

Electrocatalytic Decomposition of Lithium Oxalate-Based Composite Microspheres as a Prelithiation Additive in Lithium-Ion Batteries

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
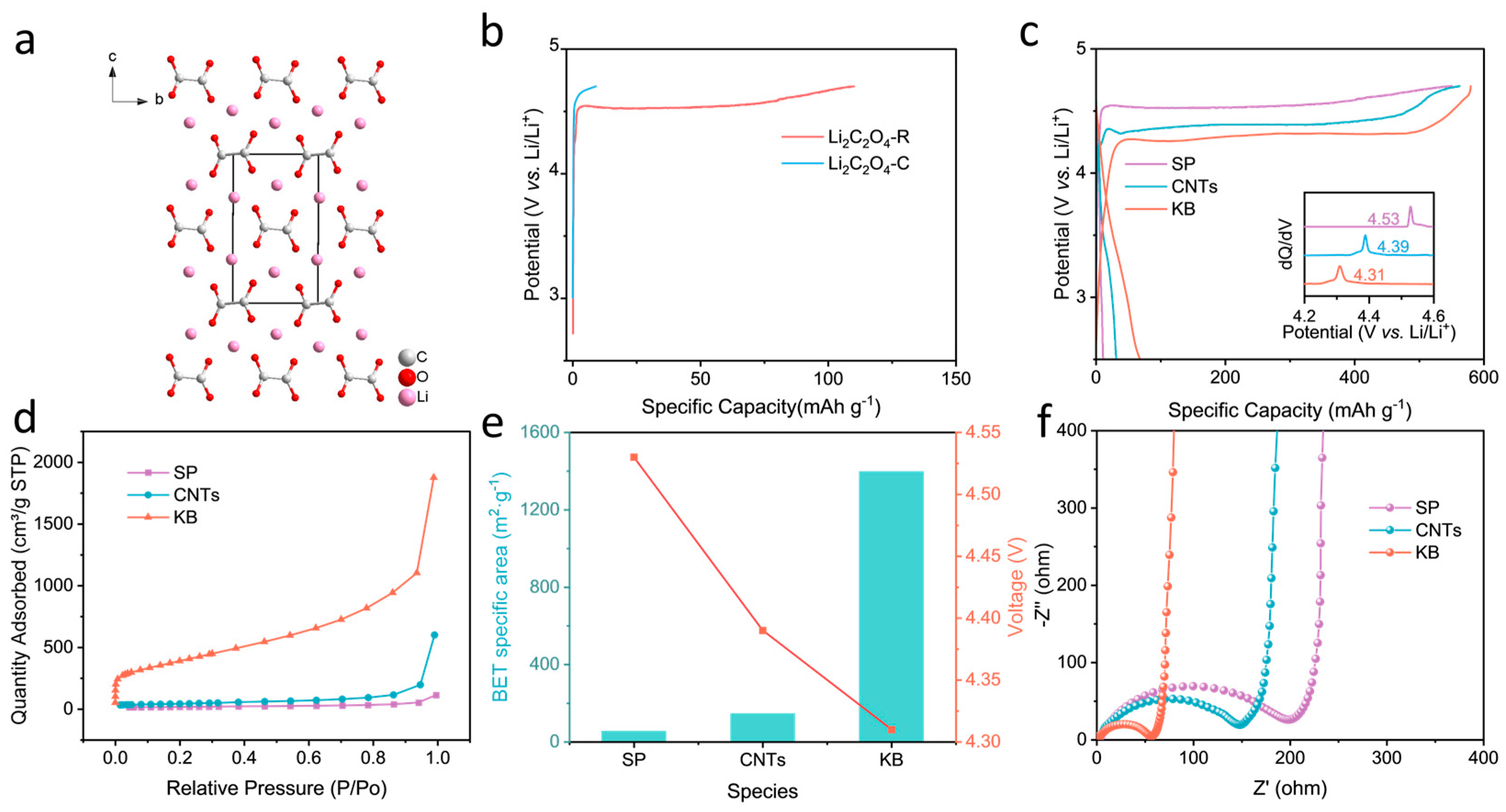
2.1. Recrystallization and Optimization of the Conductive Agent
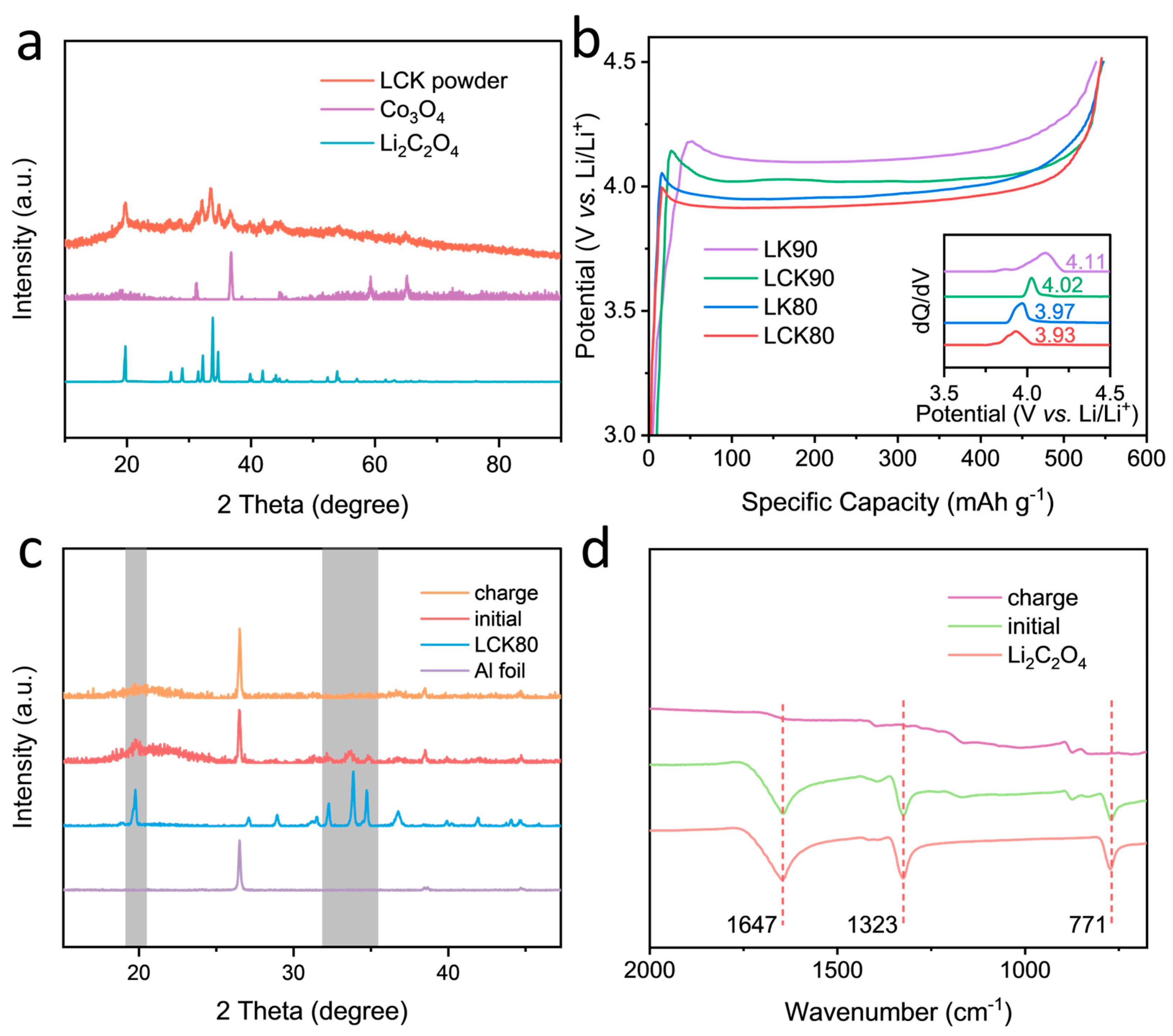
2.2. Characterization and Formulation Optimization of the Lithium Oxalate-Based Composite Microsphere as a Prelithiation Agent
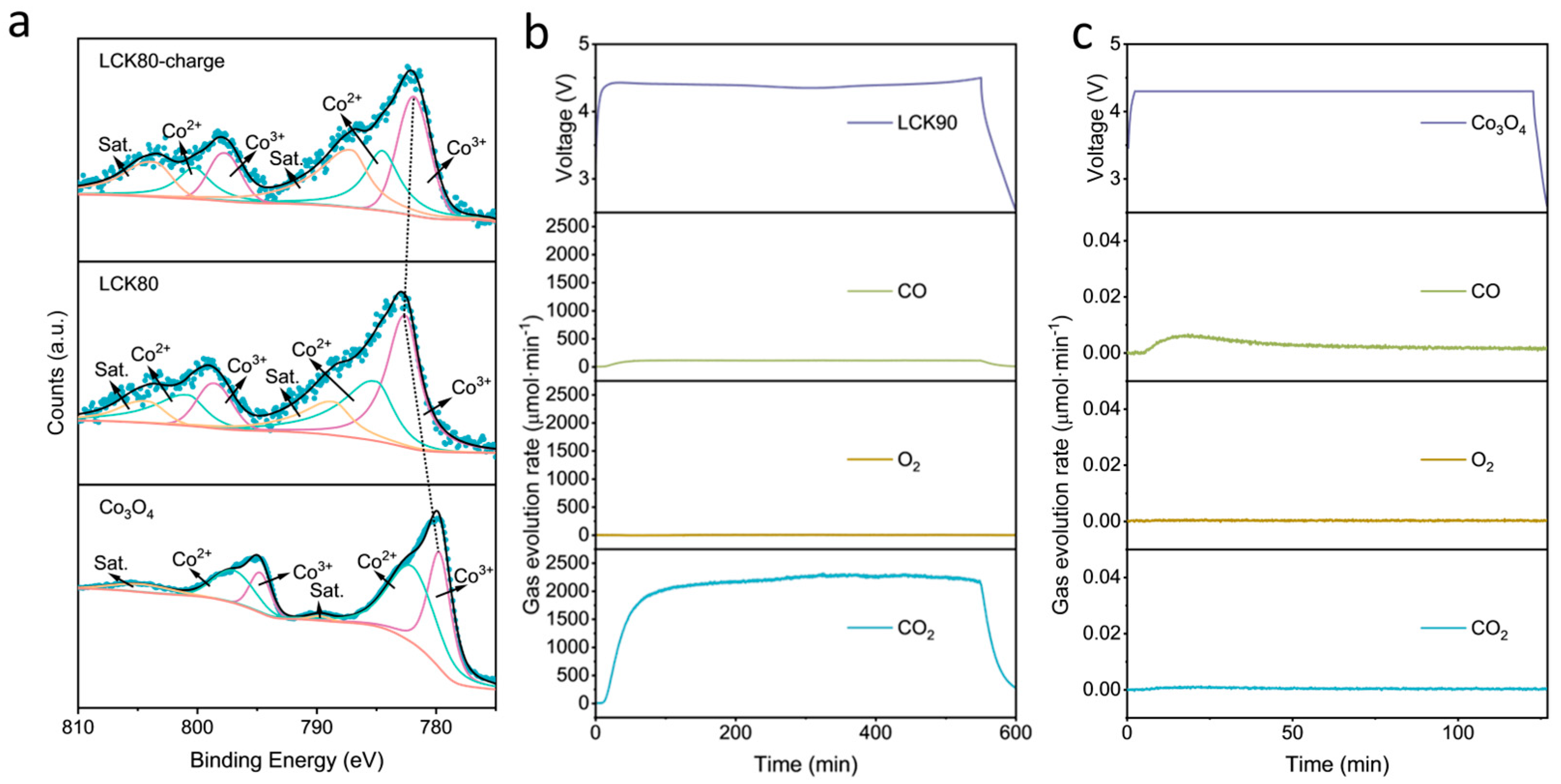
2.3. Application of the Lithium Oxalate-Based Composite Microsphere as a Prelithiation Agent
3. Materials and Methods
3.1. Materials
3.2. Recrystallization of Li2C2O4
3.3. Synthesis of the Lithium Oxalate-Based Composite Microsphere
3.4. Material Characterization
3.5. Electrochemical Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Armand, M.; Tarascon, J.-M. Building better batteries. Nature 2008, 451, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Cui, Y.; Liu, N. The path towards sustainable energy. Nat. Mater. 2017, 16, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Blomgren, G.E. The development and future of lithium ion batteries. J. Electrochem. Soc. 2016, 164, A5019. [Google Scholar] [CrossRef]
- Zou, K.; Deng, W.; Cai, P.; Deng, X.; Wang, B.; Liu, C.; Li, J.; Hou, H.; Zou, G.; Ji, X. Prelithiation/presodiation techniques for advanced electrochemical energy storage systems: Concepts, applications, and perspectives. Adv. Funct. Mater. 2021, 31, 2005581. [Google Scholar] [CrossRef]
- Huang, Z.; Deng, Z.; Zhong, Y.; Xu, M.; Li, S.; Liu, X.; Zhou, Y.; Huang, K.; Shen, Y.; Huang, Y. Progress and challenges of prelithiation technology for lithium-ion battery. Carbon Energy 2022, 4, 1107–1132. [Google Scholar] [CrossRef]
- Choi, J.W.; Aurbach, D. Promise and reality of post-lithium-ion batteries with high energy densities. Nat. Rev. Mater. 2016, 1, 16013. [Google Scholar] [CrossRef]
- Manthiram, A. An outlook on lithium ion battery technology. ACS Cent. Sci. 2017, 3, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Andreas, N.S.; Li, R.; Zhang, N.; Sun, C.; Lu, D.; Gao, T.; Chen, L.; Fan, X. Mitigating irreversible capacity loss for higher-energy lithium batteries. Energy Storage Mater. 2022, 48, 44–73. [Google Scholar] [CrossRef]
- Wang, F.; Wang, B.; Li, J.; Wang, B.; Zhou, Y.; Wang, D.; Liu, H.; Dou, S. Prelithiation: A Crucial Strategy for Boosting the Practical Application of Next-Generation Lithium Ion Battery. ACS Nano 2021, 15, 2197–2218. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, X.; Li, C.; Wang, K.; Sun, X.; Ma, Y. Recent advances in prelithiation materials and approaches for lithium-ion batteries and capacitors. Energy Storage Mater. 2020, 32, 497–516. [Google Scholar] [CrossRef]
- Saana Amiinu, I.; Imtiaz, S.; Geaney, H.; Kennedy, T.; Kapuria, N.; Singh, S.; Ryan, K.M. A thin Si nanowire network anode for high volumetric capacity and long-life lithium-ion batteries. J. Energy Chem. 2023, 81, 20–27. [Google Scholar] [CrossRef]
- Huang, G.; Han, J.; Lu, Z.; Wei, D.; Kashani, H.; Watanabe, K.; Chen, M. Ultrastable Silicon Anode by Three-Dimensional Nanoarchitecture Design. ACS Nano 2020, 14, 4374–4382. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Li, X.; Song, J.; Zhang, X.; Luo, L.; He, Y.; Li, B.; Cai, Y.; Hu, S.; Xiao, X.; et al. Hierarchical porous silicon structures with extraordinary mechanical strength as high-performance lithium-ion battery anodes. Nat. Commun. 2020, 11, 1474. [Google Scholar] [CrossRef] [PubMed]
- Mu, T.; Shen, B.; Lou, S.; Zhang, Z.; Ren, Y.; Zhou, X.; Zuo, P.; Du, C.; Ma, Y.; Huo, H.; et al. Scalable mesoporous silicon microparticles composed of interconnected nanoplates for superior lithium storage. Chem. Eng. J. 2019, 375, 121923. [Google Scholar] [CrossRef]
- Goodenough, J.B.; Kim, Y. Challenges for rechargeable Li batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Verma, P.; Maire, P.; Novák, P. A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries. Electrochim. Acta 2010, 55, 6332–6341. [Google Scholar] [CrossRef]
- Wu, H.; Cui, Y. Designing nanostructured Si anodes for high energy lithium ion batteries. Nano Today 2012, 7, 414–429. [Google Scholar] [CrossRef]
- Chae, S.; Ko, M.; Kim, K.; Ahn, K.; Cho, J. Confronting issues of the practical implementation of Si anode in high-energy lithium-ion batteries. Joule 2017, 1, 47–60. [Google Scholar] [CrossRef]
- Xu, Q.; Li, J.Y.; Sun, J.K.; Yin, Y.X.; Wan, L.J.; Guo, Y.G. Watermelon-inspired Si/C microspheres with hierarchical buffer structures for densely compacted lithium-ion battery anodes. Adv. Energy Mater. 2017, 7, 1601481. [Google Scholar] [CrossRef]
- Zhan, R.; Wang, X.; Chen, Z.; Seh, Z.W.; Wang, L.; Sun, Y. Promises and Challenges of the Practical Implementation of Prelithiation in Lithium-Ion Batteries. Adv. Energy Mater. 2021, 11, 2101565. [Google Scholar] [CrossRef]
- Abouimrane, A.; Cui, Y.; Chen, Z.; Belharouak, I.; Yahia, H.B.; Wu, H.; Assary, R.; Curtiss, L.A.; Amine, K. Enabling high energy density Li-ion batteries through Li2O activation. Nano Energy 2016, 27, 196–201. [Google Scholar] [CrossRef]
- Bie, Y.; Yang, J.; Wang, J.; Zhou, J.; Nuli, Y. Li2O2 as a cathode additive for the initial anode irreversibility compensation in lithium-ion batteries. Chem. Commun. 2017, 53, 8324–8327. [Google Scholar] [CrossRef]
- Sun, Y.; Li, Y.; Sun, J.; Li, Y.; Pei, A.; Cui, Y. Stabilized Li3N for efficient battery cathode prelithiation. Energy Storage Mater. 2017, 6, 119–124. [Google Scholar] [CrossRef]
- Park, H.; Yoon, T.; Kim, Y.-U.; Ryu, J.H.; Oh, S.M. Li2NiO2 as a sacrificing positive additive for lithium-ion batteries. Electrochim. Acta 2013, 108, 591–595. [Google Scholar] [CrossRef]
- Johnson, C.S.; Kang, S.H.; Vaughey, J.T.; Pol, S.V.; Balasubramanian, M.; Thackeray, M.M. Li2O Removal from Li5FeO4: A Cathode Precursor for Lithium-Ion Batteries. Chem. Mater. 2010, 22, 1263–1270. [Google Scholar] [CrossRef]
- Sun, Y.; Lee, H.-W.; Seh, Z.W.; Liu, N.; Sun, J.; Li, Y.; Cui, Y. High-capacity battery cathode prelithiation to offset initial lithium loss. Nat. Energy 2016, 1, 15008. [Google Scholar] [CrossRef]
- Sun, Y.; Lee, H.-W.; Zheng, G.; Seh, Z.W.; Sun, J.; Li, Y.; Cui, Y. In situ chemical synthesis of lithium fluoride/metal nanocomposite for high capacity prelithiation of cathodes. Nano Lett. 2016, 16, 1497–1501. [Google Scholar] [CrossRef]
- Sun, Y.; Lee, H.W.; Seh, Z.W.; Zheng, G.; Sun, J.; Li, Y.; Cui, Y. Lithium sulfide/metal nanocomposite as a high-capacity cathode prelithiation material. Adv. Energy Mater. 2016, 6, 1600154. [Google Scholar] [CrossRef]
- Du, J.; Wang, W.; Sheng Eng, A.Y.; Liu, X.; Wan, M.; Seh, Z.W.; Sun, Y. Metal/LiF/Li2O nanocomposite for battery cathode prelithiation: Trade-off between capacity and stability. Nano Lett. 2019, 20, 546–552. [Google Scholar] [CrossRef]
- Shanmukaraj, D.; Grugeon, S.; Laruelle, S.; Douglade, G.; Tarascon, J.-M.; Armand, M. Sacrificial salts: Compensating the initial charge irreversibility in lithium batteries. Electrochem. Commun. 2010, 12, 1344–1347. [Google Scholar] [CrossRef]
- Schomburg, F.; Heidrich, B.; Wennemar, S.; Drees, R.; Roth, T.; Kurrat, M.; Heimes, H.; Jossen, A.; Winter, M.; Cheong, J.Y.; et al. Lithium-ion battery cell formation: Status and future directions towards a knowledge-based process design. Energy Environ. Sci. 2024, 17, 2686–2733. [Google Scholar] [CrossRef]
- Solchenbach, S.; Wetjen, M.; Pritzl, D.; Schwenke, K.U.; Gasteiger, H.A. Lithium oxalate as capacity and cycle-life enhancer in LNMO/graphite and LNMO/SiG full cells. J. Electrochem. Soc. 2018, 165, A512. [Google Scholar] [CrossRef]
- Li, J.; Dai, A.; Amine, K.; Lu, J. Correlating Catalyst Design and Discharged Product to Reduce Overpotential in Li-CO2 Batteries. Small 2021, 17, 2007760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qin, C.; Wang, K.; Han, X.; Li, J.; Sui, M.; Yan, P. Deciphering the critical effect of cathode-electrolyte interphase by revealing its dynamic evolution. J. Energy Chem. 2023, 81, 192–199. [Google Scholar] [CrossRef]
- Huang, G.; Liang, J.; Zhong, X.; Liang, H.; Cui, C.; Zeng, C.; Wang, S.; Liao, M.; Shen, Y.; Zhai, T. Boosting the capability of Li2C2O4 as cathode pre-lithiation additive for lithium-ion batteries. Nano Res. 2023, 16, 3872–3878. [Google Scholar] [CrossRef]
- Zhong, W.; Zhang, C.; Li, S.; Zhang, W.; Zeng, Z.; Cheng, S.; Xie, J. Mo2C catalyzed low-voltage prelithiation using nano-Li2C2O4 for high-energy lithium-ion batteries. Sci. China Mater. 2023, 66, 903–912. [Google Scholar] [CrossRef]
- Shen, B.; Zhang, H.; Wu, Y.; Jiang, H.; Hu, Y.; Li, C. Co3O4 quantum dot-catalyzed lithium oxalate as a capacity and cycle-life enhancer in lithium-ion full cells. ACS Appl. Energy Mater. 2022, 5, 2112–2120. [Google Scholar] [CrossRef]
- Zhong, W.; Li, S.; Liu, M.; Wu, Q.; Zeng, Z.; Cheng, S.; Xie, J. Hierarchical spherical Mo2C/N-doped graphene catalyst facilitates low-voltage Li2C2O4 prelithiation. Nano Energy 2023, 115, 108757. [Google Scholar] [CrossRef]
- Haynes, W.M. CRC Handbook of Chemistry and Physics; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Cheng, X.B.; Yang, S.J.; Liu, Z.; Guo, J.X.; Jiang, F.N.; Jiang, F.; Xiong, X.; Tang, W.B.; Yuan, H.; Huang, J.Q. Electrochemically and Thermally Stable Inorganics–Rich Solid Electrolyte Interphase for Robust Lithium Metal Batteries. Adv. Mater. 2024, 36, 2307370. [Google Scholar] [CrossRef]
- Bai, S.; Bao, W.; Qian, K.; Han, B.; Li, W.; Sayahpour, B.; Sreenarayanan, B.; Tan, D.H.; Ham, S.Y.; Meng, Y.S. Elucidating the Role of Prelithiation in Si-based Anodes for Interface Stabilization. Adv. Energy Mater. 2023, 13, 2301041. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Lin, J.; Yin, Z.; Tong, Z.; Liu, J.; Wang, Z.; Zhou, Y.; Li, J. Electrocatalytic Decomposition of Lithium Oxalate-Based Composite Microspheres as a Prelithiation Additive in Lithium-Ion Batteries. Molecules 2024, 29, 2975. https://doi.org/10.3390/molecules29132975
Liu J, Lin J, Yin Z, Tong Z, Liu J, Wang Z, Zhou Y, Li J. Electrocatalytic Decomposition of Lithium Oxalate-Based Composite Microspheres as a Prelithiation Additive in Lithium-Ion Batteries. Molecules. 2024; 29(13):2975. https://doi.org/10.3390/molecules29132975
Chicago/Turabian StyleLiu, Jian, Jingyi Lin, Zuwei Yin, Zhen Tong, Junke Liu, Zhen Wang, Yao Zhou, and Juntao Li. 2024. "Electrocatalytic Decomposition of Lithium Oxalate-Based Composite Microspheres as a Prelithiation Additive in Lithium-Ion Batteries" Molecules 29, no. 13: 2975. https://doi.org/10.3390/molecules29132975
APA StyleLiu, J., Lin, J., Yin, Z., Tong, Z., Liu, J., Wang, Z., Zhou, Y., & Li, J. (2024). Electrocatalytic Decomposition of Lithium Oxalate-Based Composite Microspheres as a Prelithiation Additive in Lithium-Ion Batteries. Molecules, 29(13), 2975. https://doi.org/10.3390/molecules29132975