
Applications of the Cellular Thermal Shift Assay to Drug Discovery in Natural Products: A Review


Abstract
1. Introduction
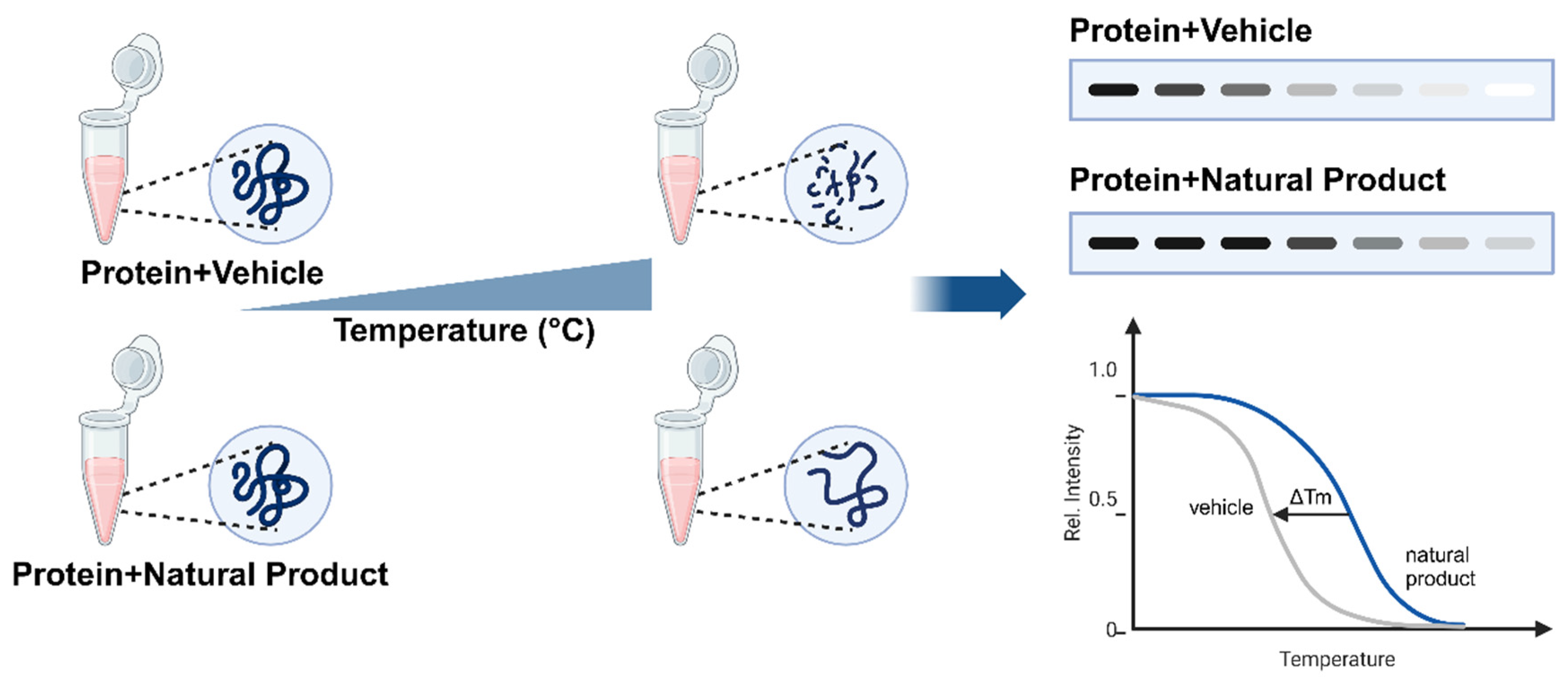
2. Overview
3. CETSA-Based Strategies and Their Applications
3.1. Target Binding
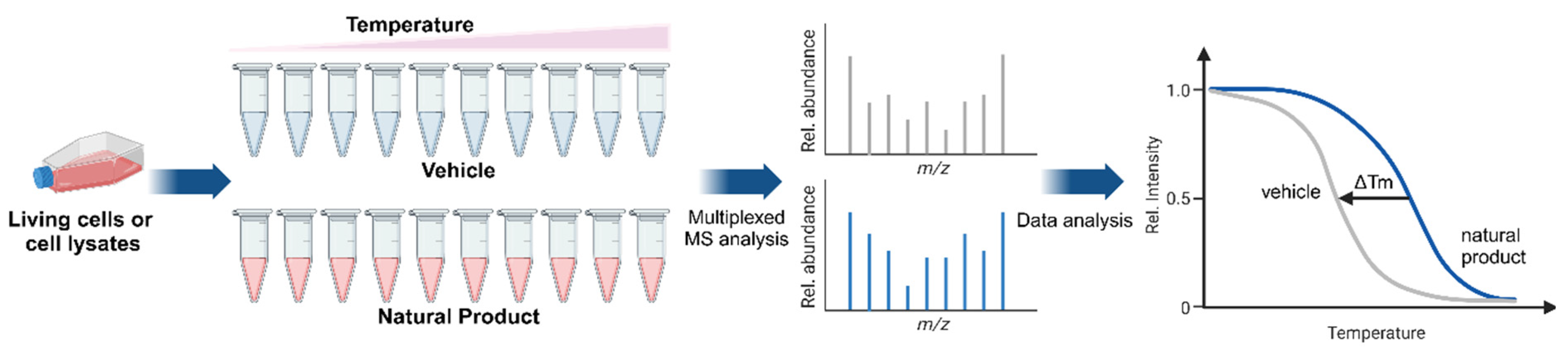
3.2. Drug–Target Engagement
3.3. Comparative Analysis of CETSA Applications: Natural Products vs. Synthetic Compounds
3.4. Integration of Multiple Techniques
3.5. Focus on High-Throughput CETSA
4. Advances in CETSA: From Tissue Applications to Multiomic Integrations
5. Challenges and Future Improvements
Funding
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.; Park, H.; Park, J.; Park, S.B. Recent advances in identifying protein targets in drug discovery. Cell Chem. Biol. 2021, 28, 394–423. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Shang, L.; Burgett, A.W.; Harran, P.G.; Wang, X. Diazonamide toxins reveal an unexpected function for ornithine delta-amino transferase in mitotic cell division. Proc. Natl. Acad. Sci. USA 2007, 104, 2068–2073. [Google Scholar] [CrossRef]
- Harding, M.W.; Galat, A.; Uehling, D.E.; Schreiber, S.L. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature 1989, 341, 758–760. [Google Scholar] [CrossRef]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef]
- Sato, S.; Murata, A.; Orihara, T.; Shirakawa, T.; Suenaga, K.; Kigoshi, H.; Uesugi, M. Marine natural product aurilide activates the OPA1-mediated apoptosis by binding to prohibitin. Chem. Biol. 2011, 18, 131–139. [Google Scholar] [CrossRef]
- Drahl, C.; Cravatt, B.F.; Sorensen, E.J. Protein-reactive natural products. Angew. Chem. Int. Ed. Engl. 2005, 44, 5788–5809. [Google Scholar] [CrossRef]
- Chang, J.; Kim, Y.; Kwon, H.J. Advances in identification and validation of protein targets of natural products without chemical modification. Nat. Prod. Rep. 2016, 33, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Tolvanen, T.A. Current Advances in CETSA. Front. Mol. Biosci. 2022, 9, 866764. [Google Scholar] [CrossRef]
- Martinez Molina, D.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef]
- Franken, H.; Mathieson, T.; Childs, D.; Sweetman, G.M.; Werner, T.; Tögel, I.; Doce, C.; Gade, S.; Bantscheff, M.; Drewes, G.; et al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 2015, 10, 1567–1593. [Google Scholar] [CrossRef] [PubMed]
- Martinez Molina, D.; Nordlund, P. The Cellular Thermal Shift Assay: A Novel Biophysical Assay for In Situ Drug Target Engagement and Mechanistic Biomarker Studies. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundbäck, T.; Nordlund, P.; Martinez Molina, D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. [Google Scholar] [CrossRef]
- Savitski, M.M.; Reinhard, F.B.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Martinez Molina, D.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Tan, L.; Tao, H.; Li, Y.; Liu, H. CETSA and thermal proteome profiling strategies for target identification and drug discovery of natural products. Phytomedicine 2023, 116, 154862. [Google Scholar] [CrossRef]
- Dai, L.; Li, Z.; Chen, D.; Jia, L.; Guo, J.; Zhao, T.; Nordlund, P. Target identification and validation of natural products with label-free methodology: A critical review from 2005 to 2020. Pharmacol. Ther. 2020, 216, 107690. [Google Scholar] [CrossRef]
- Wang, J.; Wu, J.; Li, X.; Liu, H.; Qin, J.; Bai, Z.; Chi, B.; Chen, X. Identification and validation nucleolin as a target of curcumol in nasopharyngeal carcinoma cells. J. Proteom. 2018, 182, 1–11. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, H.; Bi, X.; Hou, G.; Liu, A.; Zhao, Y.; Wang, G.; Cao, X. Src acts as the target of matrine to inhibit the proliferation of cancer cells by regulating phosphorylation signaling pathways. Cell Death Dis. 2021, 12, 931. [Google Scholar] [CrossRef]
- Liao, L.X.; Song, X.M.; Wang, L.C.; Lv, H.N.; Chen, J.F.; Liu, D.; Fu, G.; Zhao, M.B.; Jiang, Y.; Zeng, K.W.; et al. Highly selective inhibition of IMPDH2 provides the basis of antineuroinflammation therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E5986–E5994. [Google Scholar] [CrossRef]
- Li, G.; Boyle, J.W.; Ko, C.N.; Zeng, W.; Wong, V.K.W.; Wan, J.B.; Chan, P.W.H.; Ma, D.L.; Leung, C.H. Aurone derivatives as Vps34 inhibitors that modulate autophagy. Acta Pharm. Sin. B 2019, 9, 537–544. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Kim, Y.H.; Lee, Y.J.; Choi, J.; Kim, C.H.; Han, D.C.; Kwon, B.M. 2′-Hydroxycinnamaldehyde inhibits proliferation and induces apoptosis via signal transducer and activator of transcription 3 inactivation and reactive oxygen species generation. Cancer Sci. 2019, 110, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Vasaturo, M.; Cotugno, R.; Fiengo, L.; Vinegoni, C.; Dal Piaz, F.; De Tommasi, N. The anti-tumor diterpene oridonin is a direct inhibitor of Nucleolin in cancer cells. Sci. Rep. 2018, 8, 16735. [Google Scholar] [CrossRef] [PubMed]
- Dziekan, J.M.; Yu, H.; Chen, D.; Dai, L.; Wirjanata, G.; Larsson, A.; Prabhu, N.; Sobota, R.M.; Bozdech, Z.; Nordlund, P. Identifying purine nucleoside phosphorylase as the target of quinine using cellular thermal shift assay. Sci. Transl. Med. 2019, 11, eaau3174. [Google Scholar] [CrossRef]
- Kirsch, V.C.; Orgler, C.; Braig, S.; Jeremias, I.; Auerbach, D.; Müller, R.; Vollmar, A.M.; Sieber, S.A. The Cytotoxic Natural Product Vioprolide A Targets Nucleolar Protein 14, Which Is Essential for Ribosome Biogenesis. Angew. Chem. Int. Ed. Engl. 2020, 59, 1595–1600. [Google Scholar] [CrossRef]
- Xu, H.; Van der Jeught, K.; Zhou, Z.; Zhang, L.; Yu, T.; Sun, Y.; Li, Y.; Wan, C.; So, K.M.; Liu, D.; et al. Atractylenolide I enhances responsiveness to immune checkpoint blockade therapy by activating tumor antigen presentation. J. Clin. Investig. 2021, 131, e146832. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Li, S.; Liu, J.; Zhang, C.; Jian, C.; Wang, L.; Zhang, Y.; Shi, C. Natural Product Alantolactone Targeting AKR1C1 Suppresses Cell Proliferation and Metastasis in Non-Small-Cell Lung Cancer. Front. Pharmacol. 2022, 13, 847906. [Google Scholar] [CrossRef]
- Lv, C.; Huang, Y.; Wang, Q.; Wang, C.; Hu, H.; Zhang, H.; Lu, D.; Jiang, H.; Shen, R.; Zhang, W.; et al. Ainsliadimer A induces ROS-mediated apoptosis in colorectal cancer cells via directly targeting peroxiredoxin 1 and 2. Cell Chem. Biol. 2023, 30, 295–307.e5. [Google Scholar] [CrossRef]
- Islam, A.; Su, A.J.; Zeng, Z.M.; Chueh, P.J.; Lin, M.H. Capsaicin Targets tNOX (ENOX2) to Inhibit G1 Cyclin/CDK Complex, as Assessed by the Cellular Thermal Shift Assay (CETSA). Cells 2019, 8, 1275. [Google Scholar] [CrossRef]
- Almqvist, H.; Axelsson, H.; Jafari, R.; Dan, C.; Mateus, A.; Haraldsson, M.; Larsson, A.; Martinez Molina, D.; Artursson, P.; Lundbäck, T.; et al. CETSA screening identifies known and novel thymidylate synthase inhibitors and slow intracellular activation of 5-fluorouracil. Nat. Commun. 2016, 7, 11040. [Google Scholar] [CrossRef]
- Cui, Z.; Li, C.; Chen, P.; Yang, H. An update of label-free protein target identification methods for natural active products. Theranostics 2022, 12, 1829–1854. [Google Scholar] [CrossRef]
- Wu, Q.; Zheng, J.; Sui, X.; Fu, C.; Cui, X.; Liao, B.; Ji, H.; Luo, Y.; He, A.; Lu, X.; et al. High-throughput drug target discovery using a fully automated proteomics sample preparation platform. Chem. Sci. 2024, 15, 2833–2847. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.J.; Holbert, M.A.; Simeonov, A.; Kallal, L.A. High-Throughput Cellular Thermal Shift Assays in Research and Drug Discovery. SLAS Discov. 2020, 25, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Dale, I.; Hemsley, P.; Leach, L.; Dekki, N.; Orme, J.P.; Talbot, V.; Narvaez, A.J.; Bista, M.; Martinez Molina, D.; et al. Positioning High-Throughput CETSA in Early Drug Discovery through Screening against B-Raf and PARP1. SLAS Discov. 2019, 24, 121–132. [Google Scholar] [CrossRef]
- Martinez, N.J.; Asawa, R.R.; Cyr, M.G.; Zakharov, A.; Urban, D.J.; Roth, J.S.; Wallgren, E.; Klumpp-Thomas, C.; Coussens, N.P.; Rai, G.; et al. A widely-applicable high-throughput cellular thermal shift assay (CETSA) using split Nano Luciferase. Sci. Rep. 2018, 8, 9472. [Google Scholar] [CrossRef] [PubMed]
- Dart, M.L.; Machleidt, T.; Jost, E.; Schwinn, M.K.; Robers, M.B.; Shi, C.; Kirkland, T.A.; Killoran, M.P.; Wilkinson, J.M.; Hartnett, J.R.; et al. Homogeneous Assay for Target Engagement Utilizing Bioluminescent Thermal Shift. ACS Med. Chem. Lett. 2018, 9, 546–551. [Google Scholar] [CrossRef]
- McNulty, D.E.; Bonnette, W.G.; Qi, H.; Wang, L.; Ho, T.F.; Waszkiewicz, A.; Kallal, L.A.; Nagarajan, R.P.; Stern, M.; Quinn, A.M.; et al. A High-Throughput Dose-Response Cellular Thermal Shift Assay for Rapid Screening of Drug Target Engagement in Living Cells, Exemplified Using SMYD3 and IDO1. SLAS Discov. 2018, 23, 34–46. [Google Scholar] [CrossRef]
- Hendricks, J.A.; Beaton, N.; Chernobrovkin, A.; Miele, E.; Hamza, G.M.; Ricchiuto, P.; Tomlinson, R.C.; Friman, T.; Borenstain, C.; Barlaam, B.; et al. Mechanistic Insights into a CDK9 Inhibitor Via Orthogonal Proteomics Methods. ACS Chem. Biol. 2022, 17, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Zhang, Y.; Hellner, J.; Giannini, C.; Xu, X.; Pauwels, J.; Ma, Q.; Dejonghe, W.; Han, H.; Van de Cotte, B.; et al. Proteome-wide cellular thermal shift assay reveals unexpected cross-talk between brassinosteroid and auxin signaling. Proc. Natl. Acad. Sci. USA 2022, 119, e2118220119. [Google Scholar] [CrossRef] [PubMed]
- Al-Amin, R.A.; Gallant, C.J.; Muthelo, P.M.; Landegren, U. Sensitive Measurement of Drug-Target Engagement by a Cellular Thermal Shift Assay with Multiplex Proximity Extension Readout. Anal. Chem. 2021, 93, 10999–11009. [Google Scholar] [CrossRef]
- Sun, S.; Zheng, Z.; Wang, J.; Li, F.; He, A.; Lai, K.; Zhang, S.; Lu, J.H.; Tian, R.; Tan, C.S.H. Improved in situ characterization of protein complex dynamics at scale with thermal proximity co-aggregation. Nat. Commun. 2023, 14, 7697. [Google Scholar] [CrossRef]
- Sanchez, T.W.; Ronzetti, M.H.; Owens, A.E.; Antony, M.; Voss, T.; Wallgren, E.; Talley, D.; Balakrishnan, K.; Leyes Porello, S.E.; Rai, G.; et al. Real-Time Cellular Thermal Shift Assay to Monitor Target Engagement. ACS Chem. Biol. 2022, 17, 2471–2482. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, T.W.; Owens, A.; Martinez, N.J.; Wallgren, E.; Simeonov, A.; Henderson, M.J. High-Throughput Detection of Ligand-Protein Binding Using a SplitLuc Cellular Thermal Shift Assay. Methods Mol. Biol. 2021, 2365, 21–41. [Google Scholar] [PubMed]
- Friman, T. Mass spectrometry-based Cellular Thermal Shift Assay (CETSA®) for target deconvolution in phenotypic drug discovery. Bioorg Med. Chem. 2020, 28, 115174. [Google Scholar] [CrossRef] [PubMed]
- Mateus, A.; Kurzawa, N.; Becher, I.; Sridharan, S.; Helm, D.; Stein, F.; Typas, A.; Savitski, M.M. Thermal proteome profiling for interrogating protein interactions. Mol. Syst. Biol. 2020, 16, e9232. [Google Scholar] [CrossRef]
- Tan, C.S.H.; Go, K.D.; Bisteau, X.; Dai, L.; Yong, C.H.; Prabhu, N.; Ozturk, M.B.; Lim, Y.T.; Sreekumar, L.; Lengqvist, J.; et al. Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science 2018, 359, 1170–1177. [Google Scholar] [CrossRef]
- Mateus, A.; Kurzawa, N.; Perrin, J.; Bergamini, G.; Savitski, M.M. Drug Target Identification in Tissues by Thermal Proteome Profiling. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 465–482. [Google Scholar] [CrossRef]
- Lyu, J.; Wang, K.; Ye, M. Modification-free approaches to screen drug targets at proteome level. TrAC Trends Anal. Chem. 2020, 124, 115574. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Method | Sensitivity | Throughput | Application Scope | Advantages | Limitations |
|---|---|---|---|---|---|
| CETSA | High (thermal stabilization) | Medium-(Western blot)-to-High (SplitLuc/HTS) | Physiological conditions (intact cells), target engagement, off-target effects, drug resistance analysis | Operates in native cellular environments; detects membrane proteins | Requires protein-specific antibodies for WB; limited to soluble proteins in HTS formats |
| DARTS | Moderate (protease-dependent) | Low-to-Medium | Cell lysates/purified proteins, novel target discovery, validation of known targets | Label-free; no compound modification; cost-effective | Sensitivity depends on protease choice; challenges with low-abundance targets |
| SPROX | High (domain-level stability shifts) | Medium-to-High (OnePot 2D) | Lysates, weak binders, domain-specific interaction analysis | Provides binding site information via methionine oxidation | Limited to methionine-containing peptides; requires MS expertise |
| Affinity-Based | High (if reagents are available) | Low | Purified proteins/lysates, validated target analysis | High specificity; compatible with MS or fluorescence | Requires compound modification (e.g., biotinization); may alter binding properties |
| Natural Product | Source | Molecular Target | Study Objective | Key Finding |
|---|---|---|---|---|
| Curcumol [17] | Curcuma wenyujin | Nucleolin (NCL) | Validate anticancer mechanism in nasopharyngeal carcinoma | Induces apoptosis by inhibiting NCL-mediated ribosome biogenesis |
| Matrine [18] | Sophora flavescens | Src kinase | Investigate anti-proliferative effects in cancer cells | Inhibits Src phosphorylation, suppressing tumor growth |
| Rapanone A [19] | Ardisia japonica | IMPDH2 | Identify anti-neuroinflammatory targets | Selective inhibition of IMPDH2 reduces neuroinflammation |
| Aurone derivative 1a [20] | Synthetic (inspired by natural aurones) | Vps34 (Class III PI3K) | Screen autophagy modulators | Stabilizes Vps34, enhancing autophagic flux |
| 2′-Hydroxycinnamaldehyde [21] | Cinnamomum cassia | STAT3 | Uncover STAT3 inhibition in cancer | Direct binding inhibits STAT3 phosphorylation and downstream signaling |
| Oridonin [22] | Rabdosia rubescens | Nucleolin | Study anticancer effects in leukemia | Disrupts nucleolin-RNA interactions, inducing apoptosis |
| Staurosporine [14] | Streptomyces staurosporeus | Pan-kinase (51 kinases) | Profile kinase inhibitor activity | Broad-spectrum kinase inhibition confirmed via proteome-wide thermal shifts |
| Quinine [23] | Cinchona tree bark | Plasmodium falciparum PNP (PfPNP) | Elucidate antimalarial mechanism | Induces conformational changes in PfPNP, blocking purine salvage in malaria |
| Vioprolide A (VioA) [24] | Cystobacter violaceus | Nucleolar protein 14 (NOP14) | Identify anticancer targets in ribosome biogenesis | Disrupts NOP14-NEP1 interaction, halting ribosome assembly |
| Atractylenolide I [25] | Atractylodes macrocephala | PSMD4 (Proteasome subunit) | Study immunomodulatory effects in colorectal cancer | Enhances immunoproteasome activity, boosting antigen presentation and antitumor immunity |
| Alantolactone [26] | Inula helenium | AKR1C1 | Investigate anticancer activity in non-small-cell lung cancer | Inhibits AKR1C1, suppressing metastasis and proliferation |
| Ainsliadimer A (AIN) [27] | Ainsliaea macrocephala | PRDX1/PRDX2 | Uncover ROS-mediated apoptosis mechanism in colorectal cancer | Covalently binds to PRDX1/2 cysteine residues, inhibiting peroxidase activity and inducing oxidative stress |
| Capsaicin [28] | Capsicum annuum | tNOX (ENOX2) | Validate anticancer mechanism in bladder cancer | Binds tNOX, induces proteasomal degradation, and suppresses SIRT1-mediated G1 cyclin/CDK activation |
| Method | Detection | Scope | Throughput | Application |
|---|---|---|---|---|
| WB-CETSA | Western Blot | Target-specific | Low | Target validation |
| ITDR-CETSA | Western Blot | Target-specific, dose-dependent | Low | Binding affinity assessment |
| MS-CETSA (TPP) | Mass Spectrometry | Proteome-wide | High | Target discovery |
| 2D-TPP | Mass Spectrometry | Proteome-wide, multidimensional | High | Comprehensive interaction profiling |
| HT-CETSA | Immunoassay/Reporter System | High-throughput target screening | Very High | High-throughput drug screening and target identification |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J. Applications of the Cellular Thermal Shift Assay to Drug Discovery in Natural Products: A Review. Int. J. Mol. Sci. 2025, 26, 3940. https://doi.org/10.3390/ijms26093940
Song J. Applications of the Cellular Thermal Shift Assay to Drug Discovery in Natural Products: A Review. International Journal of Molecular Sciences. 2025; 26(9):3940. https://doi.org/10.3390/ijms26093940
Chicago/Turabian StyleSong, Jayoung. 2025. "Applications of the Cellular Thermal Shift Assay to Drug Discovery in Natural Products: A Review" International Journal of Molecular Sciences 26, no. 9: 3940. https://doi.org/10.3390/ijms26093940
APA StyleSong, J. (2025). Applications of the Cellular Thermal Shift Assay to Drug Discovery in Natural Products: A Review. International Journal of Molecular Sciences, 26(9), 3940. https://doi.org/10.3390/ijms26093940