
Cell-Free Systems: Ideal Platforms for Accelerating the Discovery and Production of Peptide-Based Antibiotics

Abstract
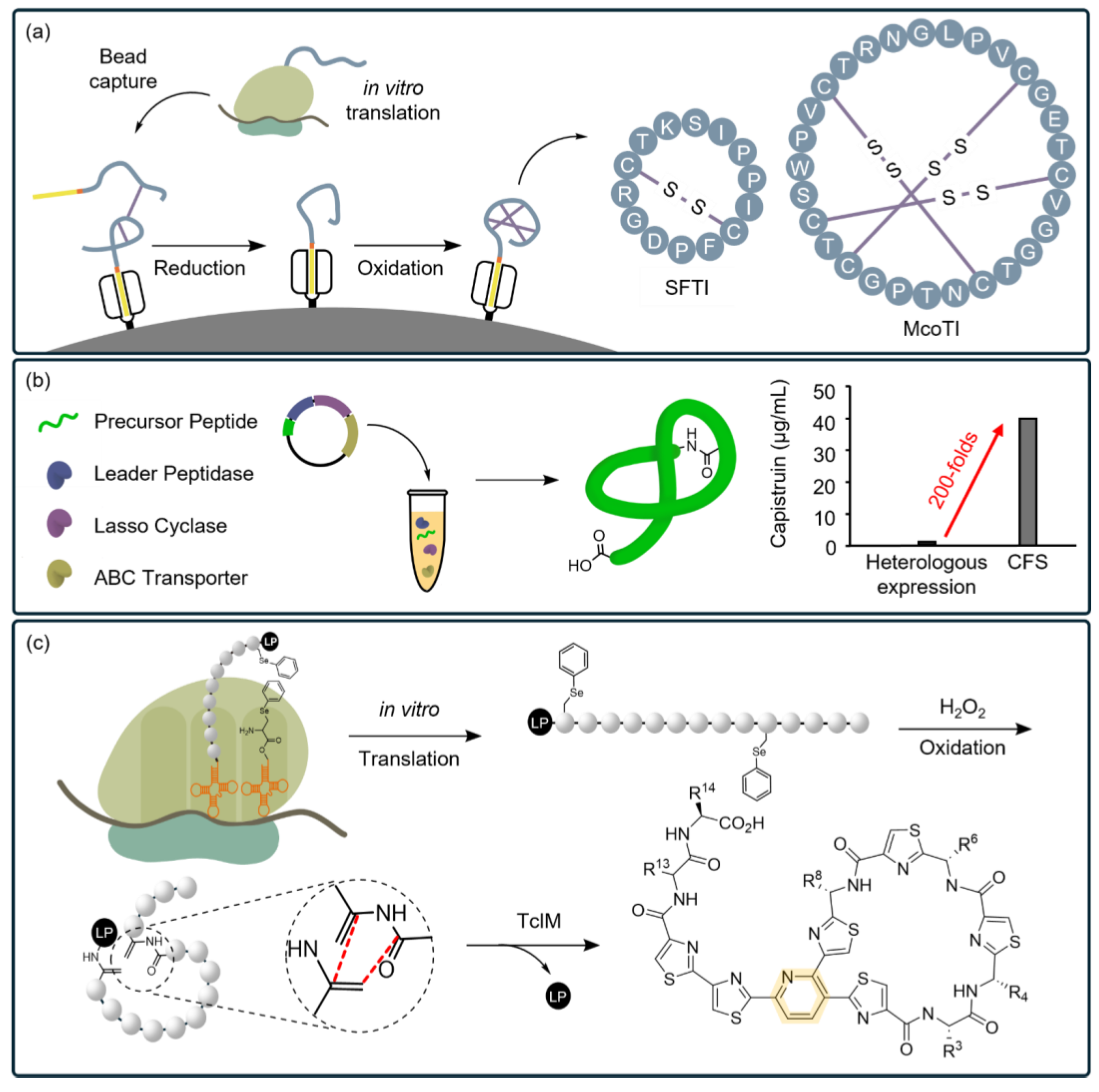
:1. Introduction
2. PBAs Are Produced through Various Biosynthetic Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Machinery for Biosynthesis | Types of Peptides | Representative Compounds |
|---|---|---|---|
| I | (a) Translational apparatus | Peptides only containing the 20 canonical amino acids | α-Helical peptides [9], PrAMPs [63,65], WrAMPs [66], RrAMPs [67], disulfide-rich peptides [69,70] |
| (b) Translational apparatus and post-translational enzymes | Ribosomally synthesized and post-translationally modified peptides (RiPPs) |
| |
| II | (c) Non-ribosomal peptide synthetases (NRPSs) | Non-ribosomal peptides (NRPs) | Lipopeptide surfactants [87], cyclodecapeptides [88], glycopeptide antibiotics [89], mycobactins [90], malleobactins [91] |
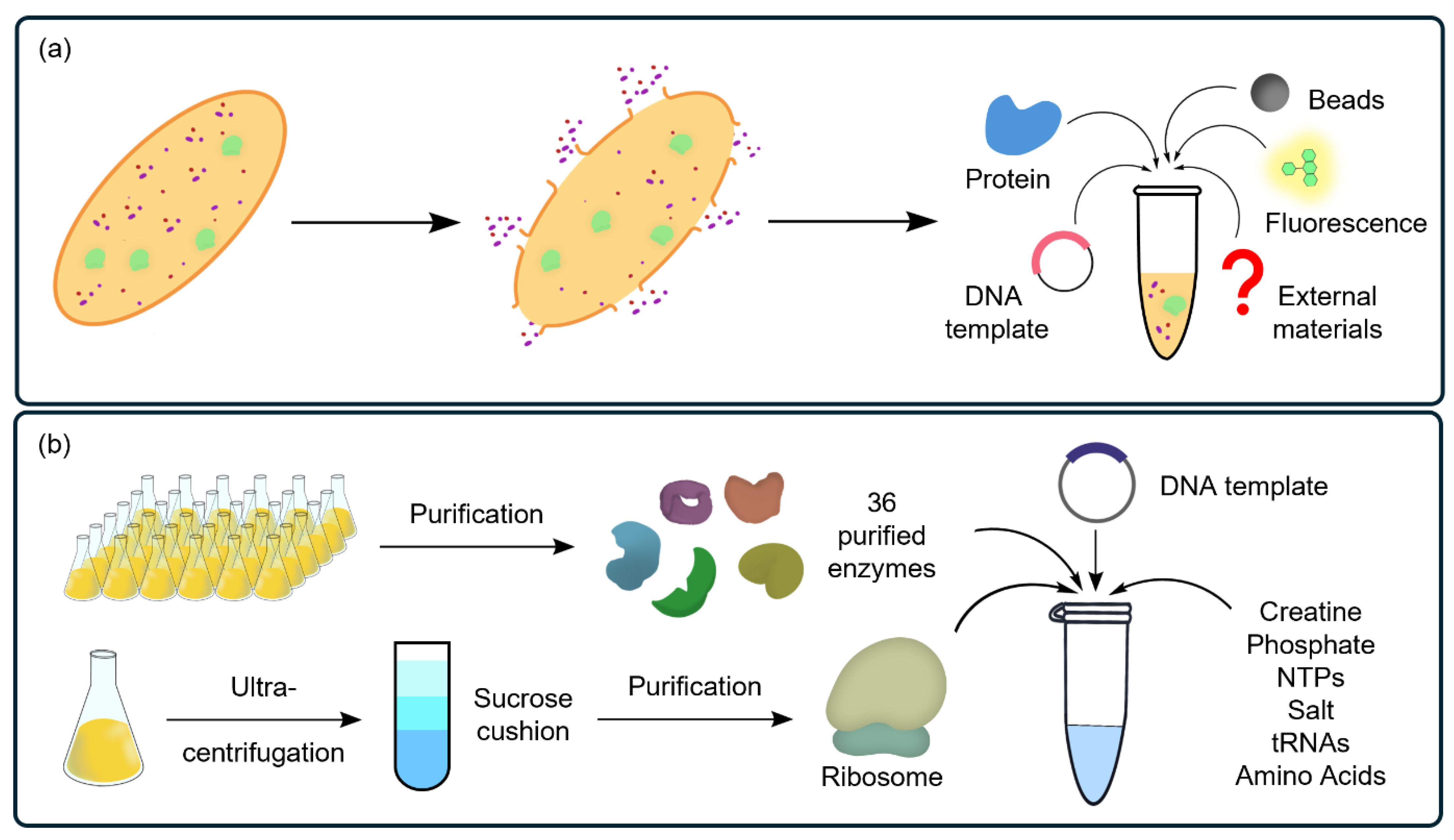
3. CFSs as Ideal Platforms for PBA Research
4. Main Research on PBAs Using CFSs
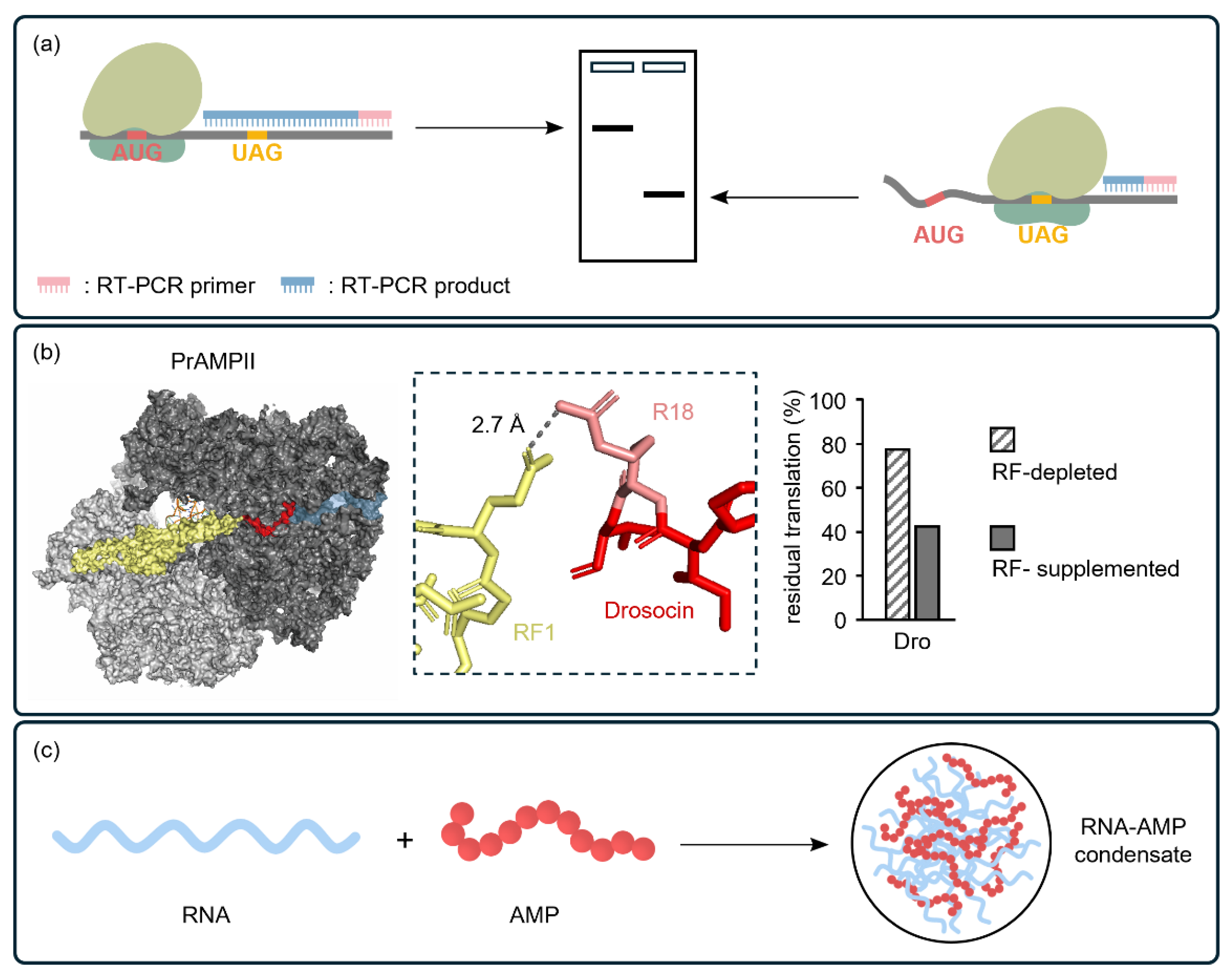
4.1. CFSs Enable the Elucidation of the Mechanism of Action of PBAs
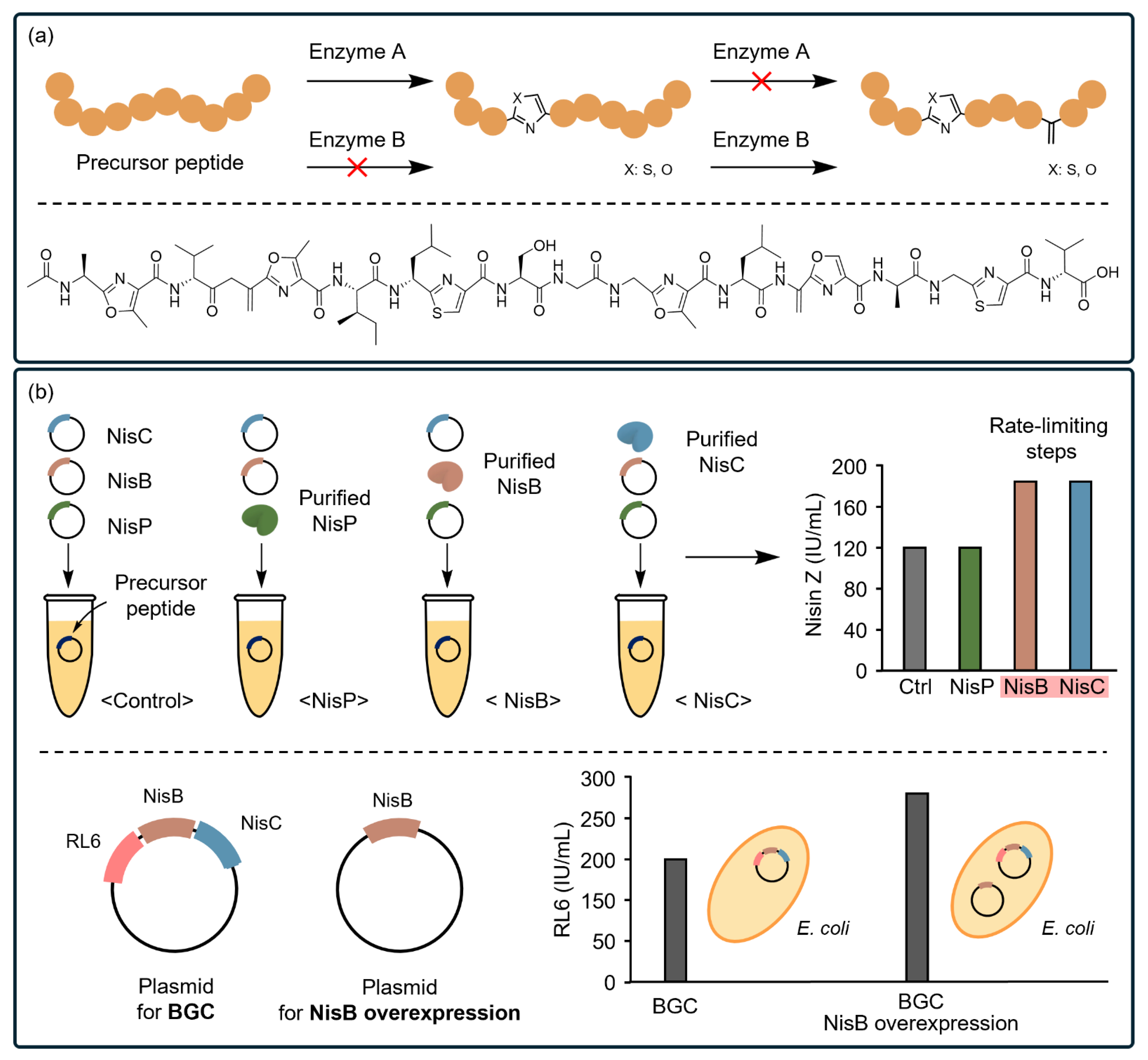
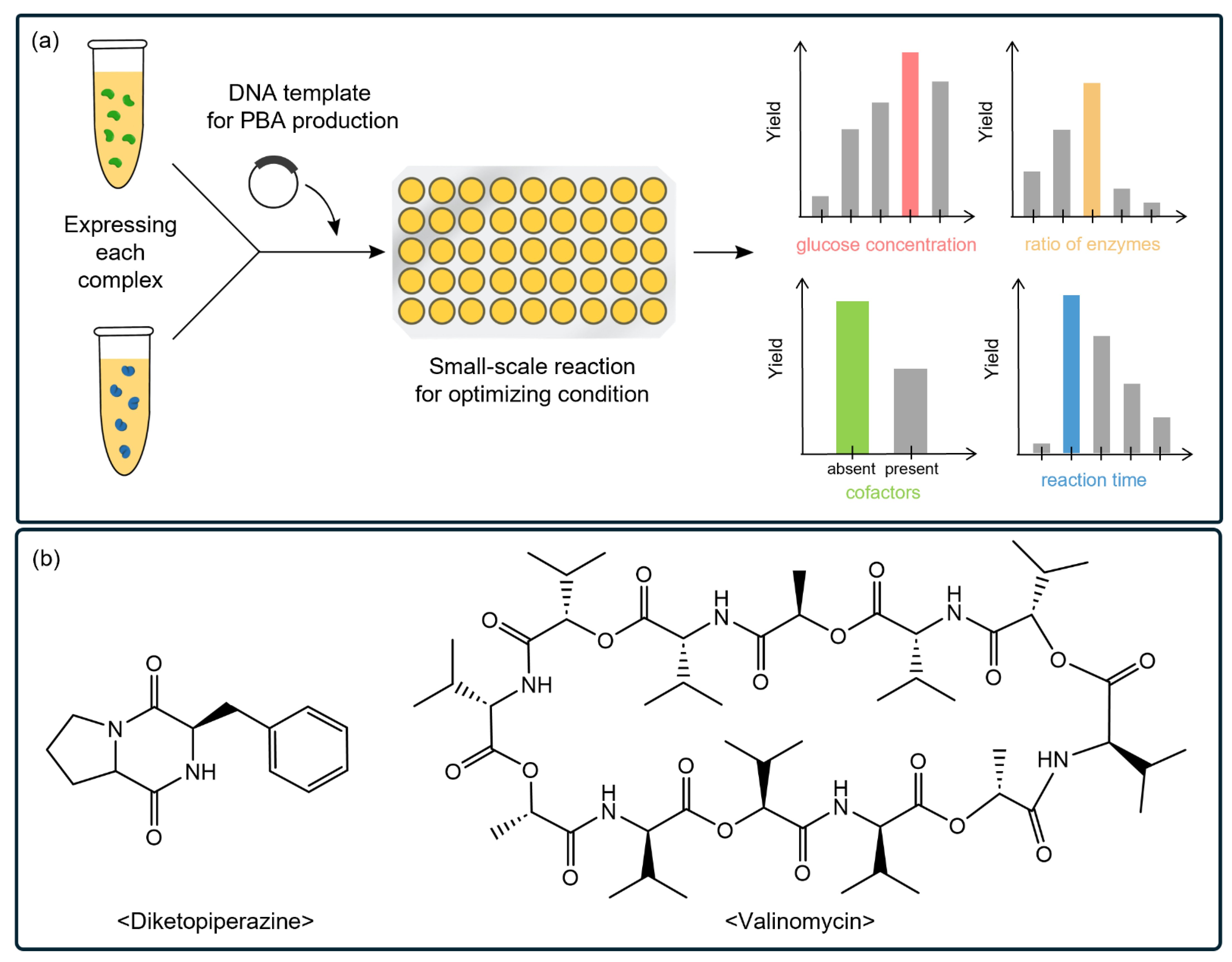
4.2. CFSs Facilitate the Characterization of Biosynthetic Pathways of PBA
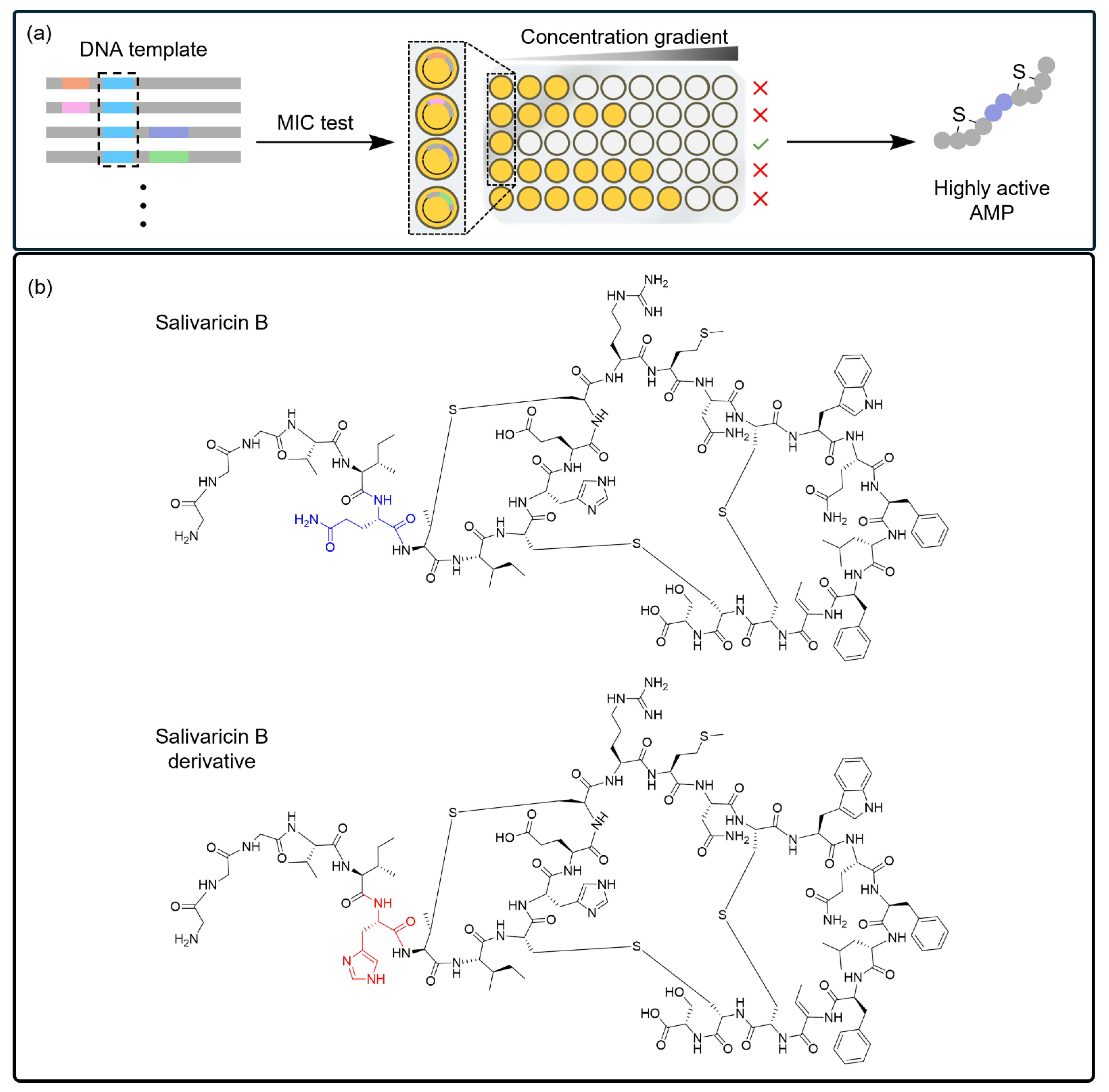
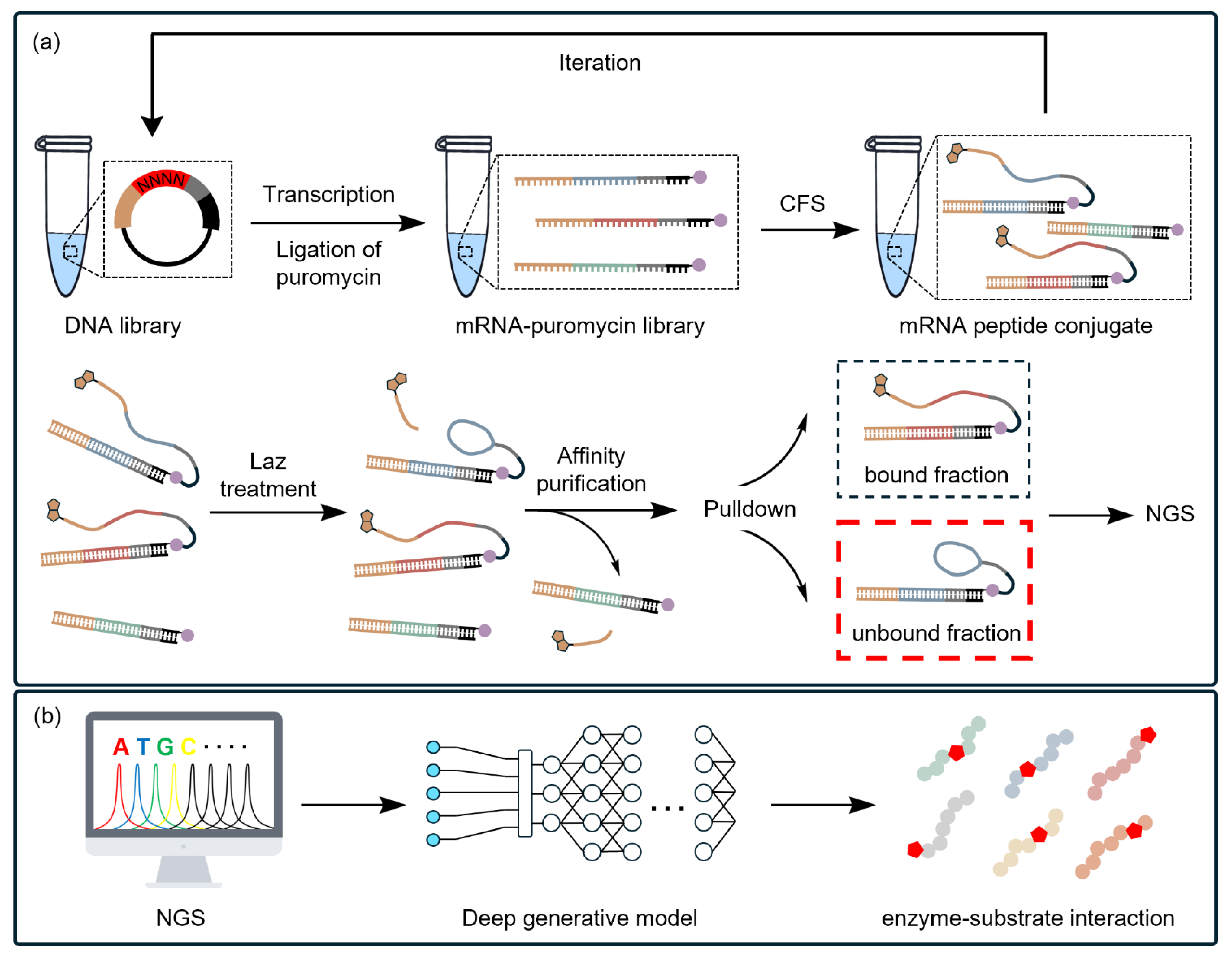
4.3. CFSs Enable the Selection of PBAs from a Diverse Library
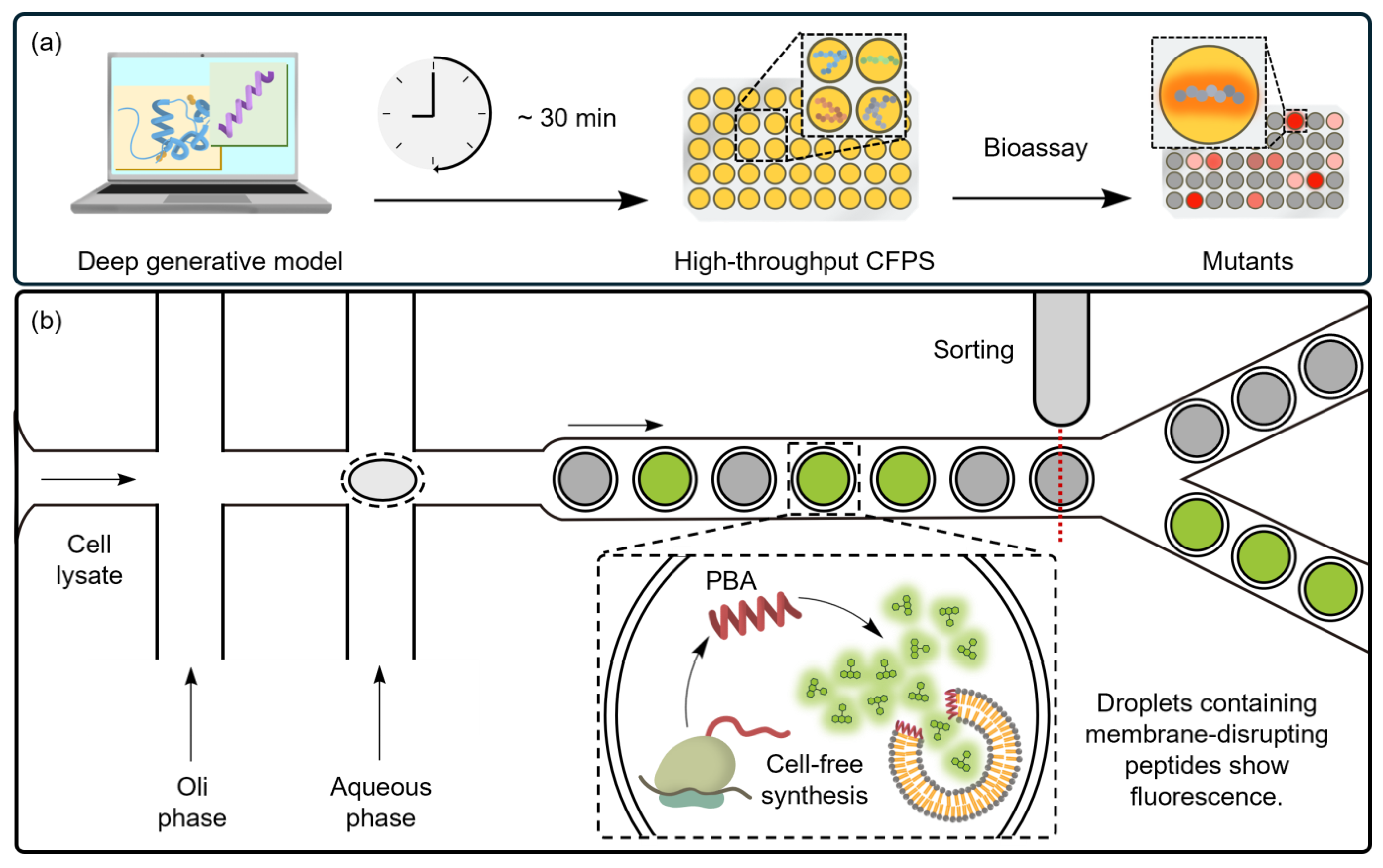
4.4. CFSs Enable the Synthesis of PBAs with Complex Structures
| Section | PBAs | Structural Features | Target Microorganisms |
|---|---|---|---|
| Section 4.1 | Drosocin |
| Gram-negative bacteria (E. coli) |
| Os-C | - | Bacteria | |
| P113 |
| Fungi, Bacteria, Yeast | |
| Buforin-2 | - | Bacteria | |
| Section 4.2 | Goadsporin |
| |
| Nisin Z |
| E. coli, B. subtilis | |
| Section 4.3 | Salivaricin B |
| Bacteria (S. aureus RN4220) |
| Valinomycin |
| Fungi, Gram-positive bacteria | |
| Gramicidin S |
| Fungi, Bacteria | |
| De novo designed PBAs | - | E. coli (MG1655), B. subtilis (PY79), E. faecium S. aureus (DSM 11729), K. pneumoniae (DSM 30104), A. baumannii, P. aeruginosa (DSM 1117), Enterobacter spp., Y. pestis (EV76), B. anthracis Sterne, S. pneumoniae (D39) | |
| Meucin-25 | - | Fungi, Bacteria | |
| Cathelicidin-BF | - | Fungi, Bacteria | |
| δ-lysin | - | Gram-positive bacteria | |
| Section 4.4 | SFTI-1 |
| Fungi |
| Kalata B1 |
| Gram-positive bacteria | |
| AA139 |
| Gram-negative bacteria | |
| HT-1 |
| Bacteria | |
| Capistruin |
| Gram-negative bacteria | |
| Thiocillin |
| Fungi, Gram-positive bacteria |
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Miller, W.R.; Arias, C.A. Eskape pathogens: Antimicrobial resistance, epidemiology, clinical impact and therapeutics. Nat. Rev. Microbiol. 2024, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Reardon, S. Who warns against ‘post-antibiotic’ era. Nature 2014, 15, 135–138. [Google Scholar] [CrossRef]
- Kwon, J.H.; Powderly, W.G. The post-antibiotic era is here. Science 2021, 373, 471. [Google Scholar] [CrossRef]
- World Health Organization. 2021 Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Baldwin, J.E.; Abraham, E. The biosynthesis of penicillins and cephalosporins. Nat. Prod. Rep. 1988, 5, 129–145. [Google Scholar] [CrossRef]
- Gunsior, M.; Breazeale, S.D.; Lind, A.J.; Ravel, J.; Janc, J.W.; Townsend, C.A. The biosynthetic gene cluster for a monocyclic beta-하lactam antibiotic, nocardicin A. Chem. Biol. 2004, 11, 927–938. [Google Scholar] [CrossRef]
- Fleming, A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. Influenzæ. Br. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar] [CrossRef]
- MacNair, C.R.; Rutherford, S.T.; Tan, M.W. Alternative therapeutic strategies to treat antibiotic-resistant pathogens. Nat. Rev. Microbiol. 2024, 22, 262–275. [Google Scholar] [CrossRef]
- Chen, E.H.; Wang, C.H.; Liao, Y.T.; Chan, F.Y.; Kanaoka, Y.; Uchihashi, T.; Kato, K.; Lai, L.; Chang, Y.W.; Ho, M.C.; et al. Visualizing the membrane disruption action of antimicrobial peptides by cryo-electron tomography. Nat. Commun. 2023, 14, 5464. [Google Scholar] [CrossRef]
- Pierre, J.F.; Peters, B.M.; La Torre, D.; Sidebottom, A.M.; Tao, Y.; Zhu, X.; Cham, C.M.; Wang, L.; Kambal, A.; Harris, K.G.; et al. Peptide yy: A paneth cell antimicrobial peptide that maintains candida gut commensalism. Science 2023, 381, 502–508. [Google Scholar] [CrossRef]
- Gidalevitz, D.; Ishitsuka, Y.; Muresan, A.S.; Konovalov, O.; Waring, A.J.; Lehrer, R.I.; Lee, K.Y. Interaction of antimicrobial peptide protegrin with biomembranes. Proc. Natl. Acad. Sci. USA 2003, 100, 6302–6307. [Google Scholar] [CrossRef]
- Stephani, J.C.; Gerhards, L.; Khairalla, B.; Solov’yov, I.A.; Brand, I. How do antimicrobial peptides interact with the outer membrane of gram-negative bacteria? Role of lipopolysaccharides in peptide binding, anchoring, and penetration. ACS Infect. Dis. 2024, 10, 763–778. [Google Scholar] [CrossRef]
- Melcrova, A.; Maity, S.; Melcr, J.; de Kok, N.A.W.; Gabler, M.; van der Eyden, J.; Stensen, W.; Svendsen, J.S.M.; Driessen, A.J.M.; Marrink, S.J.; et al. Lateral membrane organization as target of an antimicrobial peptidomimetic compound. Nat. Commun. 2023, 14, 4038. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, J.; Sineva, E.; Knight, J.; Levy, R.M.; Ebright, R.H. Antibacterial peptide microcin j25 inhibits transcription by binding within and obstructing the rna polymerase secondary channel. Mol. Cell 2004, 14, 739–751. [Google Scholar] [CrossRef]
- Michalczyk, E.; Hommernick, K.; Behroz, I.; Kulike, M.; Pakosz-Stepien, Z.; Mazurek, L.; Seidel, M.; Kunert, M.; Santos, K.; von Moeller, H.; et al. Molecular mechanism of topoisomerase poisoning by the peptide antibiotic albicidin. Nat. Catal. 2023, 6, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Sneideris, T.; Erkamp, N.A.; Ausserwoger, H.; Saar, K.L.; Welsh, T.J.; Qian, D.; Katsuya-Gaviria, K.; Johncock, M.; Krainer, G.; Borodavka, A.; et al. Targeting nucleic acid phase transitions as a mechanism of action for antimicrobial peptides. Nat. Commun. 2023, 14, 7170. [Google Scholar] [CrossRef]
- Schmidt, N.W.; Jin, F.; Lande, R.; Curk, T.; Xian, W.; Lee, C.; Frasca, L.; Frenkel, D.; Dobnikar, J.; Gilliet, M.; et al. Liquid-crystalline ordering of antimicrobial peptide-DNA complexes controls tlr9 activation. Nat. Mater. 2015, 14, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Karathanasi, G.; Bojer, M.S.; Baldry, M.; Johannessen, B.A.; Wolff, S.; Greco, I.; Kilstrup, M.; Hansen, P.R.; Ingmer, H. Linear peptidomimetics as potent antagonists of staphylococcus aureus agr quorum sensing. Sci. Rep. 2018, 8, 3562. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Saito, C.; Yokoo, H.; Goto, C.; Kawano, R.; Misawa, T.; Demizu, Y. Development of antimicrobial stapled peptides based on magainin 2 sequence. Molecules 2021, 26, 444. [Google Scholar] [CrossRef]
- Giacometti, A.; Cirioni, O.; Del Prete, M.S.; Paggi, A.M.; D’Errico, M.M.; Scalise, G. Combination studies between polycationic peptides and clinically used antibiotics against gram-positive and gram-negative bacteria. Peptides 2000, 21, 1155–1160. [Google Scholar] [CrossRef]
- Galdiero, E.; Siciliano, A.; Gesuele, R.; Di Onofrio, V.; Falanga, A.; Maione, A.; Liguori, R.; Libralato, G.; Guida, M. Melittin inhibition and eradication activity for resistant polymicrobial biofilm isolated from a dairy industry after disinfection. Int. J. Microbiol. 2019, 2019, 4012394. [Google Scholar] [CrossRef]
- Lima, W.G.; de Brito, J.C.M.; Cardoso, V.N.; Fernandes, S.O.A. In-depth characterization of antibacterial activity of melittin against staphylococcus aureus and use in a model of non-surgical mrsa-infected skin wounds. Eur. J. Pharm. Sci. 2021, 156, 105592. [Google Scholar] [CrossRef] [PubMed]
- Diederen, B.M.; van Duijn, I.; Willemse, P.; Kluytmans, J.A. In vitro activity of daptomycin against methicillin-resistant staphylococcus aureus, including heterogeneously glycopeptide-resistant strains. Antimicrob. Agents Chemother. 2006, 50, 3189–3191. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Sambanthamoorthy, K.; Palys, T.; Paranavitana, C. The human antimicrobial peptide ll-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant acinetobacter baumannii. Peptides 2013, 49, 131–137. [Google Scholar] [CrossRef]
- Turner, J.; Cho, Y.; Dinh, N.N.; Waring, A.J.; Lehrer, R.I. Activities of ll-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob. Agents Chemother. 1998, 42, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Neshani, A.; Zare, H.; Eidgahi, M.R.A.; Kakhki, R.K.; Safdari, H.; Khaledi, A.; Ghazvini, K. Ll-37: Review of antimicrobial profile against sensitive and antibiotic-resistant human bacterial pathogens. Gene Rep. 2019, 17, 100519. [Google Scholar] [CrossRef]
- Kim, M.K.; Kang, N.; Ko, S.J.; Park, J.; Park, E.; Shin, D.W.; Kim, S.H.; Lee, S.A.; Lee, J.I.; Lee, S.H.; et al. Antibacterial and antibiofilm activity and mode of action of magainin 2 against drug-resistant acinetobacter baumannii. Int. J. Mol. Sci. 2018, 19, 3041. [Google Scholar] [CrossRef]
- Gu, H.; Kato, T.; Kumeta, H.; Kumaki, Y.; Tsukamoto, T.; Kikukawa, T.; Demura, M.; Ishida, H.; Vogel, H.J.; Aizawa, T. Three-dimensional structure of the antimicrobial peptide cecropin p1 in dodecylphosphocholine micelles and the role of the c-terminal residues. ACS Omega 2022, 7, 31924–31934. [Google Scholar] [CrossRef]
- Rengaraj, R.; Mariappan, S.; Sekar, U.; Kamalanadhan, A. Detection of vancomycin resistance among enterococcus faecalis and staphylococcus aureus. J. Clin. Diagn. Res. 2016, 10, DC04-6. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.L.; Marchaim, D.; Pogue, J.M.; Sunkara, B.; Bheemreddy, S.; Bathina, P.; Pulluru, H.; Chugh, N.; Wilson, M.N.; Moshos, J.; et al. Treatment of methicillin-resistant staphylococcus aureus infections with a minimal inhibitory concentration of 2 mug/ml to vancomycin: Old (trimethoprim/sulfamethoxazole) versus new (daptomycin or linezolid) agents. Ann. Pharmacother. 2012, 46, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- AlQahtani, H.; Baloch, S.; Aleanizy, F.S.; Alqahtani, F.Y.; Tabb, D. Influence of the minimum inhibitory concentration of daptomycin on the outcomes of staphylococcus aureus bacteraemia. J. Glob. Antimicrob. Resist. 2021, 24, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Karyne, R.; Curty Lechuga, G.; Almeida Souza, A.L.; Rangel da Silva Carvalho, J.P.; Simoes Villas Boas, M.H.; De Simone, S.G. Pan-drug resistant acinetobacter baumannii, but not other strains, are resistant to the bee venom peptide mellitin. Antibiotics 2020, 9, 178. [Google Scholar]
- Zavascki, A.P.; Goldani, L.Z.; Li, J.; Nation, R.L. Polymyxin B for the treatment of multidrug-resistant pathogens: A critical review. J. Antimicrob. Chemother. 2007, 60, 1206–1215. [Google Scholar] [CrossRef]
- Shariati, A.; Arshadi, M.; Khosrojerdi, M.A.; Abedinzadeh, M.; Ganjalishahi, M.; Maleki, A.; Heidary, M.; Khoshnood, S. The resistance mechanisms of bacteria against ciprofloxacin and new approaches for enhancing the efficacy of this antibiotic. Front. Public Health 2022, 10, 1025633. [Google Scholar] [CrossRef]
- Jakobsen, L.; Sandvang, D.; Jensen, V.F.; Seyfarth, A.M.; Frimodt-Moller, N.; Hammerum, A.M. Gentamicin susceptibility in escherichia coli related to the genetic background: Problems with breakpoints. Clin. Microbiol. Infect. 2007, 13, 830–832. [Google Scholar] [CrossRef]
- Grossman, T.H. Tetracycline antibiotics and resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025387. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Forde, B.M.; Phan, M.D.; Steiner, B.; Zhang, B.; Zuegg, J.; El-Deeb, I.M.; Li, G.; Keller, N.; Brouwer, S.; et al. Rescuing tetracycline class antibiotics for the treatment of multidrug-resistant acinetobacter baumannii pulmonary infection. MBio 2022, 13, e0351721. [Google Scholar] [CrossRef]
- Sadybekov, A.V.; Katritch, V. Computational approaches streamlining drug discovery. Nature 2023, 616, 673–685. [Google Scholar] [CrossRef]
- Montalban-Lopez, M.; van Heel, A.J.; Kuipers, O.P. Employing the promiscuity of lantibiotic biosynthetic machineries to produce novel antimicrobials. Fems Microbiol. Rev. 2017, 41, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Hemmerling, F.; Piel, J. Strategies to access biosynthetic novelty in bacterial genomes for drug discovery. Nat. Rev. Drug Discov. 2022, 21, 359–378. [Google Scholar] [CrossRef]
- Antimicrobial Resistance, C. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef]
- Sayburn, A. Only 12 new antibiotics entered the market in five years, who warns in review. Pharm. J. 2023, 310, 7971. [Google Scholar]
- PhRMA. Medicines in Development for Cancer 2020 Report; PhRMA: Washington, DC, USA, 2020. [Google Scholar]
- Garenne, D.; Haines, M.C.; Romantseva, E.F.; Freemont, P.; Strychalski, E.A.; Noireaux, V. Cell-free gene expression. Nat. Rev. Method Prim. 2021, 1, 49. [Google Scholar] [CrossRef]
- Silverman, A.D.; Karim, A.S.; Jewett, M.C. Cell-free gene expression: An expanded repertoire of applications. Nat. Rev. Genet. 2020, 21, 151–170. [Google Scholar] [CrossRef]
- Kofman, C.; Lee, J.; Jewett, M.C. Engineering molecular translation systems. Cell Syst. 2021, 12, 593–607. [Google Scholar] [CrossRef]
- Inoue, S.; Nguyen, D.T.; Hamada, K.; Okuma, R.; Okada, C.; Okada, M.; Abe, I.; Sengoku, T.; Goto, Y.; Suga, H. De novo discovery of pseudo-natural prenylated macrocyclic peptide ligands. Angew. Chem. Int. Ed. 2024, e202409973. [Google Scholar] [CrossRef]
- Kawakami, T.; Ohta, A.; Ohuchi, M.; Ashigai, H.; Murakami, H.; Suga, H. Diverse backbone-cyclized peptides via codon reprogramming. Nat. Chem. Biol. 2009, 5, 888–890. [Google Scholar] [CrossRef]
- Lee, J.; Schwarz, K.J.; Kim, D.S.; Moore, J.S.; Jewett, M.C. Ribosome-mediated polymerization of long chain carbon and cyclic amino acids into peptides in vitro. Nat. Commun. 2020, 11, 4304. [Google Scholar] [CrossRef]
- Lee, K.; Willi, J.A.; Cho, N.; Kim, I.; Jewett, M.C.; Lee, J. Cell-free biosynthesis of peptidomimetics. Biotechnol. Bioprocess Eng. 2023, 28, 905–921. [Google Scholar] [CrossRef]
- Walsh, C. Antibiotics: Actions, Origins, Resistance; Harvard Medical School: Boston, MA, USA, 2003. [Google Scholar]
- Schwarzer, D.; Finking, R.; Marahiel, M.A. Nonribosomal peptides: From genes to products. Nat. Prod. Rep. 2003, 20, 275–287. [Google Scholar] [CrossRef]
- Marahiel, M.A.; Stachelhaus, T.; Mootz, H.D. Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem. Rev. 1997, 97, 2651–2674. [Google Scholar] [CrossRef]
- Zasloff, M. Magainins, a class of antimicrobial peptides from xenopus skin: Isolation, characterization of two active forms, and partial cdna sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef]
- Alalwani, S.M.; Sierigk, J.; Herr, C.; Pinkenburg, O.; Gallo, R.; Vogelmeier, C.; Bals, R. The antimicrobial peptide ll-37 modulates the inflammatory and host defense response of human neutrophils. Eur. J. Immunol. 2010, 40, 1118–1126. [Google Scholar] [CrossRef]
- Nizet, V.; Ohtake, T.; Lauth, X.; Trowbridge, J.; Rudisill, J.; Dorschner, R.A.; Pestonjamasp, V.; Piraino, J.; Huttner, K.; Gallo, R.L. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 2001, 414, 454–457. [Google Scholar] [CrossRef]
- Hoelscher, M.P.; Forner, J.; Calderone, S.; Kramer, C.; Taylor, Z.; Loiacono, F.V.; Agrawal, S.; Karcher, D.; Moratti, F.; Kroop, X.; et al. Expression strategies for the efficient synthesis of antimicrobial peptides in plastids. Nat. Commun. 2022, 13, 5856. [Google Scholar] [CrossRef]
- Deo, S.; Turton, K.L.; Kainth, T.; Kumar, A.; Wieden, H.J. Strategies for improving antimicrobial peptide production. Biotechnol. Adv. 2022, 59, 107968. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- King, A.M.; Zhang, Z.; Glassey, E.; Siuti, P.; Clardy, J.; Voigt, C.A. Systematic mining of the human microbiome identifies antimicrobial peptides with diverse activity spectra. Nat. Microbiol. 2023, 8, 2420–2434. [Google Scholar] [CrossRef]
- Mangano, K.; Klepacki, D.; Ohanmu, I.; Baliga, C.; Huang, W.; Brakel, A.; Krizsan, A.; Polikanov, Y.S.; Hoffmann, R.; Vazquez-Laslop, N.; et al. Inhibition of translation termination by the antimicrobial peptide drosocin. Nat. Chem. Biol. 2023, 19, 1082–1090. [Google Scholar] [CrossRef]
- Engelberg, Y.; Landau, M. The human ll-37(17-29) antimicrobial peptide reveals a functional supramolecular structure. Nat. Commun. 2020, 11, 3894. [Google Scholar] [CrossRef] [PubMed]
- Florin, T.; Maracci, C.; Graf, M.; Karki, P.; Klepacki, D.; Berninghausen, O.; Beckmann, R.; Vazquez-Laslop, N.; Wilson, D.N.; Rodnina, M.V.; et al. An antimicrobial peptide that inhibits translation by trapping release factors on the ribosome. Nat. Struct. Mol. Biol. 2017, 24, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [PubMed]
- Lòpez-Oyama, A.B. Interaction of the cationic peptide bactencin with ddpc/dmpg phospholipid mixtures at the air-water interface. Biophys. J. 2010, 98, 84a. [Google Scholar] [CrossRef]
- Robinson, P.J.; Bulleid, N.J. Mechanisms of disulfide bond formation in nascent polypeptides entering the secretory pathway. Cells 2020, 9, 1994. [Google Scholar] [CrossRef] [PubMed]
- Nawae, W.; Hannongbua, S.; Ruengjitchatchawalya, M. Defining the membrane disruption mechanism of kalata b1 via coarse-grained molecular dynamics simulations. Sci. Rep. 2014, 4, 3933. [Google Scholar] [CrossRef]
- Korsinczky, M.L.; Clark, R.J.; Craik, D.J. Disulfide bond mutagenesis and the structure and function of the head-to-tail macrocyclic trypsin inhibitor sfti-1. Biochemistry 2005, 44, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Mattiuzzo, M.; Bandiera, A.; Gennaro, R.; Benincasa, M.; Pacor, S.; Antcheva, N.; Scocchi, M. Role of the escherichia coli sbma in the antimicrobial activity of proline-rich peptides. Mol. Microbiol. 2007, 66, 151–163. [Google Scholar] [CrossRef]
- Lointier, M.; Aisenbrey, C.; Marquette, A.; Tan, J.H.; Kichler, A.; Bechinger, B. Membrane pore-formation correlates with the hydrophilic angle of histidine-rich amphipathic peptides with multiple biological activities. Biochim. Biophys. Acta-Biomembr. 2020, 1862, 183212. [Google Scholar] [CrossRef]
- Seefeldt, A.C.; Nguyen, F.; Antunes, S.; Perebaskine, N.; Graf, M.; Arenz, S.; Inampudi, K.K.; Douat, C.; Guichard, G.; Wilson, D.N.; et al. The proline-rich antimicrobial peptide onc112 inhibits translation by blocking and destabilizing the initiation complex. Nat. Struct. Mol. Biol. 2015, 22, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, M.; Perebaskine, N.; Benincasa, M.; Gambato, S.; Hofmann, S.; Huter, P.; Muller, C.; Hilpert, K.; Innis, C.A.; Tossi, A.; et al. The dolphin proline-rich antimicrobial peptide tur1a inhibits protein synthesis by targeting the bacterial ribosome. Cell Chem. Biol. 2018, 25, 530–539.e7. [Google Scholar] [CrossRef] [PubMed]
- Cabalteja, C.C.; Lin, Q.; Harmon, T.W.; Rao, S.R.; Di, Y.P.; Horne, W.S. Heterogeneous-backbone proteomimetic analogues of lasiocepsin, a disulfide-rich antimicrobial peptide with a compact tertiary fold. ACS Chem. Biol. 2022, 17, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Mourenza, A.; Ganesan, R.; Camarero, J.A. Resistance is futile: Targeting multidrug-resistant bacteria with de novo cys-rich cyclic polypeptides. RSC Chem. Biol. 2023, 4, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Gongora-Benitez, M.; Tulla-Puche, J.; Albericio, F. Multifaceted roles of disulfide bonds. Peptides as therapeutics. Chem. Rev. 2014, 114, 901–926. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.K.; Craik, D.J. Designing macrocyclic disulfide-rich peptides for biotechnological applications. Nat. Chem. Biol. 2018, 14, 417–427. [Google Scholar] [CrossRef]
- Colgrave, M.L.; Craik, D.J. Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: The importance of the cyclic cystine knot. Biochemistry 2004, 43, 5965–5975. [Google Scholar] [CrossRef]
- Clark, R.J.; Jensen, J.; Nevin, S.T.; Callaghan, B.P.; Adams, D.J.; Craik, D.J. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. 2010, 49, 6545–6548. [Google Scholar] [CrossRef]
- Travin, D.Y.; Watson, Z.L.; Metelev, M.; Ward, F.R.; Osterman, I.A.; Khven, I.M.; Khabibullina, N.F.; Serebryakova, M.; Mergaert, P.; Polikanov, Y.S.; et al. Structure of ribosome-bound azole-modified peptide phazolicin rationalizes its species-specific mode of bacterial translation inhibition. Nat. Commun. 2019, 10, 4563. [Google Scholar] [CrossRef]
- Parajuli, A.; Kwak, D.H.; Dalponte, L.; Leikoski, N.; Galica, T.; Umeobika, U.; Trembleau, L.; Bent, A.; Sivonen, K.; Wahlsten, M. A unique tryptophan c-prenyltransferase from the kawaguchipeptin biosynthetic pathway. Angew. Chem. Int. Ed. 2016, 55, 3596–3599. [Google Scholar] [CrossRef]
- Claesen, J.; Bibb, M. Genome mining and genetic analysis of cypemycin biosynthesis reveal an unusual class of posttranslationally modified peptides. Proc. Natl. Acad. Sci. USA 2010, 107, 16297–16302. [Google Scholar] [CrossRef]
- Ayikpoe, R.S.; Shi, C.; Battiste, A.J.; Eslami, S.M.; Ramesh, S.; Simon, M.A.; Bothwell, I.R.; Lee, H.; Rice, A.J.; Ren, H. A scalable platform to discover antimicrobials of ribosomal origin. Nat. Commun. 2022, 13, 6135. [Google Scholar] [CrossRef]
- Haste, N.M.; Thienphrapa, W.; Tran, D.N.; Loesgen, S.; Sun, P.; Nam, S.-J.; Jensen, P.R.; Fenical, W.; Sakoulas, G.; Nizet, V. Activity of the thiopeptide antibiotic nosiheptide against contemporary strains of methicillin-resistant staphylococcus aureus. J. Antibiot. 2012, 65, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Hegemann, J.D.; Zimmermann, M.; Xie, X.; Marahiel, M.A. Lasso peptides: An intriguing class of bacterial natural products. Acc. Chem. Res. 2015, 48, 1909–1919. [Google Scholar] [CrossRef]
- Rimal, B.; Chang, J.; Liu, C.; Rashid, R.; Singh, M.; Kim, S.J. The effects of daptomycin on cell wall biosynthesis in enterococcal faecalis. Sci. Rep. 2023, 13, 12227. [Google Scholar] [CrossRef]
- Rivera-Pérez, C.; Ponce González, X.P.; Hernández-Savedra, N.Y. Antimicrobial and anticarcinogenic activity of bioactive peptides derived from abalone viscera (haliotis fulgens and haliotis corrugata). Sci. Rep. 2023, 13, 15185. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.; Boddy, C.N.; Bräse, S.; Winssinger, N. Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew. Chem. Int. Ed. 1999, 38, 2096–2152. [Google Scholar] [CrossRef]
- Baltz, R.H. Function of mbth homologs in nonribosomal peptide biosynthesis and applications in secondary metabolite discovery. J. Ind. Microbiol. Biotechnol. 2011, 38, 1747. [Google Scholar] [CrossRef]
- Franke, J.; Ishida, K.; Ishida-Ito, M.; Hertweck, C. Nitro versus hydroxamate in siderophores of pathogenic bacteria: Effect of missing hydroxylamine protection in malleobactin biosynthesis. Angew. Chem. Int. Ed. 2013, 125, 8429. [Google Scholar] [CrossRef]
- Repka, L.M.; Chekan, J.R.; Nair, S.K.; van der Donk, W.A. Mechanistic understanding of lanthipeptide biosynthetic enzymes. Chem. Rev. 2017, 117, 5457–5520. [Google Scholar] [CrossRef]
- Ortega, M.A.; van der Donk, W.A. New insights into the biosynthetic logic of ribosomally synthesized and post-translationally modified peptide natural products. Cell Chem. Biol. 2016, 23, 31–44. [Google Scholar] [CrossRef]
- Shi, C.; Patel, V.A.; Mitchell, D.A.; Zhao, H. Enterolyin s, a polythiazole-containing hemolytic peptide from enterococcus caccae. ChemBioChem 2024, 25, e202400212. [Google Scholar] [CrossRef] [PubMed]
- Letzel, A.C.; Pidot, S.J.; Hertweck, C. Genome mining for ribosomally synthesized and post-translationally modified peptides (ripps) in anaerobic bacteria. BMC Genom. 2014, 15, 983. [Google Scholar] [CrossRef]
- Bednarska, N.G.; Wren, B.W.; Willcocks, S.J. The importance of the glycosylation of antimicrobial peptides: Natural and synthetic approaches. Drug Discov. Today 2017, 22, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, A.Y.; Mariasina, S.S.; Sergiev, P.V.; Polshakov, V.I. Analogs of s-adenosyl-l-methionine in studies of methyltransferases. Mol. Biol. 2022, 56, 229–250. [Google Scholar] [CrossRef]
- Winn, M.; Fyans, J.K.; Zhuo, Y.; Micklefield, J. Recent advances in engineering nonribosomal peptide assembly lines. Nat. Prod. Rep. 2016, 33, 317–347. [Google Scholar] [CrossRef] [PubMed]
- Brytan, W.; Padrela, L. Structural modifications for the conversion of proteins and peptides into stable dried powder formulations: A review. J. Drug Deliv. Sci. Technol. 2023, 89, 104992. [Google Scholar] [CrossRef]
- Sussmuth, R.D.; Mainz, A. Nonribosomal peptide synthesis-principles and prospects. Angew. Chem. Int. Ed. 2017, 56, 3770–3821. [Google Scholar] [CrossRef]
- Weissman, K.J. The structural biology of biosynthetic megaenzymes. Nat. Chem. Biol. 2015, 11, 660–670. [Google Scholar] [CrossRef]
- Brakhage, A.A. Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 2013, 11, 21–32. [Google Scholar] [CrossRef]
- Sieber, S.A.; Marahiel, M.A. Molecular mechanisms underlying nonribosomal peptide synthesis: Approaches to new antibiotics. Chem. Rev. 2005, 105, 715–738. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef]
- Folger, I.B.; Frota, N.F.; Pistofidis, A.; Niquille, D.L.; Hansen, D.A.; Schmeing, T.M.; Hilvert, D. High-throughput reprogramming of an nrps condensation domain. Nat. Chem. Biol. 2024, 20, 761–769. [Google Scholar] [CrossRef]
- Scott, T.A.; Heine, D.; Qin, Z.; Wilkinson, B. An l-threonine transaldolase is required for l-threo-beta-hydroxy-alpha-amino acid assembly during obafluorin biosynthesis. Nat. Commun. 2017, 8, 15935. [Google Scholar] [CrossRef]
- Awakawa, T.; Fujioka, T.; Zhang, L.; Hoshino, S.; Hu, Z.; Hashimoto, J.; Kozone, I.; Ikeda, H.; Shin-Ya, K.; Liu, W.; et al. Reprogramming of the antimycin nrps-pks assembly lines inspired by gene evolution. Nat. Commun. 2018, 9, 3534. [Google Scholar] [CrossRef] [PubMed]
- Niquille, D.L.; Hansen, D.A.; Mori, T.; Fercher, D.; Kries, H.; Hilvert, D. Nonribosomal biosynthesis of backbone-modified peptides. Nat. Chem. 2018, 10, 282–287. [Google Scholar] [CrossRef]
- Wang, Z.; Koirala, B.; Hernandez, Y.; Zimmerman, M.; Park, S.; Perlin, D.S.; Brady, S.F. A naturally inspired antibiotic to target multidrug-resistant pathogens. Nature 2022, 601, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, D.A.; Schmeing, T.M. Biosynthesis of depsipeptides, or depsi: The peptides with varied generations. Protein Sci. 2020, 29, 2316–2347. [Google Scholar] [CrossRef] [PubMed]
- Konz, D.; Klens, A.; Schorgendorfer, K.; Marahiel, M.A. The bacitracin biosynthesis operon of bacillus licheniformis atcc 10716: Molecular characterization of three multi-modular peptide synthetases. Chem. Biol. 1997, 4, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Khojasteh, S.C.; Su, D. Biosynthetic strategies for macrocyclic peptides. Molecules 2021, 26, 3338. [Google Scholar] [CrossRef]
- Bonjorno, A.F.; Pavan, A.R.; Fernandes, G.F.S.; Scarim, C.B.; Castagnolo, D.; Dos Santos, J.L. Bacprotacs targeting clp protease: A promising strategy for anti-mycobacterial drug discovery. Front. Chem. 2024, 12, 1358539. [Google Scholar] [CrossRef]
- Upert, G.; Luther, A.; Obrecht, D.; Ermert, P. Emerging peptide antibiotics with therapeutic potential. Med. Drug. Discov. 2021, 9, 100078. [Google Scholar] [CrossRef]
- Shi, J.; Chen, C.; Wang, D.; Wang, Z.; Liu, Y. The antimicrobial peptide li14 combats multidrug-resistant bacterial infections. Commun. Biol. 2022, 5, 926. [Google Scholar] [CrossRef]
- Randall, J.R.; Vieira, L.C.; Wilke, C.O.; Davies, B.W. Deep mutational scanning and machine learning for the analysis of antimicrobial-peptide features driving membrane selectivity. Nat. Biomed. Eng. 2024, 8, 842–853. [Google Scholar] [CrossRef]
- Levine, M.Z.; Gregorio, N.E.; Jewett, M.C.; Watts, K.R.; Oza, J.P. Escherichia coli-based cell-free protein synthesis: Protocols for a robust, flexible, and accessible platform technology. J. Vis. Exp. 2019, 144, e58882. [Google Scholar]
- Kwon, Y.C.; Jewett, M.C. High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Sci. Rep. 2015, 5, 8663. [Google Scholar] [CrossRef] [PubMed]
- Jaroentomeechai, T.; Stark, J.C.; Natarajan, A.; Glasscock, C.J.; Yates, L.E.; Hsu, K.J.; Mrksich, M.; Jewett, M.C.; DeLisa, M.P. Single-pot glycoprotein biosynthesis using a cell-free transcription-translation system enriched with glycosylation machinery. Nat. Commun. 2018, 9, 2686. [Google Scholar] [CrossRef] [PubMed]
- Rasor, B.J.; Karim, A.S.; Alper, H.S.; Jewett, M.C. Cell extracts from bacteria and yeast retain metabolic activity after extended storage and repeated thawing. ACS Synth. Biol. 2023, 12, 904–908. [Google Scholar] [CrossRef]
- Didovyk, A.; Tonooka, T.; Tsimring, L.; Hasty, J. Rapid and scalable preparation of bacterial lysates for cell-free gene expression. ACS Synth. Biol. 2017, 6, 2198–2208. [Google Scholar] [CrossRef]
- Shin, J.; Noireaux, V. Efficient cell-free expression with the endogenous E. Coli RNA polymerase and sigma factor 70. J. Biol. Eng. 2010, 4, 8. [Google Scholar] [CrossRef]
- Pardee, K.; Slomovic, S.; Nguyen, P.Q.; Lee, J.W.; Donghia, N.; Burrill, D.; Ferrante, T.; McSorley, F.R.; Furuta, Y.; Vernet, A. Portable, on-demand biomolecular manufacturing. Cell 2016, 167, 248–259.e12. [Google Scholar] [CrossRef] [PubMed]
- Des Soye, B.J.; Davidson, S.R.; Weinstock, M.T.; Gibson, D.G.; Jewett, M.C. Establishing a high-yielding cell-free protein synthesis platform derived from vibrio natriegens. ACS Synth. Biol. 2018, 7, 2245–2255. [Google Scholar] [CrossRef] [PubMed]
- Kelwick, R.; Webb, A.J.; MacDonald, J.T.; Freemont, P.S. Development of a bacillus subtilis cell-free transcription-translation system for prototyping regulatory elements. Metab. Eng. 2016, 38, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Des Soye, B.J.; Gerbasi, V.R.; Thomas, P.M.; Kelleher, N.L.; Jewett, M.C. A highly productive, one-pot cell-free protein synthesis platform based on genomically recoded escherichia coli. Cell Chem. Biol. 2019, 26, 1743–1754.e9. [Google Scholar] [CrossRef] [PubMed]
- Bowie, J.U.; Sherkhanov, S.; Korman, T.P.; Valliere, M.A.; Opgenorth, P.H.; Liu, H. Synthetic biochemistry: The bio-inspired cell-free approach to commodity chemical production. Trends Biotechnol. 2020, 38, 766–778. [Google Scholar] [CrossRef]
- Thoring, L.; Dondapati, S.K.; Stech, M.; Wustenhagen, D.A.; Kubick, S. High-yield production of "difficult-to-express" proteins in a continuous exchange cell-free system based on cho cell lysates. Sci. Rep. 2017, 7, 11710. [Google Scholar] [CrossRef]
- Takahashi, H.; Nozawa, A.; Seki, M.; Shinozaki, K.; Endo, Y.; Sawasaki, T. A simple and high-sensitivity method for analysis of ubiquitination and polyubiquitination based on wheat cell-free protein synthesis. BMC Plant Biol. 2009, 9, 39. [Google Scholar] [CrossRef]
- Ramadan, A.; Nemoto, K.; Seki, M.; Shinozaki, K.; Takeda, H.; Takahashi, H.; Sawasaki, T. Wheat germ-based protein libraries for the functional characterisation of the arabidopsis e2 ubiquitin conjugating enzymes and the ring-type e3 ubiquitin ligase enzymes. BMC Plant Biol. 2015, 15, 275. [Google Scholar] [CrossRef]
- Cheng, Z.; He, B.B.; Lei, K.; Gao, Y.; Shi, Y.; Zhong, Z.; Liu, H.; Liu, R.; Zhang, H.; Wu, S.; et al. Rule-based omics mining reveals antimicrobial macrocyclic peptides against drug-resistant clinical isolates. Nat. Commun. 2024, 15, 4901. [Google Scholar] [CrossRef]
- Cheng, Y.; Qin, J.; Huang, Y.; Wang, T. The antimicrobial effects of plga microspheres containing the antimicrobial peptide op-145 on clinically isolated pathogens in bone infections. Sci. Rep. 2022, 12, 14541. [Google Scholar] [CrossRef]
- Hilpert, K.; Volkmer-Engert, R.; Walter, T.; Hancock, R.E. High-throughput generation of small antibacterial peptides with improved activity. Nat. Biotechnol. 2005, 23, 1008–1012. [Google Scholar] [CrossRef]
- Schoborg, J.A.; Hodgman, C.E.; Anderson, M.J.; Jewett, M.C. Substrate replenishment and byproduct removal improve yeast cell-free protein synthesis. Biotechnol. J. 2014, 9, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, J.; Zheng, Y.; Li, J.; Wang, H.; Zhou, Y.; Qi, M.; Yu, H.; Tang, W.; Zhao, W.M. An optimized yeast cell-free system: Sufficient for translation of human papillomavirus 58 L1 mRNA and assembly of virus-like particles. J. Biosci. Bioeng. 2008, 106, 8–15. [Google Scholar] [CrossRef]
- Madin, K.; Sawasaki, T.; Ogasawara, T.; Endo, Y. A highly efficient and robust cell-free protein synthesis system prepared from wheat embryos: Plants apparently contain a suicide system directed at ribosomes. Proc. Natl. Acad. Sci. USA 2000, 97, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.W.; Majewska, N.I.; Chen, C.X.; Albanetti, T.E.; Jimenez, R.B.C.; Schmelzer, A.E.; Jewett, M.C.; Roy, V. Development of a cho-based cell-free platform for synthesis of active monoclonal antibodies. ACS Synth. Biol. 2017, 6, 1370–1379. [Google Scholar] [CrossRef]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.Q.; Ji, X.; Ba, F.; Zhang, Y.; Xu, H.; Huang, S.; Zheng, X.; Liu, Y.; Ling, S.; Jewett, M.C.; et al. Cell-free biosynthesis and engineering of ribosomally synthesized lanthipeptides. Nat. Commun. 2024, 15, 4336. [Google Scholar] [CrossRef]
- Sin, S.T.; Deng, J.; Ji, L.; Yukawa, M.; Chan, R.W.; Volpi, S.; Vaglio, A.; Fenaroli, P.; Bocca, P.; Cheng, S.H.; et al. Effects of nucleases on cell-free extrachromosomal circular DNA. JCI Insight 2022, 7, e156070. [Google Scholar] [CrossRef]
- Tuckey, C.; Asahara, H.; Zhou, Y.; Chong, S. Protein synthesis using a reconstituted cell-free system. Curr. Protoc. Mol. Biol. 2014, 108, 16.31.1–16.31.22. [Google Scholar] [CrossRef]
- Gabant, P.; Borrero, J. Paragen 1.0: A standardized synthetic gene library for fast cell-free bacteriocin synthesis. Front. Bioeng. Biotechnol. 2019, 7, 213. [Google Scholar] [CrossRef]
- Passioura, T.; Katoh, T.; Goto, Y.; Suga, H. Selection-based discovery of druglike macrocyclic peptides. Annu. Rev. Biochem. 2014, 83, 727–752. [Google Scholar] [CrossRef]
- Lee, J.; Coronado, J.N.; Cho, N.; Lim, J.; Hosford, B.M.; Seo, S.; Kim, D.S.; Kofman, C.; Moore, J.S.; Ellington, A.D. Ribosome-mediated biosynthesis of pyridazinone oligomers in vitro. Nat. Commun. 2022, 13, 6322. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Schwieter, K.E.; Watkins, A.M.; Kim, D.S.; Yu, H.; Schwarz, K.J.; Lim, J.; Coronado, J.; Byrom, M.; Anslyn, E.V. Expanding the limits of the second genetic code with ribozymes. Nat. Commun. 2019, 10, 5097. [Google Scholar] [CrossRef]
- Goto, Y.; Suga, H. The rapid platform for the discovery of pseudo-natural macrocyclic peptides. Acc. Chem. Res. 2021, 54, 3604–3617. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jaitzig, J.; Theuer, L.; Legala, O.E.; Sussmuth, R.D.; Neubauer, P. Type ii thioesterase improves heterologous biosynthesis of valinomycin in escherichia coli. J. Biotechnol. 2015, 193, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Krizsan, A.; Volke, D.; Weinert, S.; Strater, N.; Knappe, D.; Hoffmann, R. Insect-derived proline-rich antimicrobial peptides kill bacteria by inhibiting bacterial protein translation at the 70s ribosome. Angew. Chem. Int. Ed. 2014, 53, 12236–12239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Goto, Y.; Suga, H. Discovery, biochemical characterization, and bioengineering of cyanobactin prenyltransferases. Trends Biochem. Sci. 2023, 48, 360–374. [Google Scholar] [CrossRef]
- Alfi, A.; Popov, A.; Kumar, A.; Zhang, K.Y.J.; Dubiley, S.; Severinov, K.; Tagami, S. Cell-free mutant analysis combined with structure prediction of a lasso peptide biosynthetic protease b2. ACS Synth. Biol. 2022, 11, 2022–2028. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Suga, H. Engineering of ripp pathways for the production of artificial peptides bearing various non-proteinogenic structures. Curr. Opin. Chem. Biol. 2018, 46, 82–90. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, Y.; Zhai, G.; Fu, S.; Xia, Y.; Hu, B.; Cai, X.; Zhang, Y.; Li, Y.; Deng, Z.; et al. A cell-free platform based on nisin biosynthesis for discovering novel lanthipeptides and guiding their overproduction in vivo. Adv. Sci. 2020, 7, 2001616. [Google Scholar] [CrossRef]
- Wu, Y.; Cui, Z.; Huang, Y.H.; de Veer, S.J.; Aralov, A.V.; Guo, Z.; Moradi, S.V.; Hinton, A.O.; Deuis, J.R.; Guo, S.; et al. Towards a generic prototyping approach for therapeutically-relevant peptides and proteins in a cell-free translation system. Nat. Commun. 2022, 13, 260. [Google Scholar] [CrossRef]
- Si, Y.; Kretsch, A.M.; Daigh, L.M.; Burk, M.J.; Mitchell, D.A. Cell-free biosynthesis to evaluate lasso peptide formation and enzyme-substrate tolerance. J. Am. Chem. Soc. 2021, 143, 5917–5927. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Ochiai, A.; Kondo, H.; Fukuda, S.; Ishiyama, Y.; Saitoh, E.; Kato, T.; Tanaka, T. Pyrrhocoricin, a proline-rich antimicrobial peptide derived from insect, inhibits the translation process in the cell-free escherichia coli protein synthesis system. J. Biosci. Bioeng. 2016, 121, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Koller, T.O.; Morici, M.; Berger, M.; Safdari, H.A.; Lele, D.S.; Beckert, B.; Kaur, K.J.; Wilson, D.N. Structural basis for translation inhibition by the glycosylated drosocin peptide. Nat. Chem. Biol. 2023, 19, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.A.; Shimomura, M.; Goto, Y.; Ozaki, T.; Asamizu, S.; Sugai, Y.; Suga, H.; Onaka, H. Minimal lactazole scaffold for in vitro thiopeptide bioengineering. Nat. Commun. 2020, 11, 2272. [Google Scholar] [CrossRef]
- Ozaki, T.; Yamashita, K.; Goto, Y.; Shimomura, M.; Hayashi, S.; Asamizu, S.; Sugai, Y.; Ikeda, H.; Suga, H.; Onaka, H. Dissection of goadsporin biosynthesis by in vitro reconstitution leading to designer analogues expressed in vivo. Nat. Commun. 2017, 8, 14207. [Google Scholar] [CrossRef]
- Vinogradov, A.A.; Shimomura, M.; Kano, N.; Goto, Y.; Onaka, H.; Suga, H. Promiscuous enzymes cooperate at the substrate level en route to lactazole a. J. Am. Chem. Soc. 2020, 142, 13886–13897. [Google Scholar] [CrossRef]
- Goto, Y.; Suga, H. In vitro biosynthesis of peptides containing exotic azoline analogues. ChemBioChem 2020, 21, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Ito, Y.; Kato, Y.; Tsunoda, S.; Suga, H. One-pot synthesis of azoline-containing peptides in a cell-free translation system integrated with a posttranslational cyclodehydratase. Chem. Biol. 2014, 21, 766–774. [Google Scholar] [CrossRef]
- Goto, Y.; Suga, H. A post-translational cyclodehydratase, patd, tolerates sequence variation in the c-terminal region of substrate peptides. Chem. Lett. 2016, 45, 1247–1249. [Google Scholar] [CrossRef]
- Fleming, S.R.; Himes, P.M.; Ghodge, S.V.; Goto, Y.; Suga, H.; Bowers, A.A. Exploring the post-translational enzymology of paaa by mrna display. J. Am. Chem. Soc. 2020, 142, 5024–5028. [Google Scholar] [CrossRef]
- Zhuang, L.; Huang, S.; Liu, W.Q.; Karim, A.S.; Jewett, M.C.; Li, J. Total in vitro biosynthesis of the nonribosomal macrolactone peptide valinomycin. Metab. Eng. 2020, 60, 37–44. [Google Scholar] [CrossRef]
- Li, J.; Zhang, L.; Liu, W. Cell-free synthetic biology for in vitro biosynthesis of pharmaceutical natural products. Synth. Syst. Biotechnol. 2018, 3, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Goering, A.W.; Li, J.; McClure, R.A.; Thomson, R.J.; Jewett, M.C.; Kelleher, N.L. In vitro reconstruction of nonribosomal peptide biosynthesis directly from DNA using cell-free protein synthesis. ACS Synth. Biol. 2017, 6, 39–44. [Google Scholar] [CrossRef]
- Fleming, S.R.; Bartges, T.E.; Vinogradov, A.A.; Kirkpatrick, C.L.; Goto, Y.; Suga, H.; Hicks, L.M.; Bowers, A.A. Flexizyme-enabled benchtop biosynthesis of thiopeptides. J. Am. Chem. Soc. 2019, 141, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Pandi, A.; Adam, D.; Zare, A.; Trinh, V.T.; Schaefer, S.L.; Burt, M.; Klabunde, B.; Bobkova, E.; Kushwaha, M.; Foroughijabbari, Y.; et al. Cell-free biosynthesis combined with deep learning accelerates de novo-development of antimicrobial peptides. Nat. Commun. 2023, 14, 7197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, L.-H.; Xu, B.; Zhang, Z.; Yang, M.; He, Y.; Chen, J.; Zhang, Y.; Hu, Y.; Chen, X. Screening antimicrobial peptides and probiotics using multiple deep learning and directed evolution strategies. Acta Pharm. Sin. B 2024, 14, 3476–3492. [Google Scholar] [CrossRef]
- Nuti, N.; Rottmann, P.; Stucki, A.; Koch, P.; Panke, S.; Dittrich, P.S. A multiplexed cell-free assay to screen for antimicrobial peptides in double emulsion droplets. Angew. Chem. Int. Ed. 2022, 61, e202114632. [Google Scholar] [CrossRef]
- Vinogradov, A.A.; Nagai, E.; Chang, J.S.; Narumi, K.; Onaka, H.; Goto, Y.; Suga, H. Accurate broadcasting of substrate fitness for lactazole biosynthetic pathway from reactivity-profiling mRNA display. J. Am. Chem. Soc. 2020, 142, 20329–20334. [Google Scholar] [CrossRef]
- Vinogradov, A.A.; Chang, J.S.; Onaka, H.; Goto, Y.; Suga, H. Accurate models of substrate preferences of post-translational modification enzymes from a combination of mrna display and deep learning. ACS Cent. Sci. 2022, 8, 814–824. [Google Scholar] [CrossRef]
- Henzler Wildman, K.A.; Lee, D.K.; Ramamoorthy, A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, ll-37. Biochemistry 2003, 42, 6545–6558. [Google Scholar] [CrossRef]
- Jean-Francois, F.; Elezgaray, J.; Berson, P.; Vacher, P.; Dufourc, E.J. Pore formation induced by an antimicrobial peptide: Electrostatic effects. Biophys. J. 2008, 95, 5748–5756. [Google Scholar] [CrossRef]
- Graf, M.; Wilson, D.N. Intracellular antimicrobial peptides targeting the protein synthesis machinery. Adv. Exp. Med. Biol. 2019, 1117, 73–89. [Google Scholar] [PubMed]
- Patil, N.A.; Thombare, V.J.; Li, R.; He, X.; Lu, J.; Yu, H.H.; Wickremasinghe, H.; Pamulapati, K.; Azad, M.A.K.; Velkov, T.; et al. An efficient approach for the design and synthesis of antimicrobial peptide-peptide nucleic acid conjugates. Front. Chem. 2022, 10, 843163. [Google Scholar] [CrossRef]
- Xie, Y.; Wan, H.; Zeng, X.; Zhang, Z.; Wang, Y. Characterization and antimicrobial evaluation of a new spgly-amp, glycine-rich antimicrobial peptide from the mud crab scylla paramamosain. Fish Shellfish Immunol. 2020, 106, 384–392. [Google Scholar] [CrossRef]
- Park, S.C.; Park, Y.; Hahm, K.S. The role of antimicrobial peptides in preventing multidrug-resistant bacterial infections and biofilm formation. Int. J. Mol. Sci. 2011, 12, 5971–5992. [Google Scholar] [CrossRef]
- Cho, J.; Lee, D.G. The antimicrobial peptide arenicin-1 promotes generation of reactive oxygen species and induction of apoptosis. Biochim. Biophys. Acta 2011, 1810, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Cheng, Q.; Yi, L.; Shui Yee Leung, S.; Chen, S.; Chan, K.F.; Wong, K.Y. C-terminal modification of a de novo designed antimicrobial peptide via capping of macrolactam rings. Bioorg. Chem. 2023, 130, 106251. [Google Scholar] [CrossRef]
- Hall, K.; Aguilar, M.I. Surface plasmon resonance spectroscopy for studying the membrane binding of antimicrobial peptides. Methods Mol. Biol. 2010, 627, 213–223. [Google Scholar] [PubMed]
- Uyterhoeven, E.T.; Butler, C.H.; Ko, D.; Elmore, D.E. Investigating the nucleic acid interactions and antimicrobial mechanism of buforin ii. FEBS Lett. 2008, 582, 1715–1718. [Google Scholar] [CrossRef]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef]
- Baek, M.H.; Kamiya, M.; Kushibiki, T.; Nakazumi, T.; Tomisawa, S.; Abe, C.; Kumaki, Y.; Kikukawa, T.; Demura, M.; Kawano, K.; et al. Lipopolysaccharide-bound structure of the antimicrobial peptide cecropin p1 determined by nuclear magnetic resonance spectroscopy. J. Pept. Sci. 2016, 22, 214–221. [Google Scholar] [CrossRef]
- Fernandez, D.I.; Le Brun, A.P.; Whitwell, T.C.; Sani, M.A.; James, M.; Separovic, F. The antimicrobial peptide aurein 1.2 disrupts model membranes via the carpet mechanism. Phys. Chem. Chem. Phys. 2012, 14, 15739–15751. [Google Scholar] [CrossRef]
- Wilson, D.N.; Arenz, S.; Beckmann, R. Translation regulation via nascent polypeptide-mediated ribosome stalling. Curr. Opin. Struct. Biol. 2016, 37, 123–133. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef]
- Gould, P.S.; Bird, H.; Easton, A.J. Translation toeprinting assays using fluorescently labeled primers and capillary electrophoresis. Biotechniques 2005, 38, 397–400. [Google Scholar] [CrossRef]
- Egorova, T.; Sokolova, E.; Shuvalova, E.; Matrosova, V.; Shuvalov, A.; Alkalaeva, E. Fluorescent toeprinting to study the dynamics of ribosomal complexes. Methods 2019, 162–163, 54–59. [Google Scholar] [CrossRef]
- Correia Santos, S.; Bischler, T.; Westermann, A.J.; Vogel, J. Maps integrates regulation of actin-targeting effector stec into the virulence control network of salmonella small rna pint. Cell Rep. 2021, 34, 108722. [Google Scholar] [CrossRef]
- Li, Y.M.; Milne, J.C.; Madison, L.L.; Kolter, R.; Walsh, C.T. From peptide precursors to oxazole and thiazole-containing peptide antibiotics: Microcin b17 synthase. Science 1996, 274, 1188–1193. [Google Scholar] [CrossRef]
- Dunbar, K.L.; Dell, M.; Gude, F.; Hertweck, C. Reconstitution of polythioamide antibiotic backbone formation reveals unusual thiotemplated assembly strategy. Proc. Natl. Acad. Sci. USA 2020, 117, 8850–8858. [Google Scholar] [CrossRef]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef]
- Huang, S.; Liu, Y.; Liu, W.Q.; Neubauer, P.; Li, J. The nonribosomal peptide valinomycin: From discovery to bioactivity and biosynthesis. Microorganisms 2021, 9, 780. [Google Scholar] [CrossRef]
- Li, L.; Li, J.; Yu, X.; Cao, R.; Hong, M.; Xu, Z.; Ren Lu, J.; Wang, Y.; Zhu, H. Antimicrobial peptides fight against pseudomonas aeruginosa at a sub-inhibitory concentration via anti-qs pathway. Bioorg. Chem. 2023, 141, 106922. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Wang, L.; Yang, C.; Yan, R.; Zhang, P.; Jin, M.; Du, H.; Wang, Y. Specifically targeted antimicrobial peptides synergize with bacterial-entrapping peptide against systemic mrsa infections. J. Adv. Res. 2024. [Google Scholar] [CrossRef] [PubMed]
- Dhople, V.; Krukemeyer, A.; Ramamoorthy, A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim. Biophys. Acta 2006, 1758, 1499–1512. [Google Scholar] [CrossRef]
- Zimmermann, M.; Hegemann, J.D.; Xie, X.; Marahiel, M.A. The astexin-1 lasso peptides: Biosynthesis, stability, and structural studies. Chem. Biol. 2013, 20, 558–569. [Google Scholar] [CrossRef]
- Bagley, M.C.; Dale, J.W.; Merritt, E.A.; Xiong, X. Thiopeptide antibiotics. Chem. Rev. 2005, 105, 685–714. [Google Scholar] [CrossRef]
- Tan, S.; Ludwig, K.C.; Muller, A.; Schneider, T.; Nodwell, J.R. The lasso peptide siamycin-i targets lipid ii at the gram-positive cell surface. ACS Chem. Biol. 2019, 14, 966–974. [Google Scholar] [CrossRef]
- Cao, L.; Do, T.; Link, A.J. Mechanisms of action of ribosomally synthesized and posttranslationally modified peptides (ripps). J. Ind. Microbiol. Biotechnol. 2021, 48, kuab005. [Google Scholar] [CrossRef]
- Cheng, C.; Hua, Z.C. Lasso peptides: Heterologous production and potential medical application. Front. Bioeng. Biotechnol. 2020, 8, 571165. [Google Scholar] [CrossRef] [PubMed]
- Digal, L.; Samson, S.C.; Stevens, M.A.; Ghorai, A.; Kim, H.; Mifflin, M.C.; Carney, K.R.; Williamson, D.L.; Um, S.; Nagy, G.; et al. Nonthreaded isomers of sungsanpin and ulleungdin lasso peptides inhibit h1299 cancer cell migration. ACS Chem. Biol. 2024, 19, 81–88. [Google Scholar] [CrossRef]
- Mathavan, I.; Zirah, S.; Mehmood, S.; Choudhury, H.G.; Goulard, C.; Li, Y.; Robinson, C.V.; Rebuffat, S.; Beis, K. Structural basis for hijacking siderophore receptors by antimicrobial lasso peptides. Nat. Chem. Biol. 2014, 10, 340–342. [Google Scholar] [CrossRef]
- Martin-Gomez, H.; Tulla-Puche, J. Lasso peptides: Chemical approaches and structural elucidation. Org. Biomol. Chem. 2018, 16, 5065–5080. [Google Scholar] [CrossRef] [PubMed]
- Maksimov, M.O.; Pan, S.J.; James Link, A. Lasso peptides: Structure, function, biosynthesis, and engineering. Nat. Prod. Rep. 2012, 29, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Syroegin, E.A.; Flemmich, L.; Klepacki, D.; Vazquez-Laslop, N.; Micura, R.; Polikanov, Y.S. Structural basis for the context-specific action of the classic peptidyl transferase inhibitor chloramphenicol. Nat. Struct. Mol. Biol. 2022, 29, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef]
- Ma, B.; Fang, C.; Lu, L.; Wang, M.; Xue, X.; Zhou, Y.; Li, M.; Hu, Y.; Luo, X.; Hou, Z. The antimicrobial peptide thanatin disrupts the bacterial outer membrane and inactivates the ndm-1 metallo-beta-lactamase. Nat. Commun. 2019, 10, 3517. [Google Scholar] [CrossRef]
- Mhlongo, J.T.; Waddad, A.Y.; Albericio, F.; de la Torre, B.G. Antimicrobial peptide synergies for fighting infectious diseases. Adv. Sci. 2023, 10, e2300472. [Google Scholar] [CrossRef]
- Krizsan, A.; Prahl, C.; Goldbach, T.; Knappe, D.; Hoffmann, R. Short proline-rich antimicrobial peptides inhibit either the bacterial 70s ribosome or the assembly of its large 50s subunit. ChemBioChem 2015, 16, 2304–2308. [Google Scholar] [CrossRef]
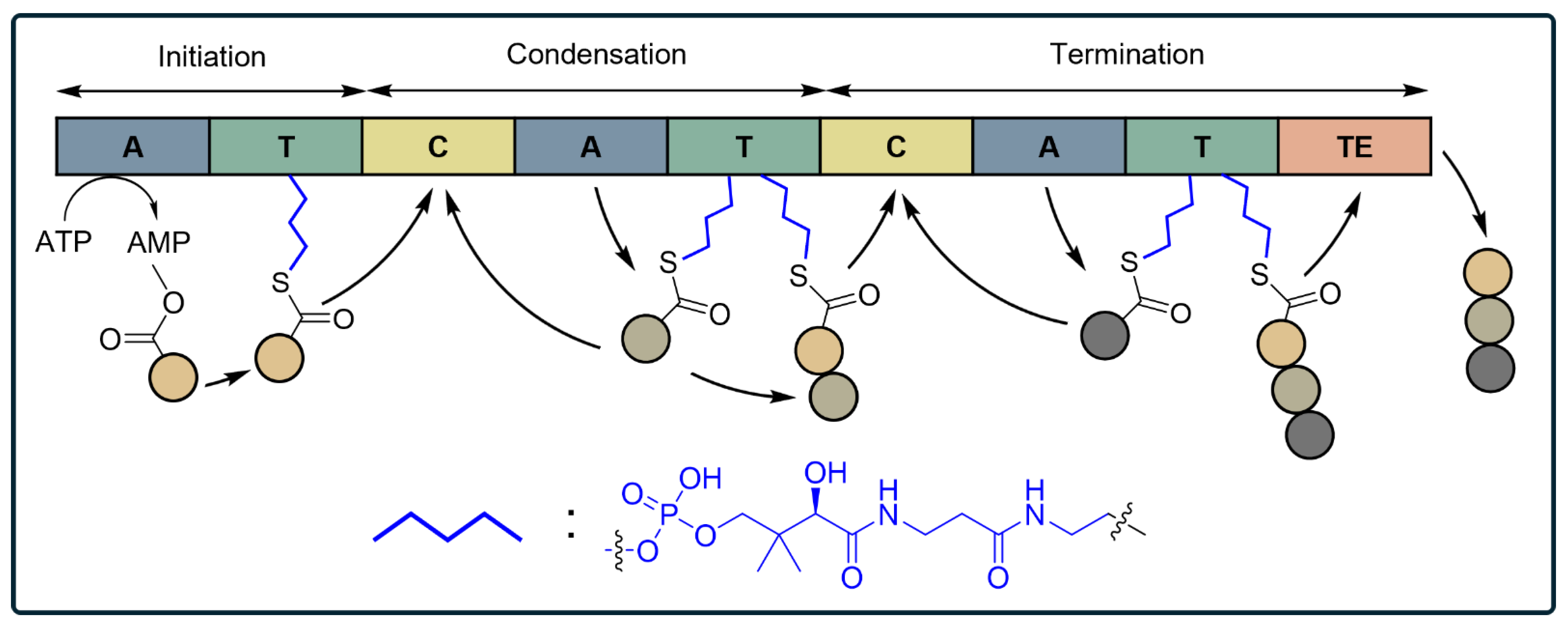
| Type | Peptide | Proposed Antibacterial Mechanisms | Target Gram-Negative Pathogens and MIC | Target Gram-Positive Pathogens and MIC | MIC for MDR |
|---|---|---|---|---|---|
| Peptides | Magainin II [19,20] |
|
|
|
|
| Melittin [21,22,23] |
|
|
|
| |
| LL-37 [24,25,26] |
|
|
|
| |
| Buforin II [20,27] |
|
|
|
| |
| Cecropin P1 [20] |
|
|
|
| |
| Indolicidin [20] |
|
|
|
| |
| PR-39 [23,28] |
|
|
|
| |
| Vancomycin [29,30] |
| - |
|
| |
| Daptomycin [23,31] |
| - |
|
| |
| Polymyxin [32,33] |
|
|
|
| |
| Small molecules | Ciprofloxacin [27,34] |
|
|
|
|
| Gentamicin [27,35] |
|
|
|
| |
| Tetracycline [36,37] |
|
|
|
|
| Types of PTMs | PTM Reactions | PBAs | ||
|---|---|---|---|---|
| Cyclization | Bridging reactions | Thioether formation |
|
|
| Disulfide bond formation (Cysteine oxidation) |
|
| ||
| Head-to-tail cyclization |
|
| ||
| Cyclodehydration | Heterocycle formation |
|
| |
| Alkylation | N-methylation |
|
| |
| Glycosylation | N-Glycosylation |
| ||
|
| |||
| S-Glycosylation |
| |||
| O-glycosylation |
|
| ||
| Mechanisms (Section 4.1) | Biosynthetic Pathways (Section 4.2) | Selections (Section 4.3) | Synthesis (Section 4.4) | |
|---|---|---|---|---|
| Methods | Mutagenesis of PBAs [148] | Identification of biosynthetic pathways [149,150,151] | Screening of multiple variants [139,152] | Inducing proper folding [153,154] |
| Qualitative and quantitative analysis on biomolecules [16,63,155,156] | Enzyme promiscuity for substrates [157,158,159,160,161,162,163] | Optimization of reaction conditions [164,165,166] | Chemo-enzymatic modification [167] | |
| Kinetic analysis of enzymes [152] | Integration of deep generative models [168,169] | |||
| Generation of diverse environments through microfluidic system [170] | ||||
| Target peptide selection using mRNA displays [171,172] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.; Jin, H.; Kim, D.; Lee, J. Cell-Free Systems: Ideal Platforms for Accelerating the Discovery and Production of Peptide-Based Antibiotics. Int. J. Mol. Sci. 2024, 25, 9109. https://doi.org/10.3390/ijms25169109
Park H, Jin H, Kim D, Lee J. Cell-Free Systems: Ideal Platforms for Accelerating the Discovery and Production of Peptide-Based Antibiotics. International Journal of Molecular Sciences. 2024; 25(16):9109. https://doi.org/10.3390/ijms25169109
Chicago/Turabian StylePark, Hyeongwoo, Haneul Jin, Dayeong Kim, and Joongoo Lee. 2024. "Cell-Free Systems: Ideal Platforms for Accelerating the Discovery and Production of Peptide-Based Antibiotics" International Journal of Molecular Sciences 25, no. 16: 9109. https://doi.org/10.3390/ijms25169109