
Risk of Potentially Neurotoxic Exposure in Infants Under High-Dose Cefepime Treatment—A Pharmacometric Simulation Study

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Pharmacometric Models
2.2.1. Pharmacometric Model 1: Neonates, Infants, and Children
2.2.2. Pharmacometric Model 2: Critically Ill Infants and Children
2.3. Virtual Patient Population
2.4. Cefepime Exposure Simulations and Outcomes
2.4.1. Dose-Exposure Simulations
2.4.2. Safety- and Efficacy-Related Exposure Outcomes
2.5. Sensitivity Analyses
3. Results
3.1. Pharmacometric Models and Virtual Patient Population
3.2. Cefepime Exposure Simulations and Outcomes
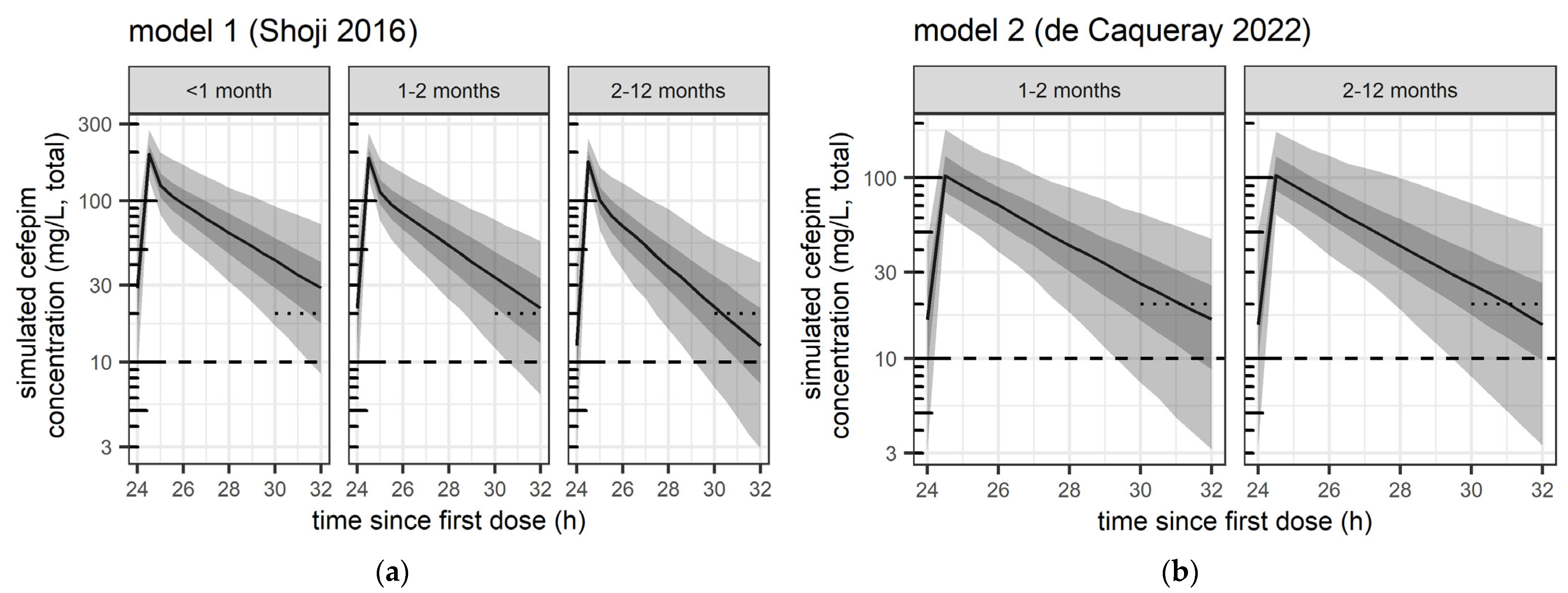
3.2.1. Dose-Exposure Simulations
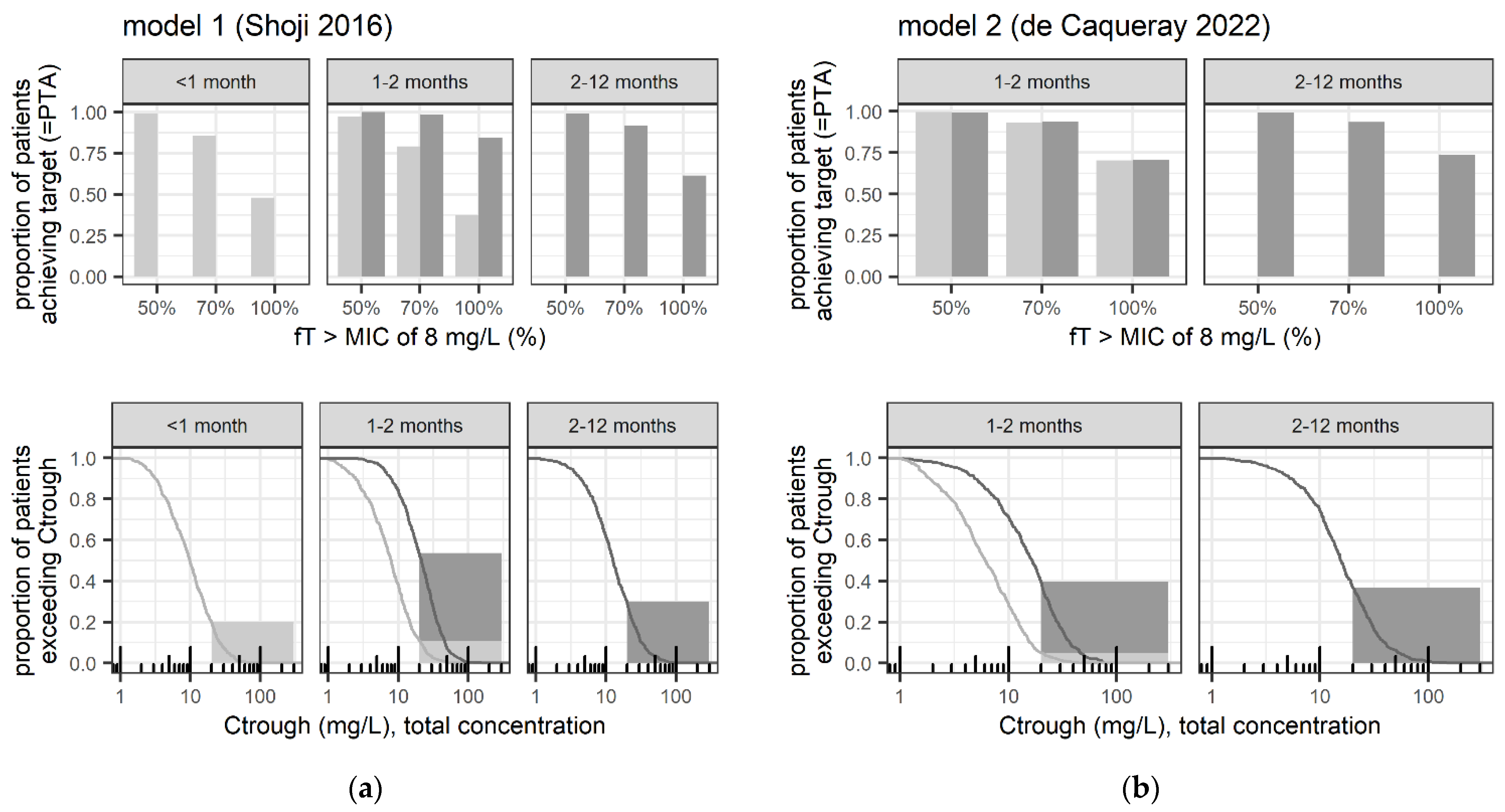
3.2.2. Safety- and Efficacy-Related Exposure Outcomes
3.3. Sensitivity Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ctrough | Trough concentration (total concentration) |
| GA | Gestational age |
| fT>MICXXmg/L | Time during which the free drug concentration (fT) remains above a defined minimal inhibitory concentration (MIC) breakpoint (XX in mg/L) |
| MIC | Minimum inhibitory concentration |
| TDM | Therapeutic drug monitoring |
| PMA | Postmenstrual age |
| PTA | Probability of target attainment in terms of a given fT>MIC |
References
- Pais, G.M.; Chang, J.; Barreto, E.F.; Stitt, G.; Downes, K.J.; Alshaer, M.H.; Lesnicki, E.; Panchal, V.; Bruzzone, M.; Bumanglag, A.V.; et al. Clinical Pharmacokinetics and Pharmacodynamics of Cefepime. Clin. Pharmacokinet. 2022, 61, 929–953. [Google Scholar] [CrossRef] [PubMed]
- Klastersky, J.; de Naurois, J.; Rolston, K.; Rapoport, B.; Maschmeyer, G.; Aapro, M.; Herrstedt, J.; ESMO Guidelines Committee. Management of Febrile Neutropaenia: ESMO Clinical Practice Guidelines. Ann. Oncol. 2016, 27, v111–v118. [Google Scholar] [CrossRef] [PubMed]
- Lehrnbecher, T.; Robinson, P.D.; Ammann, R.A.; Fisher, B.; Patel, P.; Phillips, R.; Beauchemin, M.P.; Carlesse, F.; Castagnola, E.; Davis, B.L.; et al. Guideline for the Management of Fever and Neutropenia in Pediatric Patients with Cancer and Hematopoietic Cell Transplantation Recipients: 2023 Update. J. Clin. Oncol. 2023, 41, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Deng, T.; Li, S.; Shu, Y.; Wu, Y. Efficacy, Safety, and Tolerability of Antimicrobial Agents for Complicated Intra-Abdominal Infection: A Systematic Review and Network Meta-Analysis. BMC Infect. Dis. 2023, 23, 256. [Google Scholar] [CrossRef]
- Solomkin, J.S.; Mazuski, J.E.; Bradley, J.S.; Rodvold, K.A.; Goldstein, E.J.C.; Baron, E.J.; O’Neill, P.J.; Chow, A.W.; Patchen Dellinger, E.; Eachempati, S.R.; et al. Diagnosis and Management of Complicated Intra-Abdominal Infection in Adults and Children: Guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin. Infect. Dis. 2010, 50, 133–164. [Google Scholar] [CrossRef]
- Uygun, V.; Karasu, G.T.; Ogunc, D.; Yesilipek, A.; Hazar, V. Piperacillin/Tazobactam versus Cefepime for the Empirical Treatment of Pediatric Cancer Patients with Neutropenia and Fever: A Randomized and Open-Label Study. Pediatr. Blood Cancer 2009, 53, 610–614. [Google Scholar] [CrossRef]
- Corapcioglu, F.; Sarper, N.; Zengin, E. Monotherapy with Piperacillin/Tazobactam versus Cefepime as Empirical Therapy for Febrile Neutropenia in Pediatric Cancer Patients: A Randomized Comparison. Pediatr. Hematol. Oncol. 2006, 23, 177–186. [Google Scholar] [CrossRef]
- Sano, H.; Kobayashi, R.; Suzuki, D.; Kishimoto, K.; Yasuda, K.; Kobayashi, K. Comparison between Piperacillin/Tazobactam and Cefepime Monotherapies as an Empirical Therapy for Febrile Neutropenia in Children with Hematological and Malignant Disorders: A Prospective, Randomized Study. Pediatr. Blood Cancer 2015, 62, 356–358. [Google Scholar] [CrossRef]
- Sarashina, T.; Kobayashi, R.; Yoshida, M.; Toriumi, N.; Suzuki, D.; Sano, H.; Azuma, H. A Randomized Trial of Cefozopran versus Cefepime as Empirical Antibiotic Treatment of Febrile Neutropenia in Pediatric Cancer Patients. Pediatr. Blood Cancer 2014, 61, 1992–1995. [Google Scholar] [CrossRef]
- European Committee on Antimicrobial Susceptibility Testing EUCAST Clinical Breakpoint Tables v. 14.0. 2024. Available online: https://www.eucast.org/eucast_news/news_singleview?tx_ttnews%5Btt_news%5D=566&cHash=db55f3a8829726044512a1fe74cce41b (accessed on 30 June 2024).
- Reed, M.D.; Yamashita, T.S.; Knupp, C.K.; Veazey, J.M.; Blumer, J.L. Pharmacokinetics of Intravenously and Intramuscularly Administered Cefepime in Infants and Children. Antimicrob. Agents Chemother. 1997, 41, 1783–1787. [Google Scholar] [CrossRef]
- Shoji, K.; Bradley, J.S.; Reed, M.D.; Van Den Anker, J.N.; Domonoske, C.; Capparelli, E.V. Population Pharmacokinetic Assessment and Pharmacodynamic Implications of Pediatric Cefepime Dosing for Susceptible-Dose-Dependent Organisms. Antimicrob. Agents Chemother. 2016, 60, 2150–2156. [Google Scholar] [CrossRef] [PubMed]
- de Cacqueray, N.; Hirt, D.; Zheng, Y.; Bille, E.; Leger, P.L.; Rambaud, J.; Toubiana, J.; Chosidow, A.; Vimont, S.; Callot, D.; et al. Cefepime Population Pharmacokinetics and Dosing Regimen Optimization in Critically Ill Children with Different Renal Function. Clin. Microbiol. Infect. 2022, 28, 1389.e1–1389.e7. [Google Scholar] [CrossRef] [PubMed]
- Lima-Rogel, V.; Medina-Rojas, E.L.; Del Carmen Milán-Segovia, R.; Noyola, D.E.; Nieto-Aguirre, K.; López-Delarosa, A.; Romano-Moreno, S. Population Pharmacokinetics of Cefepime in Neonates with Severe Nosocomial Infections. J. Clin. Pharm. Ther. 2008, 33, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yao, B.F.; Kou, C.; Xu, H.Y.; Tang, B.H.; Wu, Y.E.; Hao, G.X.; Zhang, X.P.; Zhao, W. Developmental Population Pharmacokinetics and Dosing Optimization of Cefepime in Neonates and Young Infants. Front. Pharmacol. 2020, 11, 14. [Google Scholar] [CrossRef]
- Capparelli, E.; Hochwald, C.; Rasmussen, M.; Parham, A.; Bradley, J.; Moya, F. Population Pharmacokinetics of Cefepime in the Neonate. Antimicrob. Agents Chemother. 2005, 49, 2760–2766. [Google Scholar] [CrossRef]
- McDonald, D.; Shah, P. Cefepime Dosing in Neonates: What Is the Evidence? Am. J. Perinatol. 2021, 38, 657–661. [Google Scholar] [CrossRef]
- Cefepime Orpha, Swiss Product Label. April 2023. Available online: https://www.swissmedicinfo.ch/ (accessed on 30 June 2024).
- FDA, U.S. Food and Drug Administration Cefepime Injection; FDA: Silver Spring, MA, USA, 2010. [Google Scholar]
- Hogg, R.J.; Furth, S.; Lemley, K.V.; Portman, R.; Schwartz, G.J.; Coresh, J.; Balk, E.; Lau, J.; Levin, A.; Kausz, A.T.; et al. National Kidney Foundation’s Kidney Disease Outcomes Quality Initiative Clinical Practice Guidelines for Chronic Kidney Disease in Children and Adolescents: Evaluation, Classification, and Stratification. Pediatrics 2003, 111, 1416–1421. [Google Scholar] [CrossRef]
- Schliamser, S.E.; Cars, O.; Norrby, S.R. Neurotoxicity of β-Lactam Antibiotics: Predisposing Factors and Pathogenesis. J. Antimicrob. Chemother. 1991, 27, 405–425. [Google Scholar] [CrossRef]
- Maan, G.; Keitoku, K.; Kimura, N.; Sawada, H.; Pham, A.; Yeo, J.; Hagiya, H.; Nishimura, Y. Cefepime-Induced Neurotoxicity: Systematic Review. J. Antimicrob. Chemother. 2022, 77, 2908–2921. [Google Scholar] [CrossRef]
- Chow, K.M.; Hui, A.C.; Szeto, C.C. Neurotoxicity Induced by Beta-Lactam Antibiotics: From Bench to Bedside. Eur. J. Clin. Microbiol. Infect. Dis. 2005, 24, 649–653. [Google Scholar] [CrossRef]
- Hambrick, H.R.; Pavia, K.; Tang Girdwood, S.; Lazear, D.; Taylor, J.M.; Benoit, S. Cefepime-Associated Neurotoxicity in a Pediatric Patient with Stage V Chronic Kidney Disease. J. Pharm. Pract. 2022, 37, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Landgrave, L.C.; Lock, J.L.; Whitmore, J.M.; Belcher, C.E. Pediatric Cefepime Neurotoxicity. Pediatr. Neurol. 2012, 47, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Paioni, P.; Jäggi, V.F.; Tilen, R.; Seiler, M.; Baumann, P.; Bräm, D.S.; Jetzer, C.; Haid, R.T.U.; Goetschi, A.N.; Goers, R.; et al. Gentamicin Population Pharmacokinetics in Pediatric Patients—A Prospective Study with Data Analysis Using the Saemix Package in R. Pharmaceutics 2021, 13, 1596. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, G.M.; Marchini, G. Clinical Pharmacology of Cefepime in Infants and Children. Int. J. Pediatr. 2017, 5, 4723–4740. [Google Scholar] [CrossRef]
- Blumer, J.L.; Reed, M.D.; Knupp, C. Review of the Pharmacokinetics of Cefepime in Children. Pediatr. Infect. Dis. J. 2001, 20, 337–342. [Google Scholar] [CrossRef]
- Saez-Llorens, X.; Castano, E.; Garcia, R.; Baez, C.; Perez, M.; Tejeira, F.; McCracken, G.H. Prospective Randomized Comparison of Cefepime and Cefotaxime for Treatment of Bacterial Meningitis in Infants and Children. Antimicrob. Agents Chemother. 1995, 39, 937–940. [Google Scholar] [CrossRef]
- Schwartz, G.J.; Haycock, G.B.; Edelmann, C.M.; Spitzer, A. A Simple Estimate of Glomerular Filtration Rate in Children Derived From Body Length and Plasma Creatinine. Pediatrics 1976, 58, 259–263. [Google Scholar] [CrossRef]
- Schwartz, G.J.; Feld, L.G.; Langford, D.J. A Simple Estimate of Glomerular Filtration Rate in Full-Term Infants during the First Year of Life. J. Pediatr. 1984, 104, 849–854. [Google Scholar] [CrossRef]
- Abdul-Aziz, M.H.; Alffenaar, J.W.C.; Bassetti, M.; Bracht, H.; Dimopoulos, G.; Marriott, D.; Neely, M.N.; Paiva, J.A.; Pea, F.; Sjovall, F.; et al. Antimicrobial Therapeutic Drug Monitoring in Critically Ill Adult Patients: A Position Paper. Intensive Care Med. 2020, 46, 1127–1153. [Google Scholar] [CrossRef]
- Allegaert, K.; Smits, A.; van Donge, T.; van den Anker, J.; Sarafidis, K.; Levtchenko, E.; Mekahli, D. Renal Precision Medicine in Neonates and Acute Kidney Injury: How to Convert a Cloud of Creatinine Observations to Support Clinical Decisions. Front. Pediatr. 2020, 8, 366. [Google Scholar] [CrossRef]
- Van Donge, T.; Staub, E.; Atkinson, A.; Gotta, V.; Van Den Anker, J.; Risch, L.; Welzel, T.; Pfister, M. Age Appropriate Reference Intervals for Eight Kidney Function and Injury Markers in Infants, Children and Adolescents. Clin. Chem. Lab. Med. 2020, 59, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Ek, C.J.; Dziegielewska, K.M.; Habgood, M.D.; Saunders, N.R. Barriers in the Developing Brain and Neurotoxicology. Neurotoxicology 2012, 33, 586–604. [Google Scholar] [CrossRef] [PubMed]
- van den Anker, J.; Reed, M.D.; Allegaert, K.; Kearns, G.L. Developmental Changes in Pharmacokinetics and Pharmacodynamics. J. Clin. Pharmacol. 2018, 58, S10–S25. [Google Scholar] [CrossRef] [PubMed]
- Boschung-Pasquier, L.; Atkinson, A.; Kastner, L.K.; Banholzer, S.; Haschke, M.; Buetti, N.; Furrer, D.I.; Hauser, C.; Jent, P.; Que, Y.A.; et al. Cefepime Neurotoxicity: Thresholds and Risk Factors. A Retrospective Cohort Study. Clin. Microbiol. Infect. 2020, 26, 333–339. [Google Scholar] [CrossRef]
- Venugopalan, V.; Casaus, D.; Kainz, L.; Slaton, C.N.; Hurst, N.; Bruzzone, M.; Hu, C.; Sword, G.; Cherabuddi, K.; Iovine, N.; et al. Use of Therapeutic Drug Monitoring to Characterize Cefepime-Related Neurotoxicity. Pharmacotherapy 2023, 43, 6–14. [Google Scholar] [CrossRef]
- Huwyler, T.; Lenggenhager, L.; Abbas, M.; Ing Lorenzini, K.; Hughes, S.; Huttner, B.; Karmime, A.; Uçkay, I.; von Dach, E.; Lescuyer, P.; et al. Cefepime Plasma Concentrations and Clinical Toxicity: A Retrospective Cohort Study. Clin. Microbiol. Infect. 2017, 23, 454–459. [Google Scholar] [CrossRef]
- Durand-Maugard, C.; Lemaire-Hurtel, A.S.; Gras-Champel, V.; Hary, L.; Maizel, J.; Prud’homme-Bernardy, A.; Andréjak, C.; Andréjak, M. Blood and CSF Monitoring of Cefepime-Induced Neurotoxicity: Nine Case Reports. J. Antimicrob. Chemother. 2012, 67, 1297–1299. [Google Scholar] [CrossRef]
- Swiss Antibiotic Resistance Report 2022; Federal Office of Public Health and Federal Food Safety and Veterinary Office: Bern, Switzerland, 2022.
- Telles, J.P.; Morales, R.; Yamada, C.H.; Marins, T.A.; D’Amaro Juodinis, V.; Sztajnbok, J.; Silva, M.; Bassetti, B.R.; Albiero, J.; Tuon, F.F. Optimization of Antimicrobial Stewardship Programs Using Therapeutic Drug Monitoring and Pharmacokinetics-Pharmacodynamics Protocols: A Cost-Benefit Review. Ther. Drug Monit. 2023, 45, 200–208. [Google Scholar] [CrossRef]
- Al-Shaer, M.H.; Alghamdi, W.A.; Graham, E.; Peloquin, C.A. Meropenem, Cefepime, and Piperacillin Protein Binding in Patient Samples. Ther. Drug Monit. 2020, 42, 129–132. [Google Scholar] [CrossRef]
- Jongmans, C.; Muller, A.E.; Van Den Broek, P.; Cruz De Almeida, B.D.M.; Van Den Berg, C.; Van Oldenrijk, J.; Bos, P.K.; Koch, B.C.P. An Overview of the Protein Binding of Cephalosporins in Human Body Fluids: A Systematic Review. Front. Pharmacol. 2022, 13, 900551. [Google Scholar] [CrossRef] [PubMed]
- Leroux, S.; Biran, V.; van den Anker, J.; Gotta, V.; Zhao, W.; Zhang, D.; Jacqz-Aigrain, E.; Pfister, M. Serum Creatinine and Serum Cystatin C Are Both Relevant Renal Markers to Estimate Vancomycin Clearance in Critically Ill Neonates. Front. Pharmacol. 2021, 12, 634686. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Kaguelidou, F.; Biran, V.; Zhang, D.; Allegaert, K.; Capparelli, E.V.; Holford, N.; Kimura, T.; Lo, Y.L.; Peris, J.E.; et al. External Evaluation of Population Pharmacokinetic Models of Vancomycin in Neonates: The Transferability of Published Models to Different Clinical Settings. Br. J. Clin. Pharmacol. 2013, 75, 1068–1080. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Population | <1 Month (<28 Days) | 1–2 Months (28–59 Days) | 2–12 Months (60–365 Days) |
| Number of patients | 131 * | 73 | 31 |
| Age, days | 9 [3, 19] | 39 [33, 47] | 157 [76, 259] |
| Weight, kg | 3.6 [3.1, 3.9] | 4.6 [4.1, 4.8] | 5.7 [4.7, 6.5] |
| Height, cm | 51.5 [49, 53] | 54.6 [53, 56] | 62 [57.7, 67.5] |
| Serum creatinine **, µmol/L | 27 [27–44.3] | 27 [27] | 27 [27–27.5] |
| Gestational age (GA), weeks | 39 [38, 40] * | 39 [38, 40] | 38 [36, 40] |
| Postmenstrual age (PMA), weeks | 41 [39.1, 42.0] | 44.7 [43.4, 46.1] | 59.6 [50.8, 72.4] |
| eGFR, Schwartz 1976 [30], mL/min/1.73 m2 | 87 [56, 95] | 97 [91, 101] | 108 [97, 117] |
| eGFR, Schwartz 1984 [31], mL/min/1.73 m2 | 71 [46, 77] | 80 [74, 82] | 88 [79, 96] |
| Mean eGFR, KDIGO, mL/min/1.73 m2 | 41 (1 week) | 66 (2–8 weeks) | 96 (>8 weeks to 2 years) |
| Mean serum creatinine, μmol/L | 44 (day 14–30) | 20 | 20 |
| Age | Dosing Regimen | Model 1 | Model 2 |
|---|---|---|---|
| Safety: Percentage of patients with predicted Ctrough > 20(> 35) mg/L | |||
| <1 month | 50 mg/kg every 8 h | 68% (38%) | X * |
| 50 mg/kg every 12 h | 20% (5%) | X * | |
| 1–2 months | 50 mg/kg every 8 h | 54% (22%) | 40% (12%) |
| 50 mg/kg every 12 h | 11% (2%) | 4.8% (0.8%) | |
| 2–12 months | 50 mg/kg every 8 h | 30 (9%) | 37% (13%) |
| Efficacy: Percentage of patients with predicted 50% fT>MIC8mg/L (Range: 70–100% fT>MIC8mg/L) | |||
| <1 month | 50 mg/kg every 8 h | 100% (99–92%) | X * |
| 50 mg/kg every 12 h | 99% (85–48%) | X * | |
| 1–2 months | 50 mg/kg every 8 h | 100% (99–85%) | 100% (94–71%) |
| 50 mg/kg every 12 h | 97% (79–37%) | 100% (93–70%) | |
| 2–12 months | 50 mg/kg every 8 h | 99% (92–61%) | 99% (94–74%) |
| Sensitivity Analysis | A | B | C | ||
|---|---|---|---|---|---|
| Model 1 | Model 2 | Model 1 | Model 2 | Model 3 (Zhao, 2020) [15] | |
| Safety: Percentage of patients with predicted Ctrough > 20(>35) mg/L | |||||
| <1 month | 40% (10%) | X * | 82% (45%) | X * | 2% (0%) |
| 1–2 months | 24% (5%) | 14% (2%) | 35% (10%) | 51% (20%) | 1% (0%) |
| 2–12 months | 10% (2%) | 15% (4%) | 15% (2%) | 38% (12%) | X * |
| Efficacy: Percentage of patients with predicted 50% fT>MIC8mg/L (Range: 70–100% fT>MIC8mg/L) | |||||
| <1 month | 100% (96–77%) | X * | 100% (100–98%) | X * | 99% (92–37%) |
| 1–2 months | 99% (91–62%) | 95% (79–49%) | 100% (96–70%) | 100% (97–79%) | 100% (89–37%) |
| 2–12 months | 94% (74–37%) | 96% (81–46%) | 98% (83–41%) | 99% (98–77%) | X * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gotta, V.; Csajka, C.; Glauser, A.; Berger, C.; Pfister, M.; Paioni, P. Risk of Potentially Neurotoxic Exposure in Infants Under High-Dose Cefepime Treatment—A Pharmacometric Simulation Study. Pharmaceutics 2025, 17, 544. https://doi.org/10.3390/pharmaceutics17050544
Gotta V, Csajka C, Glauser A, Berger C, Pfister M, Paioni P. Risk of Potentially Neurotoxic Exposure in Infants Under High-Dose Cefepime Treatment—A Pharmacometric Simulation Study. Pharmaceutics. 2025; 17(5):544. https://doi.org/10.3390/pharmaceutics17050544
Chicago/Turabian StyleGotta, Verena, Chantal Csajka, Antonia Glauser, Christoph Berger, Marc Pfister, and Paolo Paioni. 2025. "Risk of Potentially Neurotoxic Exposure in Infants Under High-Dose Cefepime Treatment—A Pharmacometric Simulation Study" Pharmaceutics 17, no. 5: 544. https://doi.org/10.3390/pharmaceutics17050544
APA StyleGotta, V., Csajka, C., Glauser, A., Berger, C., Pfister, M., & Paioni, P. (2025). Risk of Potentially Neurotoxic Exposure in Infants Under High-Dose Cefepime Treatment—A Pharmacometric Simulation Study. Pharmaceutics, 17(5), 544. https://doi.org/10.3390/pharmaceutics17050544