
Comparative Fitting of Mathematical Models to Carvedilol Release Profiles Obtained from Hypromellose Matrix Tablets
Abstract
:1. Introduction
- -
- Representation of drug release profiles;
- -
- Prediction of drug release;
- -
- Assessment of the fraction of drug released in time points, in which experimental data were not obtained (this is, for example, useful for estimating times at which 25%, 50%, 75%, 90%, etc. of the drug is released from a DDS);
- -
- As a quantitative drug profile analysis tool in designing and evaluating DDSs and formulations or studying drug release kinetics;
- -
- Comparison of drug release profiles;
- -
- Model-dependent analysis of drug release mechanism from DDSs (for example, when the Korsmeyer–Peppas or the Peppas–Sahlin model is applied to the first 60% of the drug release profile and the interpretation of the drug release mechanism is interpreted via the fitted model parameters).
2. Materials and Methods
2.1. Materials
- Water-soluble fillers
- ○
- Two grades of polyethylene glycol/PEG (Polyglykol® 4000 P, Polyglykol® 8000 P);
- ○
- Polyethylene oxide/PEO (Polyoxᵀᴹ WSR N-80);
- ○
- Two grades of povidone (Kollidon® 25, Kollidon® 90 F);
- ○
- Four grades of mannitol (C*Pharm Mannidex 16700, Pearlitol® 160C, Parteck® M 100, Parteck® M 200);
- ○
- Five grades of lactose monohydrate (Lactochem® Crystals, Lactochem® Fine Powder, SuperTab® 11SD, FlowLac® 100, Tablettose® 70);
- ○
- Sucrose (Granulated sugar N°1 600);
- ○
- Maltodextrin (Glucidex® 19).
- Water-insoluble fillers
- ○
- Two grades of anhydrous dibasic calcium phosphate/DCP (Di-Cafos® A12, Emcompress® Anhydrous);
- ○
- Two grades of microcrystalline cellulose/MCC (Avicel® PH-102, Avicel® PH-200);
- ○
- Ethylcellulose/EC (Ethocelᵀᴹ Standard 20 Premium);
- ○
- Two samples of pregelatinized starch of the same grade but different particle sizes (Starch 1500® sample with smaller particle size (↓PS), Starch 1500® sample with larger particle size (↑PS)).
2.2. Methods
2.2.1. Compression Mixtures and Tablet Preparation
2.2.2. Carvedilol Release Profiles
2.2.3. Determination of the Approximate End-Point of Carvedilol Release Using a Paired t-Test
2.2.4. Fitting of Mathematical Models to Carvedilol Release Data Using DDSolver and Overall Comparison of Model Fit
2.2.5. Selection of Best-Performing Models
- (1)
- The RSS rankingsThe RSS was used to initially rank the models, from the one which fitted the experimental dissolution data best, to the worst performing one within each formulation.
- (2)
- Criteria for visual assessment of model fitSeveral criteria were used for the visual assessment of model fit and the selection of better-suited models from less-suited ones was based on the interpretation of the fitted model parameters describing lag time or burst release in relation to experimental observations. These criteria were used on top of the initial RSS rankings to select the best-performing mathematical models from the pool of tested models. The mentioned criteria used were as follows (also see ‘Criteria for visual assessment of model fit’ in attachments, i.e., Supplementary Materials for examples):
- Progression of drug releaseA mathematical model which predicts a maximum of drug released before the last experimentally tested time point in the data set and afterwards demonstrates a significantly lower fraction of drug released in further successive time points is inferior to a model with a similar RSS result which predicts a progressively higher fraction of drug released throughout the drug release profile from t = 0 to t = max in the studied dissolution data range.
- The ability of the mathematical model to reproduce the sigmoid shape of a drug release profile from the experimental dissolution dataIf the experimental dissolution data clearly demonstrate the sigmoid shape of a dissolution profile, a mathematical model which is capable of reproducing/following this sigmoid shape is superior to a model with a similar RSS result which is not able to demonstrate a sigmoid shape.
- Matching indications of burst release or lag time (mathematical model vs. experimental dissolution data)If the experimental dissolution data indicate a possible burst release or lag time, this should be matched by the fitted mathematical model’s indication of the same two phenomena. The model which fails to do so is considered inferior to the model which matches the experimental dissolution data up to t = 60 min more accurately, where both phenomena can be observed.
- Uniformity of model fitIf two mathematical models demonstrate a similar overall fit to experimental dissolution data, i.e., RSS result, and the first one demonstrates a more uniform fit throughout the entire drug release profile than the second one (for example, the second model demonstrates a similar fit to the first one throughout the majority of the drug release profile except at the beginning, etc.), the model which demonstrates a more uniform fit throughout the entire drug release profile is considered superior.
- (3)
- Choosing representative models from model groupsWhenever possible, a single mathematical model was selected as a representative model from a model group, unless there was no clear indication of which model in a certain model group was better suited than the other one(s) based on the interpretation of fitted model parameters describing lag time or burst release in relation to experimental dissolution results or based on the visual examination of model fit. Therefore, if the experimental results showed no clear indication of burst release or lag time, a model with a negligible low absolute value of Tlag or F0 was not penalized in relation to a model without a Tlag or F0 term in it, and could still be selected as a candidate for predicting/representing carvedilol release. In the Peppas–Sahlin model group, the Peppas–Sahlin_1 and the Peppas–Sahlin_1 with Tlag models with an m value of 0.45 or lower were preferred over the Peppas–Sahlin_2 and the Peppas–Sahlin_2 with Tlag models, as the latter case is a less appropriate option for the produced tablets according to their aspect ratio, that is, the ratio between tablet diameter and tablet thickness (see Section 2.2.6 for explanation).
- -
- Green colour, bolded: the best-performing mathematical model(s) chosen according to the above criteria, i.e., the primary-choice or the first-choice model(s).
- -
- Light green colour, bolded: the near best-performing mathematical model, i.e., the near primary-choice or near first-choice model, whose performance is slightly worse but very close to the best-performing one, and is at the same time significantly better than the secondary-choice/second-choice model’s performance.
- -
- Light gold colour, bolded: secondary-choice/second-choice model, whose performance is significantly worse than the best-performing one, but is still clearly good enough to make the model practically usable for predicting carvedilol release with significant accuracy in comparison to other tested models.
- -
- Orange colour, bolded: tertiary-choice/third-choice model, whose performance is significantly worse than the secondary-choice/second-choice one, but still clearly good enough to make the model practically usable for predicting carvedilol release with reasonable accuracy in comparison to other tested models.
- -
- Grey colour, bolded: mathematical model, whose RSS result was similar to the best-performing model or near best-performing one, secondary-choice/second-choice or tertiary-choice/third-choice one, but its fitted model parameters indicate lag time or burst release are not in line with experimental dissolution results, i.e., they significantly differ from experimental observations of carvedilol release or the model is not chosen as a representative model in the model group.
- -
- No colour-coding: mathematical models whose model fit was significantly worse and/or less suited for carvedilol release profiles representation than the above ranked models according to the utilized criteria.
2.2.6. Analysis of Carvedilol Release Mechanism Using the Korsmeyer–Peppas and the Peppas–Sahlin Models
3. Results and Discussion
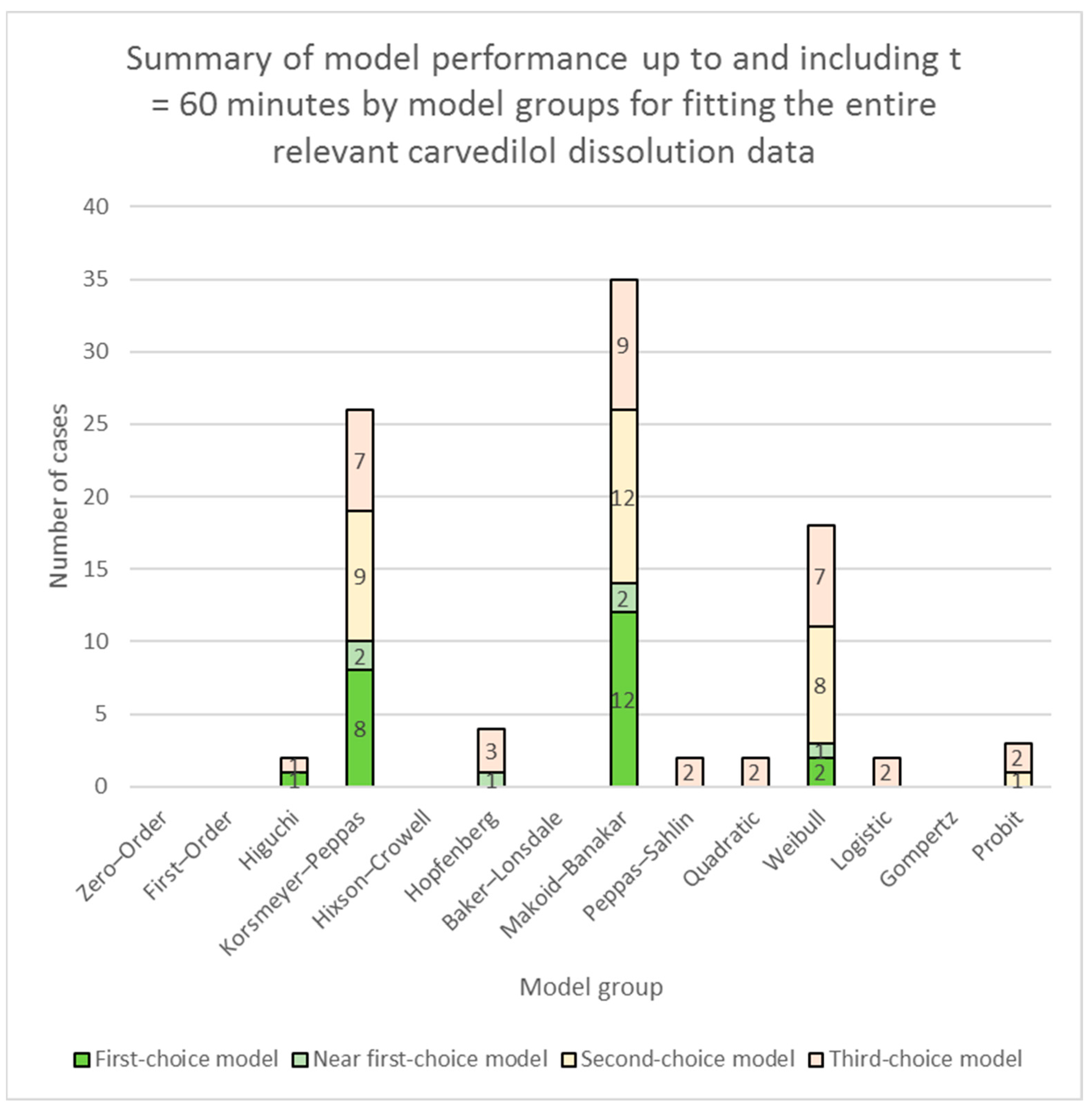
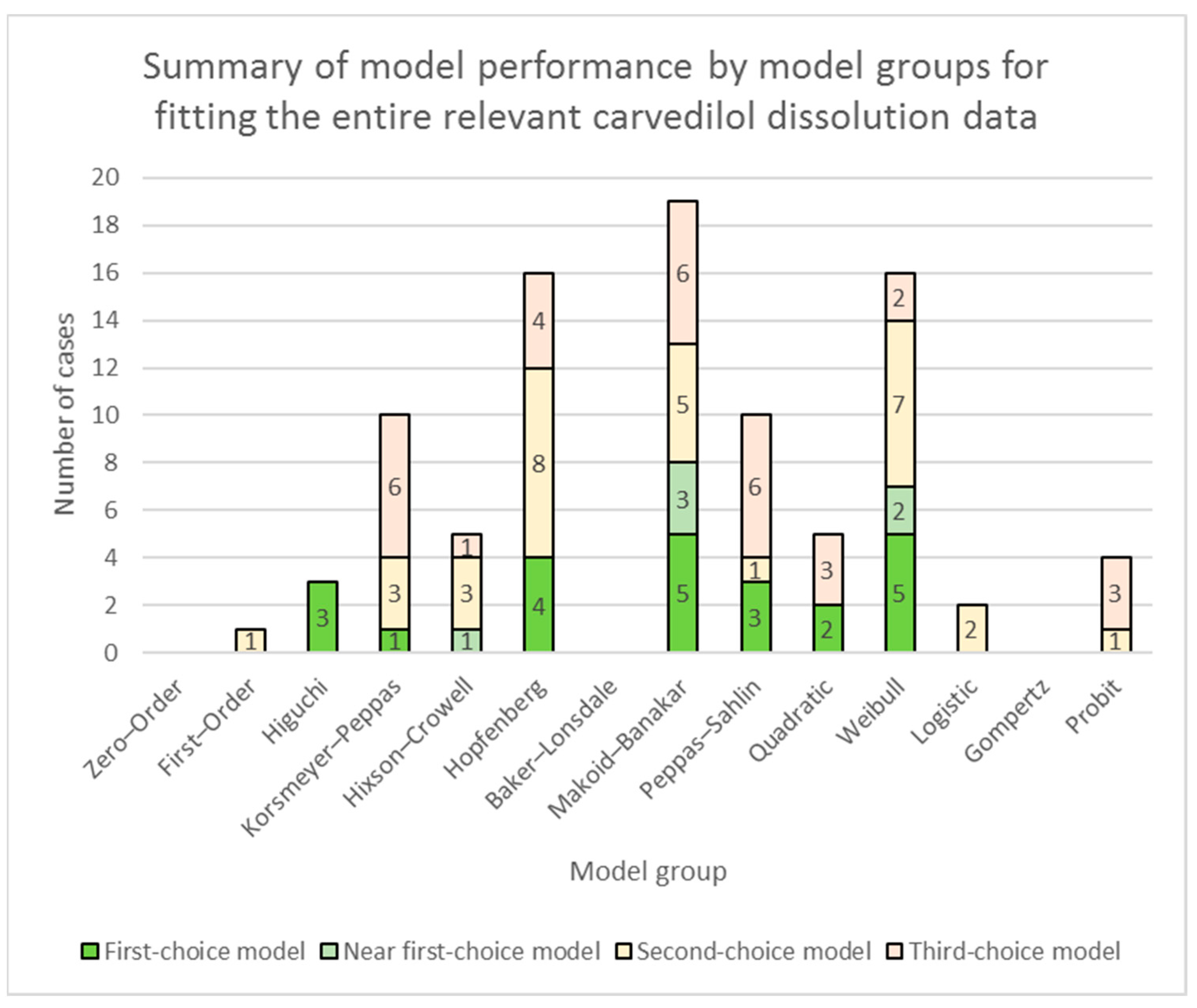
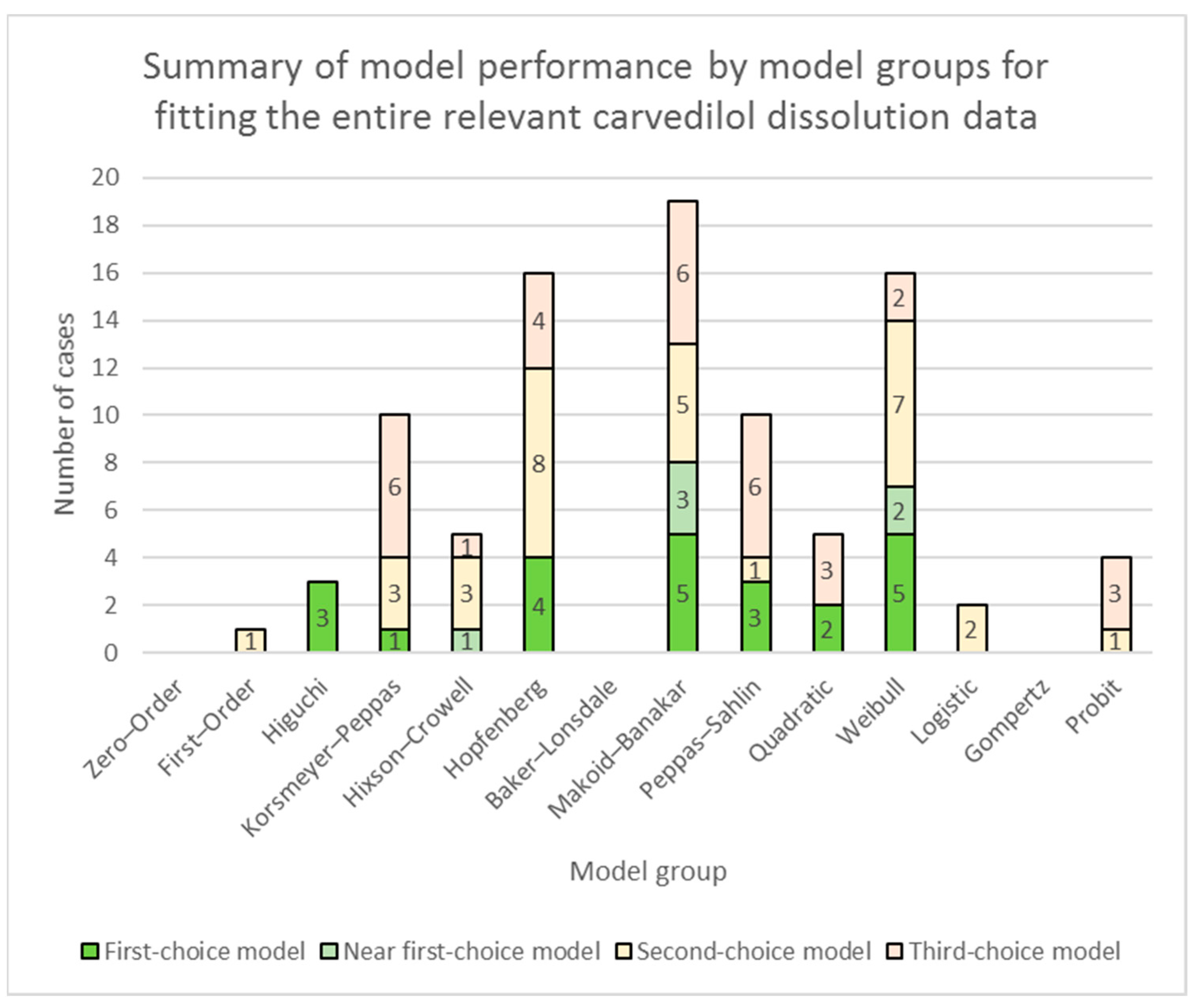
3.1. Fitting of Models to the Entire Relevant Carvedilol Dissolution Data
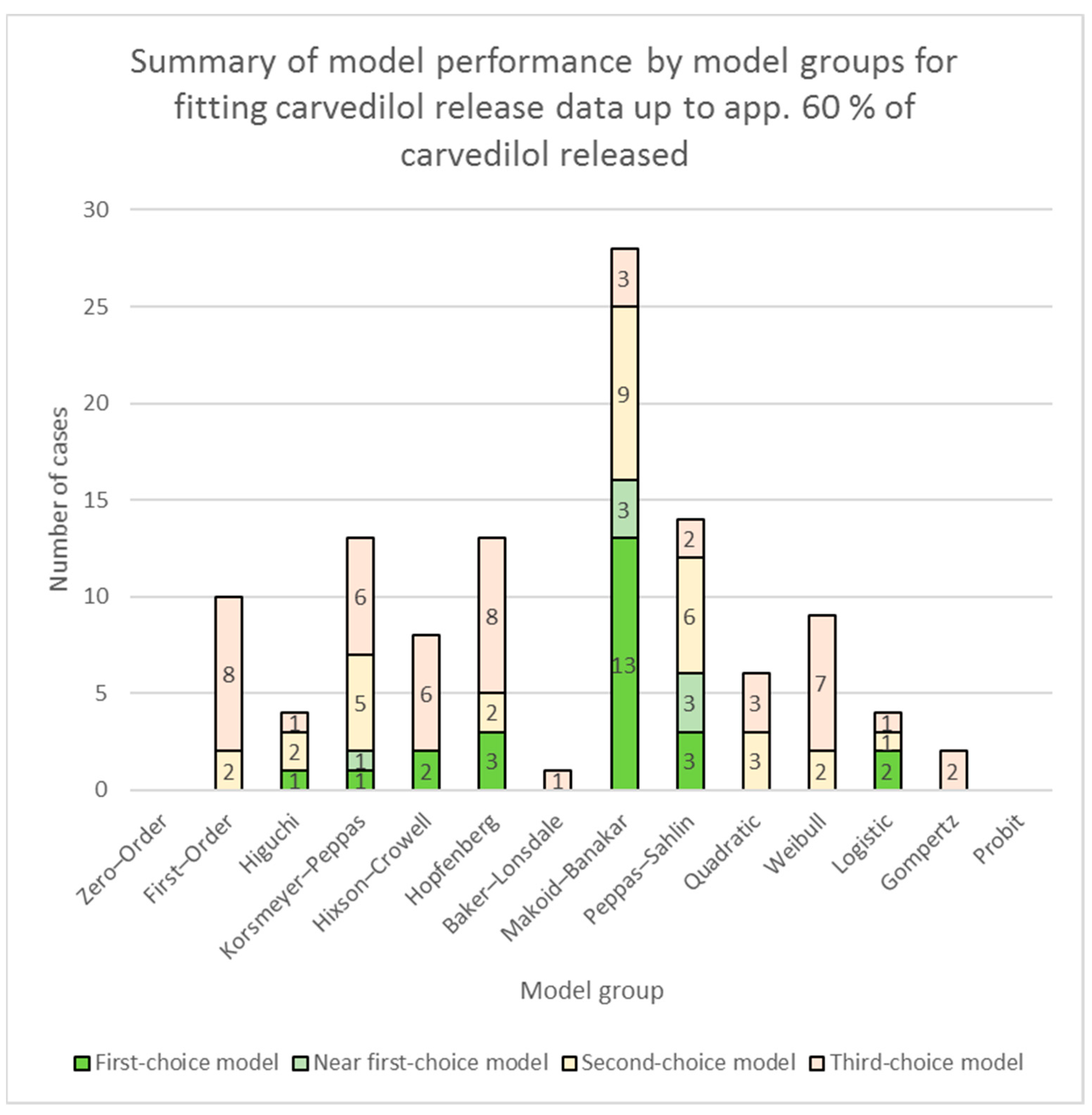
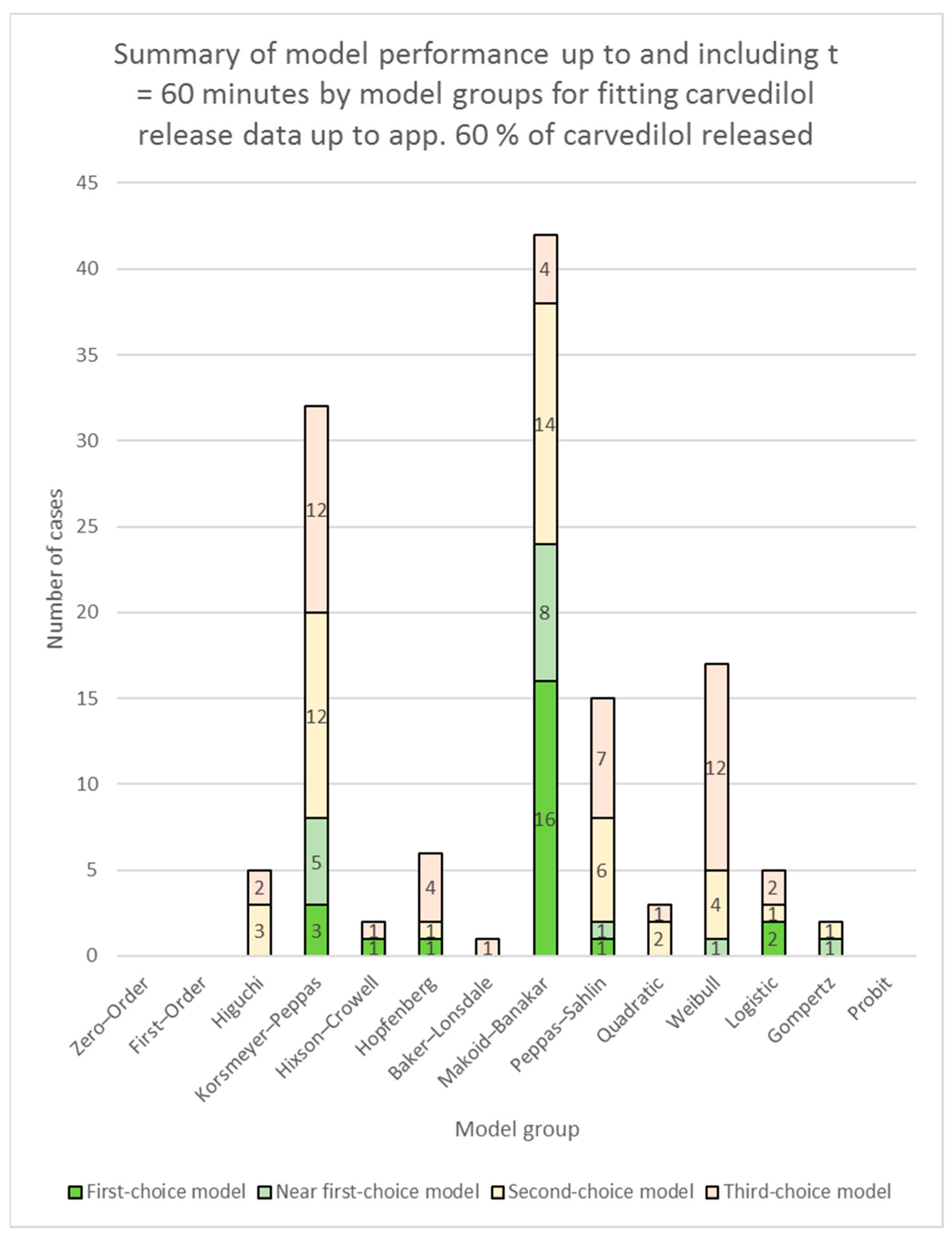
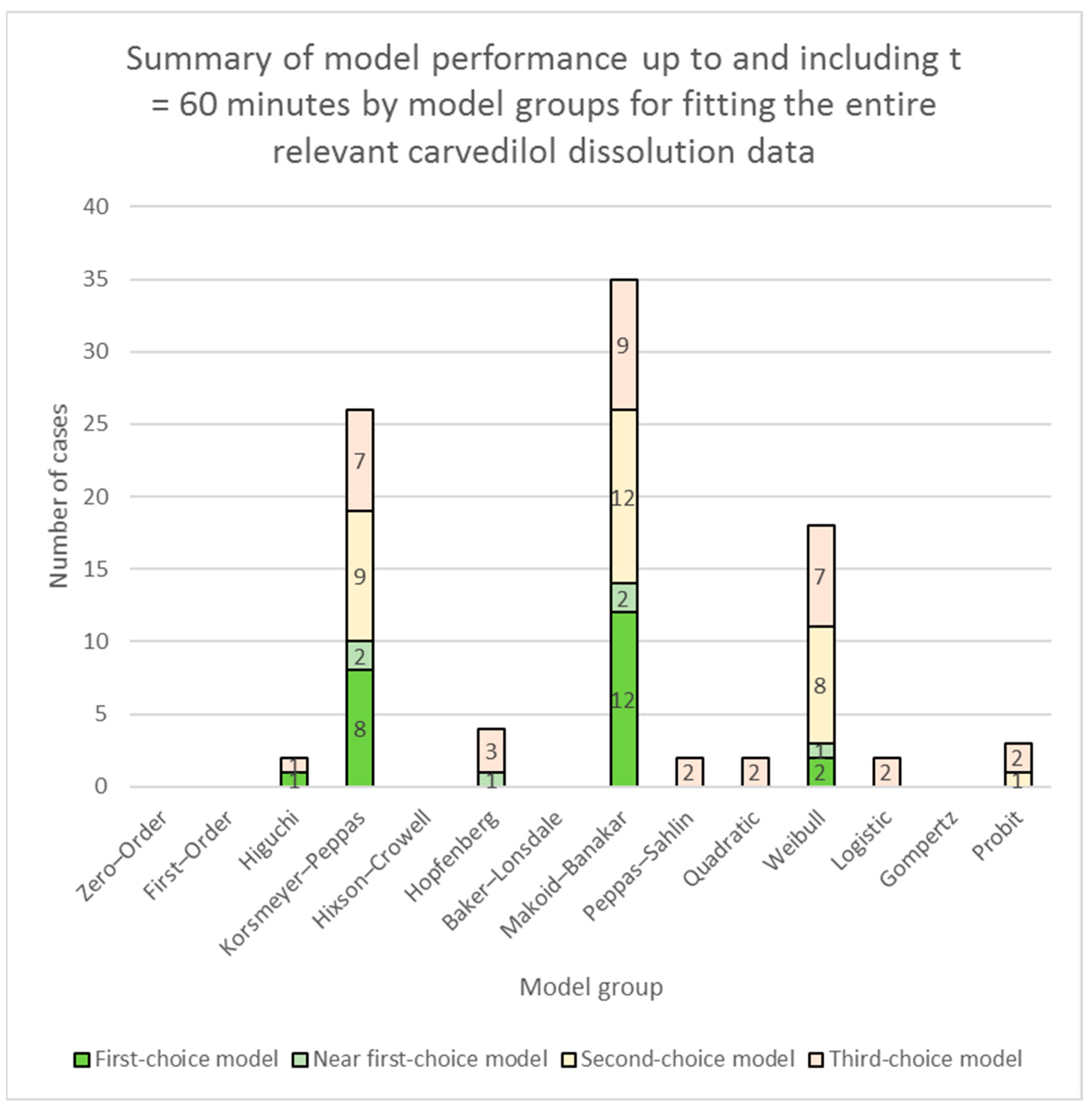
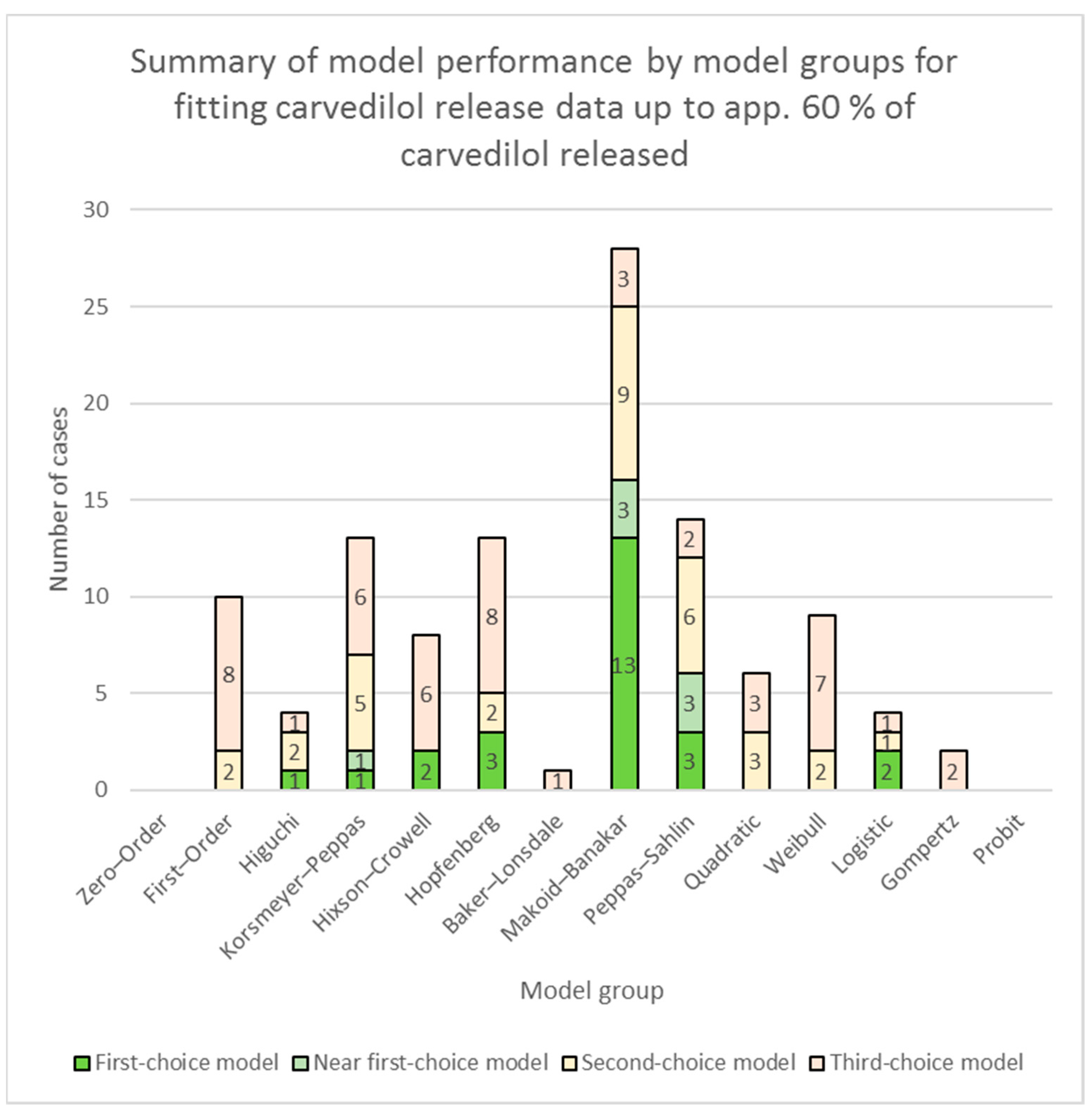
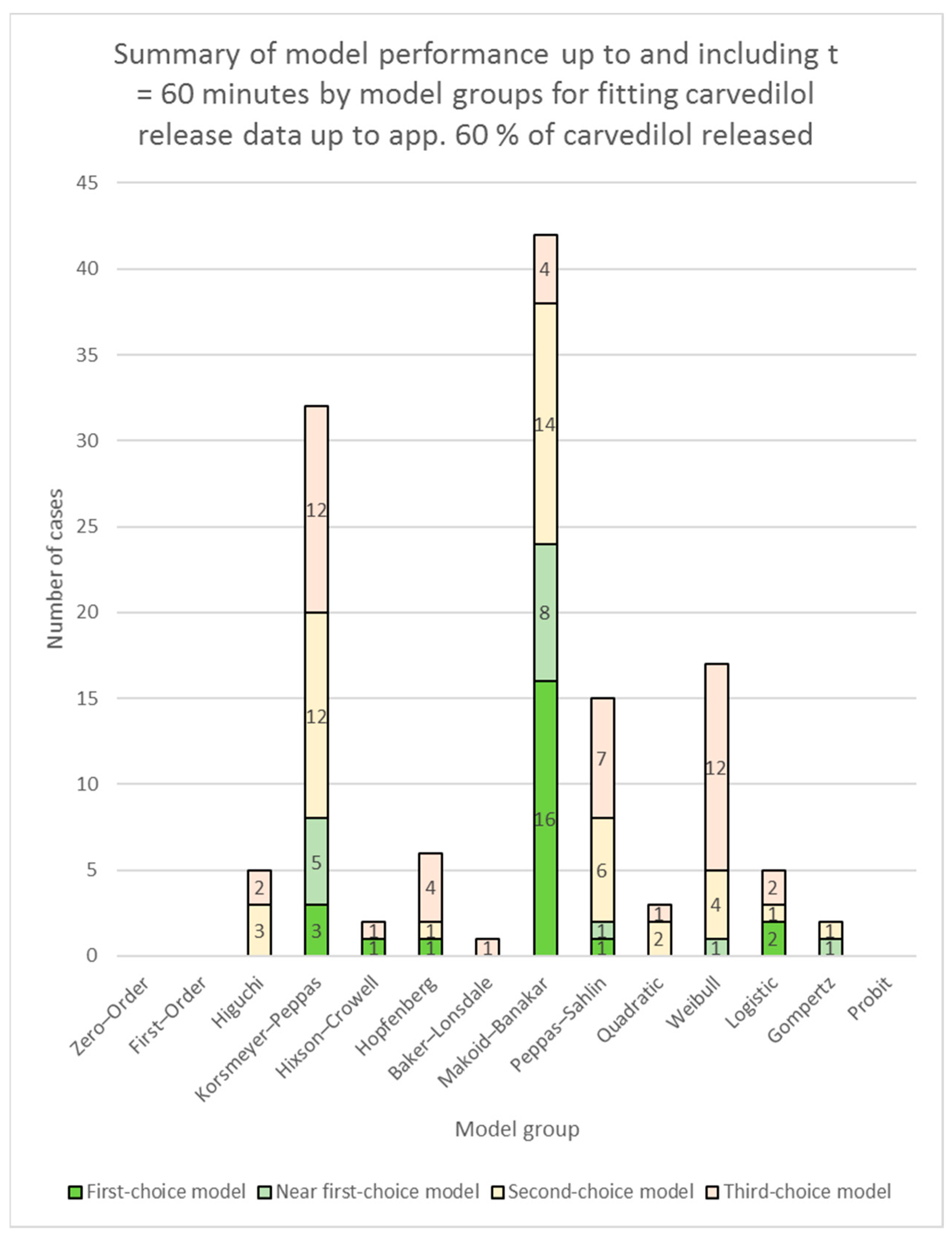
3.2. Fitting of Models to Carvedilol Release Data up to App. 60% of Carvedilol Released
3.3. Model-Dependent Estimation of the Mechanism of Carvedilol Release
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luciano, M. Mathematical models of drug release. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Woodhead Publishing: Cambridge, UK, 2015; pp. 63–86. [Google Scholar]
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.; Kaushal, A.M.; Garg, A.; Garg, S. Factors affecting mechanism and kinetics of drug release from matrix-based oral controlled drug delivery systems. Am. J. Drug Deliv. 2004, 2, 43–57. [Google Scholar] [CrossRef]
- Kalam, M.A.; Humayun, M.; Parvez, N.; Yadav, S.; Garg, A.; Amin, S.; Sultana, Y.; Ali, A. Release kinetics of modified pharmaceutical dosage forms: A review. Cont. J. Pharm. Sci. 2007, 1, 30–35. [Google Scholar]
- Peppas, N.A. Historical perspective on advanced drug delivery: How engineering design and mathematical modeling helped the field mature. Adv. Drug Deliv. Rev. 2013, 65, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N.A.; Narasimhan, B. Mathematical models in drug delivery: How modeling has shaped the way we design new drug delivery systems. J. Control. Release 2014, 190, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Caccavo, D. An overview on the mathematical modeling of hydrogels’ behavior for drug delivery systems. Int. J. Pharm. 2019, 560, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Elmas, A.; Akyüz, G.; Bergal, A.; Andaç, M.; Andaç, Ö. Mathematical modelling of drug release. Res. Eng. Struct. Mater. 2020, 6, 63–86. [Google Scholar] [CrossRef]
- Trucillo, P. Drug carriers: A review on the most used mathematical models for drug release. Processes 2022, 10, 1094. [Google Scholar] [CrossRef]
- Askarizadeh, M.; Esfandiari, N.; Honarvar, B.; Sajadian, S.A.; Azdarpour, A. Kinetic modeling to explain the release of medicine from drug delivery systems. ChemBioEng Rev. 2023, 10, 1006–1049. [Google Scholar] [CrossRef]
- Christidi, E.; Kalosakas, G. Dynamics of the fraction of drug particles near the release boundary: Justifying a stretched exponential kinetics in Fickian drug release. Eur. Phys. J. Spec. Top. 2016, 225, 1245–1254. [Google Scholar] [CrossRef]
- Papadopoulou, V.; Kosmidis, K.; Vlachou, M.; Macheras, P. On the use of the Weibull function for the discernment of drug release mechanisms. Int. J. Pharm. 2006, 309, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Paarakh, M.P.; Jose, P.A.; Setty, C.M.; Peterchristoper, G. Release kinetics–concepts and applications. Int. J. Pharm. Res. Technol. 2018, 8, 12–20. [Google Scholar]
- Muselík, J.; Komersová, A.; Kubová, K.; Matzick, K.; Skalická, B. A critical overview of FDA and EMA statistical methods to compare in vitro drug dissolution profiles of pharmaceutical products. Pharmaceutics 2021, 13, 1703. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Control. Release 1987, 5, 23–36. [Google Scholar] [CrossRef]
- Peppas, N.A.; Sahlin, J.J. A simple equation for the description of solute release. III. Coupling of diffusion and relaxation. Int. J. Pharm. 1989, 57, 169–172. [Google Scholar] [CrossRef]
- Colombo, P.; Santi, P.; Bettini, R.; Brazel, C.S.; Peppas, N.A. Drug release from swelling-controlled systems. In Handbook of Pharmaceutical Controlled Release Technology; Marcel Dekker Inc.: New York, NY, USA, 2000; Volume 9, pp. 183–209. [Google Scholar]
- Grassi, M.; Grassi, G. Mathematical modelling and controlled drug delivery: Matrix systems. Curr. Drug Deliv. 2005, 2, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug delivery. Int. J. Pharm. 2008, 364, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Samaha, D.; Shehayeb, R.; Kyriacos, S. Modeling and comparison of dissolution profiles of diltiazem modified-release formulations. Dissolution Technol. 2009, 16, 41–46. [Google Scholar] [CrossRef]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar]
- Adibkia, K.; Hamedeyazdan, S.; Javadzadeh, Y. Drug release kinetics and physicochemical characteristics of floating drug delivery systems. Expert Opin. Drug Deliv. 2011, 8, 891–903. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Higuchi equation: Derivation, applications, use and misuse. Int. J. Pharm. 2011, 418, 6–12. [Google Scholar] [CrossRef]
- Lokhandwala, H.; Deshpande, A.; Deshpande, S. Kinetic modeling and dissolution profiles comparison: An overview. Int. J. Pharm. Biol. Sci. 2013, 4, 728–773. [Google Scholar]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug dissolution. Int. J. Pharm. 2013, 453, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Yadav, G.; Bansal, M.; Thakur, N.; Khare, S.; Khare, P. Multilayer tablets and their drug release kinetic models for oral controlled drug delivery system. Middle East J. Sci. Res. 2013, 16, 782–795. [Google Scholar]
- Shah, J.C.; Deshpande, A. Kinetic modeling and comparison of invitro dissolution profiles. World J. Pharm. Sci. 2014, 2, 302–309. [Google Scholar]
- Ramteke, K.; Dighe, P.; Kharat, A.; Patil, S. Mathematical models of drug dissolution: A review. Sch. Acad. J. Pharm. 2014, 3, 388–396. [Google Scholar]
- Pascoal, A.; da Silva, P.; Pinheiro, M.C. Drug dissolution profiles from polymeric matrices: Data versus numerical solution of the diffusion problem and kinetic models. Int. Commun. Heat Mass Transf. 2015, 61, 118–127. [Google Scholar] [CrossRef]
- Siswanto, A.; Fudholi, A.; Nugroho, A.K.; Martono, S. In vitro release modeling of aspirin floating tablets using DDSolver. Indones. J. Pharm. 2015, 26, 94. [Google Scholar] [CrossRef]
- Arif, Z. A Concise review on controlled drug release devices: Models, delivery devices. Pharmstudent 2016, 27, 79–91. [Google Scholar]
- Gouda, R.; Baishya, H.; Qing, Z. Application of mathematical models in drug release kinetics of carbidopa and levodopa ER tablets. J. Dev. Drugs 2017, 6, 1–8. [Google Scholar]
- Azadi, S.; Ashrafi, H.; Azadi, A. Mathematical modeling of drug release from swellable polymeric nanoparticles. J. Appl. Pharm. Sci. 2017, 7, 125–133. [Google Scholar]
- Cascone, S. Modeling and comparison of release profiles: Effect of the dissolution method. Eur. J. Pharm. Sci. 2017, 106, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.N.; Khan, N.A. Role of mathematical modelling in controlled release drug delivery. Int. J. Med. Res. Pharm. Sci 2017, 4, 84–95. [Google Scholar]
- Nigusse, B.; Gebre-Mariam, T.; Belete, A. Design, development and optimization of sustained release floating, bioadhesive and swellable matrix tablet of ranitidine hydrochloride. PLoS ONE 2021, 16, e0253391. [Google Scholar] [CrossRef]
- Heredia, N.S.; Vizuete, K.; Flores-Calero, M.; Pazmiño, V.K.; Pilaquinga, F.; Kumar, B.; Debut, A. Comparative statistical analysis of the release kinetics models for nanoprecipitated drug delivery systems based on poly (lactic-co-glycolic acid). PLoS ONE 2022, 17, e0264825. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, C.; Massuelle, D.; Doelker, E. Towards elucidation of the drug release mechanism from compressed hydrophilic matrices made of cellulose ethers. II. Evaluation of a possible swelling-controlled drug release mechanism using dimensionless analysis. J. Control. Release 2010, 141, 223–233. [Google Scholar] [CrossRef]
- Košir, D.; Vrečer, F. The performance of HPMC matrix tablets using various agglomeration manufacturing processes. Drug Dev. Ind. Pharm. 2017, 43, 329–337. [Google Scholar] [CrossRef]
- Košir, D.; Ojsteršek, T.; Baumgartner, S.; Vrečer, F. A study of critical functionality-related characteristics of HPMC for sustained-release tablets. Pharm. Dev. Technol. 2018, 23, 865–873. [Google Scholar] [CrossRef]
- Caraballo, I. Factors affecting drug release from hydroxypropyl methylcellulose matrix systems in the light of classical and percolation theories. Expert Opin. Drug Deliv. 2010, 7, 1291–1301. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N.A. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev. 2012, 64, 163–174. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Ojsteršek, T.; Hudovornik, G.; Vrečer, F. Comparative Study of Selected Excipients’ Influence on Carvedilol Release from Hypromellose Matrix Tablets. Pharmaceutics 2023, 15, 1525. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, W.S. Robust locally weighted regression and smoothing scatterplots. J. Am. Stat. Assoc. 1979, 74, 829–836. [Google Scholar] [CrossRef]
- Cleveland, W.S.; Devlin, S.J. Locally weighted regression: An approach to regression analysis by local fitting. J. Am. Stat. Assoc. 1988, 83, 596–610. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Peppas, N. Analysis of Fickian and non-Fickian drug release from polymers. Pharm. Acta Helv. 1985, 60, 110–111. [Google Scholar]
- Ford, J.L.; Mitchell, K.; Rowe, P.; Armstrong, D.J.; Elliott, P.N.; Rostron, C.; Hogan, J.E. Mathematical modelling of drug release from hydroxypropylmethylcellulose matrices: Effect of temperature. Int. J. Pharm. 1991, 71, 95–104. [Google Scholar] [CrossRef]
- Higuchi, T. Rate of release of medicaments from ointment bases containing drugs in suspension. J. Pharm. Sci. 1961, 50, 874–875. [Google Scholar] [CrossRef]
- Tarvainen, M.; Peltonen, S.; Mikkonen, H.; Elovaara, M.; Tuunainen, M.; Paronen, P.; Ketolainen, J.; Sutinen, R. Aqueous starch acetate dispersion as a novel coating material for controlled release products. J. Control. Release 2004, 96, 179–191. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Group | Model | Equation | Parameter(s) |
|---|---|---|---|
| Zero–order | Zero–order | k0 | |
| Zero–order with Tlag | k0, Tlag | ||
| Zero–order with F0 | k0, F0 | ||
| First–order | First–order | k1 | |
| First–order with Tlag | k1, Tlag | ||
| First–order with Fmax | k1, Fmax | ||
| First–order with Tlag and Fmax | k1, Tlag, Fmax | ||
| Higuchi | Higuchi | kH | |
| Higuchi with Tlag | kH, Tlag | ||
| Higuchi with F0 | kH, F0 | ||
| Korsmeyer–Peppas | Korsmeyer–Peppas | kKP, n | |
| Korsmeyer–Peppas with Tlag | kKP, n, Tlag | ||
| Korsmeyer–Peppas with F0 | kKP, n, F0 | ||
| Hixson–Crowell | Hixson–Crowell | kHC | |
| Hixson–Crowell with Tlag | kHC, Tlag | ||
| Hopfenberg | Hopfenberg | kHB, n | |
| Hopfenberg with Tlag | kHB, n, Tlag | ||
| Baker–Lonsdale | Baker–Lonsdale | kBL | |
| Baker–Lonsdale with Tlag | kBL, Tlag | ||
| Makoid–Banakar | Makoid–Banakar | kMB, n, k | |
| Makoid–Banakar with Tlag | kMB, n, k, Tlag | ||
| Peppas–Sahlin | Peppas–Sahlin_1 | k1, k2, m | |
| Peppas–Sahlin_1 with Tlag | k1, k2, m, Tlag | ||
| Peppas–Sahlin_2 | k1, k2 | ||
| Peppas–Sahlin_2 with Tlag | k1, k2, Tlag | ||
| Quadratic | Quadratic | k1, k2 | |
| Quadratic with Tlag | k1, k2, Tlag | ||
| Weibull | Weibull_1 | α, β, Ti | |
| Weibull_2 | α, β | ||
| Weibull_3 | α, β, Fmax | ||
| Weibull_4 | α, β, Ti, Fmax | ||
| Logistic | Logistic_1 | α, β | |
| Logistic_2 | α, β, Fmax | ||
| Logistic_3 | k, γ, Fmax | ||
| Gompertz | Gompertz_1 | α, β | |
| Gompertz_2 | α, β, Fmax | ||
| Gompertz_3 | k, γ, Fmax | ||
| Gompertz_4 | k, β, Fmax | ||
| Probit | Probit_1 | α, β | |
| Probit_2 | α, β, Fmax | ||
| Explanation of symbols used | |||
| F | the fraction (%) of drug released in time t | ||
| F0 | the initial fraction of the drug in the solution resulting from a burst release | ||
| Fmax | the maximum fraction of the drug released at infinite time | ||
| t | time | ||
| Tlag, Ti | the lag time prior to drug release or the location parameter | ||
| k0 | the zero–order release constant | ||
| k1 | the first–order release constant | ||
| kH | the Higuchi release constant | ||
| kKP, n | kKP is the release constant in the Korsmeyer–Peppas model and its variations (incorporating Tlag or F0) incorporating structural and geometric characteristics of the DDS; n is the diffusional exponent in the Korsmeyer–Peppas model and its variations (incorporating Tlag or F0) indicating the drug-release mechanism | ||
| kHC | the release constant in the Hixson–Crowell model | ||
| kHB, n | kBH is the combined constant in the Hopfenberg model, kHB = k0/(C0∙a0), where k0 is the erosion rate constant, C0 is the initial concentration of drug in the matrix, and a0 is the initial radius for a sphere or cylinder or the half thickness for a slab; n is 1, 2, and 3 for a slab, cylinder, and sphere, respectively | ||
| kBL | kBL is the combined constant in the Baker–Lonsdale model, kBL = [3∙D∙Cs/(r02∙C0)], where D is the diffusion coefficient, Cs is the saturation solubility, r0 is the initial radius for a sphere or cylinder or the half-thickness for a slab, and C0 is the initial drug loading in the matrix | ||
| kMB, n, k | empirical parameters in the Makoid–Banakar model (kMB, n, k > 0) | ||
| k1, k2, m | k1 is the constant related to Fickian kinetics; k2 is the constant related to Case–II relaxation kinetics; m is the diffusional exponent for a device of any geometric shape, which exhibits controlled release | ||
| k1, k2 | k1 is the constant denoting the relative contribution of t0.5-dependent drug diffusion to drug release; k2 is the constant denoting the relative contribution of t-dependent polymer relaxation to drug release | ||
| k1, k2 | k1 is the constant in the Quadratic model denoting the relative contribution of t2-dependent drug release; k2 is the constant in the Quadratic model denoting the relative contribution of t-dependent drug release | ||
| α, β | α is the scale parameter; β is the shape parameter which characterises the curve as either exponential (β = 1; case 1), sigmoid, or S-shaped, with upward curvature followed by a turning point (β > 1; case 2), or parabolic, with a higher initial slope and after that consistent with the exponential (β < 1; case 3) | ||
| α, β | α is the scale factor in the Logistic_1 and the Logitic_2 models; β is the shape factor in the Logistic_1 and Logistic_2 models | ||
| k, γ | k is the shape factor in Logistic_3 model; γ is the time at which F = Fmax/2 | ||
| α, β | α is the scale factor in the Gompertz_1 and the Gompertz_2 models; β is the shape factor in the Gompertz_1 and the Gompertz_2 models | ||
| k, γ | k is the shape factor in the Gompertz_3 model; γ is the time at which F = Fmax/exp(1) ≈ 0.368∙Fmax | ||
| β, k | β is the scale factor in the Gompertz_4 model; k is the shape factor in the Gompertz_4 model | ||
| φ, α, β | Φ is the standard normal distribution; α is the scale factor in the Probit models; β is the shape factor in the Probit models | ||
| Value of the Diffusional Exponent n in the Fitted Korsmeyer–Peppas Model | Drug Release Mechanism |
|---|---|
| 0.45 | Fickian diffusion |
| 0.45 < n < 0.89 | Anomalous (non-Fickian) transport |
| 0.89 | Case–II transport |
| >0.89 | Super Case–II transport |
| Formulation Id | Higuchi Model | Higuchi with Tlag Model | Higuchi with F0 Model | Korsmeyer–Peppas Model | Korsmeyer–Peppas with Tlag Model | Korsmeyer–Peppas with F0 Model | |||
|---|---|---|---|---|---|---|---|---|---|
| RSS/Model Parameter | RSS | RSS | RSS | n | RSS | n | RSS | n | RSS |
| Polyglykol® 4000 P (up to app. 60% of carv. rel.) | 163.62 ± 70.65 | 22.15 ± 14.1 | 0.76 ± 0.94 | 0.847 ± 0.032 | 6.74 ± 2.71 | 0.674 ± 0.038 | 0.3 ± 0.28 | 1.123 ± 0.065 | 27.76 ± 16.1 |
| Polyglykol® 4000 P (up to app. 75% of carv. rel.) | 238.6 ± 108.06 | 67.68 ± 61.51 | 1.34 ± 1.14 | 0.82 ± 0.045 | 24.04 ± 13.55 | 0.67 ± 0.042 | 4.19 ± 3.1 | 1.06 ± 0.057 | 80.54 ± 34.94 |
| Polyglykol® 8000 P (up to app. 60% of carv. rel.) | 310.1 ± 17.94 | 168.32 ± 27.34 | 1.6 ± 0.62 | 1.039 ± 0.04 | 19.31 ± 3.06 | 0.84 ± 0.034 | 3.6 ± 0.36 | 1.394 ± 0.046 | 74.66 ± 4.25 |
| Polyglykol® 8000 P (up to app. 75% of carv. rel.) | 454.71 ± 33.45 | 169.72 ± 26.2 | 2.12 ± 0.68 | 1.031 ± 0.04 | 76.58 ± 4.85 | 0.833 ± 0.033 | 24.01 ± 2.24 | 1.273 ± 0.042 | 191.99 ± 10.55 |
| POLYOXᵀᴹ WSR N-80 (LEO NF Grade) | 651.64 ± 84.72 | 135.36 ± 20.9 | 70.08 ± 28.6 | 1.312 ± 0.081 | 68.09 ± 45.18 | 1.161 ± 0.074 | 19.61 ± 14.96 | 1.552 ± 0.084 | 230.05 ± 65.77 |
| KOLLIDON® 25 | 255.46 ± 220.44 | 119.36 ± 119.57 | 15.82 ± 10.54 | 0.881 ± 0.153 | 22.09 ± 16.63 | 0.71 ± 0.12 | 10.78 ± 10.2 | 1.087 ± 0.15 | 47.46 ± 22.69 |
| KOLLIDON® 90 F | 973.61 ± 152.58 | 259.65 ± 88.31 | 185.62 ± 91.57 | 1.15 ± 0.049 | 52.57 ± 74.56 | 1.043 ± 0.035 | 34.01 ± 30.83 | 1.267 ± 0.048 | 138.64 ± 177.25 |
| C*Pharm Mannidex 16700 | 281.42 ± 360.46 | 163.81 ± 182.16 | 104.06 ± 177.61 | 0.729 ± 0.111 | 166.52 ± 322.14 | 0.628 ± 0.083 | 132.25 ± 234.79 | 0.91 ± 0.108 | 244.31 ± 466.55 |
| PEARLITOL® 160C | 245.82 ± 373.3 | 198.16 ± 206.04 | 72.42 ± 125.69 | 0.723 ± 0.111 | 45.24 ± 82.01 | 0.6 ± 0.085 | 83.63 ± 138.26 | 0.911 ± 0.123 | 24.47 ± 44.82 |
| Parteck® M 100 (up to app. 60% of carv. rel.) | 594.97 ± 444.85 | 122.22 ± 95.88 | 63.9 ± 64.02 | 1.547 ± 0.227 | 98.83 ± 167.67 | 1.223 ± 0.157 | 66.55 ± 93.53 | 1.867 ± 0.23 | 186.5 ± 285.77 |
| Parteck® M 100 (up to app. 70% of carv. rel.) | 900.52 ± 425.2 | 169.09 ± 84.22 | 96.98 ± 57.96 | 1.433 ± 0.081 | 236.99 ± 284.79 | 1.177 ± 0.103 | 208.55 ± 313.26 | 1.717 ± 0.128 | 576.12 ± 745.48 |
| Parteck® M 200 | 599.68 ± 411.3 | 92.75 ± 34.94 | 45.9 ± 28.59 | 1.213 ± 0.102 | 116.44 ± 206.34 | 1.017 ± 0.101 | 66.78 ± 109.29 | 1.492 ± 0.151 | 227.57 ± 377.98 |
| Lactochem® Crystals | 205.22 ± 90.39 | 9.02 ± 4.1 | 0.77 ± 0.87 | 0.338 ± 0.029 | 5.88 ± 3.41 | 0.296 ± 0.026 | 19.42 ± 6.56 | 0.453 ± 0.028 | 1.3 ± 0.99 |
| Lactochem® Fine Powder | 87.03 ± 38.61 | 136.13 ± 38.16 | 17.83 ± 11.03 | 0.594 ± 0.036 | 7.29 ± 5.38 | 0.536 ± 0.033 | 29.58 ± 14.66 | 0.702 ± 0.034 | 7 ± 10.3 |
| SuperTab® 11SD | 598.31 ± 326.66 | 7.26 ± 4.39 | 0.67 ± 0.39 | 0.247 ± 0.034 | 10.38 ± 3.17 | 0.213 ± 0.029 | 23.73 ± 5.28 | 0.35 ± 0.047 | 4.36 ± 1.81 |
| FlowLac® 100 | 78.94 ± 37.09 | 213.3 ± 53.18 | 36.86 ± 5.77 | 0.538 ± 0.044 | 38.56 ± 9.48 | 0.487 ± 0.04 | 86.4 ± 12.6 | 0.667 ± 0.03 | 5.34 ± 1.78 |
| Tablettose® 70 | 406.43 ± 142.2 | 14.62 ± 6.65 | 1.67 ± 1.92 | 0.284 ± 0.032 | 3.76 ± 3.14 | 0.247 ± 0.028 | 13.96 ± 6.3 | 0.391 ± 0.037 | 0.92 ± 0.7 |
| Granulated sugar N°1 600 | 388.67 ± 26.08 | 220.83 ± 17.64 | 68.92 ± 6.67 | 0.762 ± 0.015 | 2.16 ± 1.29 | 0.691 ± 0.017 | 25.34 ± 6.81 | 0.853 ± 0.02 | 15.68 ± 6.28 |
| GLUCIDEX® 19 | 454.02 ± 157.31 | 312.58 ± 45.09 | 133.11 ± 48.18 | 0.737 ± 0.058 | 41.49 ± 10.47 | 0.673 ± 0.049 | 98.75 ± 13.98 | 0.84 ± 0.055 | 10.6 ± 11.42 |
| DI-CAFOS® A12 | 320.53 ± 77.03 | 306.29 ± 63.52 | 84.67 ± 22.01 | 0.659 ± 0.011 | 22.12 ± 6.29 | 0.608 ± 0.01 | 75.42 ± 16.14 | 0.75 ± 0.011 | 0.5 ± 0.27 |
| EMCOMPRESS® Anhydrous | 62.28 ± 41.64 | 241.08 ± 116.1 | 39.04 ± 21.61 | 0.508 ± 0.014 | 57.19 ± 27.15 | 0.468 ± 0.012 | 115.18 ± 46.28 | 0.601 ± 0.015 | 12.73 ± 9.68 |
| AVICEL® PH-102 | 517.13 ± 54.46 | 304.91 ± 44.76 | 127.2 ± 6.52 | 0.73 ± 0.047 | 12.39 ± 15.85 | 0.688 ± 0.051 | 39.37 ± 42.59 | 0.802 ± 0.05 | 29.69 ± 28.12 |
| AVICEL® PH-200 | 411.99 ± 106.09 | 259.37 ± 35.91 | 86.38 ± 31.85 | 0.731 ± 0.026 | 2.98 ± 2 | 0.675 ± 0.019 | 23.52 ± 21.65 | 0.795 ± 0.025 | 26.54 ± 18.42 |
| ETHOCELᵀᴹ Standard 20 Premium | 559.11 ± 101.29 | 257.92 ± 17.08 | 117.19 ± 28.67 | 0.767 ± 0.03 | 3.28 ± 1.46 | 0.715 ± 0.016 | 20.78 ± 10.09 | 0.834 ± 0.019 | 24.69 ± 18.73 |
| STARCH 1500® sample with ↓PS | 90.31 ± 76.42 | 149.22 ± 102.66 | 49.54 ± 16.88 | 0.415 ± 0.043 | 149.36 ± 52.78 | 0.385 ± 0.04 | 222.83 ± 64.84 | 0.505 ± 0.046 | 76.41 ± 38.97 |
| STARCH 1500® sample with ↑PS | 167.48 ± 71.61 | 419.18 ± 82.55 | 100.16 ± 34.97 | 0.52 ± 0.02 | 142.42 ± 37.83 | 0.485 ± 0.019 | 233.67 ± 51.87 | 0.607 ± 0.02 | 55.47 ± 23.08 |
| Formulation Id | Peppas–Sahlin_1 Model | Peppas–Sahlin_1 with Tlag Model | ||||
|---|---|---|---|---|---|---|
| RSS/Model Parameter | k1 | k2 | RSS | k1 | k2 | RSS |
| Polyglykol® 4000 P (up to app. 60% of carv. rel.) | 1.378 ± 0.96 | 1.849 ± 0.383 | 5.07 ± 2.17 | / | / | / |
| Polyglykol® 4000 P (up to app. 75% of carv. rel.) | 2.717 ± 0.736 | 1.528 ± 0.333 | 17.15 ± 6.32 | 5.757 ± 0.5 | 1.124 ± 0.299 | 6.25 ± 3.34 |
| Polyglykol® 8000 P (up to app. 60% of carv. rel.) | −2.008 ± 0.559 | 2.482 ± 0.065 | 7.68 ± 1.04 | / | / | / |
| Polyglykol® 8000 P (up to app. 75% of carv. rel.) | −0.209 ± 0.655 | 2.05 ± 0.07 | 28.83 ± 4.07 | 2.84 ± 0.742 | 1.666 ± 0.083 | 15.79 ± 2.47 |
| POLYOXᵀᴹ WSR N-80 (LEO NF Grade) | −2.429 ± 0.547 | 1.05 ± 0.062 | 4.54 ± 3.42 | −1.692 ± 0.594 | 0.989 ± 0.068 | 4.17 ± 3.25 |
| KOLLIDON® 25 | 1.21 ± 2.823 | 1.353 ± 0.858 | 14.33 ± 13.01 | 3.572 ± 2.494 | 1.046 ± 0.819 | 8.1 ± 7.87 |
| KOLLIDON® 90 F | −1.19 ± 0.504 | 0.499 ± 0.036 | 18.71 ± 16.88 | −0.928 ± 0.566 | 0.483 ± 0.035 | 18.55 ± 15.92 |
| C*Pharm Mannidex 16700 | 2.441 ± 1.025 | 0.548 ± 0.238 | 132.3 ± 258.2 | 3.476 ± 1.297 | 0.441 ± 0.209 | 117.35 ± 227.85 |
| PEARLITOL® 160C | 1.08 ± 2.32 | 0.787 ± 0.549 | 19.8 ± 36.7 | 2.228 ± 2.178 | 0.662 ± 0.547 | 25.39 ± 43.31 |
| Parteck® M 100 (up to app. 60% of carv. rel.) | −6.833 ± 3.787 | 3.049 ± 1.212 | 37.22 ± 49.09 | / | / | / |
| Parteck® M 100 (up to app. 70% of carv. rel.) | −5.072 ± 1.341 | 2.626 ± 0.573 | 101.13 ± 118.56 | −2.53 ± 1.081 | 2.344 ± 0.509 | 94.82 ± 107.63 |
| Parteck® M 200 | −3.645 ± 1.461 | 2.181 ± 0.678 | 35.28 ± 54.65 | −1.407 ± 0.994 | 1.928 ± 0.601 | 32.36 ± 47.47 |
| Lactochem® Crystals | 7.452 ± 0.539 | −0.17 ± 0.059 | 16.77 ± 11.02 | 7.804 ± 0.768 | −0.233 ± 0.057 | 49.14 ± 20.82 |
| Lactochem® Fine Powder | 2.233 ± 0.309 | 0.138 ± 0.023 | 2.17 ± 2.96 | 2.487 ± 0.319 | 0.121 ± 0.023 | 3.27 ± 1.31 |
| SuperTab® 11SD | 12.175 ± 3.512 | −0.373 ± 0.108 | 43.64 ± 17 | 10.736 ± 1.736 | −0.502 ± 0.143 | 127.08 ± 48.45 |
| FlowLac® 100 | 2.369 ± 0.52 | 0.112 ± 0.028 | 5.71 ± 4.91 | 2.584 ± 0.534 | 0.098 ± 0.029 | 14.95 ± 9.71 |
| Tablettose® 70 | 10.606 ± 1.644 | −0.326 ± 0.083 | 21.9 ± 14.62 | 10.054 ± 0.81 | −0.425 ± 0.081 | 74.42 ± 27.43 |
| Granulated sugar N°1 600 | 0.784 ± 0.099 | 0.189 ± 0.006 | 4.09 ± 0.39 | 0.941 ± 0.105 | 0.179 ± 0.006 | 2.77 ± 0.62 |
| GLUCIDEX® 19 | 0.541 ± 0.277 | 0.196 ± 0.027 | 8.64 ± 13.09 | 0.68 ± 0.278 | 0.188 ± 0.027 | 11.41 ± 12.9 |
| DI-CAFOS® A12 | 1.056 ± 0.039 | 0.11 ± 0.012 | 0.55 ± 0.37 | 1.148 ± 0.039 | 0.105 ± 0.011 | 1.17 ± 0.3 |
| EMCOMPRESS® Anhydrous | 1.953 ± 0.052 | 0.058 ± 0.015 | 5.8 ± 2.63 | 2.052 ± 0.053 | 0.052 ± 0.015 | 13.47 ± 4.34 |
| AVICEL® PH-102 | 0.697 ± 0.149 | 0.094 ± 0.004 | 2.77 ± 1.62 | 0.756 ± 0.151 | 0.092 ± 0.004 | 1.96 ± 1.09 |
| AVICEL® PH-200 | 0.822 ± 0.158 | 0.1 ± 0.011 | 3.7 ± 2.14 | 0.895 ± 0.161 | 0.097 ± 0.011 | 1.54 ± 1.21 |
| ETHOCELᵀᴹ Standard 20 Premium | 0.609 ± 0.135 | 0.113 ± 0.009 | 7.09 ± 2.46 | 0.68 ± 0.137 | 0.11 ± 0.009 | 5.07 ± 1.94 |
| STARCH 1500® sample with ↓PS | 2.267 ± 0.467 | 0.02 ± 0.017 | 54.07 ± 43.49 | 2.337 ± 0.472 | 0.016 ± 0.017 | 76.64 ± 55.88 |
| STARCH 1500® sample with ↑PS | 1.427 ± 0.127 | 0.055 ± 0.01 | 15.35 ± 7.53 | 1.484 ± 0.128 | 0.052 ± 0.01 | 23.68 ± 8.71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ojsteršek, T.; Vrečer, F.; Hudovornik, G. Comparative Fitting of Mathematical Models to Carvedilol Release Profiles Obtained from Hypromellose Matrix Tablets. Pharmaceutics 2024, 16, 498. https://doi.org/10.3390/pharmaceutics16040498
Ojsteršek T, Vrečer F, Hudovornik G. Comparative Fitting of Mathematical Models to Carvedilol Release Profiles Obtained from Hypromellose Matrix Tablets. Pharmaceutics. 2024; 16(4):498. https://doi.org/10.3390/pharmaceutics16040498
Chicago/Turabian StyleOjsteršek, Tadej, Franc Vrečer, and Grega Hudovornik. 2024. "Comparative Fitting of Mathematical Models to Carvedilol Release Profiles Obtained from Hypromellose Matrix Tablets" Pharmaceutics 16, no. 4: 498. https://doi.org/10.3390/pharmaceutics16040498