
Tumor Microenvironment Lactate: Is It a Cancer Progression Marker, Immunosuppressant, and Therapeutic Target?
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. TME Lactate Sources
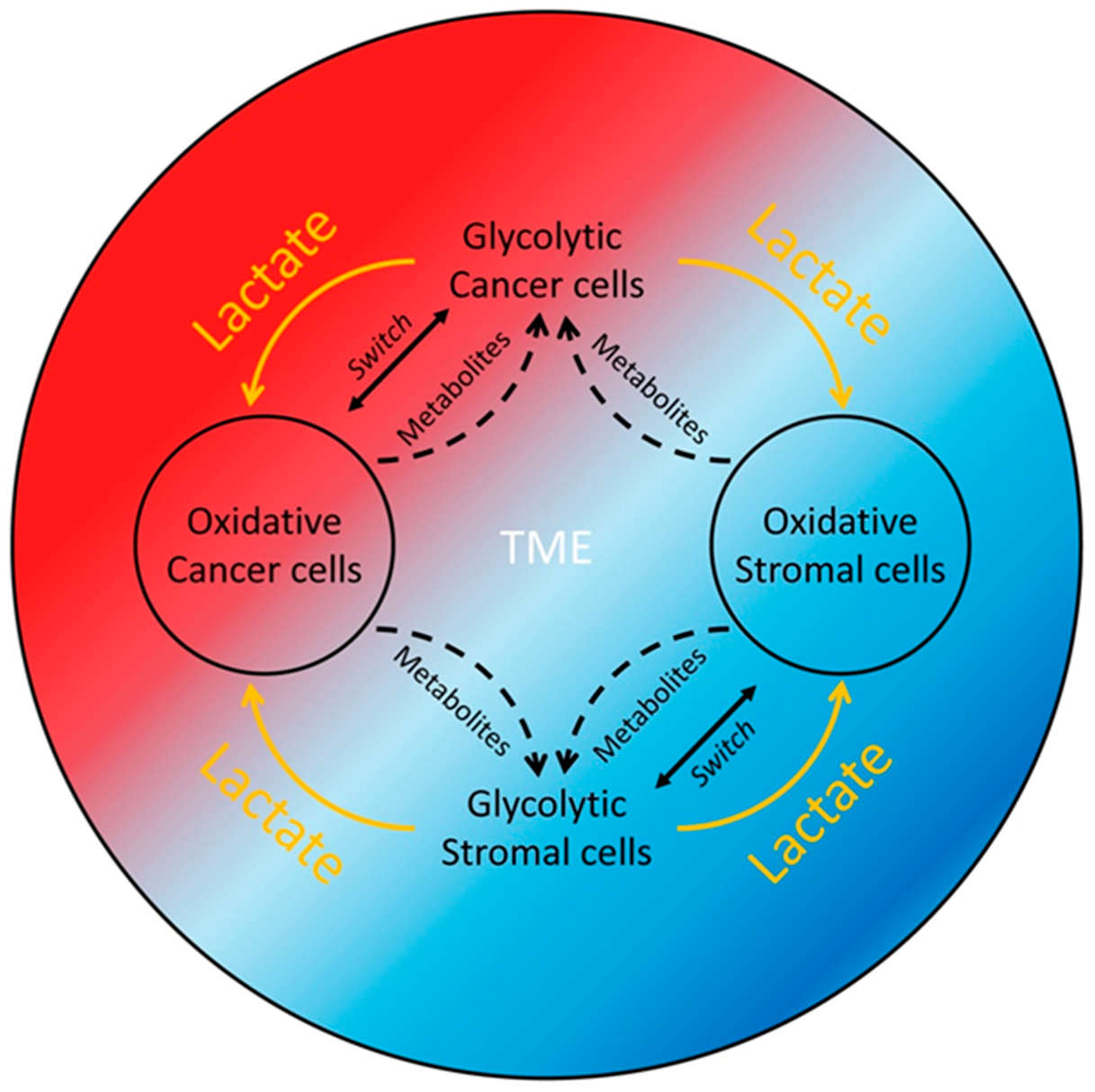
1.1. The Warburg Effect
1.2. The Reverse Warburg Effect
1.3. Glutaminolysis or Activation of Lactate-Generating Enzymes
1.4. Tumor Microbiome
1.5. Systemic Lactate Generation
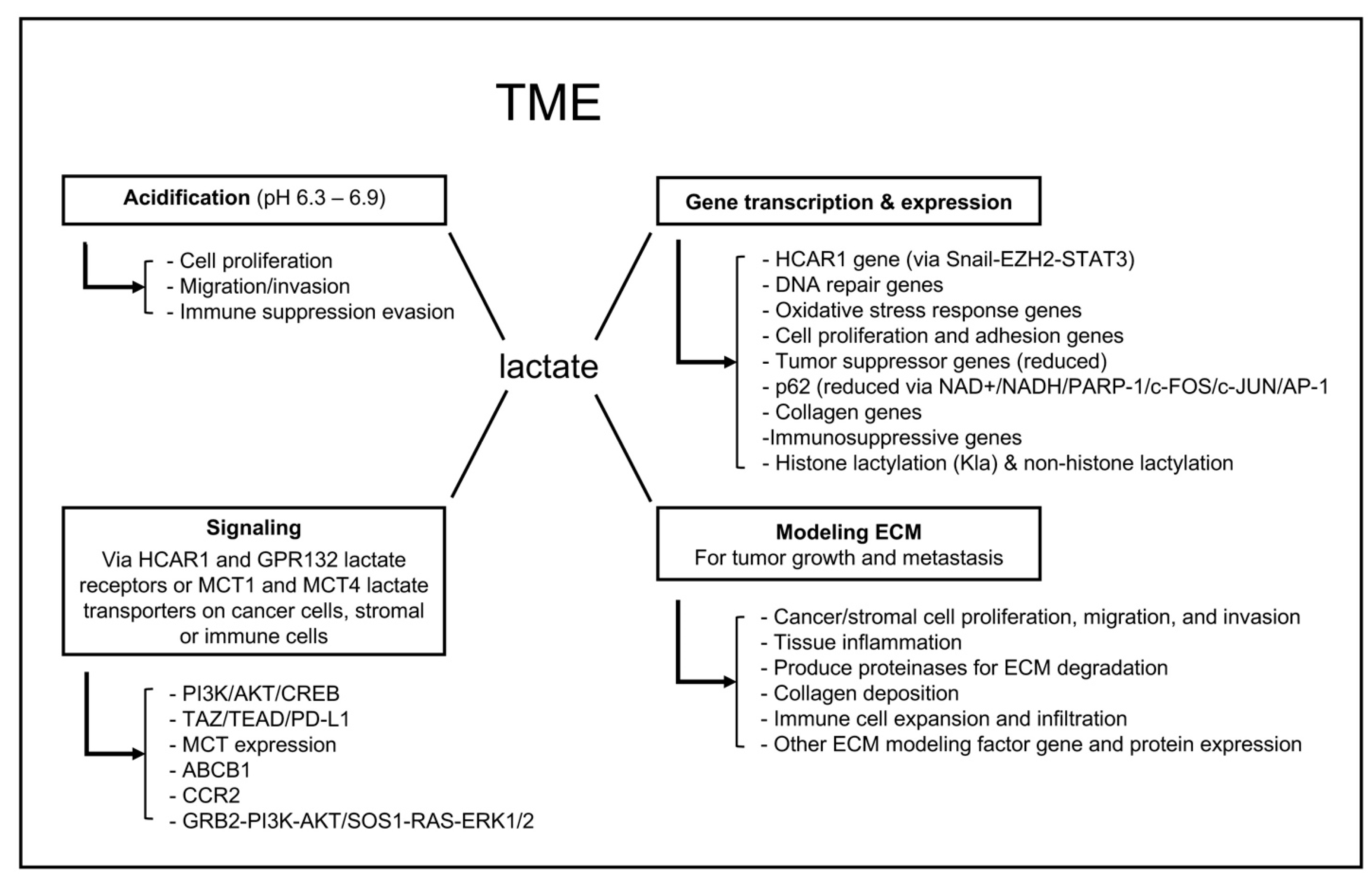
2. TME Lactate Functions
2.1. Acidification of the TME
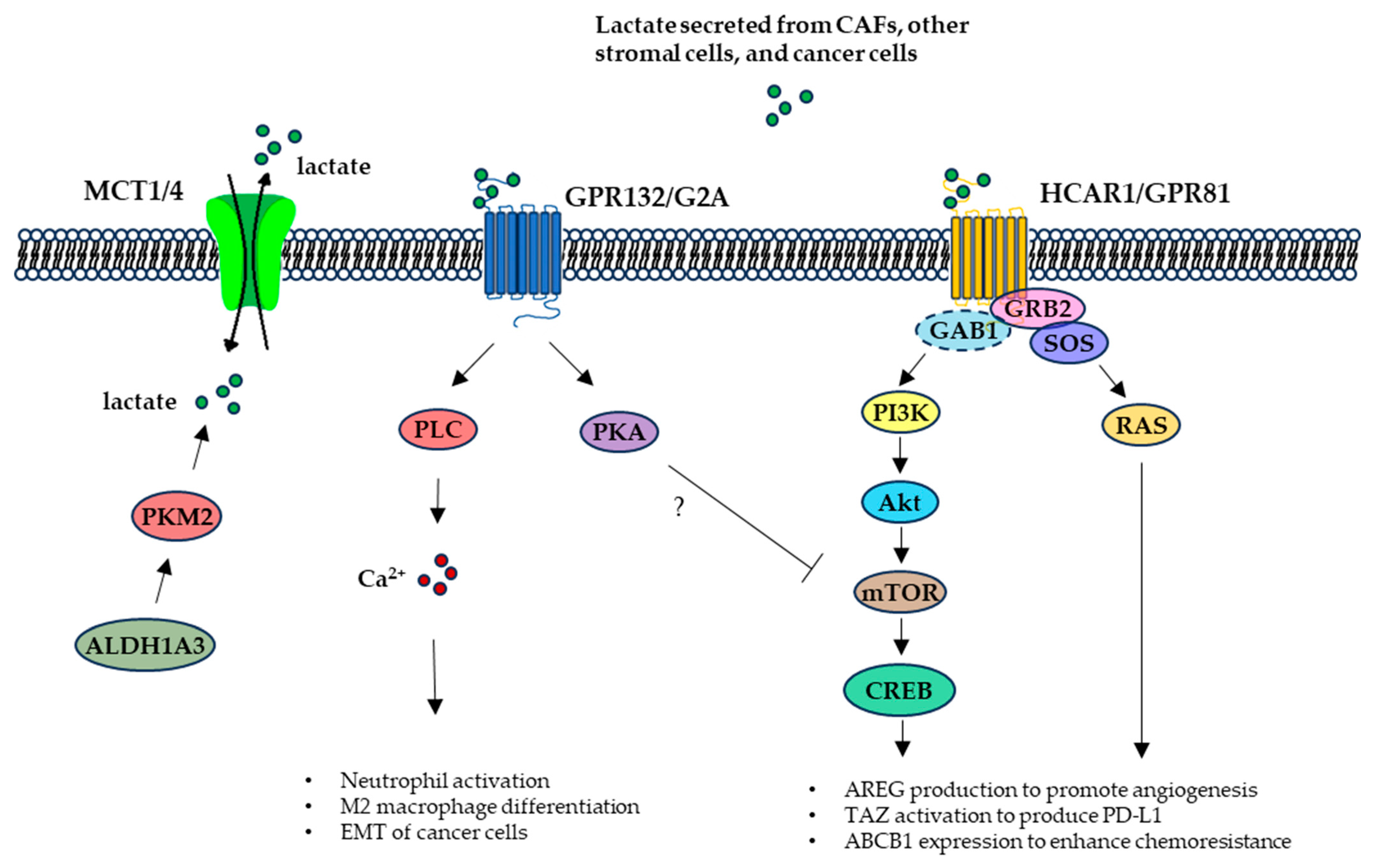
2.2. TME Lactate Signaling
2.3. TME Lactate Regulation of Gene Transcription and Expression
2.4. Lactate Modification of the TME
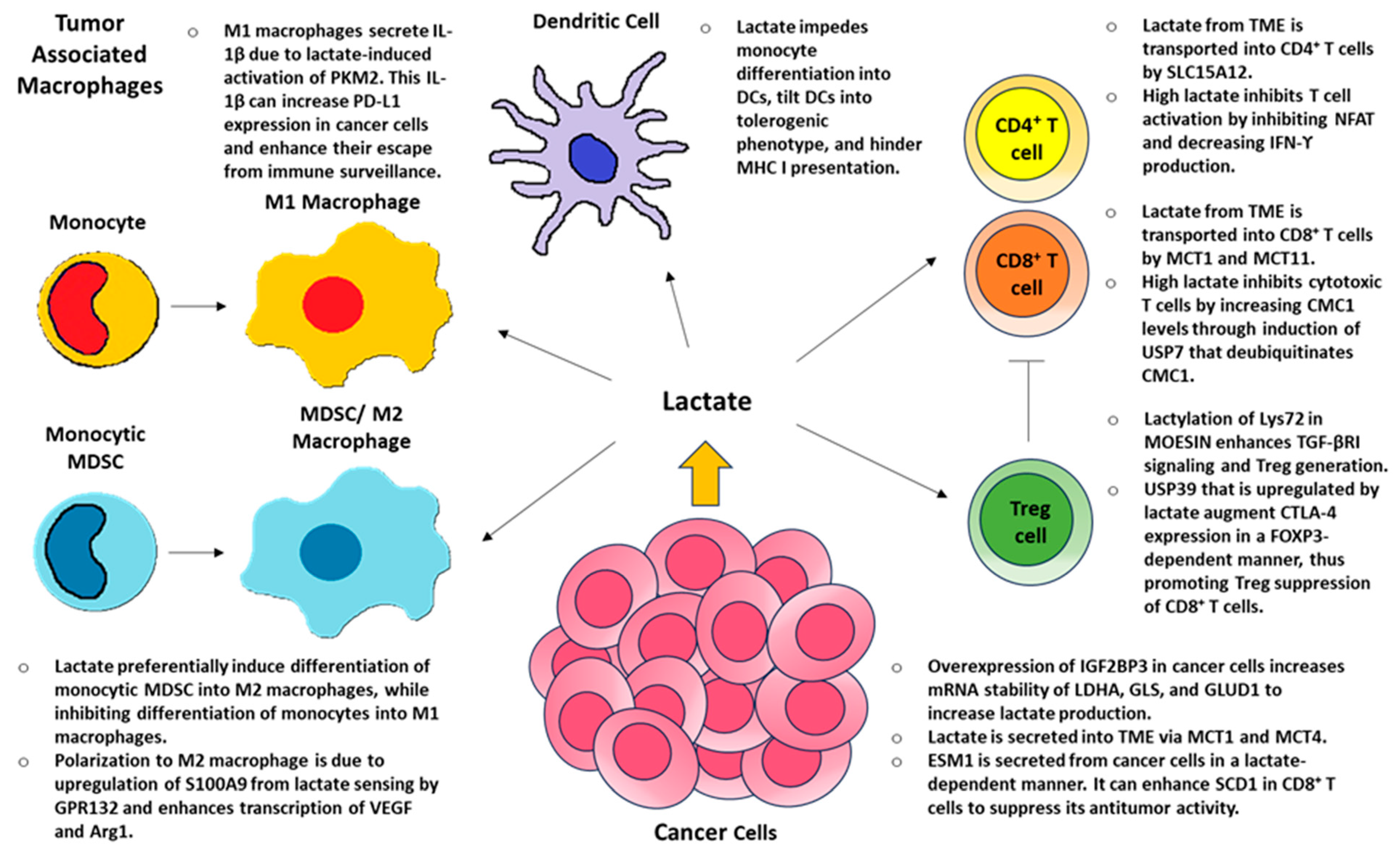
3. Lactate and Local Immunity
4. TME Lactate and Influences on Therapy
4.1. TME Lactate Effect on Therapeutic Resistance
4.2. Pre-Clinical Studies and Novel Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AARS1 | Alanyl-tRNA synthetase 1 |
| ALDH1A3 | Aldehyde dehydrogenase 1 family member A3 |
| ABCB1 | ATP-binding cassette sub-family B member 1 |
| ACAT1 | Acetyl-CoA acetyltransferase 1 |
| ALDOA | Aldolase A |
| AML | Acute myeloid leukemia |
| APOC2 | Apolipoprotein C2 |
| AREG | Amphiregulin |
| Arg1 | Arginase 1 |
| ASOs | Antisense oligonucleotides |
| BRD4 | Bromodomain-containing protein 4 |
| CAFs | Cancer-associated fibroblasts |
| CAV1 | Caveolin 1 |
| CCR2 | C-C motif chemokine receptor 2 |
| CDK7 | Cyclin-dependent kinase 7 |
| CMC1 | C-X9-C Motif Containing 1 |
| CRC | Colorectal carcinoma |
| CREB | cAMP response element-binding protein |
| CSCs | Cancer stem cells |
| CSDS | Chronic social defeat stress |
| CTLA-4 | Cytotoxic T-lymphocyte antigen 4 |
| DC | Dendritic cell |
| DCA | Dichloroacetate |
| DDR1 | Discoidin domain receptor 1 |
| DDX17 | Dead box deconjugate enzyme 17 |
| DLG5 | Discs large homolog 5 |
| DLL4 | Delta-like canonical Notch ligand 4 |
| ECM | Extracellular matrix |
| EPHA7 | EPH receptor A7 |
| ESM1 | Endothelial cell-specific molecule 1 |
| EZH2 | Enhancer of zeste homolog 2 |
| FFA | Free fatty acid |
| FOSCC | Feline oral squamous cell carcinoma |
| FOXP3 | Forkhead box P3 |
| GAB1 | GRB2-associated binding protein 1 |
| GCLC | Glutamate–cysteine ligase |
| GLS | Glutaminase |
| GLUD1 | Glutamate dehydrogenase 1 |
| GLUT1 | Glucose transporter 1 |
| GPCRs | G-protein-coupled receptors |
| GPR132 | G protein-coupled receptor 132 (aka G2A) |
| GRB2 | Growth factor receptor-bound protein 2 |
| GRN | Progranulin |
| HCAR1 | Hydroxycarboxylic acid receptor 1 (aka GPR81) |
| HGF | Hepatocyte growth factor |
| HIF1-α | Hypoxia-inducible factor 1 subunit alpha |
| HK-1 | Hexokinase 1 |
| HNSCC | Head and neck squamous cell carcinoma |
| HYOU1 | Hypoxia-upregulated 1 |
| IDH3G | Isocitrate dehydrogenase (NAD(+)) 3 non-catalytic subunit gamma |
| IGF2BP3 | Insulin-like growth factor 2 mRNA-binding protein 3 |
| ILA | Indole-3-lactic acid |
| Kla | Histone lactylation |
| KRAS | Kirsten rat sarcoma viral oncogene homologue |
| L. iners | Lactobacillus iners |
| LDHA | Lactate dehydrogenase A |
| LGSH | Lactoylglutathione |
| LMS | Lung-resident microbial score |
| LOX | Lactate oxidase |
| LRGS | Lactate-related gene signature |
| LS | Lactate shuttle |
| LUAD | Lung adenocarcinoma |
| lyso-PLs | Lysophospholipids |
| MCT | Monocarboxylate transporters |
| MDSC | Myeloid-derived suppressor cell |
| MGAT1 | α-1,3-Mannosyl-Glycoprotein 2-β-N-Acetylglucosaminyltransferase |
| MMP-9 | Matrix metalloproteinase 9 |
| MOESIN | Membrane-organizing extension spike protein |
| MRN | MRE11-RAD50-NBS1 |
| MSC | Mesenchymal stromal cell |
| MST1 | Mammalian Ste20-like kinase 1 |
| NBS1 | Nijmegen breakage syndrome protein 1 |
| NFAT | Nuclear factor of activated T cells |
| NSCLC | Non-small cell lung cancer |
| OSCC | Oral squamous cell carcinoma |
| OXPHOS | Oxidative phosphorylation |
| p62 | Sequestosome 1 (aka SQSTM1) |
| PARP-1 | Poly(ADP-ribose)-polymerase 1 |
| PCa | Prostate cancer |
| PCDH7 | Protocadherin 7 |
| PD-1 | Programmed cell death protein 1 |
| PDAC | Pancreatic ductal adenocarcinoma |
| PD-L1 | Programmed death-ligand 1 |
| PFKFB4 | 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 |
| PI3K | Phosphatidylinositol 3-kinase |
| PKM2 | Pyruvate kinase M2 isoform |
| PLC | Phospholipase C |
| PLS | Postprandial lactate shuttle |
| PMF | Primary myelofibrosis |
| PMN-MDSCs | Polymorphonuclear myeloid-derived suppressor cells |
| PTBP1 | Polypyrimidine tract-binding protein 1 |
| PTM | Posttranslational modification |
| RORγt | Retinoic acid receptor-related orphan receptor-γt |
| ROS | Reactive oxygen species |
| S100A9 | S100 calcium-binding protein A9 |
| SCD1 | Stearoyl-CoA desaturase 1 |
| SLC1A5 | Sodium-dependent solute carrier family 1 member 5 |
| SMAD3 | Mothers against decapentaplegic homolog 3 |
| SOS1 | SOS Ras/Rac guanine nucleotide exchange factor 1 |
| SOX2 | SRY-box transcription factor 2 |
| STK11/LKB1 | Serine/threonine kinase 11/liver kinase B1 |
| TAMs | Tumor-associated macrophages |
| Tcf7 | T cell factor 7 |
| TEAD | Transcriptional enhanced associate domain |
| TKIs | Tyrosine kinase inhibitors |
| TME | Tumor microenvironment |
| Tregs | Regulatory T cells |
| TRIM21 | Tripartite motif containing-21 |
| USP39 | Ubiquitin-specific peptidase 39 |
| Vegf | Vascular endothelial growth factor |
| XBP1 | X-box binding protein 1 |
| XRCC1 | X-ray repair cross-complementing protein 1 |
| YAP1 | Yes-associated protein 1 |
| YTHDF2 | YTH N6-methyladenosine RNA-binding protein 2 |
References
- Apostolova, P.; Pearce, E.L. Lactic acid and lactate: Revisiting the physiological roles in the tumor microenvironment. Trends Immunol. 2022, 43, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; Enerback, S. Lactate: The ugly duckling of energy metabolism. Nat. Metab. 2020, 2, 566–571. [Google Scholar] [CrossRef]
- Warburg, O. The Metabolism of Carcinoma Cells. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ruud, K.F.; Hiscox, W.C.; Yu, I.; Chen, R.K.; Li, W. Distinct phenotypes of cancer cells on tissue matrix gel. Breast Cancer Res. BCR 2020, 22, 82. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef]
- Curry, J.M.; Tuluc, M.; Whitaker-Menezes, D.; Ames, J.A.; Anantharaman, A.; Butera, A.; Leiby, B.; Cognetti, D.; Sotgia, F.; Lisanti, M.P.; et al. Cancer metabolism, stemness and tumor recurrence. Cell Cycle 2013, 12, 1371–1384. [Google Scholar] [CrossRef]
- Keller, C.R.; Martinez, S.R.; Keltz, A.; Chen, M.; Li, W. Lactate Oxidase Disrupts Lactate-Activated RAS and PI3K Oncogenic Signaling. Cancers 2024, 16, 2817. [Google Scholar] [CrossRef]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Q.; Huang, X.; Yang, M.; Zhou, S.; Fang, Z.; Tang, Y.; Chen, Q.; Hou, H.; Li, L.; et al. Lactate in the tumor microenvironment: A rising star for targeted tumor therapy. Front. Nutr. 2023, 10, 1113739. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ying, M.; Hu, X. Lactic acidosis switches cancer cells from aerobic glycolysis back to dominant oxidative phosphorylation. Oncotarget 2016, 7, 40621–40629. [Google Scholar] [CrossRef]
- Damiani, C.; Colombo, R.; Gaglio, D.; Mastroianni, F.; Pescini, D.; Westerhoff, H.V.; Mauri, G.; Vanoni, M.; Alberghina, L. A metabolic core model elucidates how enhanced utilization of glucose and glutamine, with enhanced glutamine-dependent lactate production, promotes cancer cell growth: The WarburQ effect. PLoS Comput. Biol. 2017, 13, e1005758. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef]
- Lin, K.; Lin, X.; Luo, F. IGF2BP3 boosts lactate generation to accelerate gastric cancer immune evasion. Apoptosis 2024, 29, 2147–2160. [Google Scholar] [CrossRef]
- Zhou, T.; Xiao, Z.; Lu, J.; Zhang, L.; Bo, L.; Wang, J. IGF2BP3-mediated regulation of GLS and GLUD1 gene expression promotes treg-induced immune escape in human cervical cancer. Am. J. Cancer Res. 2023, 13, 5289–5305. [Google Scholar]
- Qian, Y.; Galan-Cobo, A.; Guijarro, I.; Dang, M.; Molkentine, D.; Poteete, A.; Zhang, F.; Wang, Q.; Wang, J.; Parra, E.; et al. MCT4-dependent lactate secretion suppresses antitumor immunity in LKB1-deficient lung adenocarcinoma. Cancer Cell 2023, 41, 1363–1380.e7. [Google Scholar] [CrossRef]
- Colbert, L.E.; El Alam, M.B.; Wang, R.; Karpinets, T.; Lo, D.; Lynn, E.J.; Harris, T.A.; Elnaggar, J.H.; Yoshida-Court, K.; Tomasic, K.; et al. Tumor-resident Lactobacillus iners confer chemoradiation resistance through lactate-induced metabolic rewiring. Cancer Cell 2023, 41, 1945–1962.e11. [Google Scholar] [CrossRef]
- Gu, J.; Xu, X.; Li, X.; Yue, L.; Zhu, X.; Chen, Q.; Gao, J.; Takashi, M.; Zhao, W.; Zhao, B.; et al. Tumor-resident microbiota contributes to colorectal cancer liver metastasis by lactylation and immune modulation. Oncogene 2024, 43, 2389–2404. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Tang, Q.; Yu, S.; Xie, M.; Zheng, W.; Chen, G.; Yin, Y.; Huang, X.; Wo, K.; Lei, H.; et al. F. nucleatum facilitates oral squamous cell carcinoma progression via GLUT1-driven lactate production. EBioMedicine 2023, 88, 104444. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Chen, X.; Luo, Y.; Que, J.; Chen, L. Intratumor microbiome derived glycolysis-lactate signatures depicts immune heterogeneity in lung adenocarcinoma by integration of microbiomic, transcriptomic, proteomic and single-cell data. Front. Microbiol. 2023, 14, 1202454. [Google Scholar] [CrossRef]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Duncan, S.H.; Sheridan, P.O.; Walker, A.W.; Flint, H.J. Microbial lactate utilisation and the stability of the gut microbiome. Gut Microbiome 2022, 3, e3. [Google Scholar] [CrossRef]
- Leija, R.G.; Curl, C.C.; Arevalo, J.A.; Osmond, A.D.; Duong, J.J.; Huie, M.J.; Masharani, U.; Brooks, G.A. Enteric and systemic postprandial lactate shuttle phases and dietary carbohydrate carbon flow in humans. Nat. Metab. 2024, 6, 670–677. [Google Scholar] [CrossRef]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef]
- Yu, K.; Li, Q.; Sun, X.; Peng, X.; Tang, Q.; Chu, H.; Zhou, L.; Wang, B.; Zhou, Z.; Deng, X.; et al. Bacterial indole-3-lactic acid affects epithelium-macrophage crosstalk to regulate intestinal homeostasis. Proc. Natl. Acad. Sci. USA 2023, 120, e2309032120. [Google Scholar] [CrossRef]
- Hu, J.; Cai, M.; Shang, Q.; Li, Z.; Feng, Y.; Liu, B.; Xue, X.; Lou, S. Elevated Lactate by High-Intensity Interval Training Regulates the Hippocampal BDNF Expression and the Mitochondrial Quality Control System. Front. Physiol. 2021, 12, 629914. [Google Scholar] [CrossRef]
- Gass, G.C.; Rogers, S.; Mitchell, R. Blood lactate concentration following maximum exercise in trained subjects. Br. J. Sports Med. 1981, 15, 172–176. [Google Scholar] [CrossRef]
- Hermann, R.; Lay, D.; Wahl, P.; Roth, W.T.; Petrowski, K. Effects of psychosocial and physical stress on lactate and anxiety levels. Stress 2019, 22, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zhu, T.; Huang, Y.; Fang, Z.; Luo, F. Current understanding of the contribution of lactate to the cardiovascular system and its therapeutic relevance. Front. Endocrinol. 2023, 14, 1205442. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.X.; Wang, Z.; Yu, T. Lactate metabolism in human health and disease. Signal Transduct. Target. Ther. 2022, 7, 305. [Google Scholar] [CrossRef]
- Meert, K.L.; McCaulley, L.; Sarnaik, A.P. Mechanism of lactic acidosis in children with acute severe asthma. Pediatr. Crit. Care Med. 2012, 13, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Heinig, R.E.; Clarke, E.F.; Waterhouse, C. Lactic acidosis and liver disease. Arch. Intern. Med. 1979, 139, 1229–1232. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.A. Cell-cell and intracellular lactate shuttles. J. Physiol. 2009, 587, 5591–5600. [Google Scholar] [CrossRef]
- Hopkins, E.; Sanvictores, T.; Sharma, S. Physiology, Acid Base Balance. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Hosonuma, M.; Yoshimura, K. Association between pH regulation of the tumor microenvironment and immunological state. Front. Oncol. 2023, 13, 1175563. [Google Scholar] [CrossRef]
- Fournier, P.A.; Fairchild, T.J.; Ferreira, L.D.; Brau, L. Post-exercise muscle glycogen repletion in the extreme: Effect of food absence and active recovery. J. Sports Sci. Med. 2004, 3, 139–146. [Google Scholar]
- Zhang, Y.; Peng, Q.; Zheng, J.; Yang, Y.; Zhang, X.; Ma, A.; Qin, Y.; Qin, Z.; Zheng, X. The function and mechanism of lactate and lactylation in tumor metabolism and microenvironment. Genes Dis. 2023, 10, 2029–2037. [Google Scholar] [CrossRef]
- Toft, N.J.; Axelsen, T.V.; Pedersen, H.L.; Mele, M.; Burton, M.; Balling, E.; Johansen, T.; Thomassen, M.; Christiansen, P.M.; Boedtkjer, E. Acid-base transporters and pH dynamics in human breast carcinomas predict proliferative activity, metastasis, and survival. eLife 2021, 10, e68447. [Google Scholar] [CrossRef]
- Bogdanov, A.; Bogdanov, A.; Chubenko, V.; Volkov, N.; Moiseenko, F.; Moiseyenko, V. Tumor acidity: From hallmark of cancer to target of treatment. Front. Oncol. 2022, 12, 979154. [Google Scholar] [CrossRef] [PubMed]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Busco, G.; Cardone, R.A.; Greco, M.R.; Bellizzi, A.; Colella, M.; Antelmi, E.; Mancini, M.T.; Dell’Aquila, M.E.; Casavola, V.; Paradiso, A.; et al. NHE1 promotes invadopodial ECM proteolysis through acidification of the peri-invadopodial space. FASEB J. 2010, 24, 3903–3915. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Singleton, P.A.; Diedrich, F.; Stern, R.; Gilad, E. CD44 interaction with Na+-H+ exchanger (NHE1) creates acidic microenvironments leading to hyaluronidase-2 and cathepsin B activation and breast tumor cell invasion. J. Biol. Chem. 2004, 279, 26991–27007. [Google Scholar] [CrossRef]
- Shuman Moss, L.A.; Jensen-Taubman, S.; Stetler-Stevenson, W.G. Matrix Metalloproteinases: Changing Roles in Tumor Progression and Metastasis. Am. J. Pathol. 2012, 181, 1895–1899. [Google Scholar] [CrossRef]
- Kumar, V.B.S.; Viji, R.I.; Kiran, M.S.; Sudhakaran, P.R. Endothelial cell response to lactate: Implication of PAR modification of VEGF. J. Cell. Physiol. 2007, 211, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Choi, S.Y.C.; Niu, X.; Kang, N.; Xue, H.; Killam, J.; Wang, Y. Lactic Acid and an Acidic Tumor Microenvironment suppress Anticancer Immunity. Int. J. Mol. Sci. 2020, 21, 8363. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Ferrari, P. Involvement of tumor immune microenvironment metabolic reprogramming in colorectal cancer progression, immune escape, and response to immunotherapy. Front. Immunol. 2024, 15, 1353787. [Google Scholar] [CrossRef]
- Quinn, W.J., 3rd; Jiao, J.; TeSlaa, T.; Stadanlick, J.; Wang, Z.; Wang, L.; Akimova, T.; Angelin, A.; Schafer, P.M.; Cully, M.D.; et al. Lactate Limits T Cell Proliferation via the NAD(H) Redox State. Cell Rep. 2020, 33, 108500. [Google Scholar] [CrossRef]
- Grzes, K.M.; Field, C.S.; Pearce, E.J. Treg Cells Survive and Thrive in Inhospitable Environments. Cell Metab. 2017, 25, 1213–1215. [Google Scholar] [CrossRef] [PubMed]
- Wada, H.; Hamaguchi, R.; Narui, R.; Morikawa, H. Meaning and Significance of “Alkalization Therapy for Cancer”. Front. Oncol. 2022, 12, 920843. [Google Scholar] [CrossRef]
- Goodwin, M.L.; Harris, J.E.; Hernandez, A.; Gladden, L.B. Blood lactate measurements and analysis during exercise: A guide for clinicians. J. Diabetes Sci. Technol. 2007, 1, 558–569. [Google Scholar] [CrossRef]
- Wideman, L.; Weltman, J.Y.; Hartman, M.L.; Veldhuis, J.D.; Weltman, A. Growth hormone release during acute and chronic aerobic and resistance exercise: Recent findings. Sports Med. 2002, 32, 987–1004. [Google Scholar] [CrossRef]
- Brooks, G.A.; Osmond, A.D.; Arevalo, J.A.; Duong, J.J.; Curl, C.C.; Moreno-Santillan, D.D.; Leija, R.G. Lactate as a myokine and exerkine: Drivers and signals of physiology and metabolism. J. Appl. Physiol. 2023, 134, 529–548. [Google Scholar] [CrossRef]
- Lee, Y.J.; Shin, K.J.; Park, S.A.; Park, K.S.; Park, S.; Heo, K.; Seo, Y.K.; Noh, D.Y.; Ryu, S.H.; Suh, P.G. G-protein-coupled receptor 81 promotes a malignant phenotype in breast cancer through angiogenic factor secretion. Oncotarget 2016, 7, 70898–70911. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.P.; Bhattacharjee, P.; Ramachandran, S.; Sivaprakasam, S.; Ristic, B.; Sikder, M.O.F.; Ganapathy, V. The lactate receptor GPR81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene 2020, 39, 3292–3304. [Google Scholar] [CrossRef]
- Roland, C.L.; Arumugam, T.; Deng, D.; Liu, S.H.; Philip, B.; Gomez, S.; Burns, W.R.; Ramachandran, V.; Wang, H.; Cruz-Monserrate, Z.; et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res. 2014, 74, 5301–5310. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Yang, H.; Zhang, Y.; Wei, H.; Zhu, Z.; Zhu, B.; Yang, M.; Cao, W.; Wang, L.; Wu, Z. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene 2017, 36, 5829–5839. [Google Scholar] [CrossRef]
- Wagner, W.; Kania, K.D.; Blauz, A.; Ciszewski, W.M. The lactate receptor (HCAR1/GPR81) contributes to doxorubicin chemoresistance via ABCB1 transporter up-regulation in human cervical cancer HeLa cells. J. Physiol. Pharmacol. 2017, 68, 555–564. [Google Scholar]
- He, J.; Chai, X.; Zhang, Q.; Wang, Y.; Wang, Y.; Yang, X.; Wu, J.; Feng, B.; Sun, J.; Rui, W.; et al. The lactate receptor HCAR1 drives the recruitment of immunosuppressive PMN-MDSCs in colorectal cancer. Nat. Immunol. 2025, 26, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Ye, R.D. The lysophospholipid receptor G2A activates a specific combination of G proteins and promotes apoptosis. J. Biol. Chem. 2003, 278, 14379–14386. [Google Scholar] [CrossRef] [PubMed]
- Frasch, S.C.; Zemski-Berry, K.; Murphy, R.C.; Borregaard, N.; Henson, P.M.; Bratton, D.L. Lysophospholipids of different classes mobilize neutrophil secretory vesicles and induce redundant signaling through G2A. J. Immunol. 2007, 178, 6540–6548. [Google Scholar] [CrossRef] [PubMed]
- Obinata, H.; Hattori, T.; Nakane, S.; Tatei, K.; Izumi, T. Identification of 9-hydroxyoctadecadienoic acid and other oxidized free fatty acids as ligands of the G protein-coupled receptor G2A. J. Biol. Chem. 2005, 280, 40676–40683. [Google Scholar] [CrossRef]
- Yi, C.; He, J.; Huang, D.; Zhao, Y.; Zhang, C.; Ye, X.; Huang, Y.; Nussinov, R.; Zheng, J.; Liu, M.; et al. Activation of orphan receptor GPR132 induces cell differentiation in acute myeloid leukemia. Cell Death Dis. 2022, 13, 1004. [Google Scholar] [CrossRef]
- Chen, P.; Zuo, H.; Xiong, H.; Kolar, M.J.; Chu, Q.; Saghatelian, A.; Siegwart, D.J.; Wan, Y. Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, 580–585. [Google Scholar] [CrossRef]
- Tong, Z.; Wang, X.; Shi, S.; Hou, T.; Gao, G.; Li, D.; Shan, Y.; Zhang, C. Development of lactate-related gene signature and prediction of overall survival and chemosensitivity in patients with colorectal cancer. Cancer Med. 2023, 12, 10105–10122. [Google Scholar] [CrossRef]
- Liu, C.; Ni, C.; Li, C.; Tian, H.; Jian, W.; Zhong, Y.; Zhou, Y.; Lyu, X.; Zhang, Y.; Xiang, X.J.; et al. Lactate-related gene signatures as prognostic predictors and comprehensive analysis of immune profiles in nasopharyngeal carcinoma. J. Transl. Med. 2024, 22, 1116. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, J.; Li, M.; Zhang, Y.; Li, Q.; Zheng, Y.; Lai, W. Identification of Lactate-Related Gene Signature for Prediction of Progression and Immunotherapeutic Response in Skin Cutaneous Melanoma. Front. Oncol. 2022, 12, 818868. [Google Scholar] [CrossRef]
- Zhang, Z.; Fang, T.; Lv, Y. A novel lactate metabolism-related signature predicts prognosis and tumor immune microenvironment of breast cancer. Front. Genet. 2022, 13, 934830. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, S.; Li, J.; Yuan, Y.; Chen, S.; Zuo, M.; Li, W.; Feng, W.; Chen, M.; Liu, Y. Prognostic value of lactate metabolism-related gene expression signature in adult primary gliomas and its impact on the tumor immune microenvironment. Front. Oncol. 2022, 12, 1008219. [Google Scholar] [CrossRef]
- Li, X.; Du, Y. Lactate Metabolism Subtypes Analysis Reveals CCDC80 as a Novel Prognostic Biomarker in Gastric Cancer. J. Cancer 2024, 15, 5557–5576. [Google Scholar] [CrossRef]
- Xie, Q.; Zhu, Z.; He, Y.; Zhang, Z.; Zhang, Y.; Wang, Y.; Luo, J.; Peng, T.; Cheng, F.; Gao, J.; et al. A lactate-induced Snail/STAT3 pathway drives GPR81 expression in lung cancer cells. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165576. [Google Scholar] [CrossRef] [PubMed]
- Govoni, M.; Rossi, V.; Di Stefano, G.; Manerba, M. Lactate Upregulates the Expression of DNA Repair Genes, Causing Intrinsic Resistance of Cancer Cells to Cisplatin. Pathol. Oncol. Res. 2021, 27, 1609951. [Google Scholar] [CrossRef] [PubMed]
- Rijal, G.; Li, W. 3D scaffolds in breast cancer research. Biomaterials 2016, 81, 135–156. [Google Scholar] [CrossRef] [PubMed]
- Rijal, G.; Li, W. Native-mimicking in vitro microenvironment: An elusive and seductive future for tumor modeling and tissue engineering. J. Biol. Eng. 2018, 12, 20. [Google Scholar] [CrossRef]
- Yang, D.; Liu, J.; Qian, H.; Zhuang, Q. Cancer-associated fibroblasts: From basic science to anticancer therapy. Exp. Mol. Med. 2023, 55, 1322–1332. [Google Scholar] [CrossRef]
- Linares, J.F.; Cid-Diaz, T.; Duran, A.; Osrodek, M.; Martinez-Ordonez, A.; Reina-Campos, M.; Kuo, H.H.; Elemento, O.; Martin, M.L.; Cordes, T.; et al. The lactate-NAD(+) axis activates cancer-associated fibroblasts by downregulating p62. Cell Rep. 2022, 39, 110792. [Google Scholar] [CrossRef]
- Ippolito, L.; Duatti, A.; Iozzo, M.; Comito, G.; Pardella, E.; Lorito, N.; Bacci, M.; Pranzini, E.; Santi, A.; Sandrini, G.; et al. Lactate supports cell-autonomous ECM production to sustain metastatic behavior in prostate cancer. EMBO Rep. 2024, 25, 3506–3531. [Google Scholar] [CrossRef]
- Shi, W.; Cassmann, T.J.; Bhagwate, A.V.; Hitosugi, T.; Ip, W.K.E. Lactic acid induces transcriptional repression of macrophage inflammatory response via histone acetylation. Cell Rep. 2024, 43, 113746. [Google Scholar] [CrossRef]
- Karnib, N.; El-Ghandour, R.; El Hayek, L.; Nasrallah, P.; Khalifeh, M.; Barmo, N.; Jabre, V.; Ibrahim, P.; Bilen, M.; Stephan, J.S.; et al. Lactate is an antidepressant that mediates resilience to stress by modulating the hippocampal levels and activity of histone deacetylases. Neuropsychopharmacology 2019, 44, 1152–1162. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, J.; Chen, Y.; Liang, W.; Liu, H.; Du, R.; Sun, Y.; Hu, C.; Shang, Z. CAFs-derived lactate enhances the cancer stemness through inhibiting the MST1 ubiquitination degradation in OSCC. Cell Biosci. 2024, 14, 144. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, D.; Zhai, Y.; Pan, C.; Zhang, J.; Wang, C.; Huang, R.; Yu, M.; Li, Y.; Liu, X.; et al. Glycometabolic reprogramming-induced XRCC1 lactylation confers therapeutic resistance in ALDH1A3-overexpressing glioblastoma. Cell Metab. 2024, 36, 1696–1710.e10. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.S.; Tseng, H.Y.; Chen, Y.A.; Shen, P.C.; Al Haq, A.T.; Chen, L.M.; Tung, Y.C.; Hsu, H.L. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol. Cancer 2019, 18, 42. [Google Scholar] [CrossRef]
- Feng, F.; Wu, J.; Chi, Q.; Wang, S.; Liu, W.; Yang, L.; Song, G.; Pan, L.; Xu, K.; Wang, C. Lactylome Analysis Unveils Lactylation-Dependent Mechanisms of Stemness Remodeling in the Liver Cancer Stem Cells. Adv. Sci. 2024, 11, e2405975. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Feng, F.; Wu, J.; Fan, S.; Han, J.; Wang, S.; Yang, L.; Liu, W.; Wang, C.; Xu, K. Demethylzeylasteral targets lactate by inhibiting histone lactylation to suppress the tumorigenicity of liver cancer stem cells. Pharmacol. Res. 2022, 181, 106270. [Google Scholar] [CrossRef]
- Zhou, Z.; Yin, X.; Sun, H.; Lu, J.; Li, Y.; Fan, Y.; Lv, P.; Han, M.; Wu, J.; Li, S.; et al. PTBP1 Lactylation Promotes Glioma Stem Cell Maintenance through PFKFB4-Driven Glycolysis. Cancer Res. 2025, 85, 739–757. [Google Scholar] [CrossRef]
- Deng, J.; Li, Y.; Yin, L.; Liu, S.; Li, Y.; Liao, W.; Mu, L.; Luo, X.; Qin, J. Histone lactylation enhances GCLC expression and thus promotes chemoresistance of colorectal cancer stem cells through inhibiting ferroptosis. Cell Death Dis. 2025, 16, 193. [Google Scholar] [CrossRef]
- Nguyen, N.T.B.; Gevers, S.; Kok, R.N.U.; Burgering, L.M.; Neikes, H.; Akkerman, N.; Betjes, M.A.; Ludikhuize, M.C.; Gulersonmez, C.; Stigter, E.C.A.; et al. Lactate controls cancer stemness and plasticity through epigenetic regulation. Cell Metab. 2025, 37, 903–919.e10. [Google Scholar] [CrossRef]
- Lv, M.; Gong, Y.; Liu, X.; Wang, Y.; Wu, Q.; Chen, J.; Min, Q.; Zhao, D.; Li, X.; Chen, D.; et al. CDK7-YAP-LDHD axis promotes D-lactate elimination and ferroptosis defense to support cancer stem cell-like properties. Signal Transduct. Target. Ther. 2023, 8, 302. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chai, P.; Xie, M.; Ge, S.; Ruan, J.; Fan, X.; Jia, R. Histone lactylation drives oncogenesis by facilitating m(6)A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 2021, 22, 85. [Google Scholar] [CrossRef]
- Jiang, J.; Huang, D.; Jiang, Y.; Hou, J.; Tian, M.; Li, J.; Sun, L.; Zhang, Y.; Zhang, T.; Li, Z.; et al. Lactate Modulates Cellular Metabolism Through Histone Lactylation-Mediated Gene Expression in Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 647559. [Google Scholar] [CrossRef]
- Shi, L.; Li, B.; Tan, J.; Zhu, L.; Zhang, S.; Zhang, Y.; Xiang, M.; Li, J.; Chen, Y.; Han, X.; et al. Exosomal lncRNA Mir100hg from lung cancer stem cells activates H3K14 lactylation to enhance metastatic activity in non-stem lung cancer cells. J. Nanobiotechnol. 2025, 23, 156. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, D.; Wang, Y.; Liu, M.; Zhang, Y.; Feng, T.; Xiao, C.; Song, H.; Miao, R.; Xu, L.; et al. Lactylated Apolipoprotein C-II Induces Immunotherapy Resistance by Promoting Extracellular Lipolysis. Adv. Sci. 2024, 11, e2406333. [Google Scholar] [CrossRef] [PubMed]
- Zong, Z.; Xie, F.; Wang, S.; Wu, X.; Zhang, Z.; Yang, B.; Zhou, F. Alanyl-tRNA synthetase, AARS1, is a lactate sensor and lactyltransferase that lactylates p53 and contributes to tumorigenesis. Cell 2024, 187, 2375–2392.e33. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, D.O.; Jennings, E.Q.; Anderson, C.C.; Marentette, J.O.; Shi, T.; Schou Oxvig, A.M.; Streeter, M.D.; Johannsen, M.; Spiegel, D.A.; Chapman, E.; et al. Non-enzymatic Lysine Lactoylation of Glycolytic Enzymes. Cell Chem. Biol. 2020, 27, 206–213.e6. [Google Scholar] [CrossRef]
- Chen, H.; Li, Y.; Li, H.; Chen, X.; Fu, H.; Mao, D.; Chen, W.; Lan, L.; Wang, C.; Hu, K.; et al. NBS1 lactylation is required for efficient DNA repair and chemotherapy resistance. Nature 2024, 631, 663–669. [Google Scholar] [CrossRef]
- Minami, E.; Sasa, K.; Yamada, A.; Kawai, R.; Yoshida, H.; Nakano, H.; Maki, K.; Kamijo, R. Lactate-induced histone lactylation by p300 promotes osteoblast differentiation. PLoS ONE 2023, 18, e0293676. [Google Scholar] [CrossRef]
- Yang, K.; Fan, M.; Wang, X.; Xu, J.; Wang, Y.; Tu, F.; Gill, P.S.; Ha, T.; Liu, L.; Williams, D.L.; et al. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022, 29, 133–146. [Google Scholar] [CrossRef]
- Moreno-Yruela, C.; Zhang, D.; Wei, W.; Baek, M.; Liu, W.; Gao, J.; Dankova, D.; Nielsen, A.L.; Bolding, J.E.; Yang, L.; et al. Class I histone deacetylases (HDAC1-3) are histone lysine delactylases. Sci. Adv. 2022, 8, eabi6696. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, Z.; Chen, Y.; Tian, H.; Chai, P.; Shen, Y.; Yao, Y.; Xu, S.; Ge, S.; Jia, R. Lactate and lactylation in cancer. Signal Transduct. Target. Ther. 2025, 10, 38. [Google Scholar] [CrossRef]
- Liu, H.; Pan, M.; Liu, M.; Zeng, L.; Li, Y.; Huang, Z.; Guo, C.; Wang, H. Lactate: A rising star in tumors and inflammation. Front. Immunol. 2024, 15, 1496390. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Tibullo, D.; Spampinato, M.; Duminuco, A.; La Spina, E.; Giallongo, S.; Romano, A.; Scandura, G.; Zuppelli, T.; Caltabiano, R.; Salvatorelli, L.; et al. Lactate Transporters Modulate Stromal Cell Remodeling in Myeloproliferative Neoplasms (MPN). Blood 2023, 142, 1790. [Google Scholar] [CrossRef]
- Lundo, K.; Dmytriyeva, O.; Spohr, L.; Goncalves-Alves, E.; Yao, J.; Blasco, L.P.; Trauelsen, M.; Ponniah, M.; Severin, M.; Sandelin, A.; et al. Lactate receptor GPR81 drives breast cancer growth and invasiveness through regulation of ECM properties and Notch ligand DLL4. BMC Cancer 2023, 23, 1136. [Google Scholar] [CrossRef]
- Certo, M.; Tsai, C.H.; Pucino, V.; Ho, P.C.; Mauro, C. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat. Rev. Immunol. 2021, 21, 151–161. [Google Scholar] [CrossRef]
- Liu, S.; Li, Y.; Yuan, M.; Song, Q.; Liu, M. Correlation between the Warburg effect and progression of triple-negative breast cancer. Front. Oncol. 2022, 12, 1060495. [Google Scholar] [CrossRef]
- Schreier, A.; Zappasodi, R.; Serganova, I.; Brown, K.A.; Demaria, S.; Andreopoulou, E. Facts and Perspectives: Implications of tumor glycolysis on immunotherapy response in triple negative breast cancer. Front. Oncol. 2022, 12, 1061789. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, Y.; Xiong, Y.; Gong, Y.; Lu, J.; Zhang, Y.; Wang, D.; Liu, Z.; Shi, X. Research Progress of Warburg Effect in Hepatocellular Carcinoma. Front. Biosci. 2024, 29, 178. [Google Scholar] [CrossRef]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Pucino, V.; Certo, M.; Bulusu, V.; Cucchi, D.; Goldmann, K.; Pontarini, E.; Haas, R.; Smith, J.; Headland, S.E.; Blighe, K.; et al. Lactate Buildup at the Site of Chronic Inflammation Promotes Disease by Inducing CD4(+) T Cell Metabolic Rewiring. Cell Metab. 2019, 30, 1055–1074.e8. [Google Scholar] [CrossRef] [PubMed]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef]
- Peralta, R.M.; Xie, B.; Lontos, K.; Nieves-Rosado, H.; Spahr, K.; Joshi, S.; Ford, B.R.; Quann, K.; Frisch, A.T.; Dean, V.; et al. Dysfunction of exhausted T cells is enforced by MCT11-mediated lactate metabolism. Nat. Immunol. 2024, 25, 2297–2307. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, J.; Ma, M.; Wang, K.; Liu, F.; Yang, F.; Zou, X.; Cheng, Z.; Wu, D. The potential role of CMC1 as an immunometabolic checkpoint in T cell immunity. Oncoimmunology 2024, 13, 2344905. [Google Scholar] [CrossRef]
- Fan, Z.; Ye, M.; Liu, D.; Zhou, W.; Zeng, T.; He, S.; Li, Y. Lactate drives the ESM1-SCD1 axis to inhibit the antitumor CD8(+) T-cell response by activating the Wnt/beta-catenin pathway in ovarian cancer cells and inducing cisplatin resistance. Int. Immunopharmacol. 2024, 137, 112461. [Google Scholar] [CrossRef]
- Sugi, T.; Katoh, Y.; Ikeda, T.; Seta, D.; Iwata, T.; Nishio, H.; Sugawara, M.; Kato, D.; Katoh, K.; Kawana, K.; et al. SCD1 inhibition enhances the effector functions of CD8(+) T cells via ACAT1-dependent reduction of esterified cholesterol. Cancer Sci. 2024, 115, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Liu, Z.; Yu, X.; Huang, T.; Chen, J.; Wang, J.; Wilhelm, J.; Li, S.; Song, J.; Li, W.; et al. Lactate increases stemness of CD8 + T cells to augment anti-tumor immunity. Nat. Commun. 2022, 13, 4981. [Google Scholar] [CrossRef] [PubMed]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; Macintyre, A.N.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E.; et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef]
- Watson, M.J.; Vignali, P.D.A.; Mullett, S.J.; Overacre-Delgoffe, A.E.; Peralta, R.M.; Grebinoski, S.; Menk, A.V.; Rittenhouse, N.L.; DePeaux, K.; Whetstone, R.D.; et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 2021, 591, 645–651. [Google Scholar] [CrossRef]
- Zhou, J.; Gu, J.; Qian, Q.; Zhang, Y.; Huang, T.; Li, X.; Liu, Z.; Shao, Q.; Liang, Y.; Qiao, L.; et al. Lactate supports Treg function and immune balance via MGAT1 effects on N-glycosylation in the mitochondria. J. Clin. Investig. 2024, 134, e175897. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gomez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Yu, X.; Hu, Z.; Dong, Y.; Huang, H.; Zhang, Y.; Han, Q.; Ni, Z.Y.; Zhao, R.; Ye, Y.; et al. Lactate modulates RNA splicing to promote CTLA-4 expression in tumor-infiltrating regulatory T cells. Immunity 2024, 57, 528–540.e6. [Google Scholar] [CrossRef]
- Gu, J.; Zhou, J.; Chen, Q.; Xu, X.; Gao, J.; Li, X.; Shao, Q.; Zhou, B.; Zhou, H.; Wei, S.; et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-beta signaling in regulatory T cells. Cell Rep. 2022, 40, 111122. [Google Scholar] [CrossRef]
- Su, J.; Mao, X.; Wang, L.; Chen, Z.; Wang, W.; Zhao, C.; Li, G.; Guo, W.; Hu, Y. Lactate/GPR81 recruits regulatory T cells by modulating CX3CL1 to promote immune resistance in a highly glycolytic gastric cancer. Oncoimmunology 2024, 13, 2320951. [Google Scholar] [CrossRef]
- Umemura, N.; Saio, M.; Suwa, T.; Kitoh, Y.; Bai, J.; Nonaka, K.; Ouyang, G.F.; Okada, M.; Balazs, M.; Adany, R.; et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J. Leukoc. Biol. 2008, 83, 1136–1144. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Weyand, C.M.; Zeisbrich, M.; Goronzy, J.J. Metabolic signatures of T-cells and macrophages in rheumatoid arthritis. Curr. Opin. Immunol. 2017, 46, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Xia, Y.; Zhang, B.W.; Drokow, E.K.; Li, H.Y.; Xu, S.; Wang, Z.; Wang, S.Y.; Jin, P.; Fang, T.; et al. Macrophages facilitate tumor cell PD-L1 expression via an IL-1beta-centered loop to attenuate immune checkpoint blockade. MedComm 2023, 4, e242. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Dyck, L.; Zaslona, Z.; Menon, D.; McGettrick, A.F.; Mills, K.H.G.; O’Neill, L.A. Pyruvate Kinase M2 Is Required for the Expression of the Immune Checkpoint PD-L1 in Immune Cells and Tumors. Front. Immunol. 2017, 8, 1300. [Google Scholar] [CrossRef] [PubMed]
- Shime, H.; Yabu, M.; Akazawa, T.; Kodama, K.; Matsumoto, M.; Seya, T.; Inoue, N. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J. Immunol. 2008, 180, 7175–7183. [Google Scholar] [CrossRef]
- Dietl, K.; Renner, K.; Dettmer, K.; Timischl, B.; Eberhart, K.; Dorn, C.; Hellerbrand, C.; Kastenberger, M.; Kunz-Schughart, L.A.; Oefner, P.J.; et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J. Immunol. 2010, 184, 1200–1209. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, D.; Xu, T.; Liu, P.; Cao, Y.; Wang, Y.; Yang, X.; Xu, X.; Wang, X.; Niu, H. Bladder cancer cells re-educate TAMs through lactate shuttling in the microfluidic cancer microenvironment. Oncotarget 2015, 6, 39196–39210. [Google Scholar] [CrossRef]
- Lin, S.; Sun, L.; Lyu, X.; Ai, X.; Du, D.; Su, N.; Li, H.; Zhang, L.; Yu, J.; Yuan, S. Lactate-activated macrophages induced aerobic glycolysis and epithelial-mesenchymal transition in breast cancer by regulation of CCL5-CCR5 axis: A positive metabolic feedback loop. Oncotarget 2017, 8, 110426–110443. [Google Scholar] [CrossRef]
- Kwak, T.; Wang, F.; Deng, H.; Condamine, T.; Kumar, V.; Perego, M.; Kossenkov, A.; Montaner, L.J.; Xu, X.; Xu, W.; et al. Distinct Populations of Immune-Suppressive Macrophages Differentiate from Monocytic Myeloid-Derived Suppressor Cells in Cancer. Cell Rep. 2020, 33, 108571. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yang, J.; Xu, J.; Pan, H.; Wang, W.; Shi, S. Histone lactylation: From tumor lactate metabolism to epigenetic regulation. Int. J. Biol. Sci. 2024, 20, 1833–1854. [Google Scholar] [CrossRef]
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C.; et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 2018, 17, 428–438. [Google Scholar] [CrossRef]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Puig-Kroger, A.; Pello, O.M.; Muniz-Pello, O.; Selgas, R.; Criado, G.; Bajo, M.A.; Sanchez-Tomero, J.A.; Alvarez, V.; del Peso, G.; Sanchez-Mateos, P.; et al. Peritoneal dialysis solutions inhibit the differentiation and maturation of human monocyte-derived dendritic cells: Effect of lactate and glucose-degradation products. J. Leukoc. Biol. 2003, 73, 482–492. [Google Scholar] [CrossRef]
- Nasi, A.; Fekete, T.; Krishnamurthy, A.; Snowden, S.; Rajnavolgyi, E.; Catrina, A.I.; Wheelock, C.E.; Vivar, N.; Rethi, B. Dendritic cell reprogramming by endogenously produced lactic acid. J. Immunol. 2013, 191, 3090–3099. [Google Scholar] [CrossRef] [PubMed]
- Caronni, N.; Simoncello, F.; Stafetta, F.; Guarnaccia, C.; Ruiz-Moreno, J.S.; Opitz, B.; Galli, T.; Proux-Gillardeaux, V.; Benvenuti, F. Downregulation of Membrane Trafficking Proteins and Lactate Conditioning Determine Loss of Dendritic Cell Function in Lung Cancer. Cancer Res. 2018, 78, 1685–1699. [Google Scholar] [CrossRef]
- Baggstrom, M.Q.; Qi, Y.; Koczywas, M.; Argiris, A.; Johnson, E.A.; Millward, M.J.; Murphy, S.C.; Erlichman, C.; Rudin, C.M.; Govindan, R.; et al. A phase II study of AT-101 (Gossypol) in chemotherapy-sensitive recurrent extensive-stage small cell lung cancer. J. Thorac. Oncol. 2011, 6, 1757–1760. [Google Scholar] [CrossRef]
- Curry, J.; Johnson, J.; Tassone, P.; Vidal, M.D.; Menezes, D.W.; Sprandio, J.; Mollaee, M.; Cotzia, P.; Birbe, R.; Lin, Z.; et al. Metformin effects on head and neck squamous carcinoma microenvironment: Window of opportunity trial. Laryngoscope 2017, 127, 1808–1815. [Google Scholar] [CrossRef]
- Dunbar, E.M.; Coats, B.S.; Shroads, A.L.; Langaee, T.; Lew, A.; Forder, J.R.; Shuster, J.J.; Wagner, D.A.; Stacpoole, P.W. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Investig. New Drugs 2014, 32, 452–464. [Google Scholar] [CrossRef]
- Garon, E.B.; Christofk, H.R.; Hosmer, W.; Britten, C.D.; Bahng, A.; Crabtree, M.J.; Hong, C.S.; Kamranpour, N.; Pitts, S.; Kabbinavar, F.; et al. Dichloroacetate should be considered with platinum-based chemotherapy in hypoxic tumors rather than as a single agent in advanced non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2014, 140, 443–452. [Google Scholar] [CrossRef]
- Halford, S.; Veal, G.J.; Wedge, S.R.; Payne, G.S.; Bacon, C.M.; Sloan, P.; Dragoni, I.; Heinzmann, K.; Potter, S.; Salisbury, B.M.; et al. A Phase I Dose-escalation Study of AZD3965, an Oral Monocarboxylate Transporter 1 Inhibitor, in Patients with Advanced Cancer. Clin. Cancer Res. 2023, 29, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Polanski, R.; Hodgkinson, C.L.; Fusi, A.; Nonaka, D.; Priest, L.; Kelly, P.; Trapani, F.; Bishop, P.W.; White, A.; Critchlow, S.E.; et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin. Cancer Res. 2014, 20, 926–937. [Google Scholar] [CrossRef]
- Powell, S.F.; Mazurczak, M.; Dib, E.G.; Bleeker, J.S.; Geeraerts, L.H.; Tinguely, M.; Lohr, M.M.; McGraw, S.C.; Jensen, A.W.; Ellison, C.A.; et al. Phase II study of dichloroacetate, an inhibitor of pyruvate dehydrogenase, in combination with chemoradiotherapy for unresected, locally advanced head and neck squamous cell carcinoma. Investig. New Drugs 2022, 40, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Renner, O.; Mayer, M.; Leischner, C.; Burkard, M.; Berger, A.; Lauer, U.M.; Venturelli, S.; Bischoff, S.C. Systematic Review of Gossypol/AT-101 in Cancer Clinical Trials. Pharmaceuticals 2022, 15, 144. [Google Scholar] [CrossRef]
- Silva, A.; Antunes, B.; Batista, A.; Pinto-Ribeiro, F.; Baltazar, F.; Afonso, J. In Vivo Anticancer Activity of AZD3965: A Systematic Review. Molecules 2021, 27, 181. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.D.; Bennett, S.K.; Coupland, L.A.; Forwood, K.; Lwin, Y.; Pooryousef, N.; Tea, I.; Truong, T.T.; Neeman, T.; Crispin, P.; et al. GSTZ1 genotypes correlate with dichloroacetate pharmacokinetics and chronic side effects in multiple myeloma patients in a pilot phase 2 clinical trial. Pharmacol. Res. Perspect. 2019, 7, e00526. [Google Scholar] [CrossRef]
- Van Poznak, C.; Seidman, A.D.; Reidenberg, M.M.; Moasser, M.M.; Sklarin, N.; Van Zee, K.; Borgen, P.; Gollub, M.; Bacotti, D.; Yao, T.J.; et al. Oral gossypol in the treatment of patients with refractory metastatic breast cancer: A phase I/II clinical trial. Breast Cancer Res. Treat. 2001, 66, 239–248. [Google Scholar] [CrossRef]
- Wang, Y.; Li, X.; Zhang, L.; Li, M.; Dai, N.; Luo, H.; Shan, J.; Yang, X.; Xu, M.; Feng, Y.; et al. A randomized, double-blind, placebo-controlled study of B-cell lymphoma 2 homology 3 mimetic gossypol combined with docetaxel and cisplatin for advanced non-small cell lung cancer with high expression of apurinic/apyrimidinic endonuclease 1. Investig. New Drugs 2020, 38, 1862–1871. [Google Scholar] [CrossRef]
- Xie, H.; Yin, J.; Shah, M.H.; Menefee, M.E.; Bible, K.C.; Reidy-Lagunes, D.; Kane, M.A.; Quinn, D.I.; Gandara, D.R.; Erlichman, C.; et al. A phase II study of the orally administered negative enantiomer of gossypol (AT-101), a BH3 mimetic, in patients with advanced adrenal cortical carcinoma. Investig. New Drugs 2019, 37, 755–762. [Google Scholar] [CrossRef]
- de la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Cappellesso, F.; Orban, M.P.; Shirgaonkar, N.; Berardi, E.; Serneels, J.; Neveu, M.A.; Di Molfetta, D.; Piccapane, F.; Caroppo, R.; Debellis, L.; et al. Targeting the bicarbonate transporter SLC4A4 overcomes immunosuppression and immunotherapy resistance in pancreatic cancer. Nat. Cancer 2022, 3, 1464–1483. [Google Scholar] [CrossRef]
- Babl, N.; Decking, S.M.; Voll, F.; Althammer, M.; Sala-Hojman, A.; Ferretti, R.; Korf, C.; Schmidl, C.; Schmidleithner, L.; Nerb, B.; et al. MCT4 blockade increases the efficacy of immune checkpoint blockade. J. Immunother. Cancer 2023, 11, e007349. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Xue, H.; Wu, R.; Fazli, L.; Lin, D.; Collins, C.C.; Gleave, M.E.; Gout, P.W.; Wang, Y. The MCT4 Gene: A Novel, Potential Target for Therapy of Advanced Prostate Cancer. Clin. Cancer Res. 2016, 22, 2721–2733. [Google Scholar] [CrossRef]
- Draoui, N.; Schicke, O.; Seront, E.; Bouzin, C.; Sonveaux, P.; Riant, O.; Feron, O. Antitumor activity of 7-aminocarboxycoumarin derivatives, a new class of potent inhibitors of lactate influx but not efflux. Mol. Cancer Ther. 2014, 13, 1410–1418. [Google Scholar] [CrossRef]
- Huang, T.; Feng, Q.; Wang, Z.; Li, W.; Sun, Z.; Wilhelm, J.; Huang, G.; Vo, T.; Sumer, B.D.; Gao, J. Tumor-Targeted Inhibition of Monocarboxylate Transporter 1 Improves T-Cell Immunotherapy of Solid Tumors. Adv. Healthc. Mater. 2021, 10, e2000549. [Google Scholar] [CrossRef] [PubMed]
- Khammanivong, A.; Saha, J.; Spartz, A.K.; Sorenson, B.S.; Bush, A.G.; Korpela, D.M.; Gopalakrishnan, R.; Jonnalagadda, S.; Mereddy, V.R.; O’Brien, T.D.; et al. A novel MCT1 and MCT4 dual inhibitor reduces mitochondrial metabolism and inhibits tumour growth of feline oral squamous cell carcinoma. Vet. Comp. Oncol. 2020, 18, 324–341. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, Y.; Wu, C.; Shi, J. Engineering lactate-modulating nanomedicines for cancer therapy. Chem. Soc. Rev. 2023, 52, 973–1000. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, Y.; Liu, Y.; Zhang, T.; Zhao, Y.; Zang, J.; Yang, Y.; He, R.; Chong, G.; Ruan, S.; et al. Dual Closed-Loop of Catalyzed Lactate Depletion and Immune Response to Potentiate Photothermal Immunotherapy. ACS Appl. Mater. Interfaces 2022, 14, 23260–23276. [Google Scholar] [CrossRef]
- Zhou, X.; Zhao, W.; Wang, M.; Zhang, S.; Li, Y.; Hu, W.; Ren, L.; Luo, S.; Chen, Z. Dual-Modal Therapeutic Role of the Lactate Oxidase-Embedded Hierarchical Porous Zeolitic Imidazolate Framework as a Nanocatalyst for Effective Tumor Suppression. ACS Appl. Mater. Interfaces 2020, 12, 32278–32288. [Google Scholar] [CrossRef]
- Choi, H.; Yeo, M.; Kang, Y.; Kim, H.J.; Park, S.G.; Jang, E.; Park, S.H.; Kim, E.; Kang, S. Lactate oxidase/catalase-displaying nanoparticles efficiently consume lactate in the tumor microenvironment to effectively suppress tumor growth. J. Nanobiotechnol. 2023, 21, 5. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, E.Y.; Abides, J.; Keller, C.R.; Martinez, S.R.; Li, W. Tumor Microenvironment Lactate: Is It a Cancer Progression Marker, Immunosuppressant, and Therapeutic Target? Molecules 2025, 30, 1763. https://doi.org/10.3390/molecules30081763
Kim EY, Abides J, Keller CR, Martinez SR, Li W. Tumor Microenvironment Lactate: Is It a Cancer Progression Marker, Immunosuppressant, and Therapeutic Target? Molecules. 2025; 30(8):1763. https://doi.org/10.3390/molecules30081763
Chicago/Turabian StyleKim, Eugene Y., Joyce Abides, Chandler R. Keller, Steve R. Martinez, and Weimin Li. 2025. "Tumor Microenvironment Lactate: Is It a Cancer Progression Marker, Immunosuppressant, and Therapeutic Target?" Molecules 30, no. 8: 1763. https://doi.org/10.3390/molecules30081763
APA StyleKim, E. Y., Abides, J., Keller, C. R., Martinez, S. R., & Li, W. (2025). Tumor Microenvironment Lactate: Is It a Cancer Progression Marker, Immunosuppressant, and Therapeutic Target? Molecules, 30(8), 1763. https://doi.org/10.3390/molecules30081763