
Non-Invasive Delivery of Negatively Charged Nanobodies by Anodal Iontophoresis: When Electroosmosis Dominates Electromigration



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
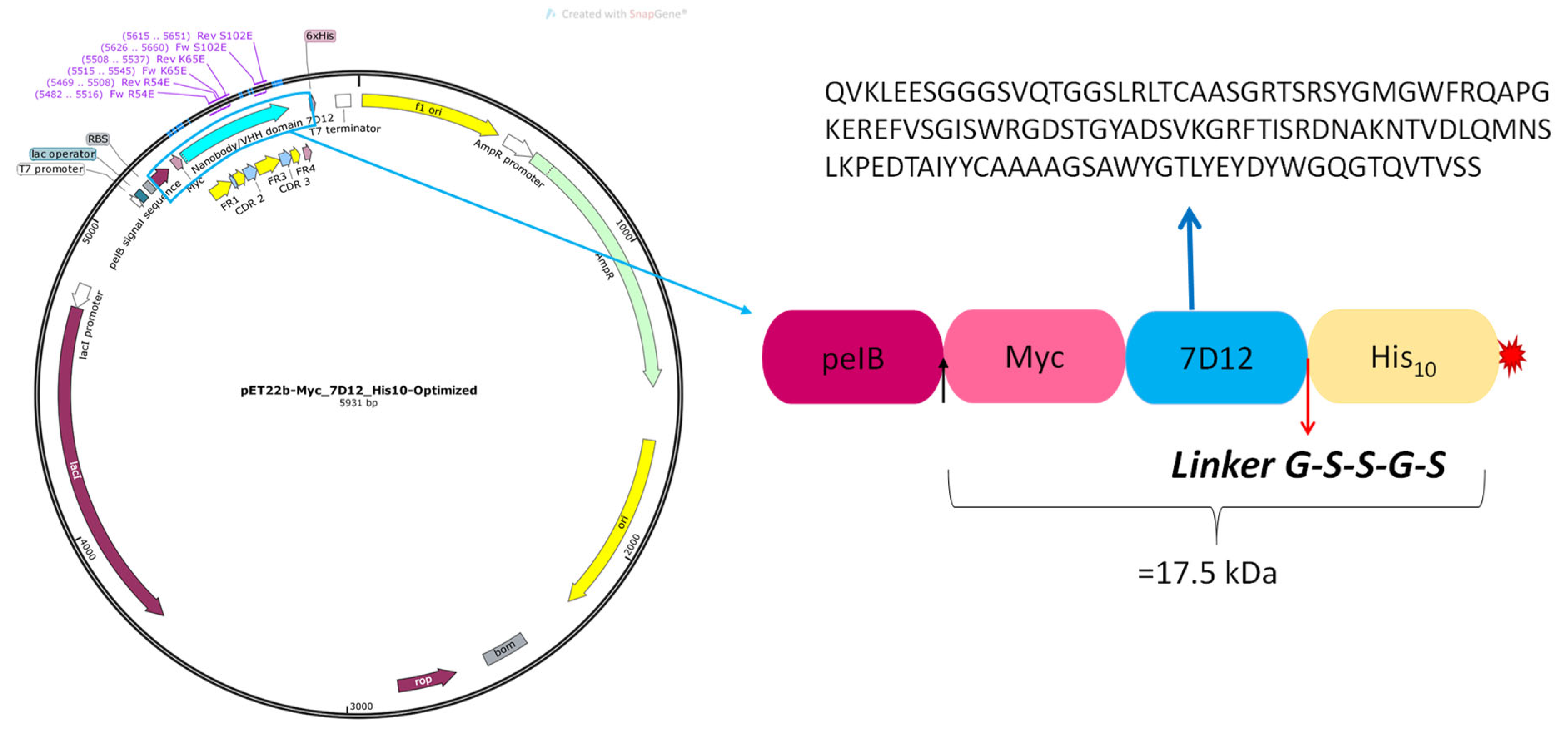
2.2. M7D12H Plasmid Design
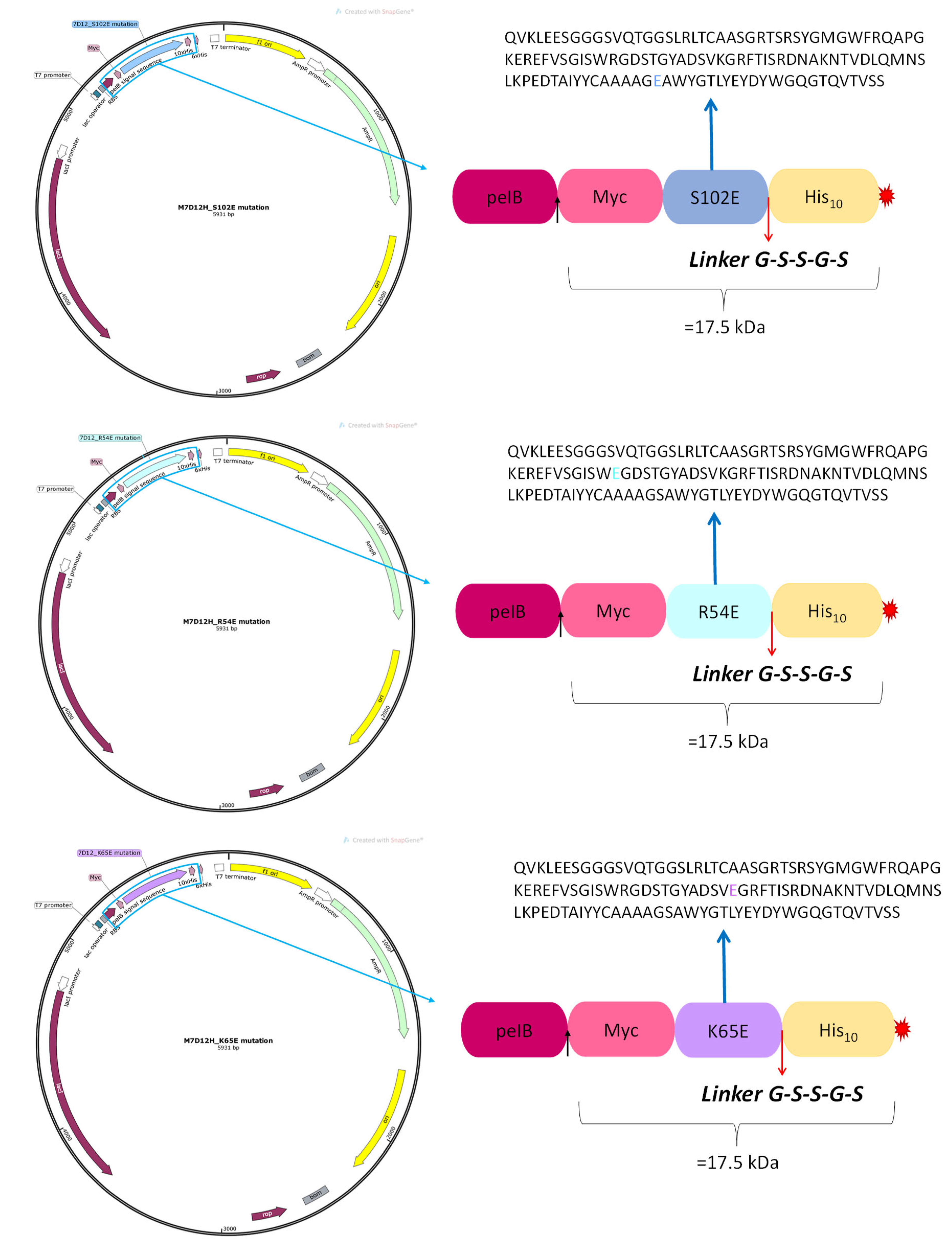
2.3. Site-Directed Mutagenesis
- R54E: Arginine (R) at position 54 substituted by glutamic acid (E);
- K65E: Lysine (K) at position 65 replaced by glutamic acid (E);
- S102E: Serine (S) at position 102 substituted by glutamic acid (E).
2.4. Nanobody Expression and Purification
2.5. Skin Source
2.6. Nanobody Quantification by Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Protein Stability in the Presence of Skin and Current
2.8. Nanobody Delivery Studies
2.8.1. Iontophoresis Setup
2.8.2. Using Acetaminophen as a Marker of Electroosmotic Solvent Flow
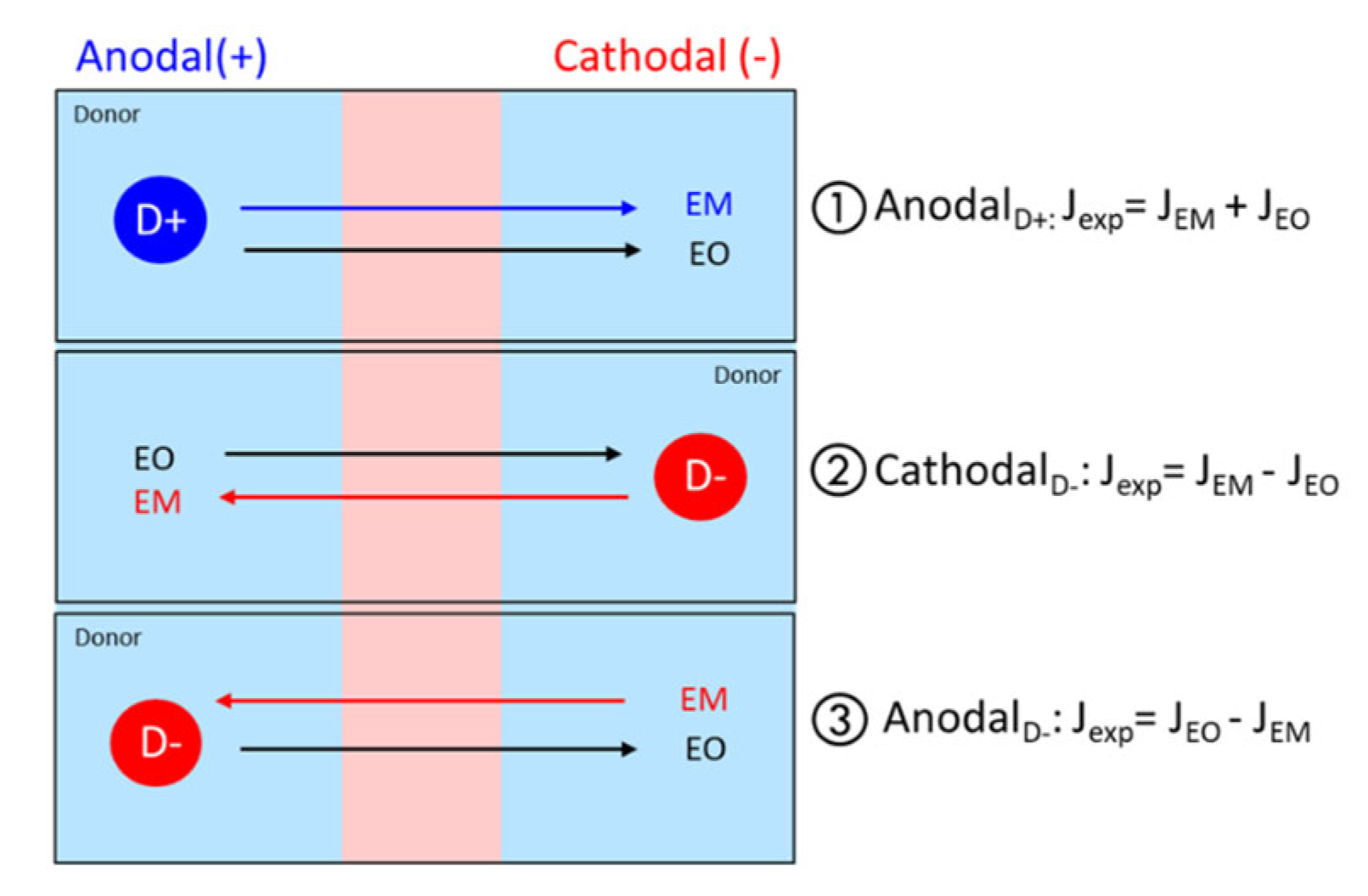
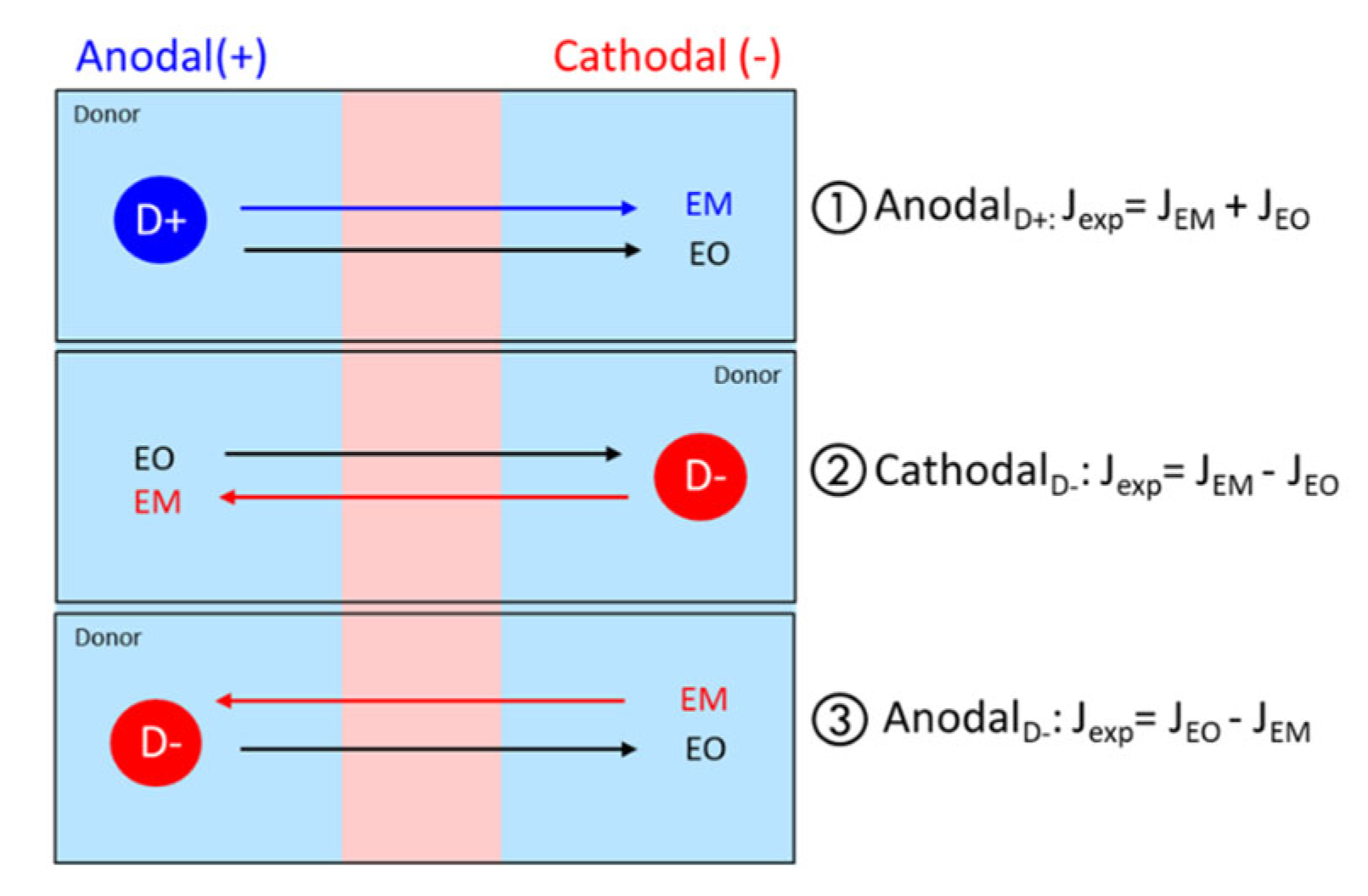
2.8.3. Estimation of EM and EO Contributions
- Anodal iontophoresis for a positively charged protein P (+):
- Cathodal iontophoresis for a negatively charged protein P (−):
- Anodal iontophoresis for a negatively charged protein P (−):
2.9. Immunohistochemistry
2.9.1. Microscope Sample Preparation
2.9.2. Microscope Images
2.10. Statistical Analysis
3. Results and Discussion
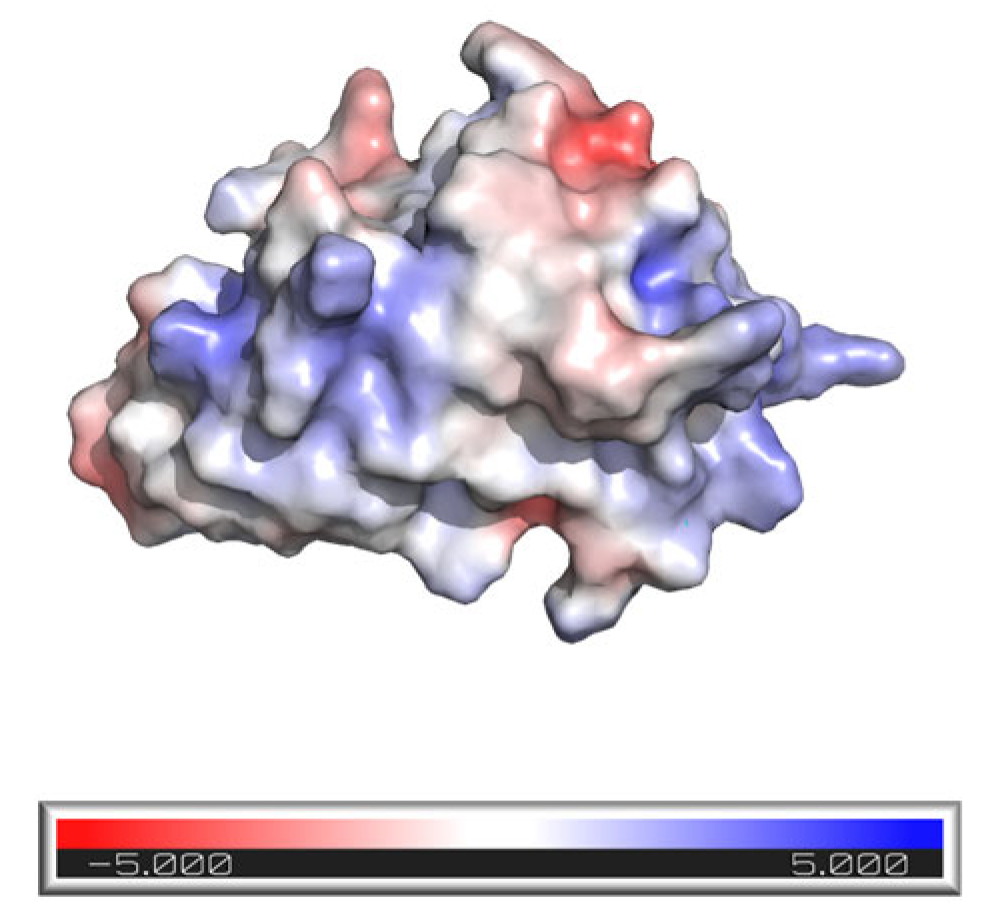
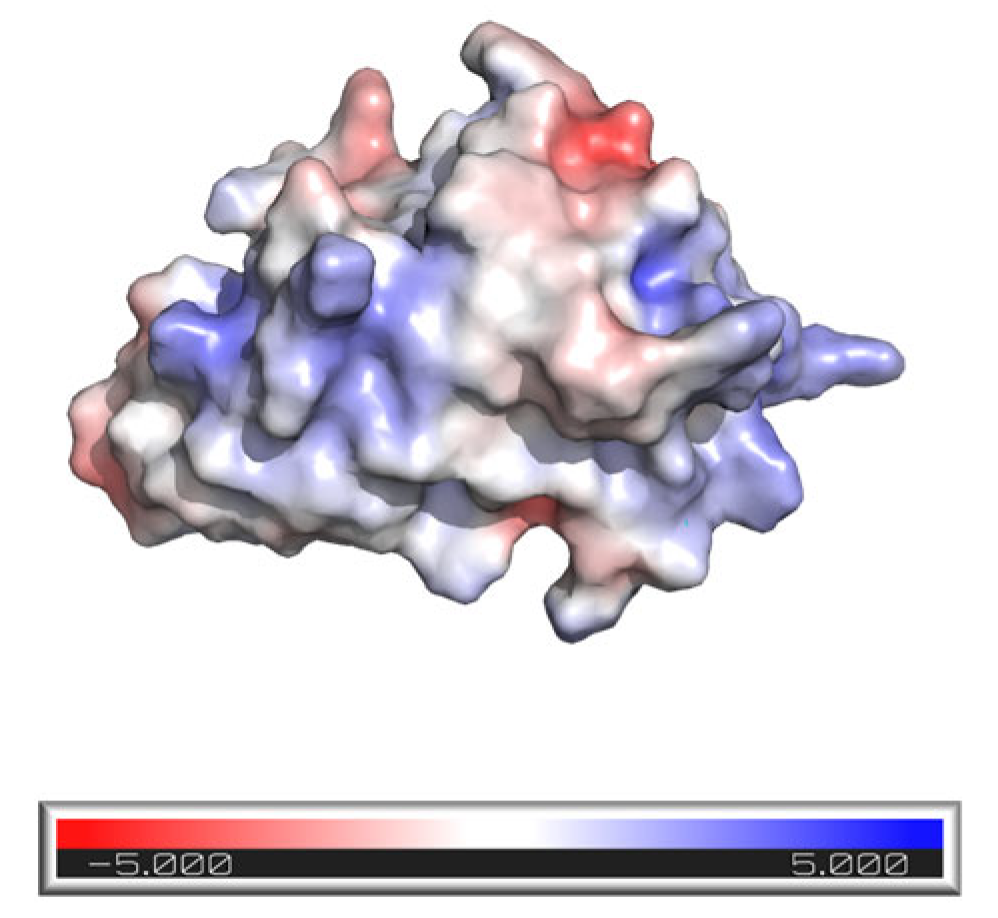
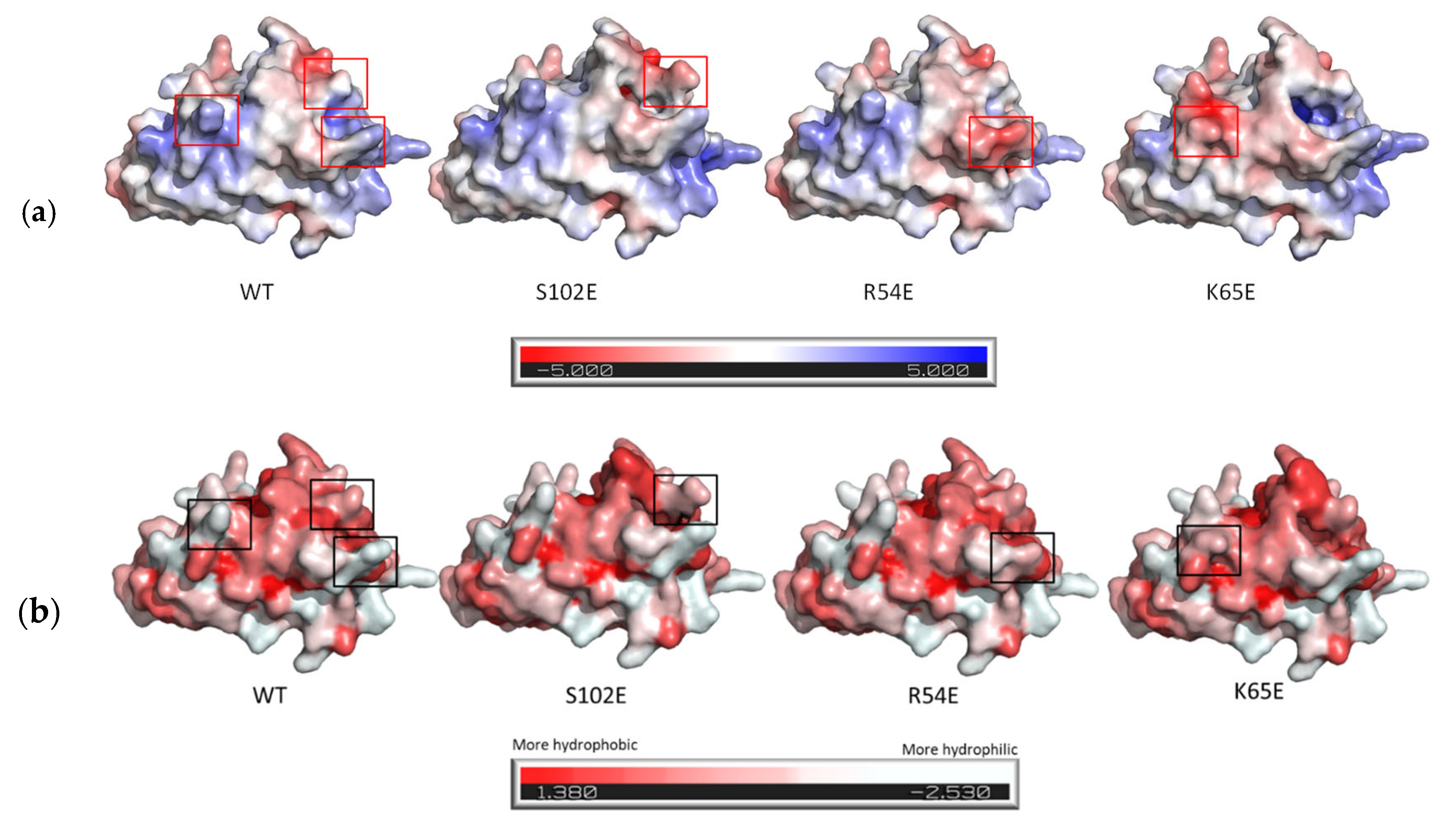
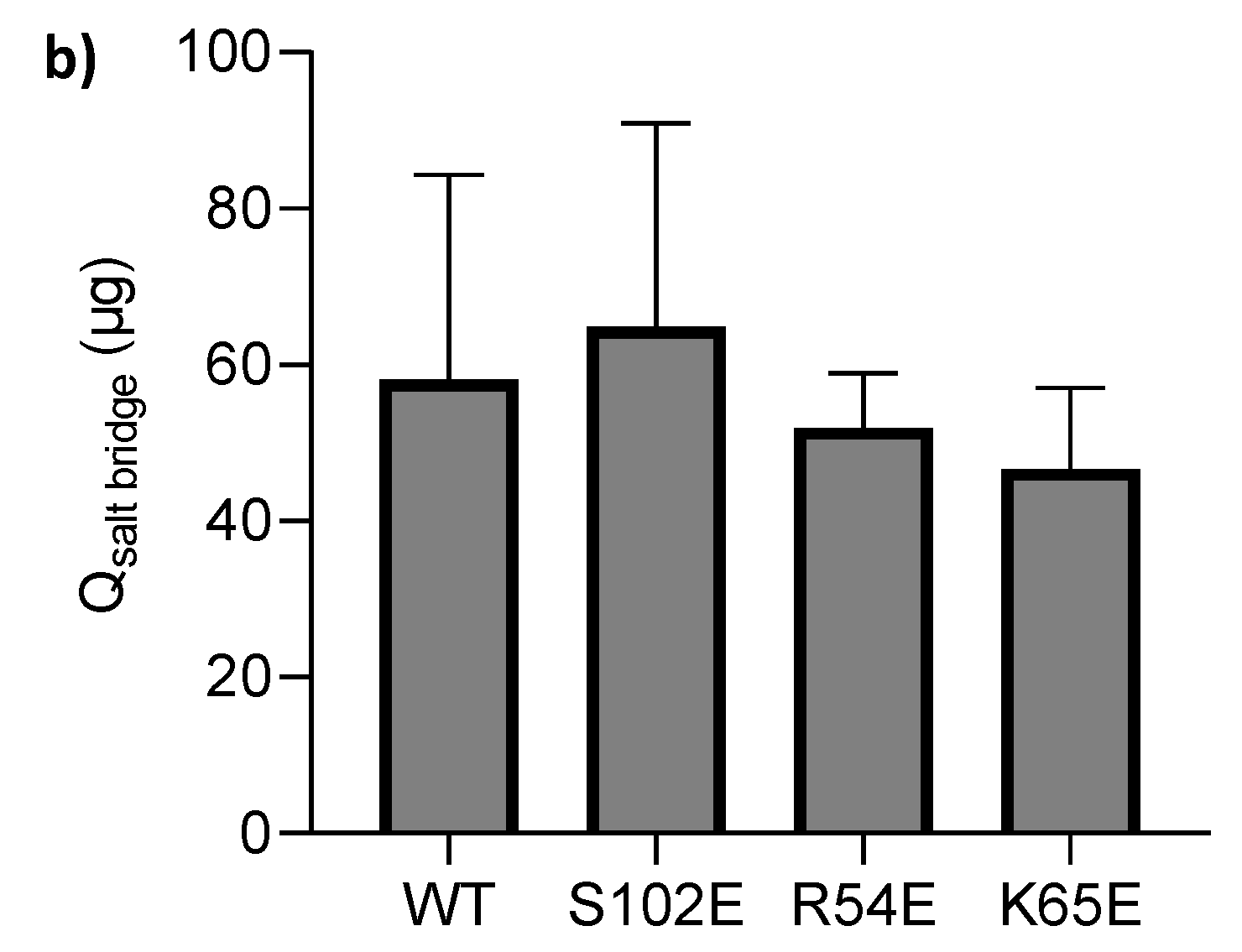
3.1. M7D12H Plasmid Design
3.2. Nanobody Expression and Purification
3.3. Protein Stability in the Presence of Skin and Current
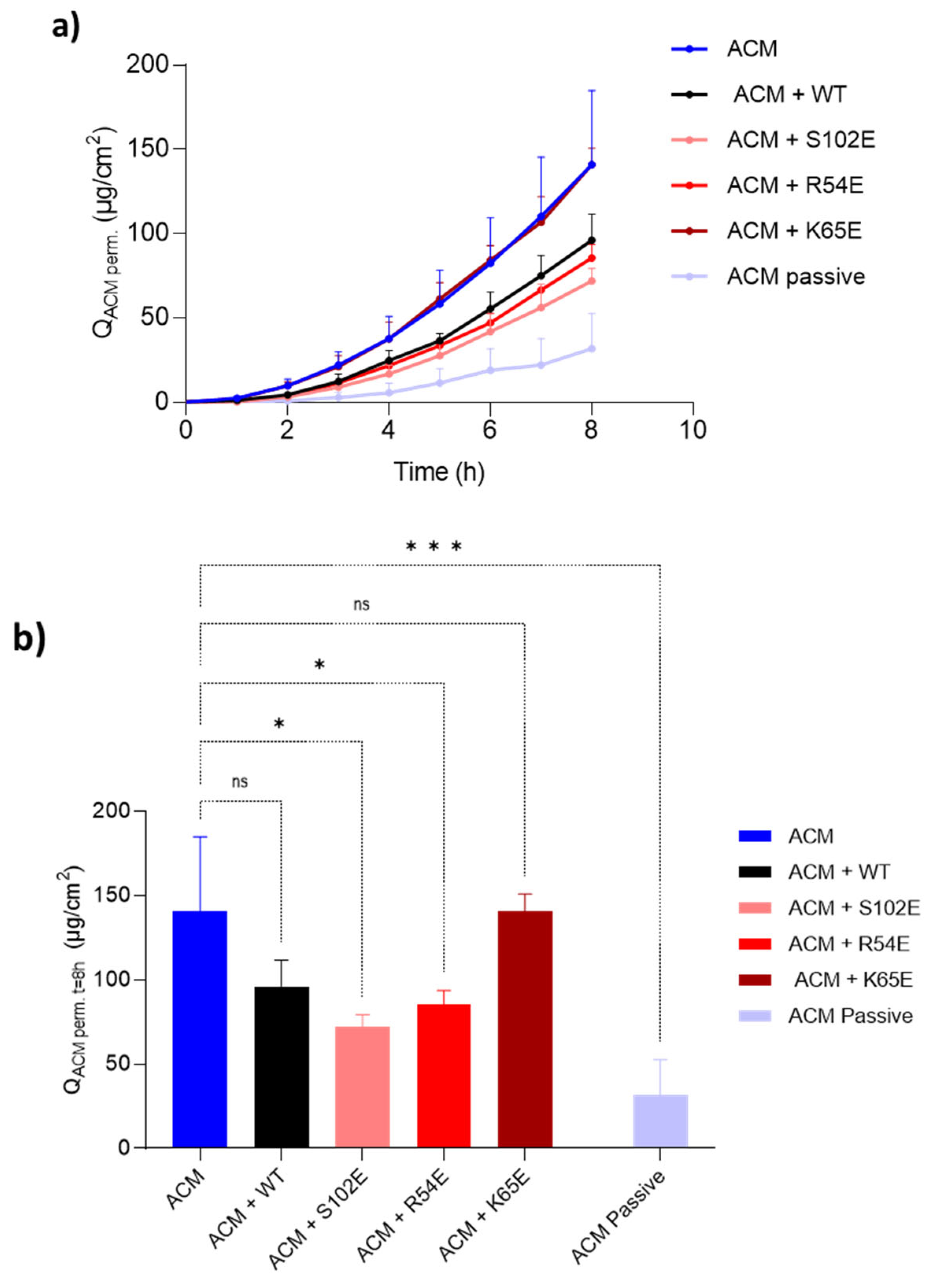
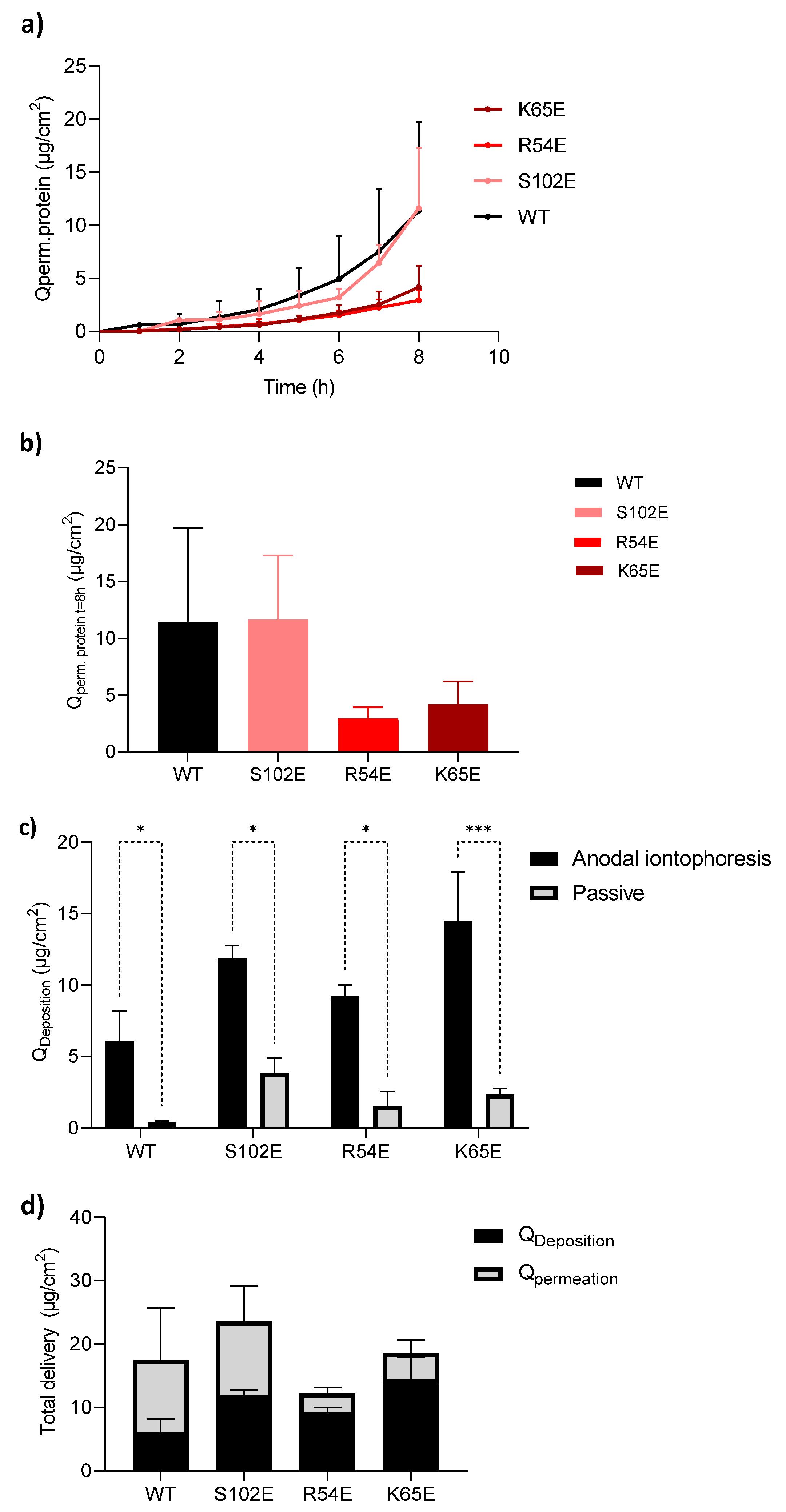
3.4. Investigation of Nanobody Iontophoretic Transport
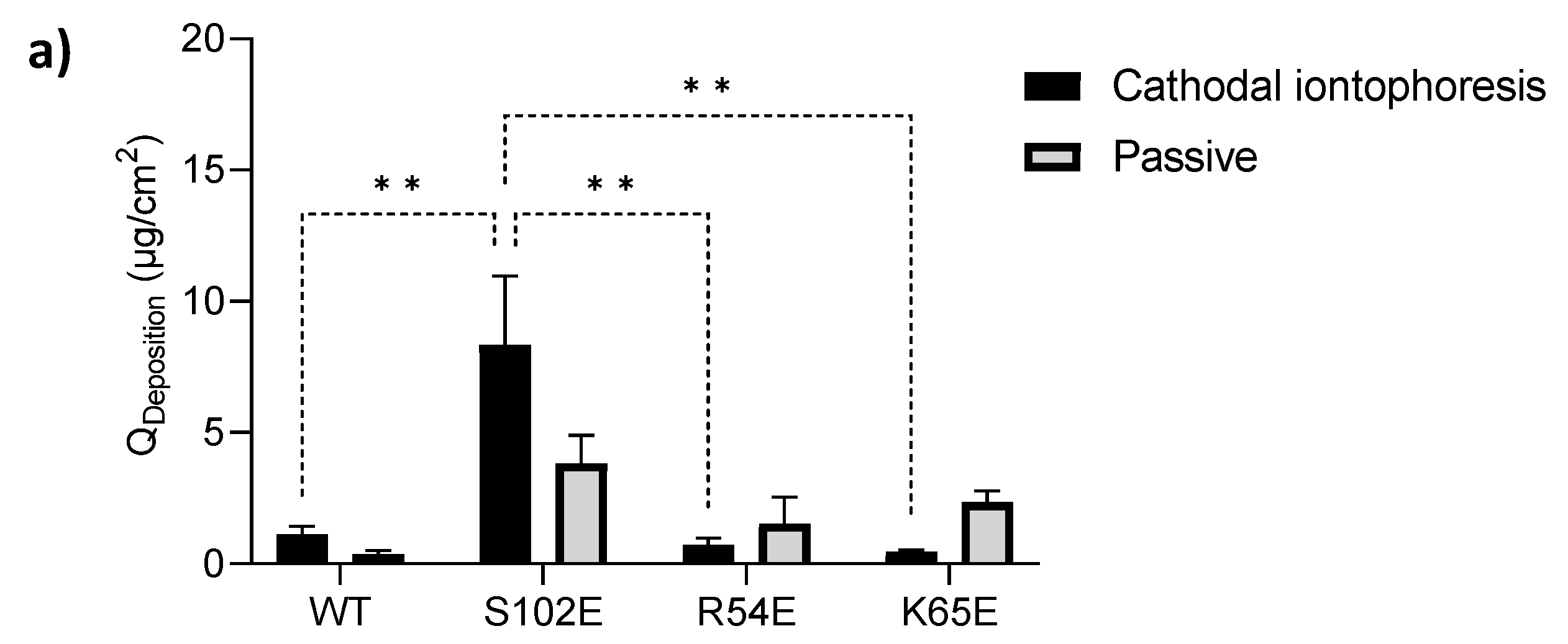
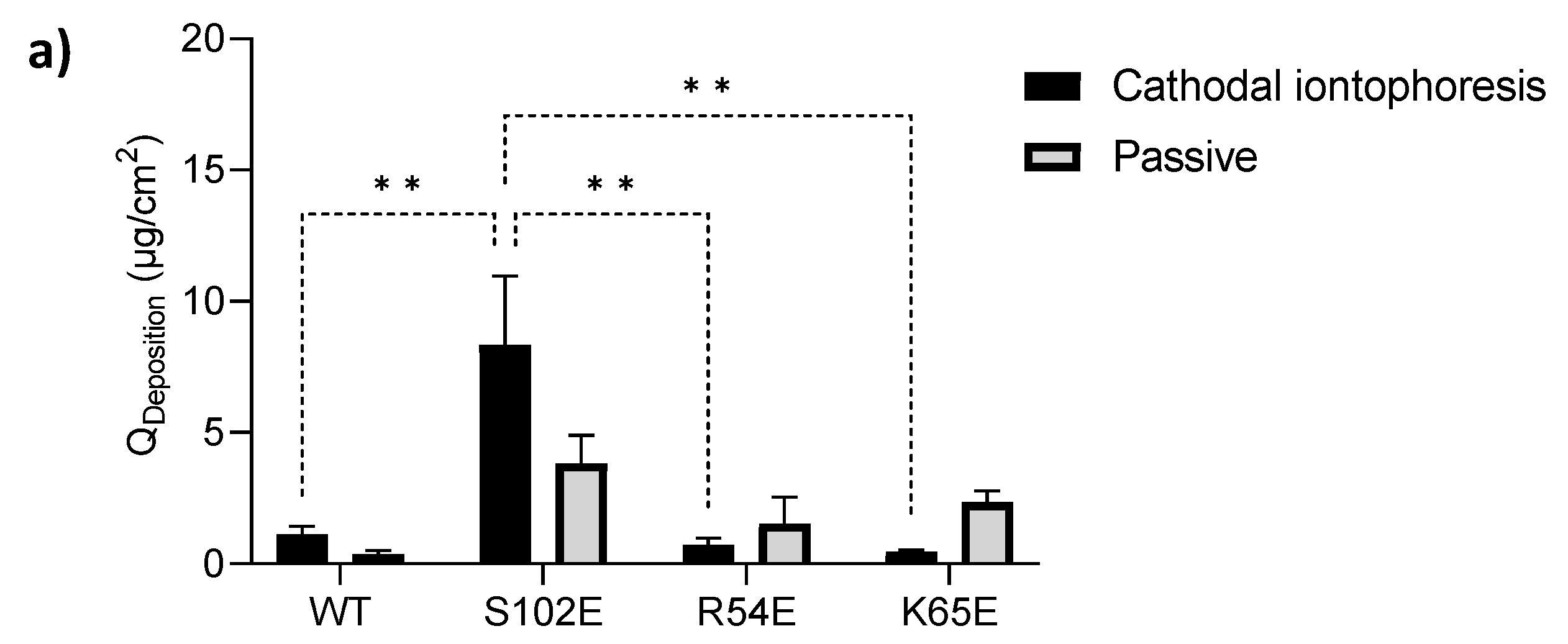
3.4.1. Cathodal Iontophoresis
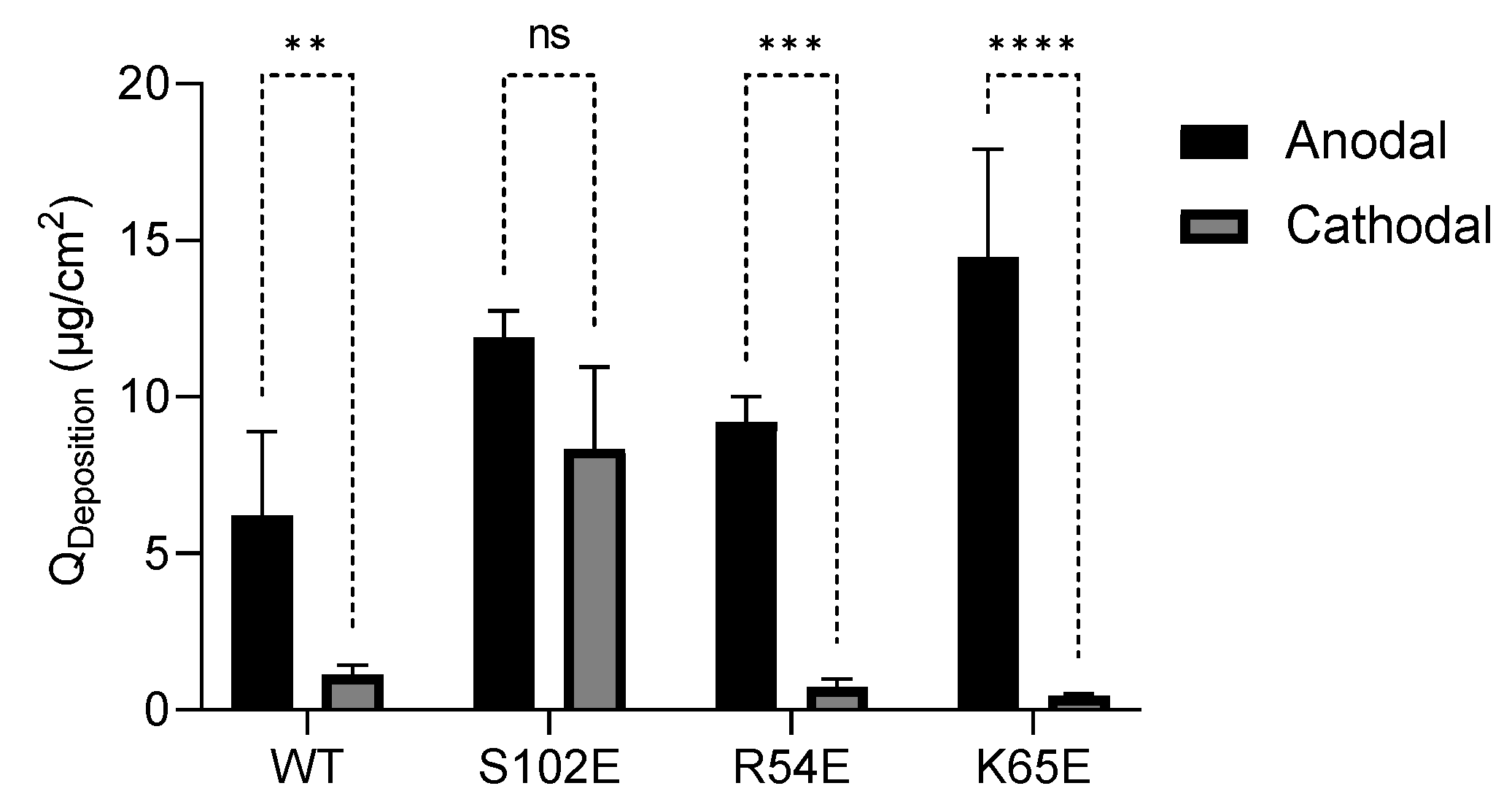
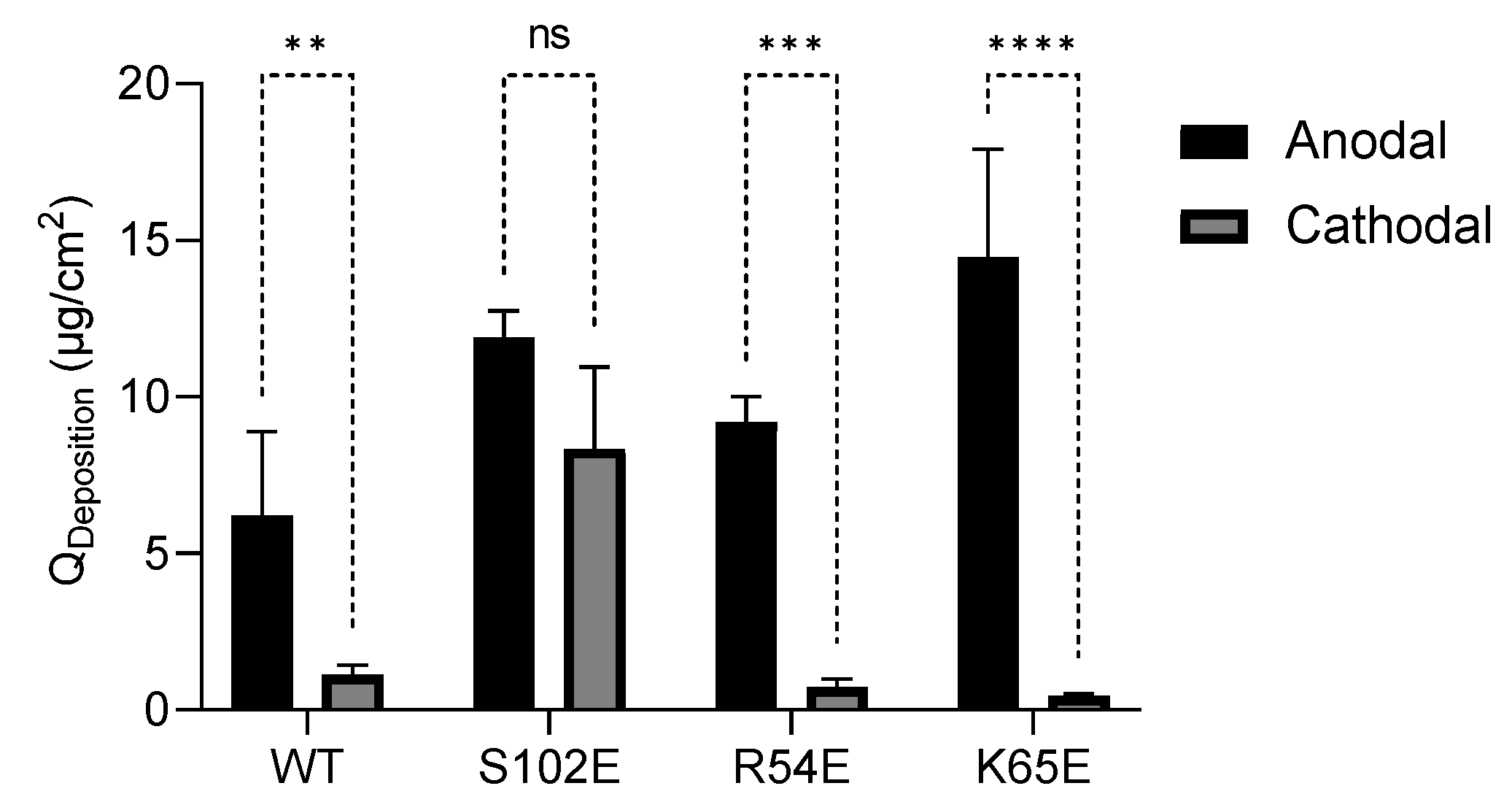
3.4.2. Anodal Iontophoresis
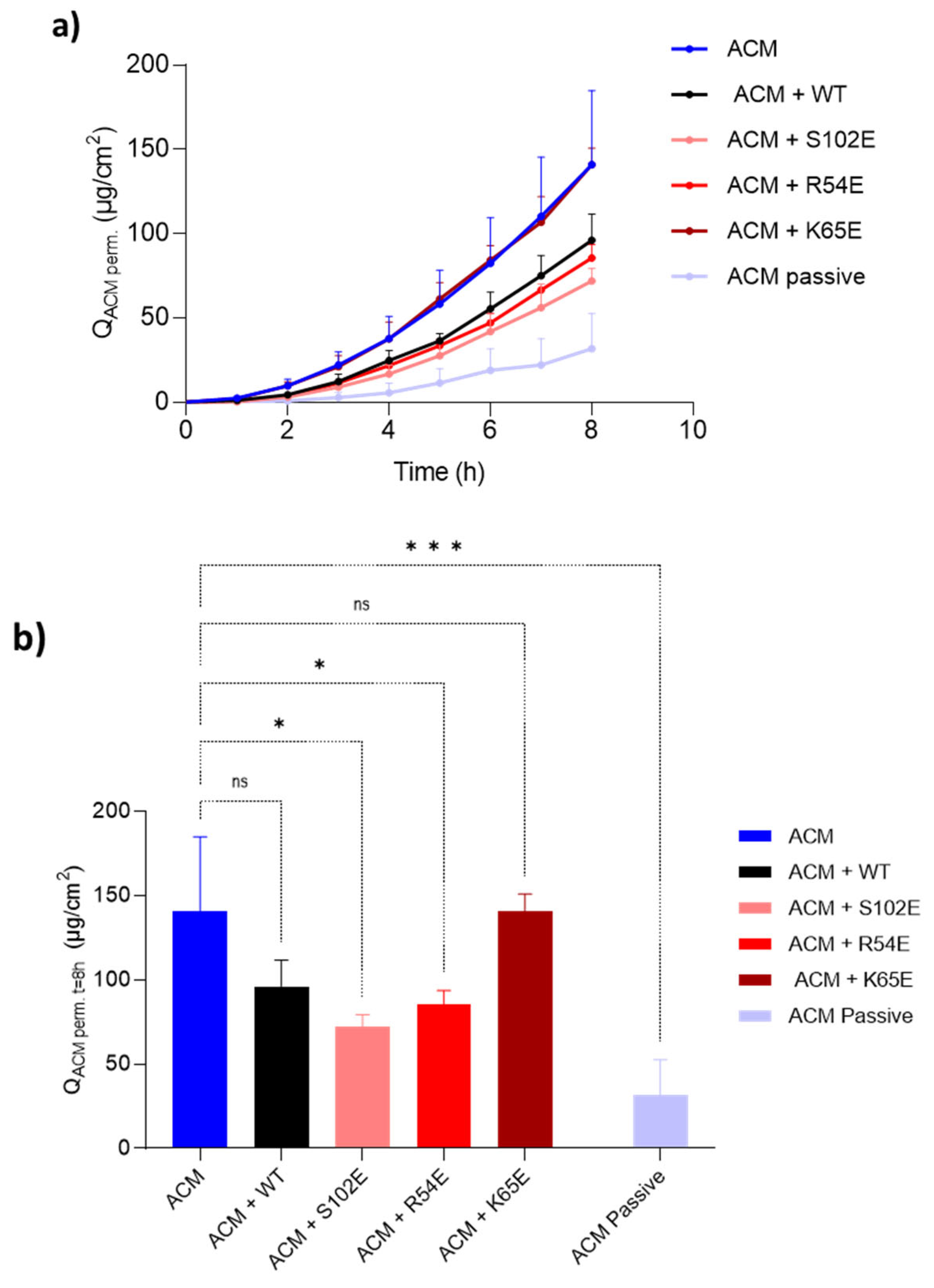
3.4.3. Acetaminophen Co-Iontophoresis
3.5. Therapeutic Relevance of the Iontophoretic Delivery of 7D12
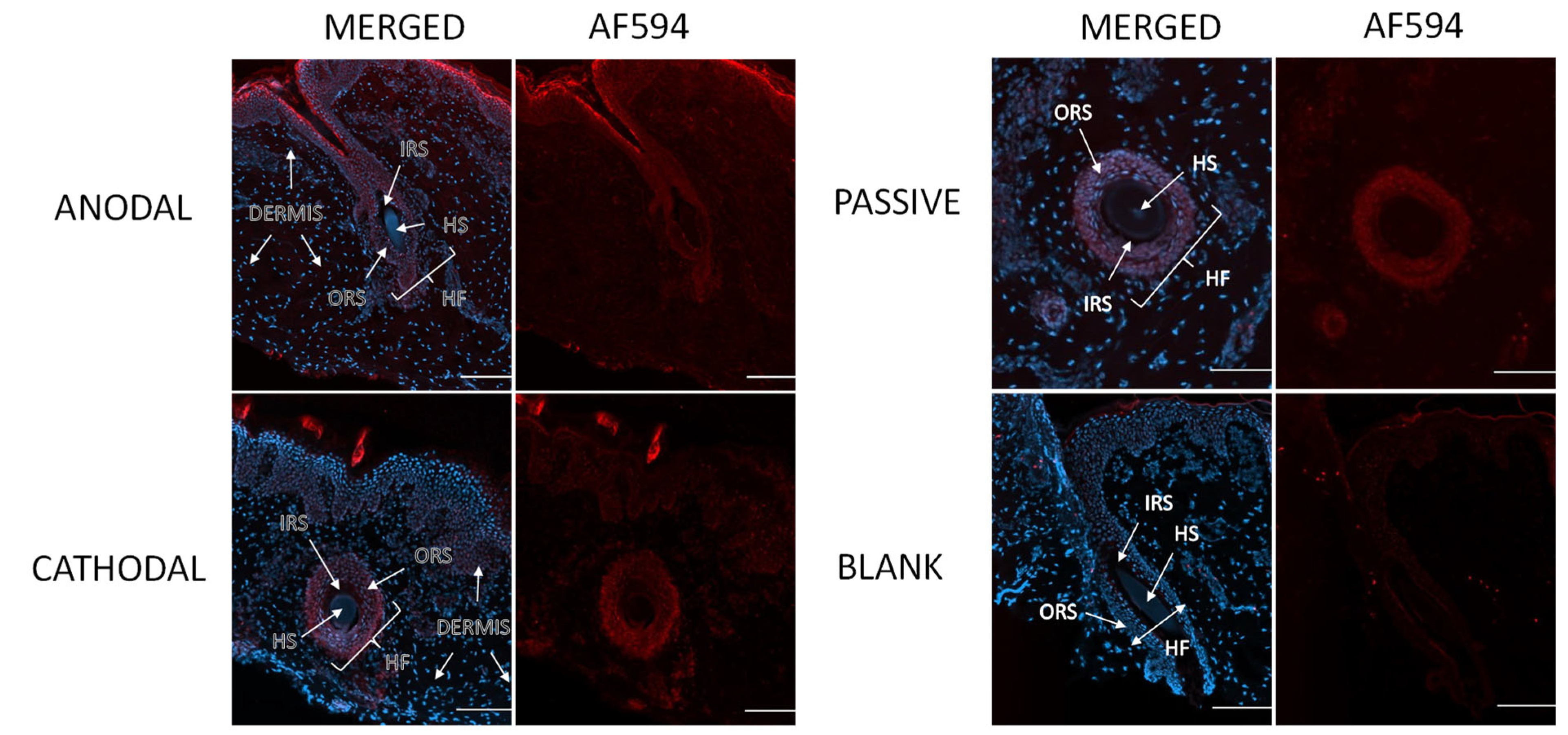
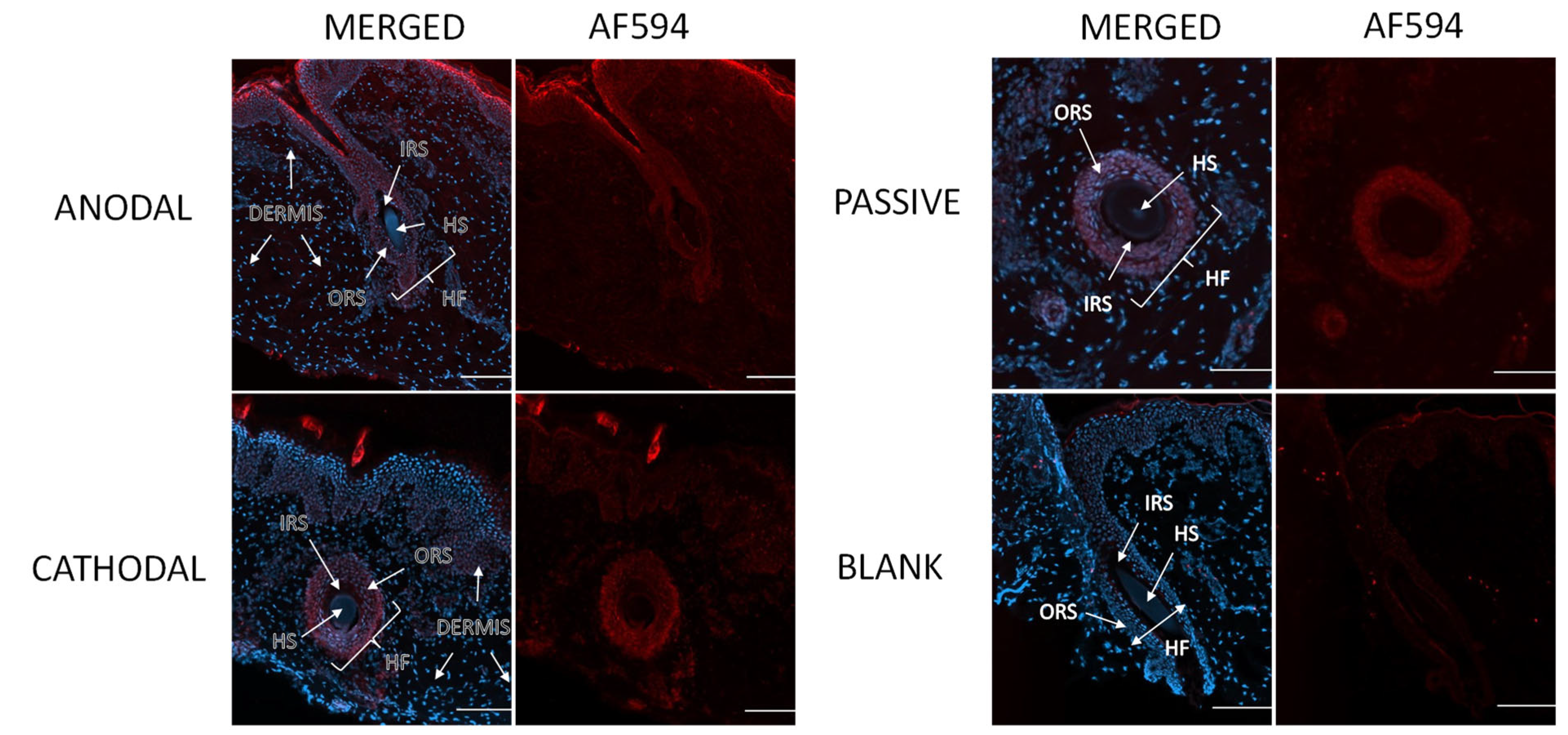
3.6. Visualization of Penetration Pathways
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hasan, M.; Khatun, A.; Kogure, K. Iontophoresis of Biological Macromolecular Drugs. Pharmaceutics 2022, 14, 525. [Google Scholar] [CrossRef] [PubMed]
- Alkilani, A.Z.; McCrudden, M.T.; Donnelly, R.F. Transdermal Drug Delivery: Innovative Pharmaceutical Developments Based on Disruption of the Barrier Properties of the stratum corneum. Pharmaceutics 2015, 7, 438–470. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Du, L.; Li, Q.; Li, Q.; Zhu, L.; Yang, M.; Wang, X.; Zhao, B.; Ma, S. How physical techniques improve the transdermal permeation of therapeutics: A review. Medicine 2022, 101, e29314. [Google Scholar] [CrossRef]
- Chaulagain, B.; Jain, A.; Tiwari, A.; Verma, A.; Jain, S.K. Passive delivery of protein drugs through transdermal route. Artif. Cells Nanomed. Biotechnol. 2018, 46, 472–487. [Google Scholar] [CrossRef]
- Bouwstra, J.A.; Dubbelaar, F.E.; Gooris, G.S.; Ponec, M. The lipid organisation in the skin barrier. Acta Derm. -Venereol. 2000, 208, 23–30. [Google Scholar]
- Naik, A.; Kalia, Y.N.; Guy, R.H. Transdermal drug delivery: Overcoming the skin’s barrier function. Pharm. Sci. Technol. Today 2000, 3, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.F.; Lee, D.; Carlson, R.; Rao, G.S.; Hui, H.W.; Adjei, L.; Herrin, M.; Sundberg, D.; Hsu, L. The Effects of Formulation Variables on Iontophoretic Transdermal Delivery of Leuprolide to Humans. Drug Dev. Ind. Pharm. 1993, 19, 1557–1571. [Google Scholar] [CrossRef]
- Cázares-Delgadillo, J.; Naik, A.; Ganem-Rondero, A.; Quintanar-Guerrero, D.; Kalia, Y.N. Transdermal delivery of cytochrome C--A 12.4 kDa protein--across intact skin by constant-current iontophoresis. Pharm. Res. 2007, 24, 1360–1368. [Google Scholar] [CrossRef]
- Dubey, S.; Kalia, Y.N. Non-invasive iontophoretic delivery of enzymatically active ribonuclease A (13.6 kDa) across intact porcine and human skins. J. Control. Release 2010, 145, 203–209. [Google Scholar] [CrossRef]
- Dubey, S.; Kalia, Y.N. Electrically-assisted delivery of an anionic protein across intact skin: Cathodal iontophoresis of biologically active ribonuclease T1. J. Control. Release 2011, 152, 356–362. [Google Scholar] [CrossRef]
- Dubey, S.; Perozzo, R.; Scapozza, L.; Kalia, Y.N. Noninvasive transdermal iontophoretic delivery of biologically active human basic fibroblast growth factor. Mol. Pharm. 2011, 8, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Marro, D.; Guy, R.H.; Delgado-Charro, M.B. Characterization of the iontophoretic permselectivity properties of human and pig skin. J. Control. Release 2001, 70, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Guy, R.H.; Kalia, Y.N.; Delgado-Charro, M.B.; Merino, V.; López, A.; Marro, D. Iontophoresis: Electrorepulsion and electroosmosis. J. Control. Release 2000, 64, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Lapteva, M.; Sallam, M.A.; Goyon, A.; Guillarme, D.; Veuthey, J.L.; Kalia, Y.N. Non-invasive targeted iontophoretic delivery of cetuximab to skin. Expert. Opin. Drug Deliv. 2020, 17, 589–602. [Google Scholar] [CrossRef]
- Abla, N.; Naik, A.; Guy, R.H.; Kalia, Y.N. Effect of charge and molecular weight on transdermal peptide delivery by iontophoresis. Pharm. Res. 2005, 22, 2069–2078. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.; Kalia, Y.N. Understanding the poor iontophoretic transport of lysozyme across the skin: When high charge and high electrophoretic mobility are not enough. J. Control. Release 2014, 183, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, Y.B.; Carrupt, P.A.; Naik, A.; Guy, R.H.; Kalia, Y.N. Structure-permeation relationships for the non-invasive transdermal delivery of cationic peptides by iontophoresis. Eur. J. Pharm. Sci. 2006, 29, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef]
- Huang, L.; Muyldermans, S.; Saerens, D. Nanobodies®: Proficient tools in diagnostics. Expert. Rev. Mol. Diagn. 2010, 10, 777–785. [Google Scholar] [CrossRef]
- Frenken, L.G.; van der Linden, R.H.; Hermans, P.W.; Bos, J.W.; Ruuls, R.C.; de Geus, B.; Verrips, C.T. Isolation of antigen specific llama VHH antibody fragments and their high level secretion by Saccharomyces cerevisiae. J. Biotechnol. 2000, 78, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Frenken, L.G.; Hessing, J.G.; Van den Hondel, C.A.; Verrips, C.T. Recent advances in the large-scale production of antibody fragments using lower eukaryotic microorganisms. Res. Immunol. 1998, 149, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Chakravarty, R.; Goel, S.; Cai, W. Nanobody: The “magic bullet” for molecular imaging? Theranostics 2014, 4, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Van Audenhove, I.; Gettemans, J. Nanobodies as Versatile Tools to Understand, Diagnose, Visualize and Treat Cancer. EBioMedicine 2016, 8, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef] [PubMed]
- Baral, T.N.; Magez, S.; Stijlemans, B.; Conrath, K.; Vanhollebeke, B.; Pays, E.; Muyldermans, S.; De Baetselier, P. Experimental therapy of African trypanosomiasis with a nanobody-conjugated human trypanolytic factor. Nat. Med. 2006, 12, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Retamozo, V.; Backmann, N.; Senter, P.D.; Wernery, U.; De Baetselier, P.; Muyldermans, S.; Revets, H. Efficient cancer therapy with a nanobody-based conjugate. Cancer Res. 2004, 64, 2853–2857. [Google Scholar] [CrossRef]
- Rothbauer, U.; Zolghadr, K.; Muyldermans, S.; Schepers, A.; Cardoso, M.C.; Leonhardt, H. A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol. Cell Proteom. 2008, 7, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Saerens, D.; Frederix, F.; Reekmans, G.; Conrath, K.; Jans, K.; Brys, L.; Huang, L.; Bosmans, E.; Maes, G.; Borghs, G.; et al. Engineering camel single-domain antibodies and immobilization chemistry for human prostate-specific antigen sensing. Anal. Chem. 2005, 77, 7547–7555. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Peng, H.; Zeng, K. Recent advances on the roles of epidermal growth factor receptor in psoriasis. Am. J. Transl. Res. 2019, 11, 520–528. [Google Scholar]
- ICH M10 on Bioanalytical Method Validation. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-guideline-m10-bioanalytical-method-validation-step-2b_en.pdf. (accessed on 13 December 2022).
- SnapGene. Available online: https://www.snapgene.com/resources/plasmid-files/?set=pet_and_duet_vectors_(novagen)&plasmid=pET-22b%28%2B%29 (accessed on 11 January 2022).
- Singh, P.; Sharma, L.; Kulothungan, S.R.; Adkar, B.V.; Prajapati, R.S.; Ali, P.S.S.; Krishnan, B.; Varadarajan, R. Effect of signal peptide on stability and folding of Escherichia coli thioredoxin. PLoS ONE 2013, 8, e63442. [Google Scholar] [CrossRef]
- Turner, N.G.; Cullander, C.; Guy, R.H. Determination of the pH gradient across the stratum corneum. J. Investig. Dermatol. Symp. Proc. 1998, 3, 110–113. [Google Scholar] [CrossRef]
- Ohman, H.; Vahlquist, A. In vivo studies concerning a pH gradient in human stratum corneum and upper epidermis. Acta Derm. -Venereol. 1994, 74, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Hanson, K.M.; Behne, M.J.; Barry, N.P.; Mauro, T.M.; Gratton, E.; Clegg, R.M. Two-photon fluorescence lifetime imaging of the skin stratum corneum pH gradient. Biophys. J. 2002, 83, 1682–1690. [Google Scholar] [CrossRef]
- Plessis, J.L.D.; Stefaniak, A.B.; Wilhelm, K.P. Measurement of Skin Surface pH. Curr. Probl. Dermatol. 2018, 54, 19–25. [Google Scholar] [PubMed]
- Pikal, M.J. The role of electroosmotic flow in transdermal iontophoresis. Adv. Drug Deliv. Rev. 2001, 46, 281–305. [Google Scholar] [CrossRef] [PubMed]
- Padula, C.; Sartori, F.; Marra, F.; Santi, P. The influence of iontophoresis on acyclovir transport and accumulation in rabbit ear skin. Pharm. Res. 2005, 22, 1519–1524. [Google Scholar] [CrossRef]
- Acetaminophen (Compound). Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Acetaminophen#section=Depositor-Supplied-Synonyms (accessed on 15 December 2022).
- Pikal, M.J.; Shah, S. Transport mechanisms in iontophoresis. III. An experimental study of the contributions of electroosmotic flow and permeability change in transport of low and high molecular weight solutes. Pharm. Res. 1990, 7, 222–229. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar]
- Kus, K.J.B.; Ruiz, E.S. Non-Surgical Treatments for Keratinocyte Carcinomas. Adv. Ther. 2021, 38, 5635–5648. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/125084s279lbl.pdf (accessed on 10 November 2022).
- Schmitz, K.R.; Bagchi, A.; Roovers, R.C.; en Henegouwen, P.M.V.B.; Ferguson, K.M. Ferguson, Structural evaluation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure 2013, 21, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Roovers, R.C.; Vosjan, M.J.; Laeremans, T.; el Khoulati, R.; de Bruin, R.C.; Ferguson, K.M.; Verkleij, A.J.; van Dongen, G.A.; van Bergen en Henegouwen, P.M. A biparatopic anti-EGFR nanobody efficiently inhibits solid tumour growth. Int. J. Cancer 2011, 129, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Hama, S.; Kimura, Y.; Mikami, A.; Shiota, K.; Toyoda, M.; Tamura, A.; Nagasaki, Y.; Kanamura, K.; Kajimoto, K.; Kogure, K. Electric Stimulus Opens Intercellular Spaces in Skin. J. Biol. Chem. 2014, 289, 2450–2456. [Google Scholar] [CrossRef] [PubMed]
- Fukuta, T.; Oshima, Y.; Michiue, K.; Tanaka, D.; Kogure, K. Non-invasive delivery of biological macromolecular drugs into the skin by iontophoresis and its application to psoriasis treatment. J. Control. Release 2020, 323, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Niderla-Bielinska, J.; Jankowska-Steifer, E.; Moskalewski, S. Keratinization of outer root sheath cells is prevented by contact with inner root sheath of rat hair follicles. Arch. Dermatol. Res. 2009, 301, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Pena, J.C.; Kelekar, A.; Fuchs, E.V.; Thompson, C.B. Manipulation of outer root sheath cell survival perturbs the hair-growth cycle. EMBO J. 1999, 18, 3596–3603. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Fuchs, E. Epidermal stem cells of the skin. Annu. Rev. Cell Dev. Biol. 2006, 22, 339–373. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-González, M.; Farías, C.; Tello, S.; Pérez-Etcheverry, D.; Romero, A.; Zúñiga, R.; Ribeiro, C.H.; Lorenzo-Ferreiro, C.; Molina, M.C. Optimization of culture conditions for the expression of three different insoluble proteins in Escherichia coli. Sci. Rep. 2019, 9, 16850. [Google Scholar] [CrossRef]
- Francis, D.M.; Page, R. Strategies to optimize protein expression in E. coli. Curr. Protoc. Protein Sci. 2010, 61, 5–24. [Google Scholar] [CrossRef]
- San-Miguel, T.; Pérez-Bermúdez, P. Gavidia, I. Production of soluble eukaryotic recombinant proteins in E. coli is favoured in early log-phase cultures induced at low temperature. Springerplus 2013, 2, 89. [Google Scholar] [CrossRef] [PubMed]
- Galloway, C.A.; Sowden, M.P.; Smith, H.C. Increasing the yield of soluble recombinant protein expressed in E. coli by induction during late log phase. Biotechniques 2003, 34, 524–530. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WB | EMTX | PMTX | |
|---|---|---|---|
| LOQ (ng/mL) | 5.18 | 5.14 | 5.07 |
| LOD (ng/mL) | 1.73 | 1.71 | 1.69 |
| M7D12HWT | S102E | R54E | K65E | |
|---|---|---|---|---|
| Approximate amount of protein per liter of culture (mg/L) | 23–28 | 22 | 17 | 16 |
| Mass of bacterial pellet (g) | 7.9 | 7.9 | 6.1 | 9.8 |
| Protein quantity (mg/g bacteria) | 3.2 | 2.8 | 2.8 | 1.6 |
| MW (Da) | pI (Isoelectric Point) | Z (Net Charge at pH 7.4) | GRAVY | |
|---|---|---|---|---|
| M7D12HWT | 17486.89 | 6.50 | −3.35 | −0.703 |
| S102E | 17528.93 | 6.33 | −4.35 | −0.720 |
| R54E | 17459.82 | 6.17 | −5.35 | −0.697 |
| K65E | 17487.84 | 6.17 | −5.34 | −0.701 |
| Anodal Iontophoresis | ||
|---|---|---|
| QDeposition (µg·cm−2) | QPermeation (µg·cm−2) | |
| M7D12HWT | 6.07 ± 2.11 | 11.39 ± 8.30 |
| S102E | 11.89 ± 0.87 | 11.63 ± 5.67 |
| R54E | 9.22 ± 0.80 | 2.94 ± 0.99 |
| K65E | 14.45 ± 3.45 | 4.18 ± 2.04 |
| QACM perm. t = 8h Cumulative Permeation (µg·cm−2) | IF (QACM Prot(−)/QACM Prot(+)) | ACM Flux JACM 5–8 h (µg·cm−2·h−1) | Vw (JACM/CACM donor ) (µL·cm−2·h−1) | |
|---|---|---|---|---|
| ACM | 140.96 ± 43.94 | ─ | 27.59 ± 8.00 | 12.17 ± 3.53 |
| ACM + M7D12HWT | 96.08 ± 15.55 | 1.47 | 19.85 ± 3.62 | 8.76 ± 1.60 |
| ACM + S102E | 71.93 ± 7.32 | 1.96 | 14.75 ± 0.84 | 6.50 ± 0.37 |
| ACM + R54E | 85.52 ± 8.05 | 1.65 | 17.52 ± 2.68 | 7.73 ± 1.18 |
| ACM + K65E | 140.83 ± 9.93 | 1.00 | 26.15 ± 1.95 | 11.53 ± 0.86 |
| Protein Flux Jexp. protein 4–8 h (µg·cm−2·h−1) | JEO protein cacluated (µg·cm−2·h−1) | JEM protein estimated (µg·cm−2·h−1) | |
|---|---|---|---|
| M7D12HWT | 3.53 ± 1.81 | 44.12 | 40.59 |
| S102E | 4.20 ± 3.24 | 29.92 | 25.72 |
| R54E | 0.56 ± 0.25 | 38.94 | 38.38 |
| K65E | 0.85 ± 0.44 | 51.78 | 50.93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahraoui, P.F.; Vadas, O.; Kalia, Y.N. Non-Invasive Delivery of Negatively Charged Nanobodies by Anodal Iontophoresis: When Electroosmosis Dominates Electromigration. Pharmaceutics 2024, 16, 539. https://doi.org/10.3390/pharmaceutics16040539
Sahraoui PF, Vadas O, Kalia YN. Non-Invasive Delivery of Negatively Charged Nanobodies by Anodal Iontophoresis: When Electroosmosis Dominates Electromigration. Pharmaceutics. 2024; 16(4):539. https://doi.org/10.3390/pharmaceutics16040539
Chicago/Turabian StyleSahraoui, Phedra Firdaws, Oscar Vadas, and Yogeshvar N. Kalia. 2024. "Non-Invasive Delivery of Negatively Charged Nanobodies by Anodal Iontophoresis: When Electroosmosis Dominates Electromigration" Pharmaceutics 16, no. 4: 539. https://doi.org/10.3390/pharmaceutics16040539
APA StyleSahraoui, P. F., Vadas, O., & Kalia, Y. N. (2024). Non-Invasive Delivery of Negatively Charged Nanobodies by Anodal Iontophoresis: When Electroosmosis Dominates Electromigration. Pharmaceutics, 16(4), 539. https://doi.org/10.3390/pharmaceutics16040539