, D.; Piaggio, P.; Musso, G.F.; Garbarino, G.; Cuniberti, C.; Dell’Erba, C.; Dellepiane, G. Vibrational properties of novel diacetylenic monomers. J. Chem. Soc. Perkin Trans. 2 1998, 2249–2254. [Google Scholar] [CrossRef]
- Baschieri, A.; Sambri, L.; Gualandi, I.; Tonelli, D.; Monti, F.; Esposti, A.D.; Armaroli, N. Carbazole-terpyridine donor-acceptor luminophores. RSC Adv. 2013, 3, 6507–6517. [Google Scholar] [CrossRef]
- Betou, M.; Durand, R.J.; Sallustrau, A.; Gousset, C.; Le Coz, E.; Leroux, Y.R.; Toupet, L.; Trzop, E.; Roisnel, T.; Trolez, Y. Reactivity of Functionalized Ynamides with Tetracyanoethylene: Scope, Limitations and Optoelectronic Properties of the Adducts. Chem. Asian J. 2017, 12, 1338–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burley, G.A.; Boutadla, Y.; Davies, D.L.; Singh, K. Triazoles from N-Alkynylheterocycles and Their Coordination to Iridium. Organometallics 2012, 31, 1112–1117. [Google Scholar] [CrossRef]
- Li, J.; Kaoud, T.S.; Laroche, C.; Dalby, K.N.; Kerwin, S.M. Synthesis and biological evaluation of p38α kinase-targeting dialkynylimidazoles. Bioorg. Med. Chem. Lett. 2009, 19, 6293–6297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Kaoud, T.S.; LeVieux, J.; Gilbreath, B.; Moharana, S.; Dalby, K.N.; Kerwin, S.M. A Fluorescence-Based Assay for p38 alpha Recruitment Site Binders: Identification of Rooperol as a Novel p38 alpha Kinase Inhibitor. ChemBioChem 2013, 14, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Laroche, C.; Li, J.; Gonzales, C.; David, W.M.; Kerwin, S.M. Cyclization kinetics and biological evaluation of an anticancer 1,2-dialkynylimidazole. Org. Biomol. Chem. 2010, 8, 1535–1539. [Google Scholar] [CrossRef] [PubMed]













































































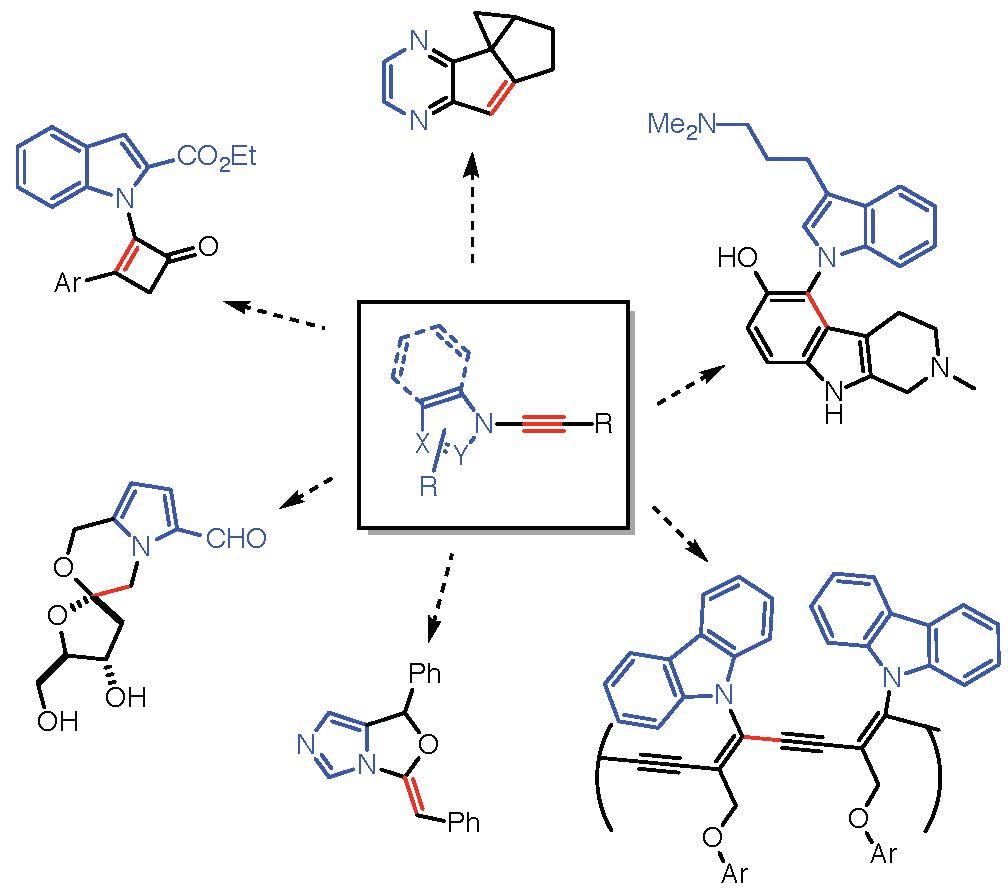{kind=link}
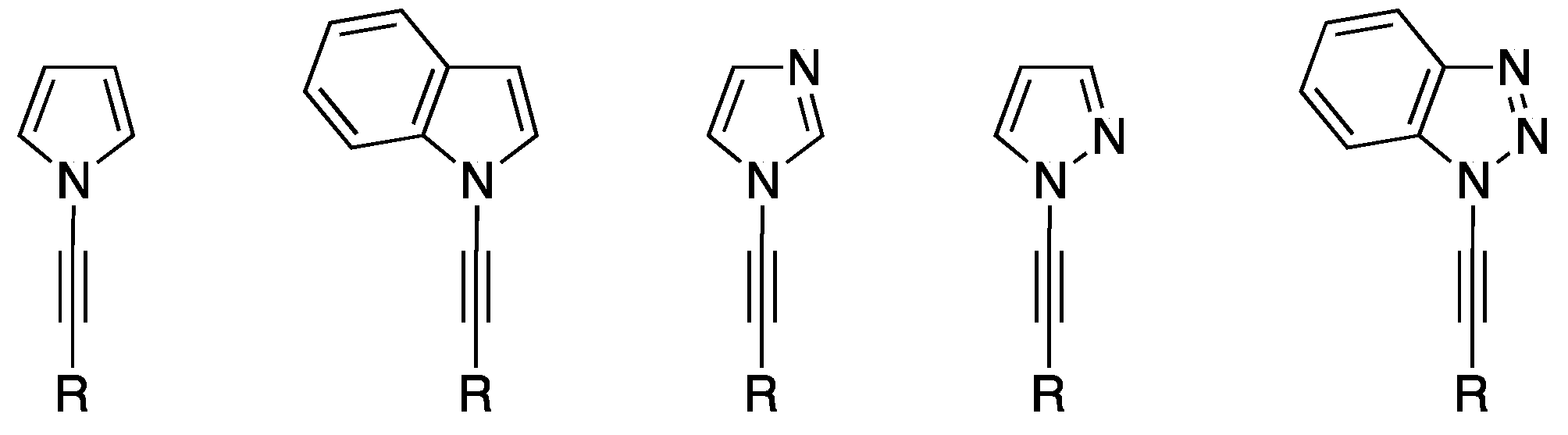{kind=link}
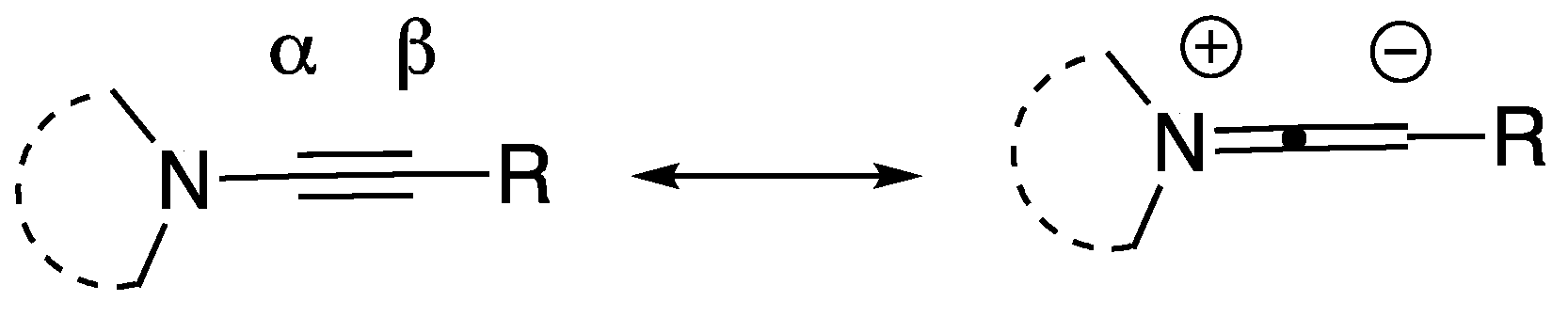{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
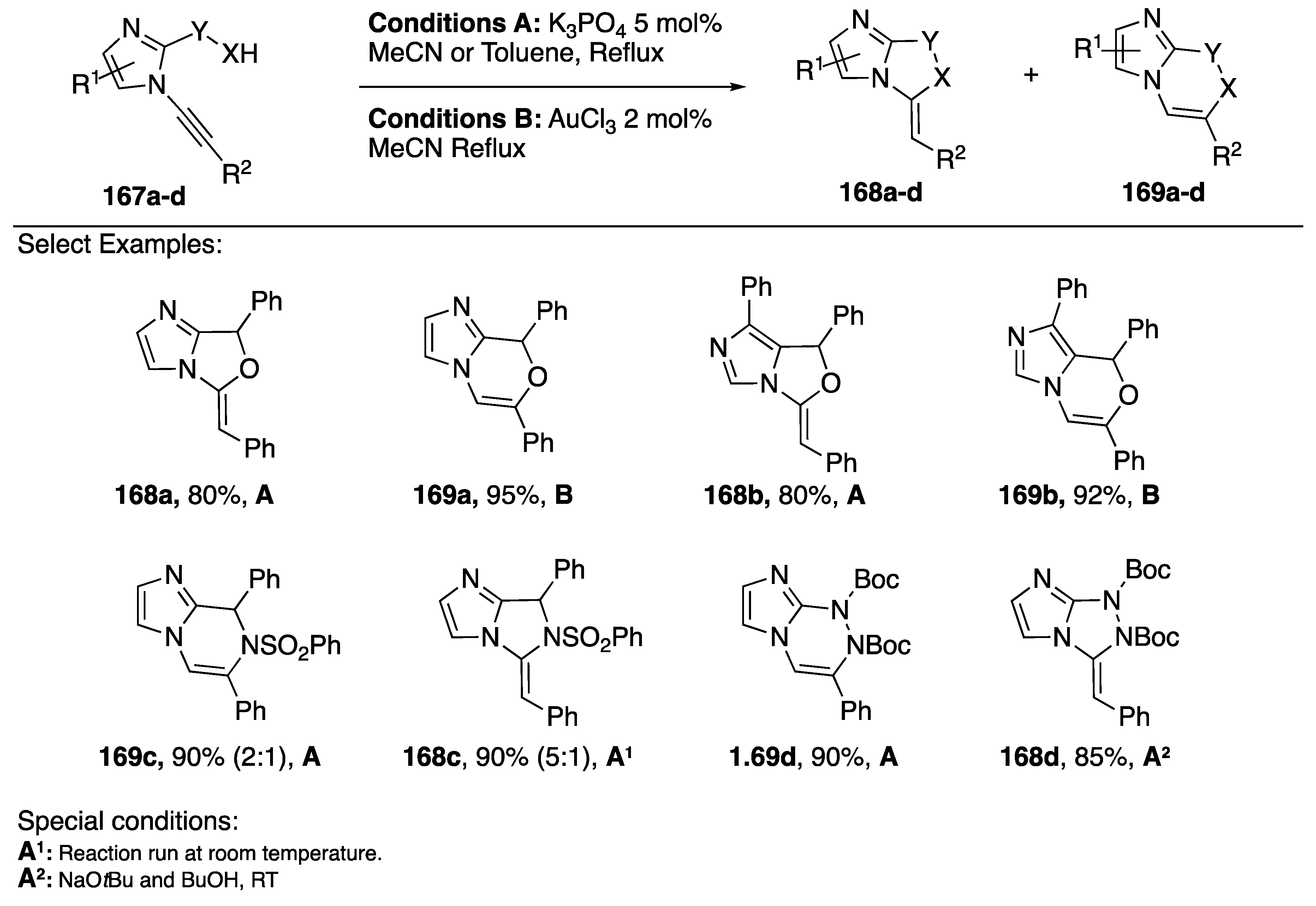{kind=link}
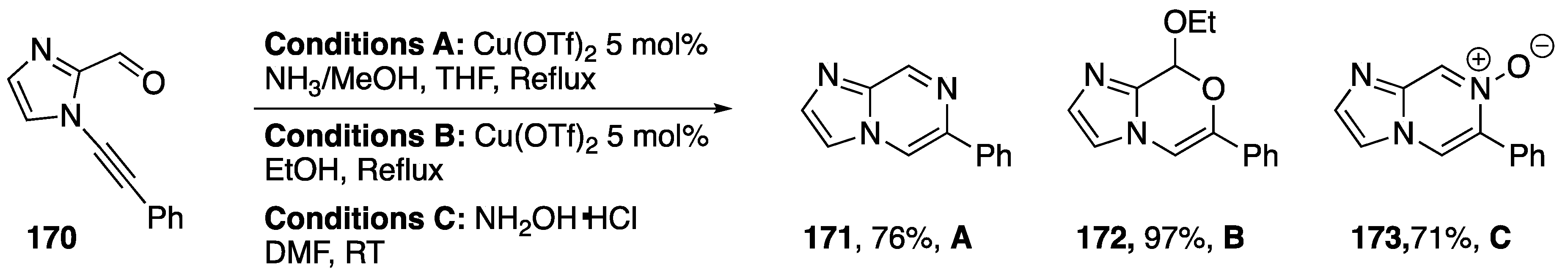{kind=link}
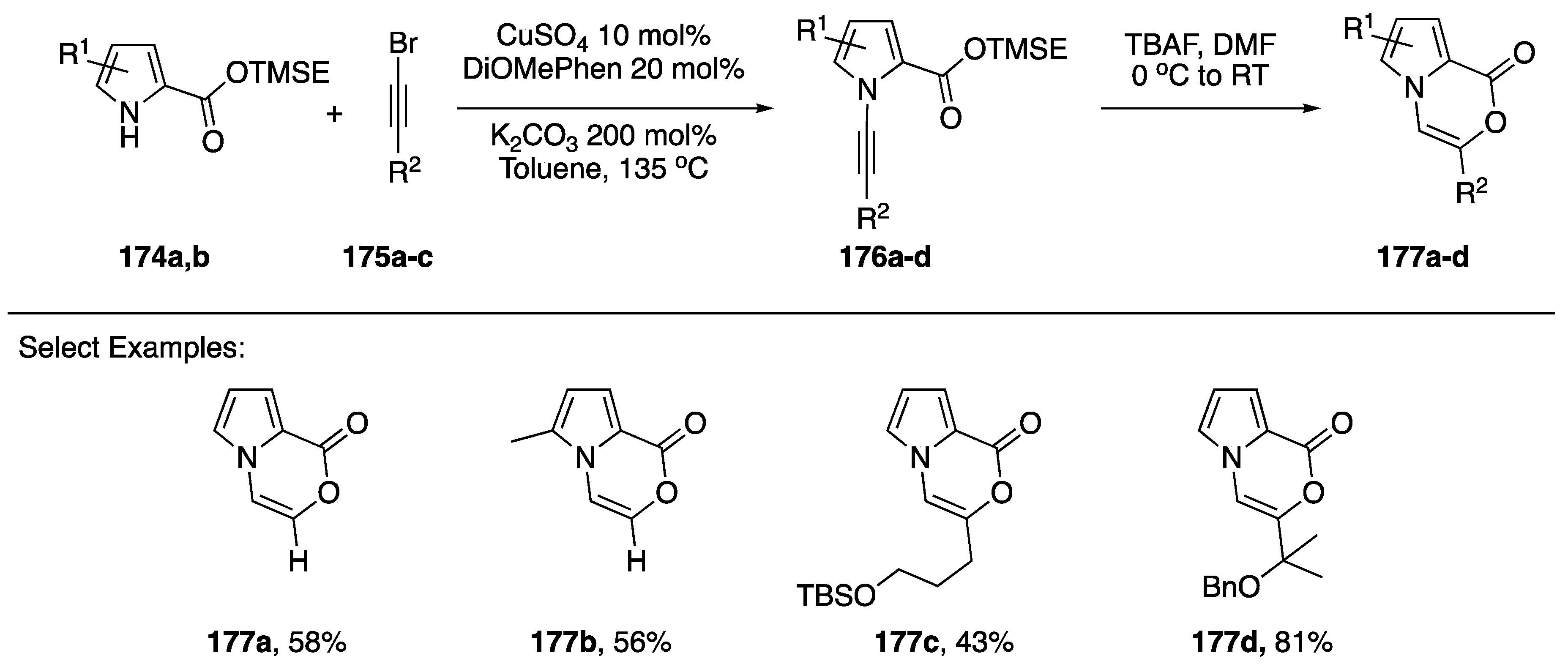{kind=link}
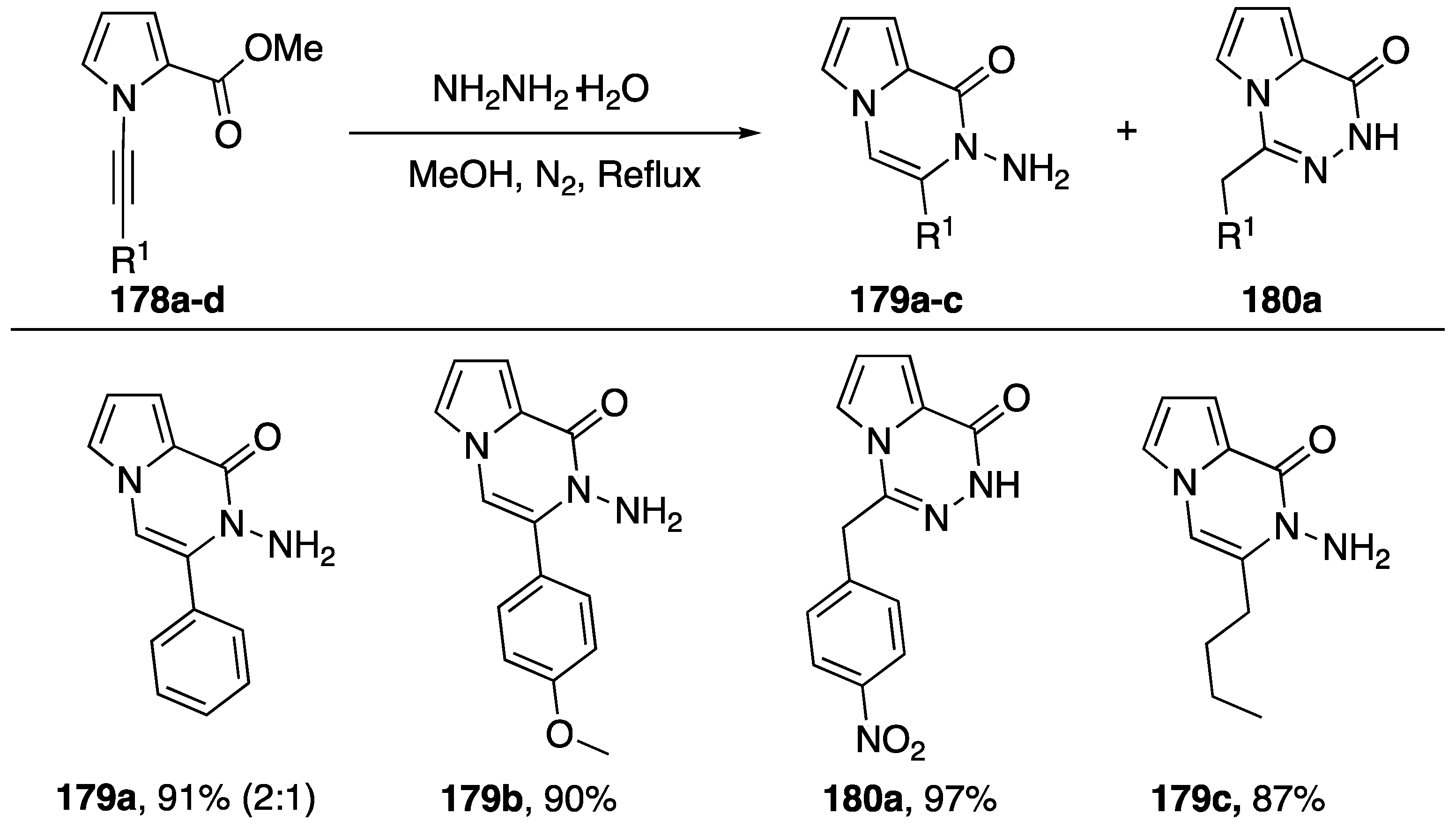{kind=link}
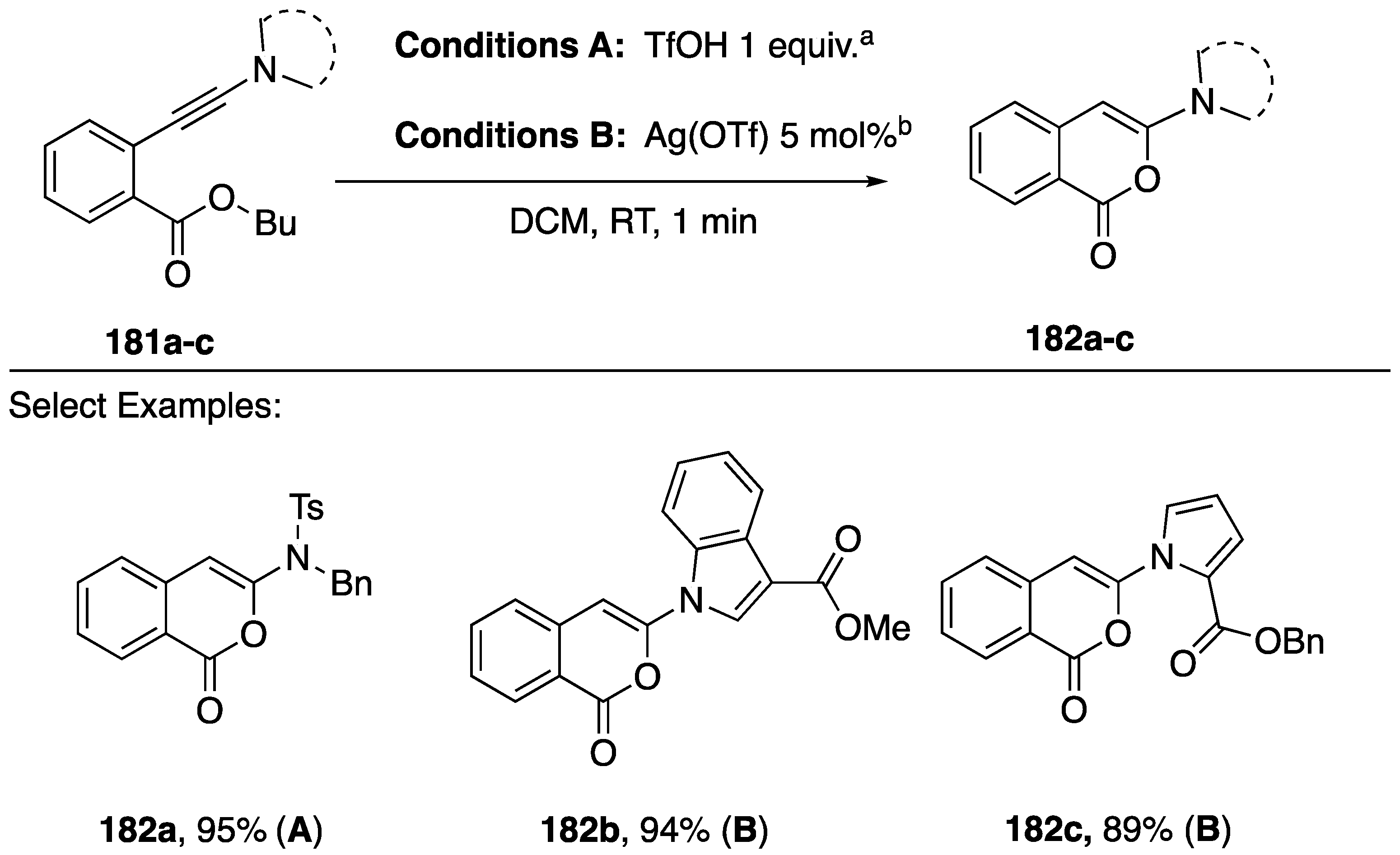{kind=link}
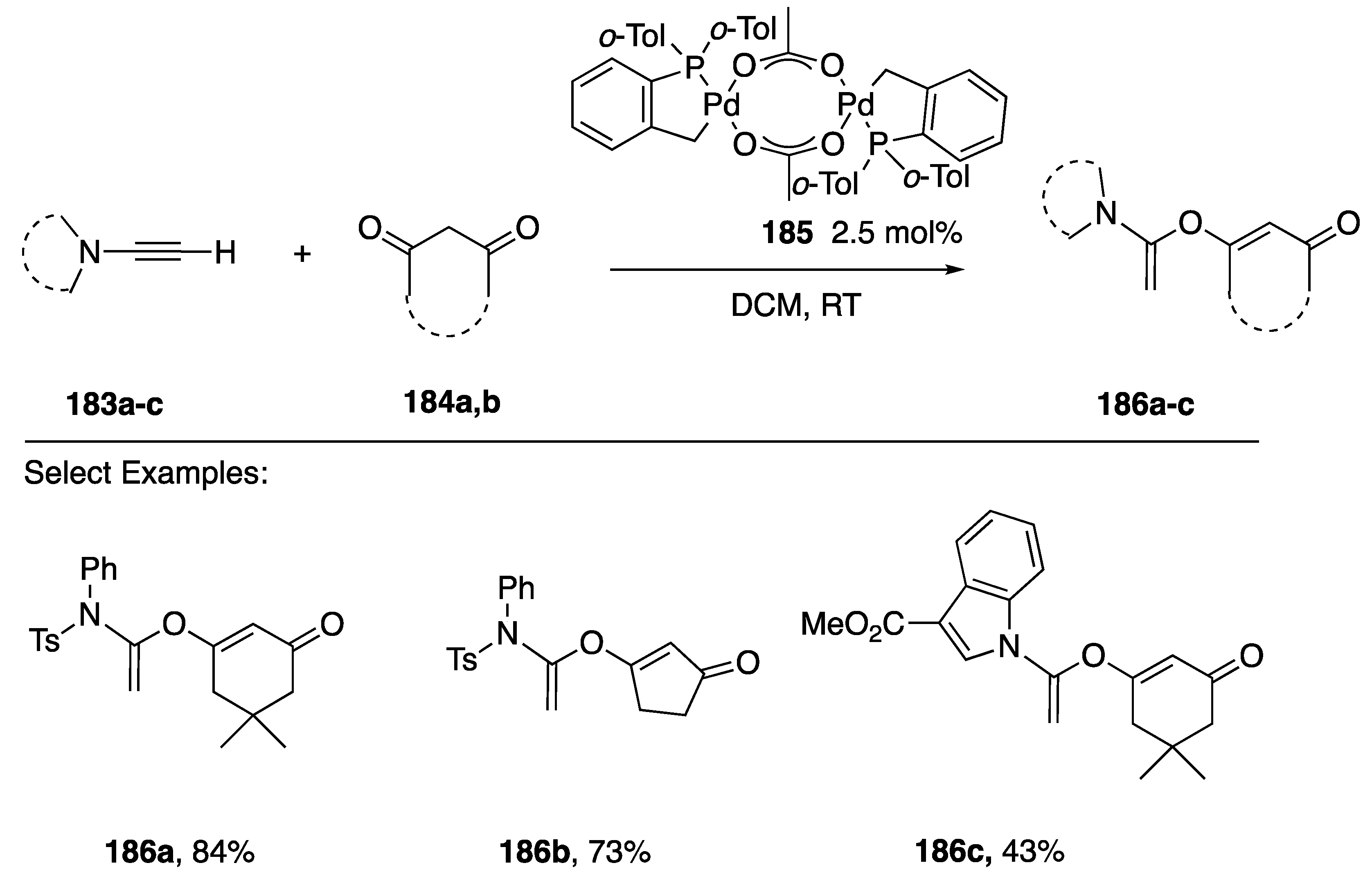{kind=link}
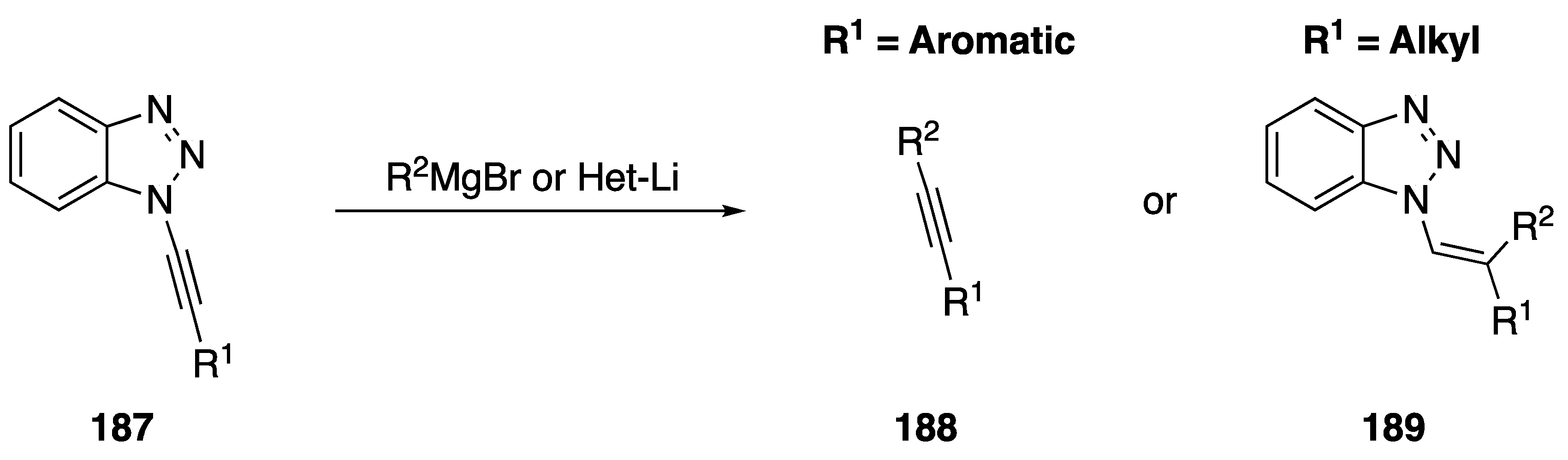{kind=link}
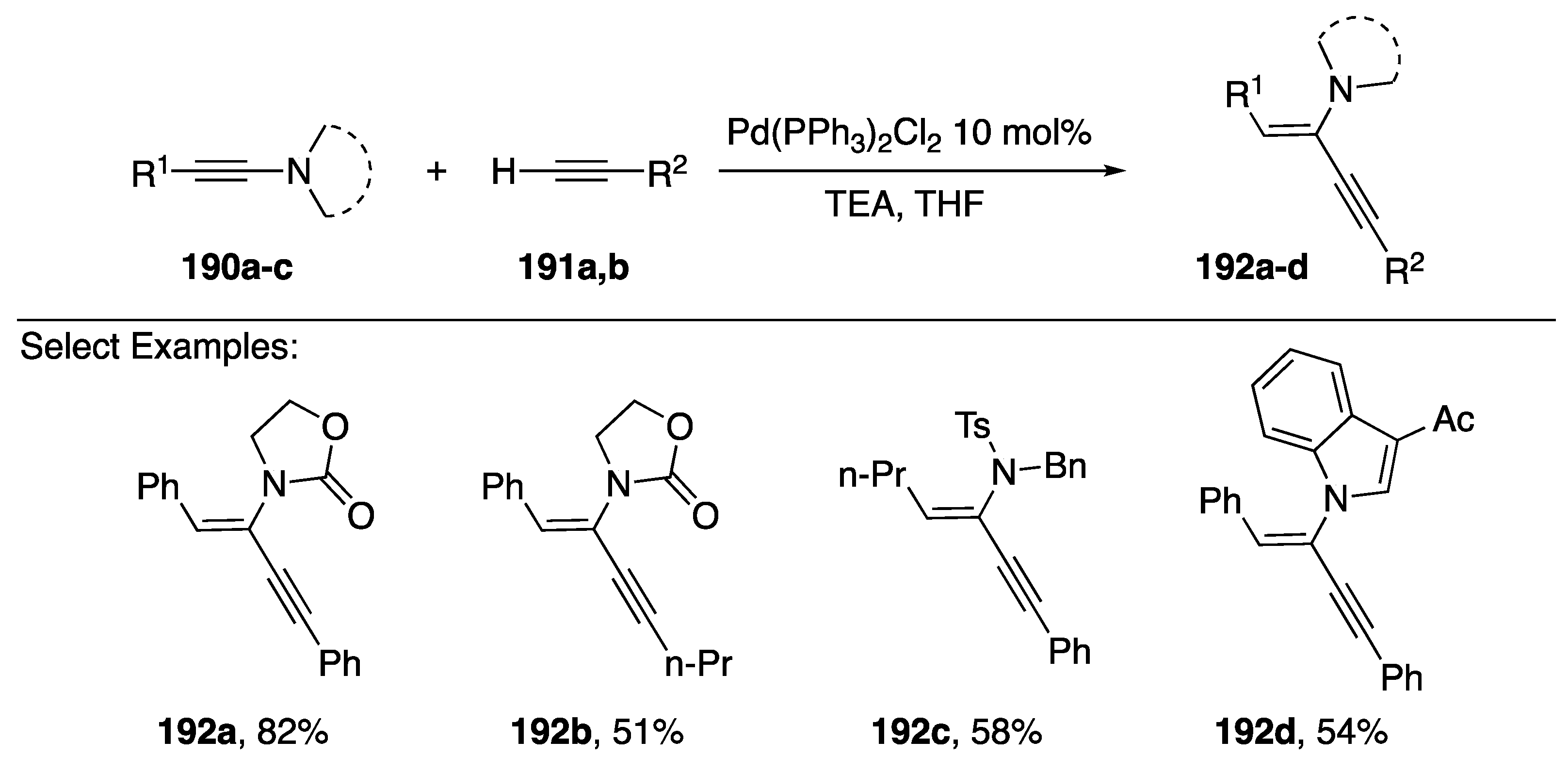{kind=link}
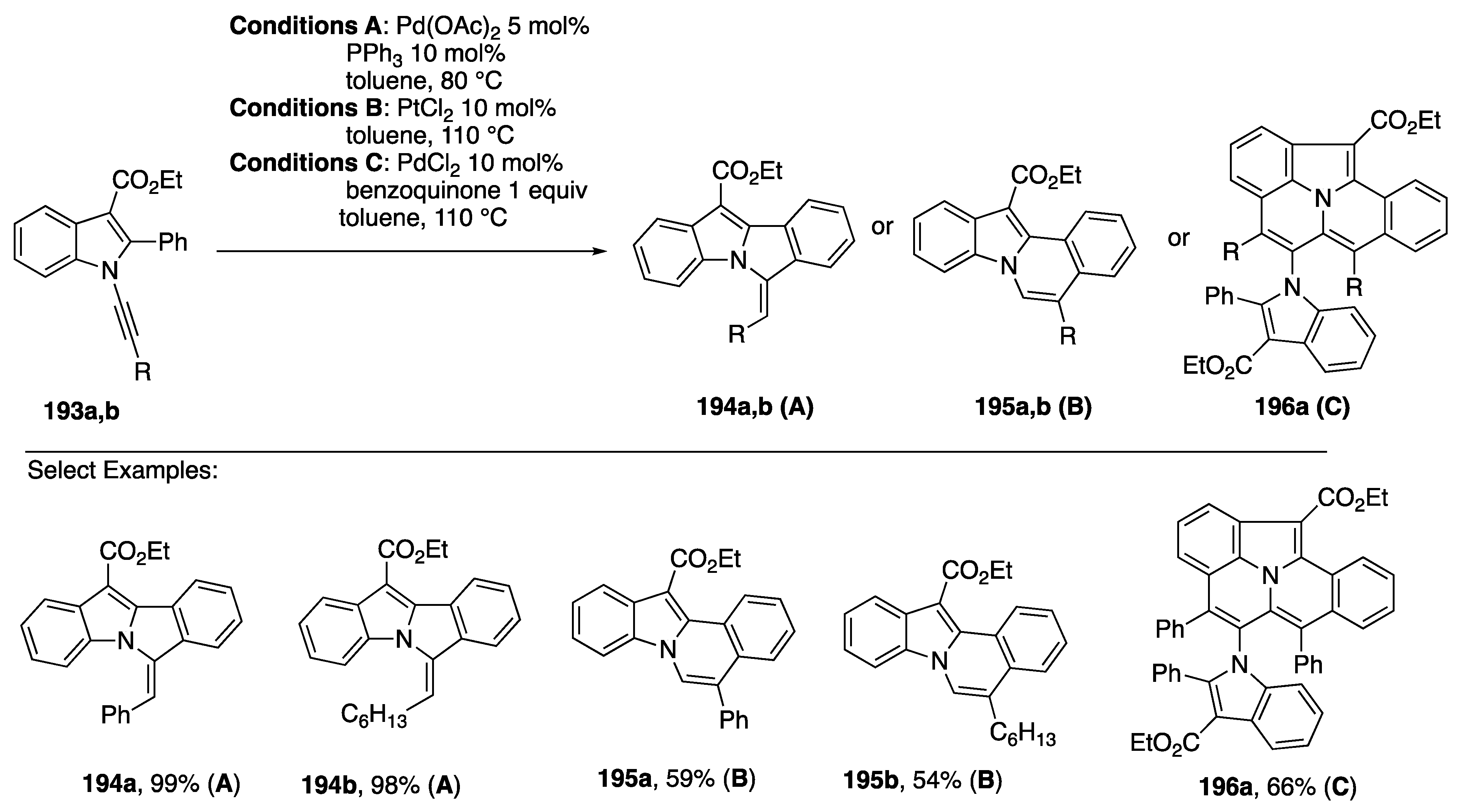{kind=link}
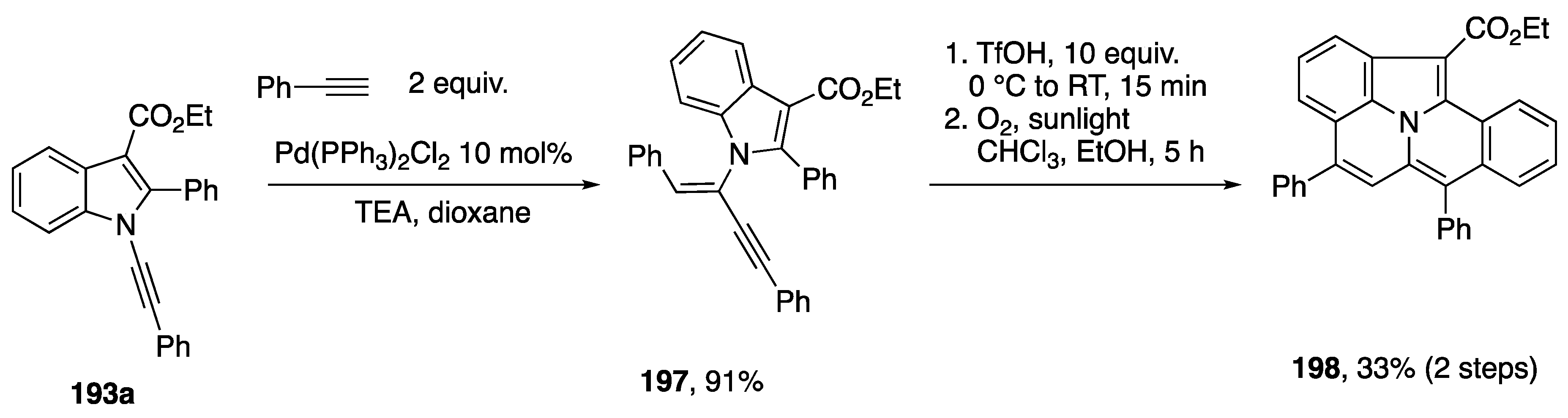{kind=link}
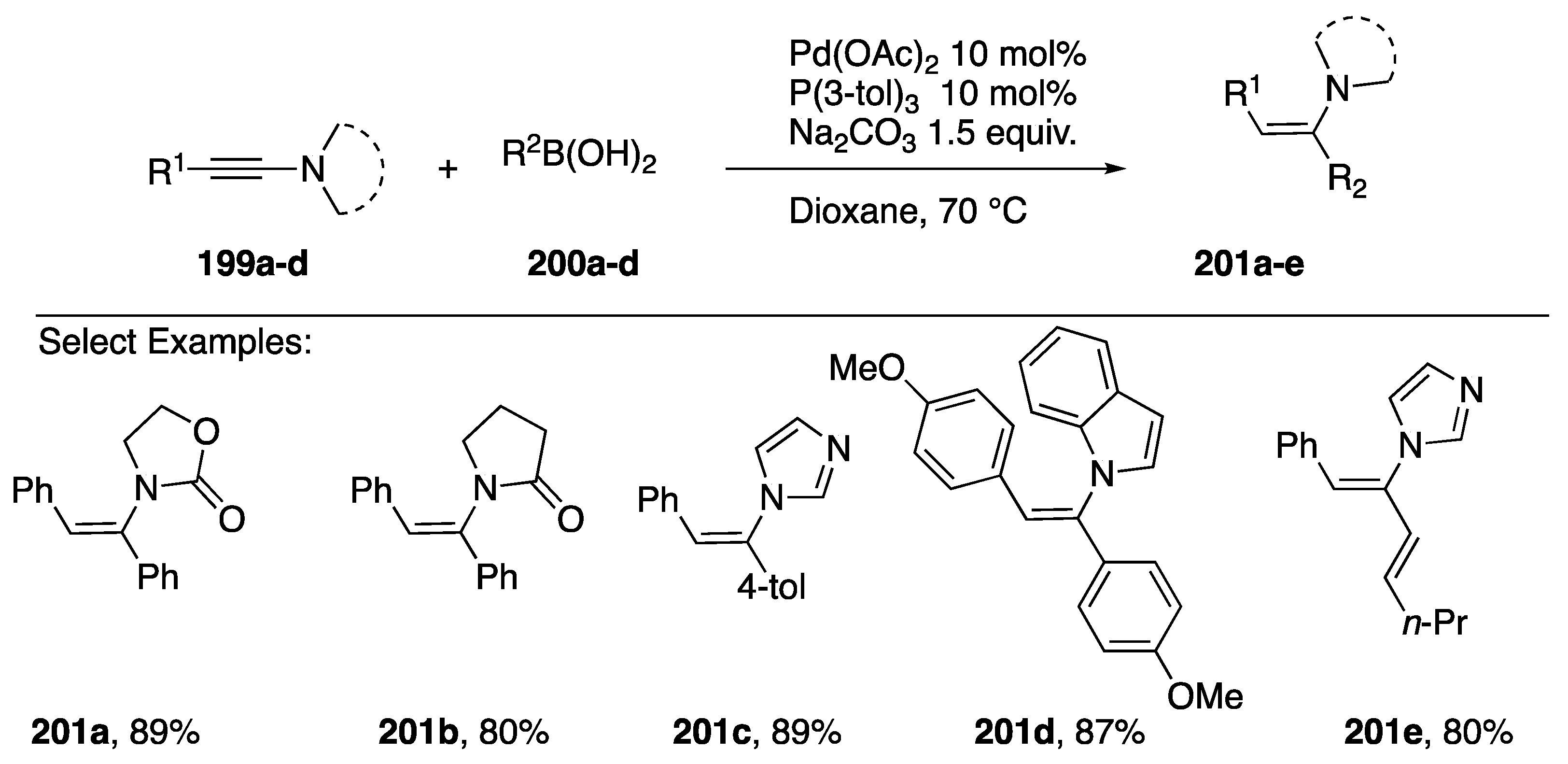{kind=link}
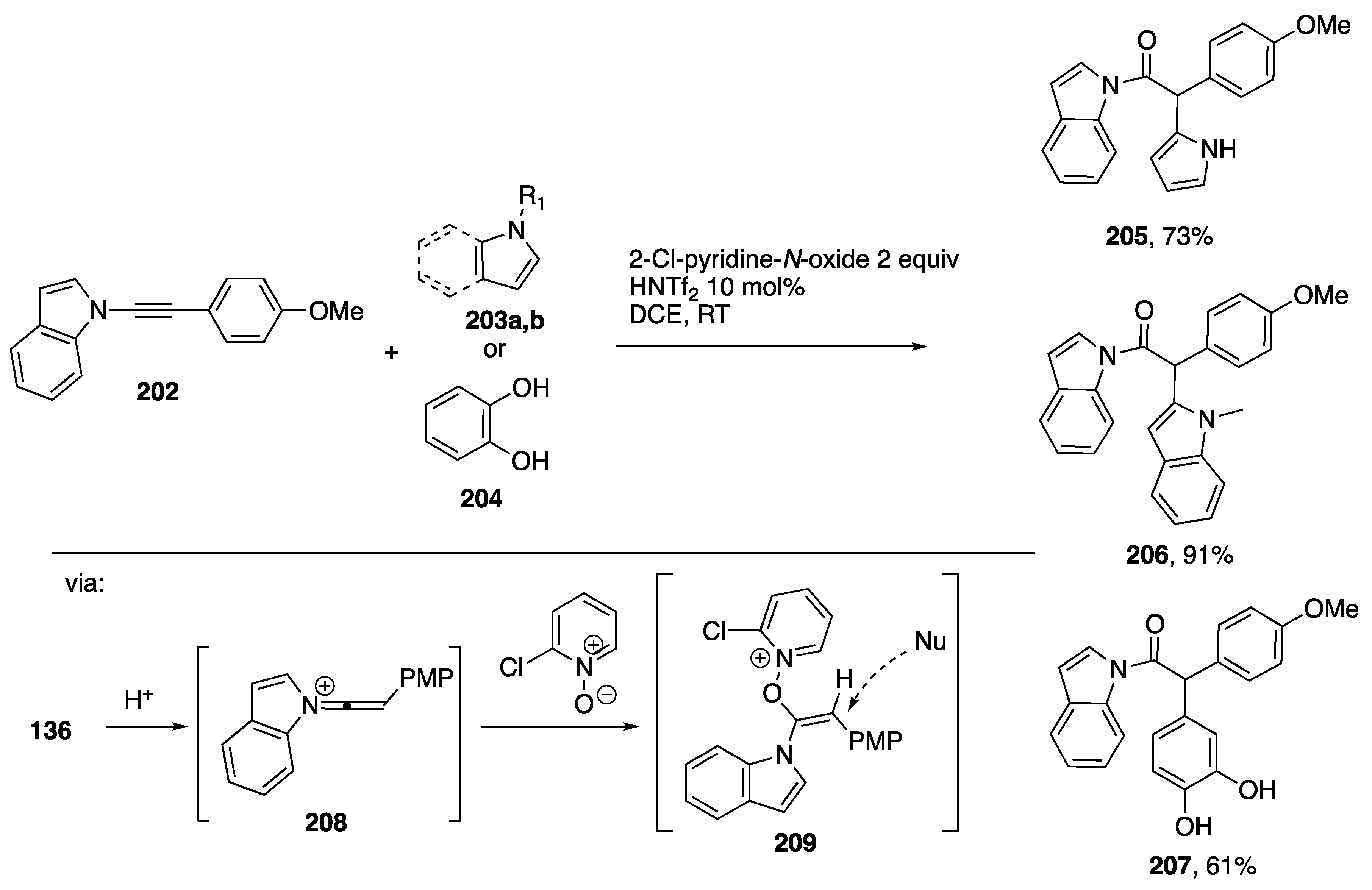{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
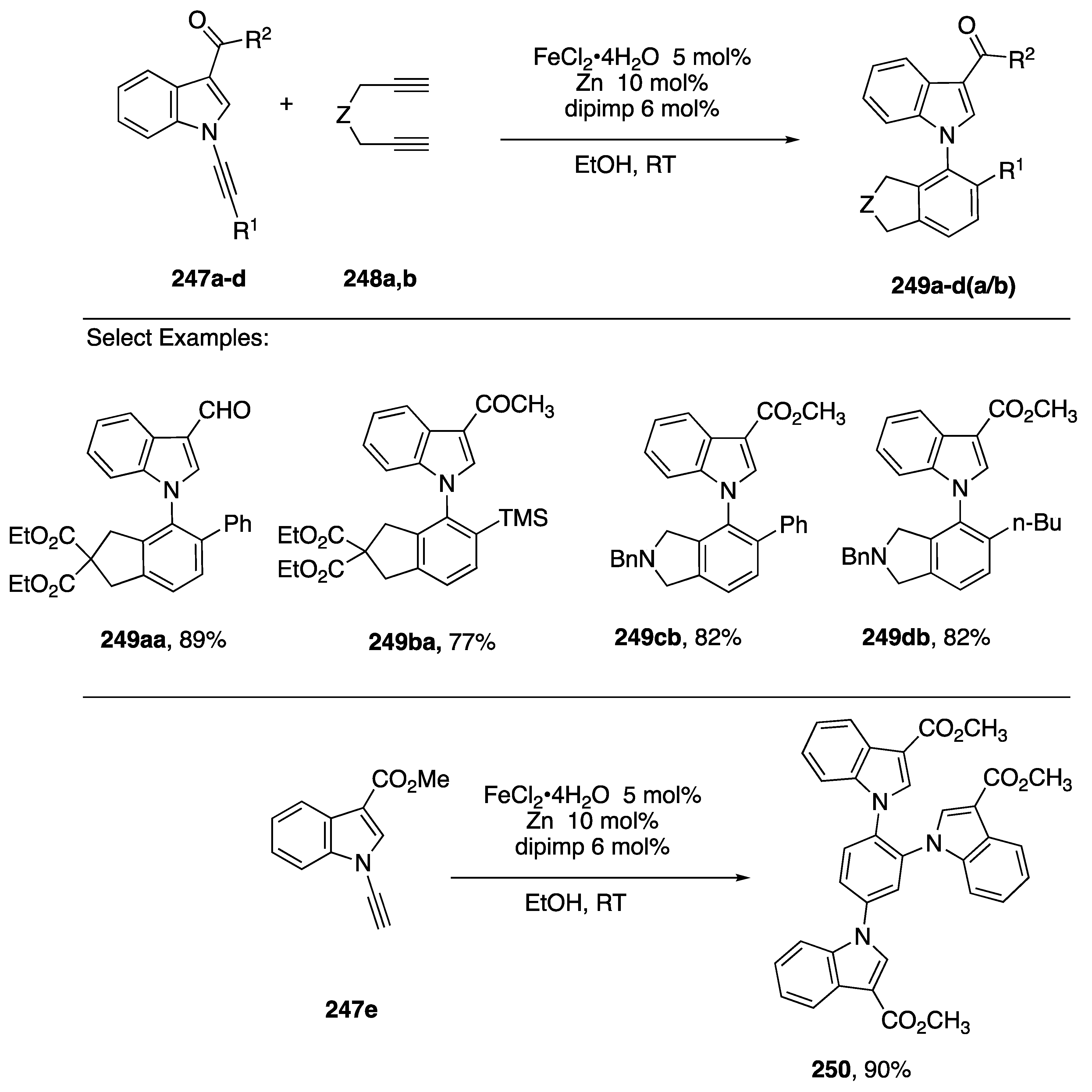{kind=link}
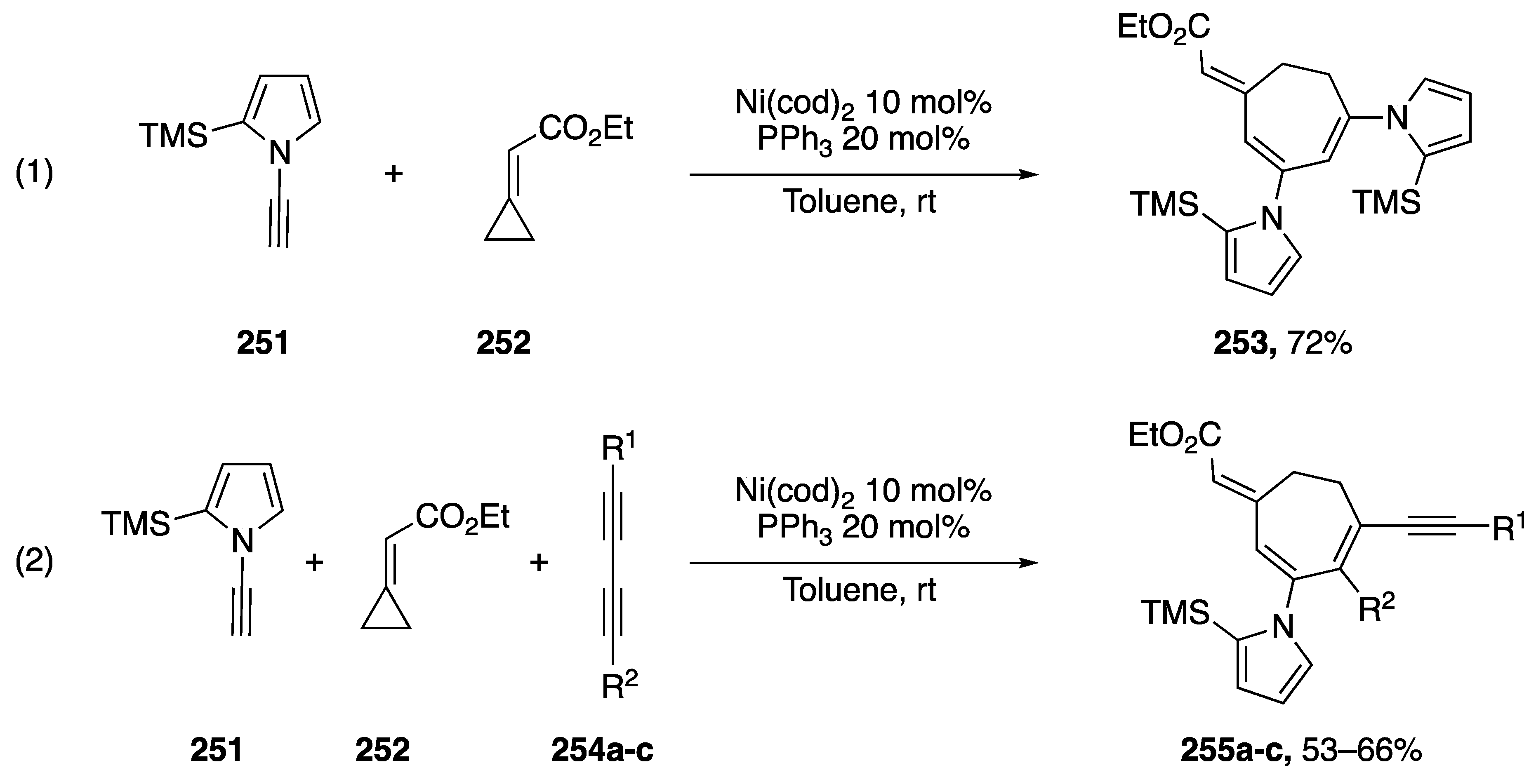{kind=link}
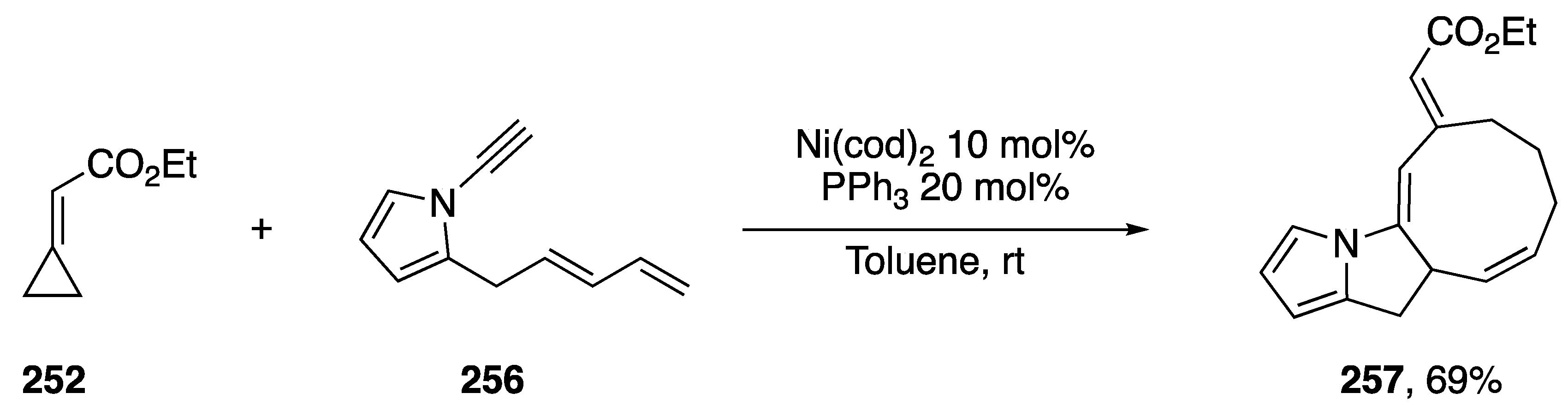{kind=link}
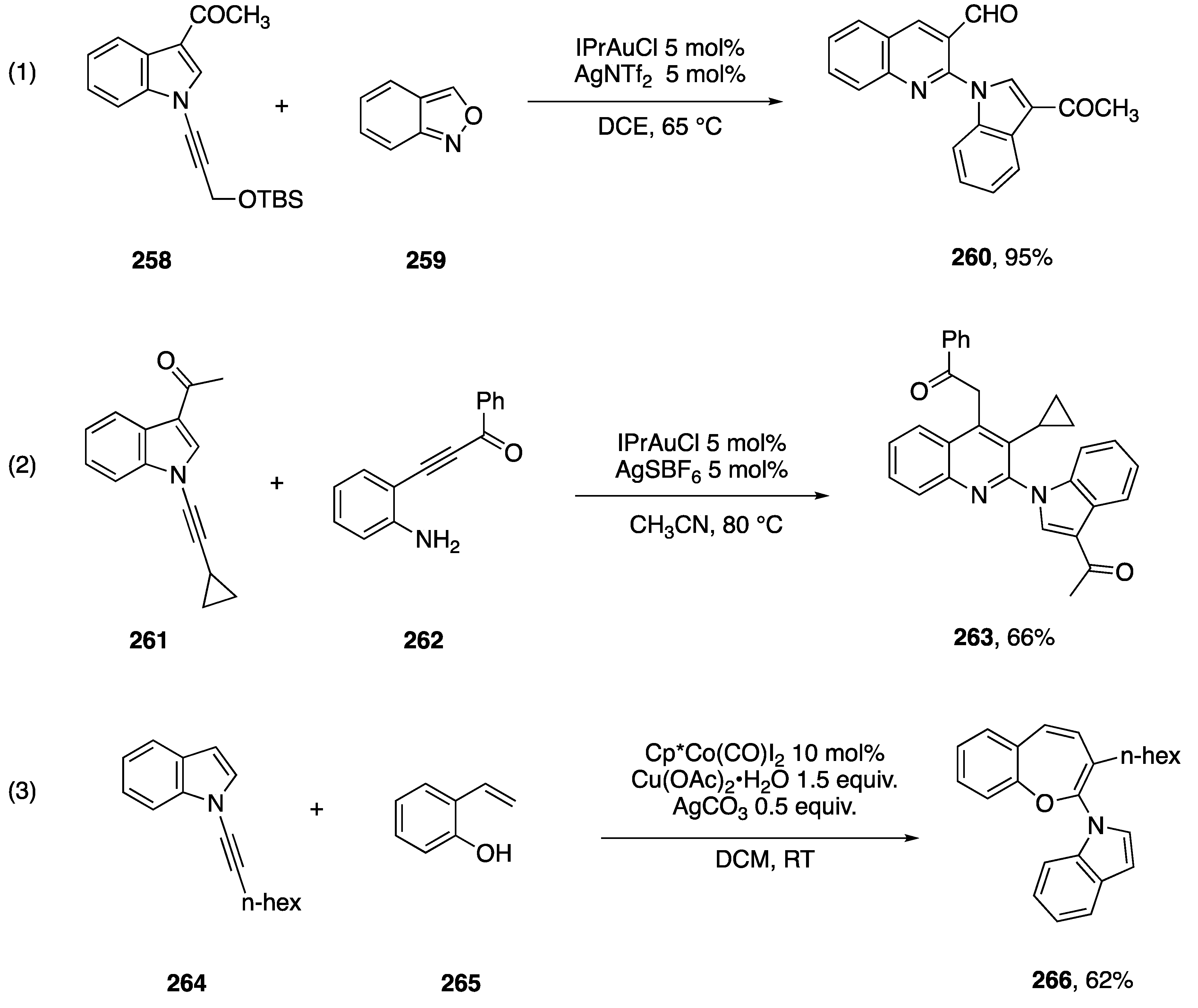{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reinus, B.; Kerwin, S.M. Preparation and Utility of N-Alkynyl Azoles in Synthesis. Molecules 2019, 24, 422. https://doi.org/10.3390/molecules24030422
Reinus B, Kerwin SM. Preparation and Utility of N-Alkynyl Azoles in Synthesis. Molecules. 2019; 24(3):422. https://doi.org/10.3390/molecules24030422
Chicago/Turabian StyleReinus, Brandon, and Sean M. Kerwin. 2019. "Preparation and Utility of N-Alkynyl Azoles in Synthesis" Molecules 24, no. 3: 422. https://doi.org/10.3390/molecules24030422
APA StyleReinus, B., & Kerwin, S. M. (2019). Preparation and Utility of N-Alkynyl Azoles in Synthesis. Molecules, 24(3), 422. https://doi.org/10.3390/molecules24030422