

6-Chlorocoumarin Conjugates with Nucleobases and Nucleosides as Potent Anti-Hepatitis C Virus Agents

 ,
,  , , , , , and
, , , , , and 
Abstract
1. Introduction
2. Results
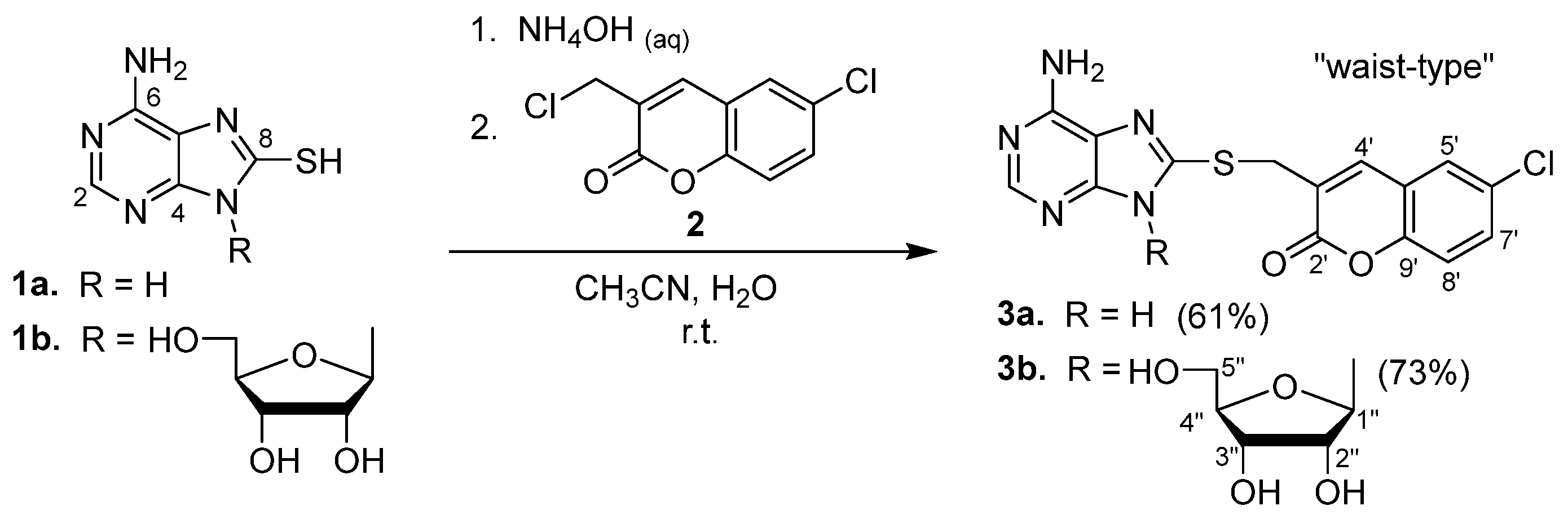
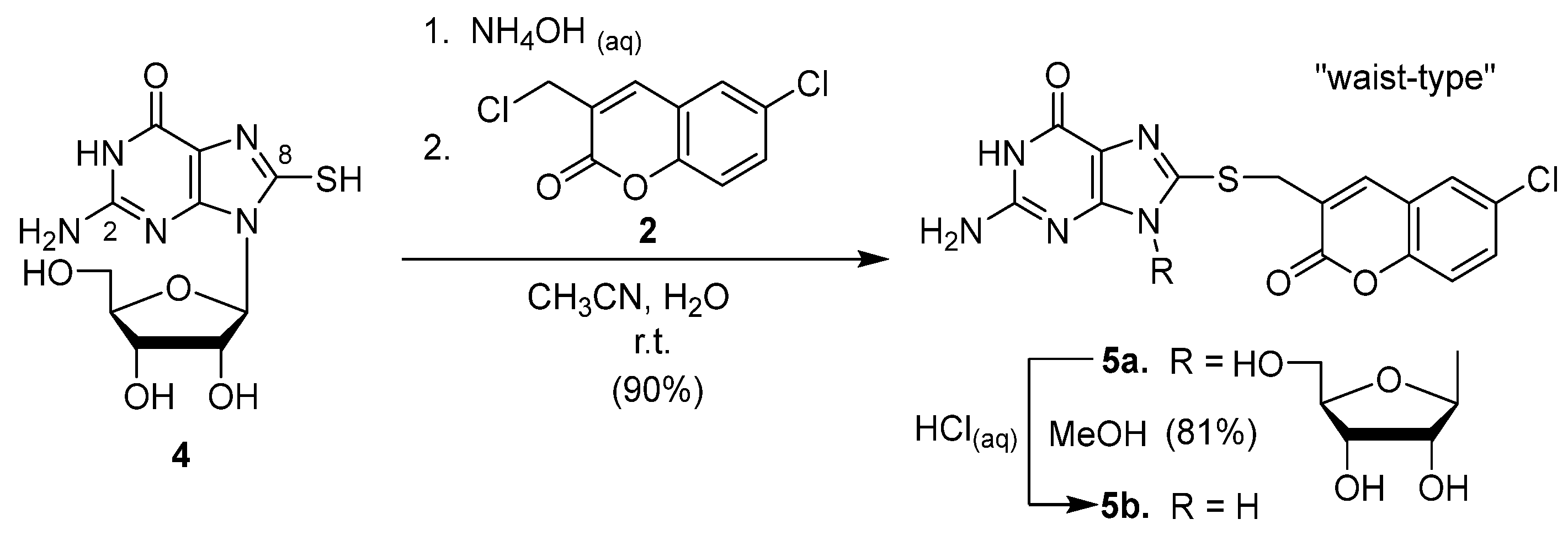
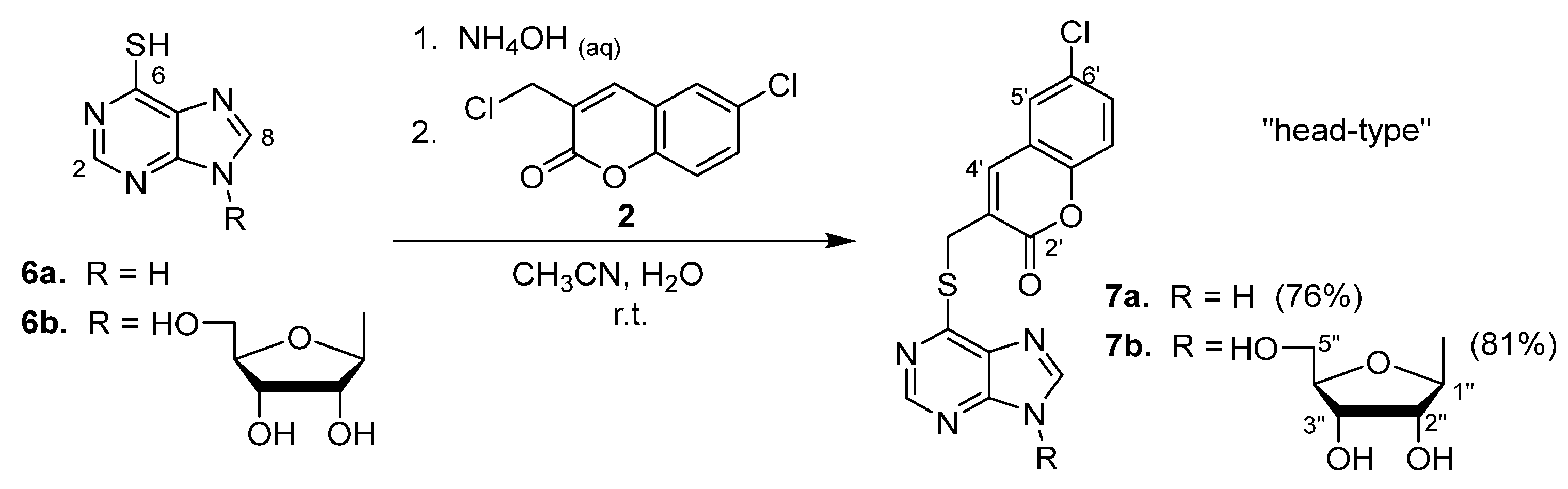
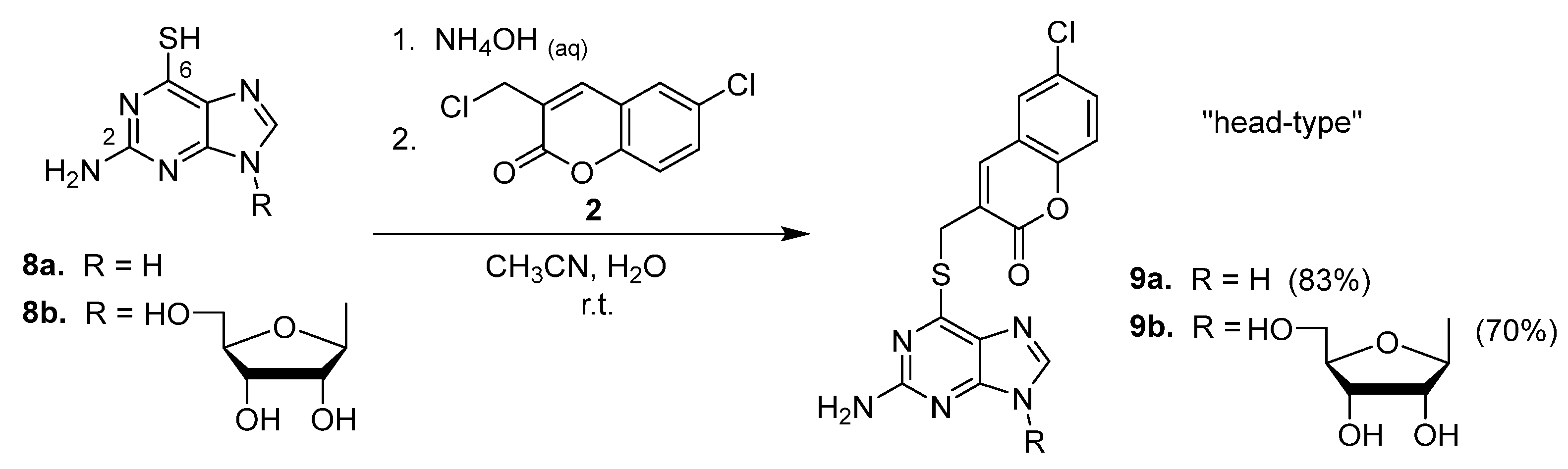
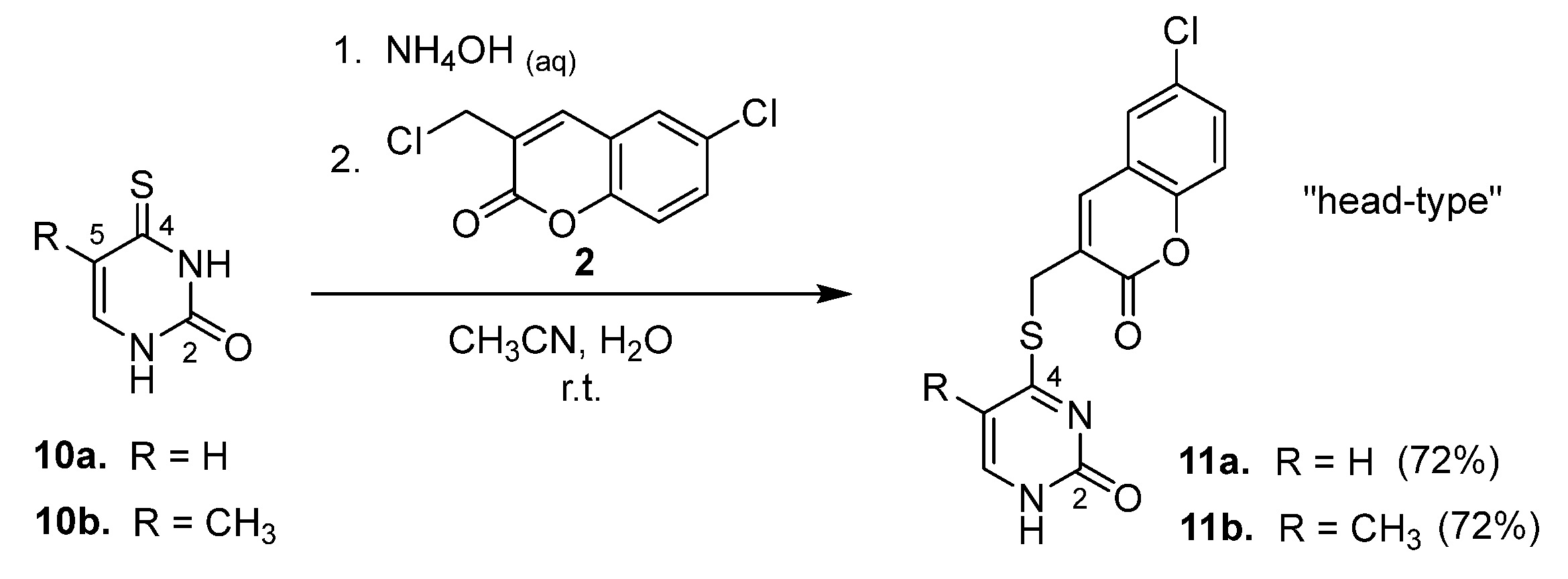
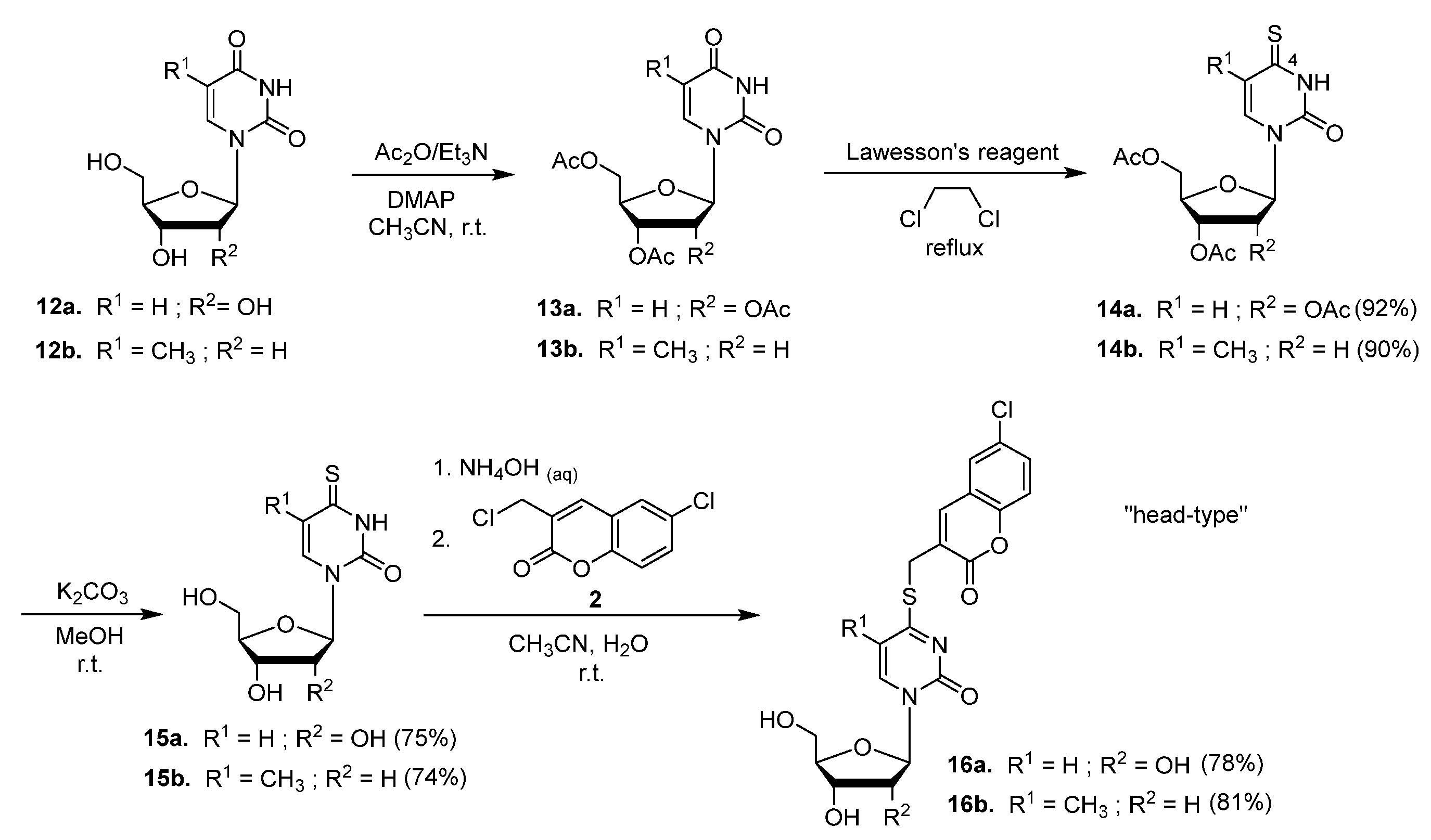
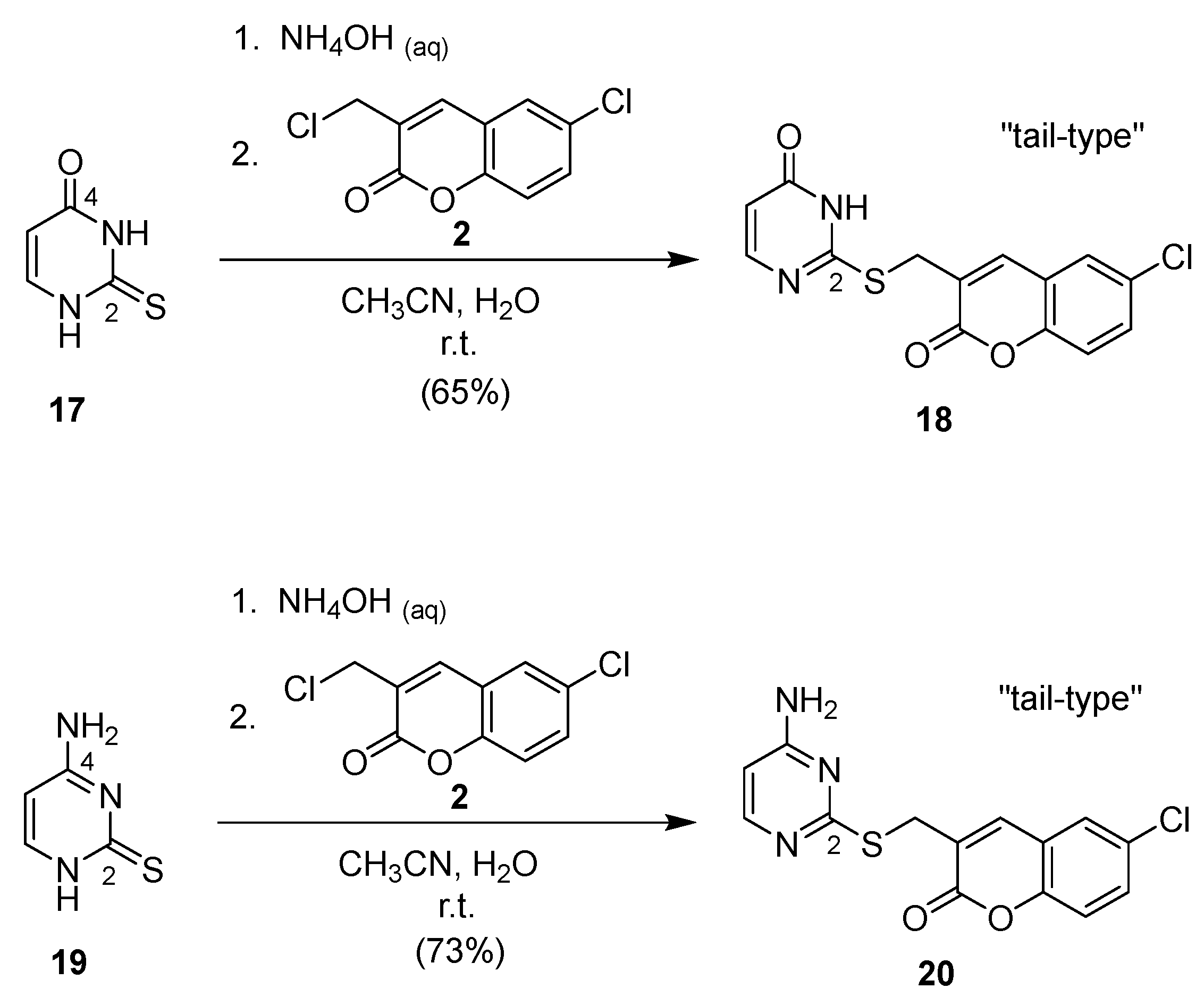
2.1. Syntheses of Coumarin-Conjugated Nucleobases and Nucleosides
2.2. Structural Identification
2.3. Evaluation of the Anti-HCV Activity
2.4. Early-Stage Safety Screening in Detroit 551 Assay (Normal Human Cells)
3. Discussion
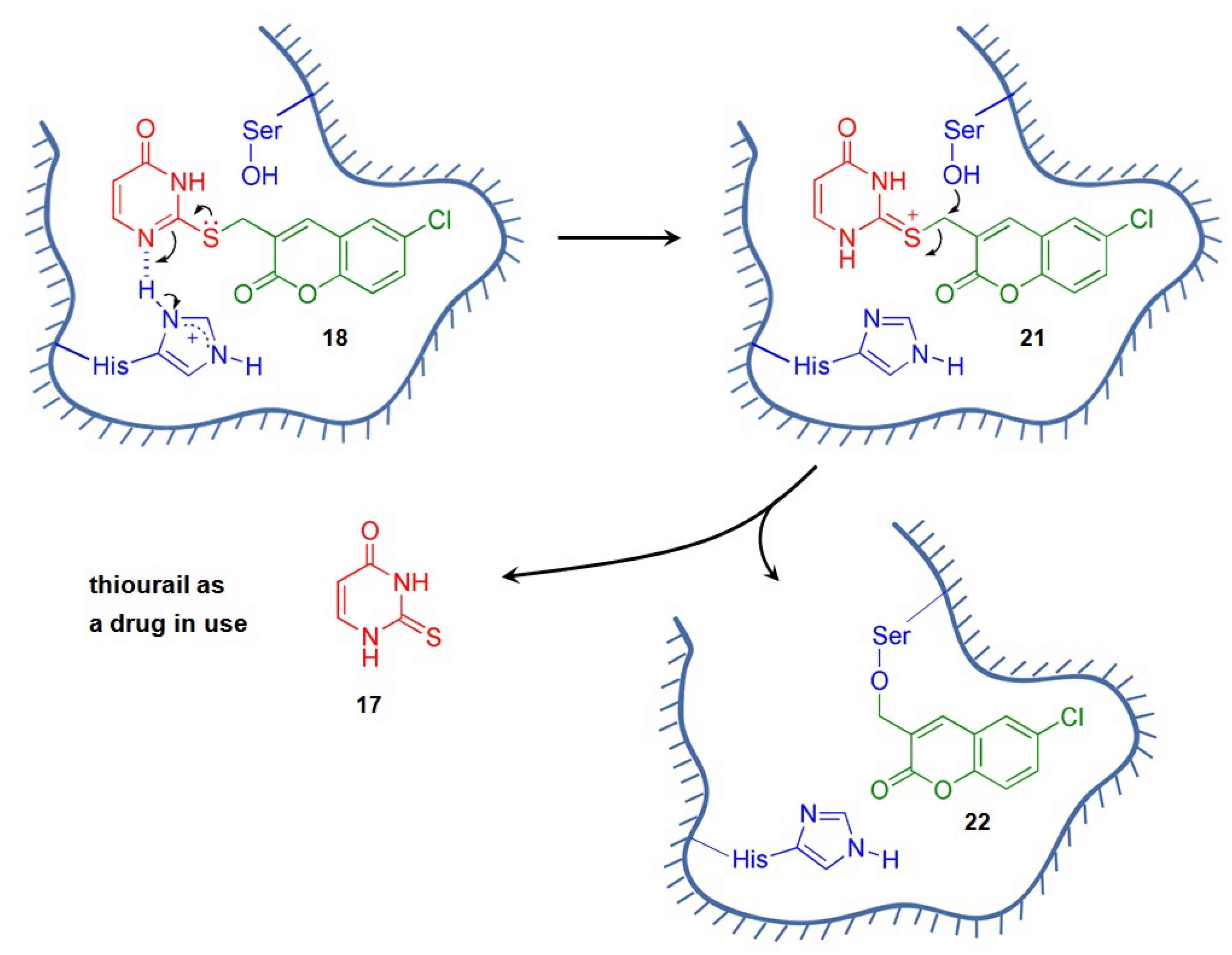
3.1. Rationale of Anti-HCV Drug Design
3.2. Structure–Activity Relationship
- (1)
- On the basis of the position of the coumarin moiety attached to the nucleobases and nucelosides of the conjugates, the three candidates selected for the Huh 9-13 assay system are “head-type” and “tail-type” conjugates. These targets, 7a, 7b, and 18, showed impressive anti-HCV activity (6.6–9.4 μM) on the same order, and, among them, the “tail-type” 18 possessed the best SI value (i.e., >41);
- (2)
- Regarding the purine nucleobases or the corresponding nucleosides, the SI values of the conjugates are better for the “head-type” than for the “waist-type” attachments (i.e., 7a, 7b, 9a, and 9b compared with 3a, 3b, 5a, and 5b, respectively). On the other hand, regarding the pyrimidine nucleobases or the corresponding nucleosides, the SI values of the conjugates are better for the “tail-type” than for the “head-type conjugates (i.e., 18 and 20 vs. 11a, 11b, 16a, and 16b);
- (3)
- Switching the coumarin moiety from the wait position of a purine (i.e., 3a,b) to the head position (i.e., 7a,b and 9a,b) resulted in the abatement of cytotoxicity;
- (4)
- The removal of an amino group from the C2 position of coumarin–thioguanine 9a gave 6-mercaptopurine conjugate 7a, which exhibited a 7.6-fold improvement in its anti-HCV activity;
- (5)
- Regarding its biological effects, the 2′,3′,5′-β-D-ribofuranosyl group plays an enigmatic role in terms of its incorporation in coumarin-purine conjugates. Its presence decreased the HCV inhibition and selectivity of the conjugates, which can be observed by comparing 3a with 3b; did not lead to a dramatic impact on the resulting activity and cytotoxicity, which can be observed by comparing 7a with 7b; and increased both the resulting HCV inhibition and selectivity, which can be observed by comparing 9a with 9b. Notwithstanding, it is certain that its presence improved the aqueous solubility of the conjugates.
4. Materials and Methods
4.1. General Information
4.2. Standard Procedure for the Preparation of Conjugated Compounds 3, 5, 7, 9, 11, 16, 18, and 20
4.2.1. 8-[(6′-Chlorocoumarin-3′-yl)methylthio]adenine (3a)
4.2.2. 8-[(6′-Chlorocoumarin-3′-yl)methylthio]adenosine (3b)
4.2.3. 8-[(6′-Chlorocoumarin-3′-yl)methylthio]guanosine (5a)
4.2.4. 8-[(6’-Chlorocoumarin-3’-yl)methylthio]guanine (5b)
4.2.5. 6-[(6′-Chlorocoumarin-3′-yl)methylthio]purine (7a)
4.2.6. 6-(6′-Chlorocoumarin-3′-yl)methylthio-9-(β-D-ribofuranos-1″-yl)purine (7b)
4.2.7. 2-Amino-6-[(6′-chlorocoumarin-3′-yl)methylthio]purine (9a)
4.2.8. 2-Amino-6-(6′-chlorocoumarin-3′-yl)methylthio-9-(β-D-ribofuranos-1″-yl)purine (9b)
4.2.9. 4-[(6′-Chlorocoumarin-3′-yl)methylthio]uracil (11a)
4.2.10. 4-(6′-Chlorocoumarin-3′-yl)methylthio-5-methyluracil (11b)
4.2.11. 1-(2′,3′,5′-Tri-O-acetyl-β-D-ribofuranosyl)-4-thiouracil (14a)
4.2.12. 1-(3′,5′-Di-O-acetyl-β-D-ribofuranosyl)-4-thiothymine (14b)
4.2.13. 4-Thiouridine (15a)
4.2.14. 4-Thiothymidine (15b)
4.2.15. 4-[(6′-Chlorocoumarin-3′-yl)methylthio]uridine (16a)
4.2.16. 4-[(6′-Chlorocoumarin-3′-yl)methylthio]thymidine (16b)
4.2.17. 2-[(6′-Chlorocoumarin-3′-yl)methylthio]uracil (18)
4.2.18. 4-Amino-2-[(6′-chlorocoumarin-3′-yl)methylthio]pyrimidine (20)
4.3. Antiviral Assay and Cell Viability Assay
4.3.1. Anti-HCV Assay in Huh 5-2 Cells
4.3.2. Anti-HCV Assay in Huh 9-13 Cells
4.3.3. Cell Viability Assay in Detroit 551 Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stroffolini, T.; Stroffolini, G. Prevalence and modes of transmission of hepatitis C virus infection: A historical worldwide review. Viruses 2024, 16, 1115. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed on 9 April 2024).
- Meshram, R.; Kolte, B.; Gacche, R. Reverse vaccinology approach for identification of epitopes from E1 protein as peptide vaccine against HCV: A proof of concept. Vaccine 2024, 42, 126106. [Google Scholar] [CrossRef]
- Bhattacharjee, C.; Singh, M.; Das, D.; Chaudhuri, S.; Mukhopadhyay, A. Current therapeutics against HCV. Virusdisease 2021, 32, 228–243. [Google Scholar] [CrossRef]
- Spengler, U. Direct antiviral agents (DAAs)—A new age in the treatment of hepatitis C virus infection. Pharmacol. Ther. 2018, 183, 118–126. [Google Scholar] [CrossRef]
- Ren, X.-D.; Fu, X.; He, Y.-Q.; Li, C.-Y.; Guo, M.; Qiao, M. Safety and efficacy of sofosbuvir-velpatasvir: A meta-analysis. Medicine 2022, 101, e31183. [Google Scholar] [CrossRef]
- Mengshetti, S.; Zhou, L.; Sari, O.; De Schutter, C.; Zhang, H.; Cho, J.H.; Tao, S.; Bassit, L.C.; Verma, K.; Domaoal, R.A.; et al. Discovery of a Series of 2′-α-Fluoro,2′-β-bromo-ribonucleosides and their phosphoramidate prodrugs as potent pan-genotypic inhibitors of hepatitis C virus. J. Med. Chem. 2019, 62, 1859–1874. [Google Scholar] [CrossRef]
- Nio, Y.; Sasai, M.; Akahori, Y.; Okamura, H.; Hasegawa, H.; Oshima, M.; Watashi, K.; Wakita, T.; Ryo, A.; Tanaka, Y.; et al. Bardoxolone methyl as a novel potent antiviral agent against hepatitis B and C viruses in human hepatocyte cell culture systems. Antiviral Res. 2019, 169, 104537. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, X.; Li, J.; Liu, Q.; Huang, X.-X.; Ding, H.; Suzuki, R.; Muramatsu, M.; Song, S.-J. Flavonoid-triazolyl hybrids as potential anti-hepatitis C virus agents: Synthesis and biological evaluation. Eur. J. Med. Chem. 2021, 218, 113395. [Google Scholar] [CrossRef]
- Pawlotsky, J.-M. New antiviral agents for hepatitis C. F1000 Biol. Rep. 2012, 4, 5. [Google Scholar] [CrossRef]
- Yeung, K.-S.; Yeung, K.-S.; Meanwell, N.A.; Qiu, Z.; Hernandez, D.; Zhang, S.; McPhee, F.; Weinheimer, S.; Clark, J.M.; Janc, J.W. Structure–activity relationship studies of a bisbenzimidazole-based, Zn2+-dependent inhibitor of HCV NS3 serine protease. Bioorg. Med. Chem. Lett. 2001, 11, 2355–2359. [Google Scholar] [CrossRef]
- Chen, C.-S.; Chiou, C.-T.; Chen, G.S.; Chen, S.-C.; Hu, C.-Y.; Chi, W.-K.; Chu, Y.-D.; Hwang, L.-H.; Chen, P.-J.; Chen, D.-S.; et al. Structure-based discovery of triphenylmethane derivatives as inhibitors of hepatitis C virus helicase. J. Med. Chem. 2009, 52, 2716–2723. [Google Scholar] [CrossRef]
- François, J.; Raymond, A. Small-Molecule Hepatitis C Virus (HCV) NS3/4A Serine Protease Inhibitors. WO Patent 128344 A1, 30 October 2008. [Google Scholar]
- De Clercq, E. The Design of Drugs for HIV and HCV. Nat. Rev. Drug Discov. 2007, 6, 1001–1018. [Google Scholar] [CrossRef]
- Njoroge, F.G.; Chen, K.X.; Shih, N.-Y.; Piwinski, J.J. Challenges in modern drug discovery: A case study of Boceprevir, an HCV protease inhibitor for the treatment of hepatitis C virus infection. Accounts Chem. Res. 2008, 41, 50–59. [Google Scholar] [CrossRef]
- Lin, C.; Kwong, A.D.; Perni, R.B. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3·D4A serine protease. Infect. Disord. Drug Targets 2006, 6, 3–16. [Google Scholar] [CrossRef]
- Paul, B.; Chen, M.F.; Paterson, A.R.P. Inhibitors of nucleoside transport. a structure–activity study using human erythrocytes. J. Med. Chem. 1975, 18, 968–973. [Google Scholar] [CrossRef]
- Yadav, V.; Chu, C.K.; Rais, R.H.; Al Safarjalani, O.N.; Guarcello, V.; Naguib, F.N.M.; el Kouni, M.H. Synthesis, biological activity and molecular modeling of 6-benzylthioinosine analogues as subversive substrates of Toxoplasma gondii adenosine kinase. J. Med. Chem. 2004, 47, 1987–1996. [Google Scholar] [CrossRef]
- Fu, Y.H.; Xu, Z.X.; Jiang, N.; Zheng, Y.P.; Rameix-Welti, M.A.; Jiao, Y.Y.; Peng, X.L.; Wang, Y.; Eleouet, J.F.; Cen, S.; et al. High-throughput screening of active compounds against human respiratory syncytial virus. Virology 2019, 535, 171–178. [Google Scholar] [CrossRef]
- Cheng, K.W.; Cheng, S.C.; Chen, W.Y.; Lin, M.H.; Chuang, S.J.; Cheng, I.H.; Sun, C.Y.; Chou, C.Y. Thiopurine analogs and mycophenolic acid synergistically inhibit the papain-like protease of Middle East respiratory syndrome coronavirus. Antiviral Res. 2015, 115, 9–16. [Google Scholar] [CrossRef]
- de Carvalho, O.V.; Felix, D.M.; de Mendonca, L.R.; de Araujo, C.; de Oliveira Franca, R.F.; Cordeiro, M.T.; Silva Junior, A.; Pena, L.J. The thiopurine nucleoside analogue 6-methylmercaptopurine riboside (6MMPr) effectively blocks Zika virus replication. Int. J. Antimicrob. Agents 2017, 50, 718–725. [Google Scholar] [CrossRef]
- Hoover, S.; Striker, R. Thiopurines inhibit bovine viral diarrhea virus production in a thiopurine methyltransferase-dependent manner. J. Gen. Virol. 2008, 89, 1000–1009. [Google Scholar] [CrossRef]
- Elion, G.B. The purine path to chemotherapy. Science 1989, 244, 41–47. [Google Scholar] [CrossRef]
- Podsiadlo, P.; Sinani, V.A.; Bahng, J.H.; Kam, N.W.S.; Lee, J.; Kotov, N.A. Gold nanoparticles enhance the anti-leukemia action of a 6-mercaptopurine chemotherapeutic agent. Langmuir 2008, 24, 568–574. [Google Scholar] [CrossRef]
- Miron, T.; Arditti, F.; Konstantinovski, L.; Rabinkov, A.; Mirelman, D.; Berrebi, A.; Wilchek, M. Novel derivatives of 6-mercaptopurine: Synthesis, characterization and antiproliferative activities of s-allylthio-mercaptopurines. Eur. J. Med. Chem. 2009, 44, 541–550. [Google Scholar] [CrossRef]
- Borrmann, T.; Abdelrahman, A.; Volpini, R.; Lambertucci, C.; Alksnis, E.; Gorzalka, S.; Knospe, M.; Schiedel, A.C.; Cristalli, G.; Müller, C.E. Structure–activity relationships of adenine and deazaadenine derivatives as ligands for adenine receptors, a new purinergic receptor family. J. Med. Chem. 2009, 52, 5974–5989. [Google Scholar] [CrossRef]
- Hocek, M.; Holý, A.; Dvořáková, H. Cytostatic 6-arylpurine nucleosides IV. synthesis of 2-substituted 6-phenylpurine ribonucleosides. Collect. Czech. Chem. Commun. 2002, 67, 325–335. [Google Scholar] [CrossRef]
- Neyts, J. Selective inhibitors of hepatitis C virus replication. Antiviral Res. 2006, 71, 363–371. [Google Scholar] [CrossRef]
- De Francesco, F.; Carfí, A. Advances in the development of new therapeutic agents targeting the NS3-4A serine protease or the NS5B RNA-dependent RNA polymerase of the hepatitis C virus. Adv. Drug Deliv. Rev. 2007, 59, 1242–1262. [Google Scholar] [CrossRef]
- Lange, C.M.; Sarrazin, C.; Zeuzem, S. Review article: Specifically targeted anti-viral therapy for hepatitis C–a new era in therapy. Aliment. Pharmacol. Ther. 2010, 32, 14–28. [Google Scholar] [CrossRef]
- Jazwinski, A.B.; Muir, A.J. Direct-acting antiviral medications for chronic hepatitis C virus infection. Gastroenterol. Hepatol. 2011, 7, 154–162. [Google Scholar]
- Tsay, S.-C.; Lin, S.-Y.; Huang, W.-C.; Hsu, M.-H.; Hwang, K.C.; Lin, C.-C.; Horng, J.-C.; Chen, I.-C.; Hwu, J.R.; Shieh, F.-K.; et al. Synthesis and structure-activity relationships of imidazole-coumarin conjugates against hepatitis C virus. Molecules 2016, 21, 228. [Google Scholar] [CrossRef]
- Hwu, J.R.; Singha, R.; Hong, S.C.; Chang, Y.H.; Das, A.R.; Vliegen, I.; De Clercq, E.; Neyts, J. Synthesis of New Benzimidazole–Coumarin Conjugates as Anti-Hepatitis C Virus Agents. Antiviral Res. 2008, 77, 157–162. [Google Scholar] [CrossRef]
- Neyts, J.; De Clercq, E.; Singha, R.; Chang, Y.H.; Das, A.R.; Chakraborty, S.K.; Hong, S.C.; Tsay, S.-C.; Hsu, M.-H.; Hwu, J.R. Structure–activity relationship of new anti-hepatitis C virus agents: Heterobicycle–coumarin conjugates. J. Med. Chem. 2009, 52, 1486–1490. [Google Scholar] [CrossRef]
- Hwu, J.R.; Lin, S.-Y.; Tsay, S.-C.; De Clercq, E.; Leyssen, P.; Neyts, J. Coumarin–purine ribofuranoside conjugates as new agents against hepatitis C virus. J. Med. Chem. 2011, 54, 2114–2126. [Google Scholar] [CrossRef]
- Kaye, P.T.; Musa, M.A. A convenient and improved Bayli–Hillman synthesis of 3-substututed 2H-1-benzopyran-2-ones. Synthesis 2002, 18, 2701–2706. [Google Scholar] [CrossRef]
- Kaneko, K.; Katayama, H.; Wakabayashi, T.; Kumonaka, T. Pyrimidine derivatives; 1. the highly regioselective 4-thionation of pyrimidine-2,4(1H,3H)-dione derivatives with Lawesson reagent. Synthesis 1988, 2, 152–154. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Webster, F.X.; Kiemle, D.J. Spectrometric Identification of Organic Compounds, 7th ed.; John Wiley & Sons: New York, NY, USA, 2005; p. 98. [Google Scholar]
- Lohmann, V.; Körner, F.; Koch, J.-O.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef]
- Paeshuyse, J.; Kaul, A.; De Clercq, E.; Rosenwirth, B.; Dumont, J.-M.; Scalfaro, P.; Bartenschlager, R.; Neyts, J. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 2006, 43, 761–770. [Google Scholar] [CrossRef]
- Vliegen, I.; Paeshuyse, J.; De Burghgraeve, T.; Lehman, L.S.; Paulson, M.; Shih, I.-H.; Mabery, E.; Boddeker, N.; De Clercq, E.; Reiser, H.; et al. Substituted imidazopyridines as potent inhibitors of HCV replication. J. Hepatol. 2009, 50, 999–1009. [Google Scholar] [CrossRef]
- Han, B.; Martin, R.; Xu, S.; Parvangada, A.; Svarovskaia, E.S.; Mo, H.; Dvory-Sobol, H. Sofosbuvir susceptibility of genotype 1 to 6 HCV from DAA-naïve subjects. Antiviral Res. 2019, 170, 104574. [Google Scholar] [CrossRef]
- Han, Z.; Liang, X.; Wang, Y.; Qing, J.; Cao, L.; Shang, L.; Yin, Z. The discovery of indole derivatives as novel hepatitis C virus inhibitors. Eur. J. Med. Chem. 2016, 116, 147–155. [Google Scholar] [CrossRef]
- Wang, G.; Dyatkina, N.; Prhavc, M.; Williams, C.; Serebryany, V.; Hu, Y.; Huang, Y.; Wan, J.; Wu, X.; Deval, J.; et al. Synthesis and anti-HCV activities of 4’-fluoro-2’-substituted uridine triphosphates and nucleotide prodrugs: Discovery of 4’-fluoro-2’-C-methyluridine 5’-phosphoramidate prodrug (AL-335) for the treatment of hepatitis C infection. J. Med. Chem. 2019, 62, 4555–4570. [Google Scholar] [CrossRef] [PubMed]
- Ito, C.; Itoigawa, M.; Mishina, Y.; Filho, V.C.; Enjo, F.; Tokuda, H.; Nishino, H.; Furukawa, H. Chemical constituents of Calophyllum brasiliense. 2. structure of three new coumarins and cancer chemopreventive activity of 4-substituted coumarins. J. Nat. Prod. 2003, 66, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Nam, N.-H.; Kim, Y.; You, Y.-J.; Hong, D.-H.; Kim, H.-M.; Ahn, B.-Z. Preliminary structure–antiangiogenic activity relationships of 4-senecioyloxymethyl-6,7- dimethoxycoumarin. Bioorg. Med. Chem. Lett. 2002, 12, 2345–2348. [Google Scholar] [CrossRef] [PubMed]
- Lin, C. Hepatitis C Viruses: Genomes and Molecular Biology; Tan, S.-L., Ed.; Horizon Bioscience: Norfolk, UK, 2006; Chapter 6. [Google Scholar]
- Wei, Y.; Huang, M.; Jiang, L. Advancements in serine protease inhibitors: From mechanistic insights to clinical applications. Catalysts 2024, 14, 787. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Wang, C.; Yu, K.; Kuzin, A.P.; Richter, F.; Lew, S.; Miklos, A.E.; Matthews, M.L.; Seetharaman, J.; Su, M.; et al. Design of activated serine–containing catalytic triads with atomic-level accuracy. Nat. Chem. Biol. 2014, 10, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Gerabek, W.E.; Haage, B.D.; Keil, G.; Wegner, W. Enzyklopädie Medizingeschichte; Walter de Gruyter GmbH & Co. KG: Berlin, Germany, 2005; p. 152. [Google Scholar]
- Bunevicius, R.; Prange, A.J., Jr. Psychiatric manifestations of graves’ hyperthyroidism: Pathophysiology and treatment options. CNS Drugs 2006, 20, 897–909. [Google Scholar] [CrossRef]
- Kurtovic, S.; Modén, O.; Shokeer, A.; Mannervik, B. Structural determinants of glutathione transferases with azathioprine activity identified by DNA shuffling of alpha class members. J. Mol. Biol. 2008, 375, 1365–1369. [Google Scholar] [CrossRef]
- Lazarević, S.; Đanic, M.; Al-Salami, H.; Mooranian, A.; Mikov, M. Gut Microbiota Metabolism of Azathioprine: A New Hallmark for Personalized Drug-Targeted Therapy of Chronic Inflammatory Bowel Disease. Front. Pharmacol. 2022, 13, 879170. [Google Scholar] [CrossRef]
- Holmes, R.E.; Robins, R.K. Purine nucleosides. VII. direct bromination of adenosine, deoxyadenosine, guanosine, and related purine nucleosides. J. Am. Chem. Soc. 1964, 86, 1242–1245. [Google Scholar] [CrossRef]
- Lin, T.-S.; Cheng, J.-C.; Ishiguro, K.; Sartorelli, A.C. 8-Substituted guanosine and 2′-deoxyguanosine derivatives as potential inducers of the differentiation of friend erythroleukemia cells. J. Med. Chem. 1985, 28, 1194–1198. [Google Scholar] [CrossRef]
- Matsuda, A.; Itoh, H.; Takenuki, K.; Sasaki, T.; Ueda, T. Alkyl addition reaction of pyrimidine 2′-ketonucleosides: Synthesis of 2′-branched-chain sugar pyrimidine nucleosides (nucleosides and nucleotides. LXXXI). Chem. Pharm. Bull. 1988, 36, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Lusic, H.; Young, D.D.; Lively, M.O.; Deiters, A. Photochemical DNA activation. Org. Lett. 2007, 9, 1903–1906. [Google Scholar] [CrossRef] [PubMed]
- Kaleta, Z.; Makowski, B.T.; Soós, T.; Dembinski, R. Thionation using fluorous Lawesson’s Reagent. Org. Lett. 2006, 8, 1625–1628. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 3032615, Thiouridine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Thiouridine (accessed on 17 February 2025).
- Palomino, E.; Meltsner, B.R.; Kessel, D.; Horwitz, J.P. Synthesis and in Vitro evaluation of some modified 4-thiopyrimidine nucleosides for prevention or reversal of AIDS-associated neurological disorders. J. Med. Chem. 1990, 33, 258–263. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Connection | 50 μg/mL a | 125 μg/mL b | ||||
|---|---|---|---|---|---|---|---|
| Type | CC50 c (μM) | EC50 d (μM) | SI e | CC50 c (μM) | EC50 d (μM) | SI e | |
| 3a | waist | 69 | 10 | 6.7 | — | — | — |
| 3b | waist | 48 | 32 | 1.5 | — | — | — |
| 5a | waist | >98 | >98 | ND f | — | — | — |
| 5b | waist | >133 | 51 | >2.6 | — | — | — |
| 7a | head | >145 | 8.6 | >17 | >363 | 4.8 | >76 |
| 7b | head | >105 | 8.8 | >12 | >262 | 5.6 | >47 |
| 9a | head | >139 | 65 | >2.1 | >347 | 48 | >7.3 |
| 9b | head | >102 | 15 | >6.9 | — | — | — |
| 11a | head | >156 | 18 | >8.6 | — | — | — |
| 11b | head | >149 | 16 | >9.1 | — | — | — |
| 16a | head | >108 | 16 | >6.8 | — | — | — |
| 16b | head | >111 | 59 | >1.9 | — | — | — |
| 18 | tail | >156 | 48 | >3.2 | >390 | 87 | >4.5 |
| 20 | tail | >156 | 53 | >2.9 | >391 | 21 | >19 |
| Compound | Connection Type | CC50 a (μM) | EC50 b (μM) | SI c |
|---|---|---|---|---|
| 7a | head | >145 | 7.3 | >20 |
| 7b | head | >105 | 6.6 | >16 |
| 18 | tail | >390 d | 9.4 | >41 |
| Compound | Connection Type | IC50 c (μM) |
|---|---|---|
| 7a | head | >20.0 |
| 7b | head | >20.0 |
| 18 | tail | >20.0 |
| Actinomycin D | — | 15.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.-Y.; Huang, W.-C.; Tsay, S.-C.; Neyts, J.; Leyssen, P.; Lin, C.-C.; Hwang, K.C.; Horng, J.-C.; Hwu, J.R. 6-Chlorocoumarin Conjugates with Nucleobases and Nucleosides as Potent Anti-Hepatitis C Virus Agents. Molecules 2025, 30, 1776. https://doi.org/10.3390/molecules30081776
Lin S-Y, Huang W-C, Tsay S-C, Neyts J, Leyssen P, Lin C-C, Hwang KC, Horng J-C, Hwu JR. 6-Chlorocoumarin Conjugates with Nucleobases and Nucleosides as Potent Anti-Hepatitis C Virus Agents. Molecules. 2025; 30(8):1776. https://doi.org/10.3390/molecules30081776
Chicago/Turabian StyleLin, Shu-Yu, Wen-Chieh Huang, Shwu-Chen Tsay, Johan Neyts, Pieter Leyssen, Chun-Cheng Lin, Kuo Chu Hwang, Jia-Cherng Horng, and Jih Ru Hwu. 2025. "6-Chlorocoumarin Conjugates with Nucleobases and Nucleosides as Potent Anti-Hepatitis C Virus Agents" Molecules 30, no. 8: 1776. https://doi.org/10.3390/molecules30081776
APA StyleLin, S.-Y., Huang, W.-C., Tsay, S.-C., Neyts, J., Leyssen, P., Lin, C.-C., Hwang, K. C., Horng, J.-C., & Hwu, J. R. (2025). 6-Chlorocoumarin Conjugates with Nucleobases and Nucleosides as Potent Anti-Hepatitis C Virus Agents. Molecules, 30(8), 1776. https://doi.org/10.3390/molecules30081776