1. Introduction
In recent years, major advances in our understanding of the tumor microenvironment and responses to therapy have led to a shift in the focus of cancer treatments from the reliance on monotherapies targeting tumor cells, to the development of rational combinatorial approaches involving new cancer immunotherapies to optimally support and unleash the endogenous anti-tumor immune response. Cancer immunotherapeutics involve numerous strategies to target different aspects of the tumor microenvironment and the innate and adaptive immune response to allow the tumor to be recognized and destroyed [
1].
The successful induction of an anti-tumor immune response is illustrated in the cancer-immunity cycle introduced by Chen et al. in 2013 [
2]. In short, tumor antigens released by dying tumor cells are taken up by antigen presenting cells (APCs), especially dendritic cells (DCs), that present these tumor antigens to naïve T cells in the lymph nodes, priming them for recognition and tumor cell elimination when they reach the tumor and infiltrate the lesion. If iterative expansion of the cancer immunity cycle is not inhibited, a broad anti-tumor immune response can achieve complete tumor clearance; however, multiple suppressive mechanisms, including masking of tumor antigens, infiltration barriers, and immunosuppressive signaling in the tumor microenvironment limit the ability of the anti-tumor immune response to effectively destroy the cancer [
3].
Combination therapy can target multiple mechanisms, thereby supporting and stimulating the immune system on different levels. A rise in combinatorial clinical trials indicates the importance of these approaches in patient treatment [
4]. Finding the right combination will depend on a profound understanding of the immune system, sophisticated bioinformatic analyses, and prediction systems as well as a multitude of well-characterized treatment options. Immunotherapeutic approaches, such as adoptive cell transfer (ACT) and oncolytic viruses (OVs) have been under preclinical development for years but have only recently proven their worth in the clinic. Chimeric antigen receptor (CAR) T cells targeting CD19 have been approved for therapy of leukemia [
5], while an engineered herpes simplex virus expressing GM-CSF (T-Vec/Imlygic) has been approved for the treatment of late-stage malignant melanoma in 2015 [
6]. Recently, their value in combination has also been under investigation, and evidence suggests the promising effects of oncolytic viruses in combination with the adoptive transfer of tumor-specific T cells, as summarized in detailed reviews [
7,
8,
9]. In short, the multifaceted mechanism of oncolytic viruses enables debulking of the tumor, release of tumor-specific neoantigens, and an inflammatory change in the tumor microenvironment (TME). These factors improve the efficiency of ACT in solid tumors by promoting infiltration and prolongation of their cytotoxic activity with the aim of breaking the tumor’s immune tolerance and inducing a broad anti-tumor immune response, resulting in tumor clearance. Optimal success, however, is dependent on the selection of a safe virus platform that is both, highly oncolytic and immunogenic in its mediated cell death, as well as the design of an informed dosing schedule that leads to optimized synergistic responses.
We have recently reported that human CD8
+ central memory T cells could be used as cell carriers to deliver oncolytic vesicular stomatitis virus (VSV) to human acute myeloid leukemia (AML) cells and exert cytotoxic effector functions to result in an effective combinatorial approach in an immune-deficient xenograft model [
10]. Here, we focus on the potential of an enhanced and safer oncolytic virus platform in combination with adoptive T cell transfer in an immune-competent model as a further step towards clinical translation. The hybrid VSV-NDV platform was recently introduced by our group as an immune-stimulatory oncolytic virus, showing substantially enhanced safety and efficacy compared to VSV after systemic administration of an aggressive murine HCC model [
11]. VSV-NDV was engineered by replacing the VSV glycoprotein within the VSV genome with the surface proteins of NDV to improve safety and introduce a fusogenic mechanism of action. In this construct, a polybasic cleavage site (F3aa) and the leucine to alanine substitution at amino acid 289 (L289A) lead to the hyperfusogenicity of VSV-NDV and efficient induction of immunogenic cell death [
11]. The aspect of fusogenicity as a strategy to enhance oncolytic viruses as immunotherapeutics has recently gained attention [
12,
13]. Therefore, we hypothesized that the highly fusogenic capacity of VSV-NDV would make it an ideal candidate for the combination with other immunotherapeutic approaches, such as ACT.
In this study, we investigated the combination of oncolytic VSV-NDV with antigen-specific OTI T cells using the B16-OVA system, in which the mouse B16 melanoma cells express chicken ovalbumin as an immunogenic model antigen to facilitate immune monitoring. In vitro combination experiments revealed increased cytotoxicity in the tumor cells, compared to treatment with either monotherapy, as well as a specific homing of antigen-specific T cells to areas of virus replication. Furthermore, combination treatment led to the upregulation of MHC-I and downregulation of PD-L1 on the tumor cells, which indicates that the strategy could lead to enhanced antigen presentation and modulation of immune suppression, leading to additional immune-mediated effects. In vivo studies in immune-competent C57BL/6 mice bearing subcutaneous, syngeneic B16-OVA tumors demonstrated that VSV-NDV could mediate direct and abscopal tumor responses, but combination therapy with OTI T cells was the only effective approach when using a clinically relevant dosing scheme. The combination resulted in significantly delayed tumor growth and prolonged overall survival in this model. These results support the combination of oncolytic viruses with ACT as a rational combination, leading to multi-mechanistic and synergistic tumor responses for further development as cancer immunotherapeutics.
2. Materials and Methods
2.1. Cells and Virus
The murine melanoma cell line, B16, expressing the ovalbumin (B16-OVA) protein was cultured in DMEM GlutaMAX
TM-I (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat inactivated fetal calf serum (FCS), MEM non-essential amino acids, sodium pyruvate, penicillin (50 U/mL) and streptomycin (50 μg/mL). The B16-F10 clone without the OVA protein was cultured analogously, and both B16 cell lines were kindly provided by Simon Heidegger (Klinik und Poliklinik für Innere Medizin III, Klinikum rechts der Isar, Munich, Germany). OTI T cells were isolated from the spleens of OTI mice (kindly provided by Melanie Kimm, Institute for Radiology, Klinikum rechts der Isar, Munich, Germany). The homozygous OTI mice produce T cells expressing transgenic Tcra-V2 and Tcrb-V5 T cell receptor chains in order to recognize the SIINFEKL peptide (ovalbumin residues 257–264) in the context of the H-2K
d MHC class I alloantigen. They were cultured and expanded in murine T cell medium (mTCM) consisting of RPMI GlutaMAX
TM-I medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat inactivated FCS, MEM non-essential amino acids, sodium pyruvate, β-mercaptoethanol (50 mM) and activated with CD3/CD28 beads (Dynabeads, Thermo Fisher Scientific), as well as 40 U/mL IL-2, and 50 ng/mL IL-15. The green fluorescence protein (GFP) reporter gene was cloned as an additional transcription unit using unique restriction sites between the HN and L genes of VSV-NDV to generate the recombinant VSV-NDV-GFP construct. The virus was rescued and produced as previously described [
11]. Virus stocks used for all experiments were produced in AGE1.CR.pIX cells (ProBioGen AG, Berlin, Germany) and purified by ultracentrifugation over a sucrose gradient. For animal experiments, sucrose was removed by ultracentrifugation and the concentrated virus was resuspended in PBS.
2.2. Growth Curves
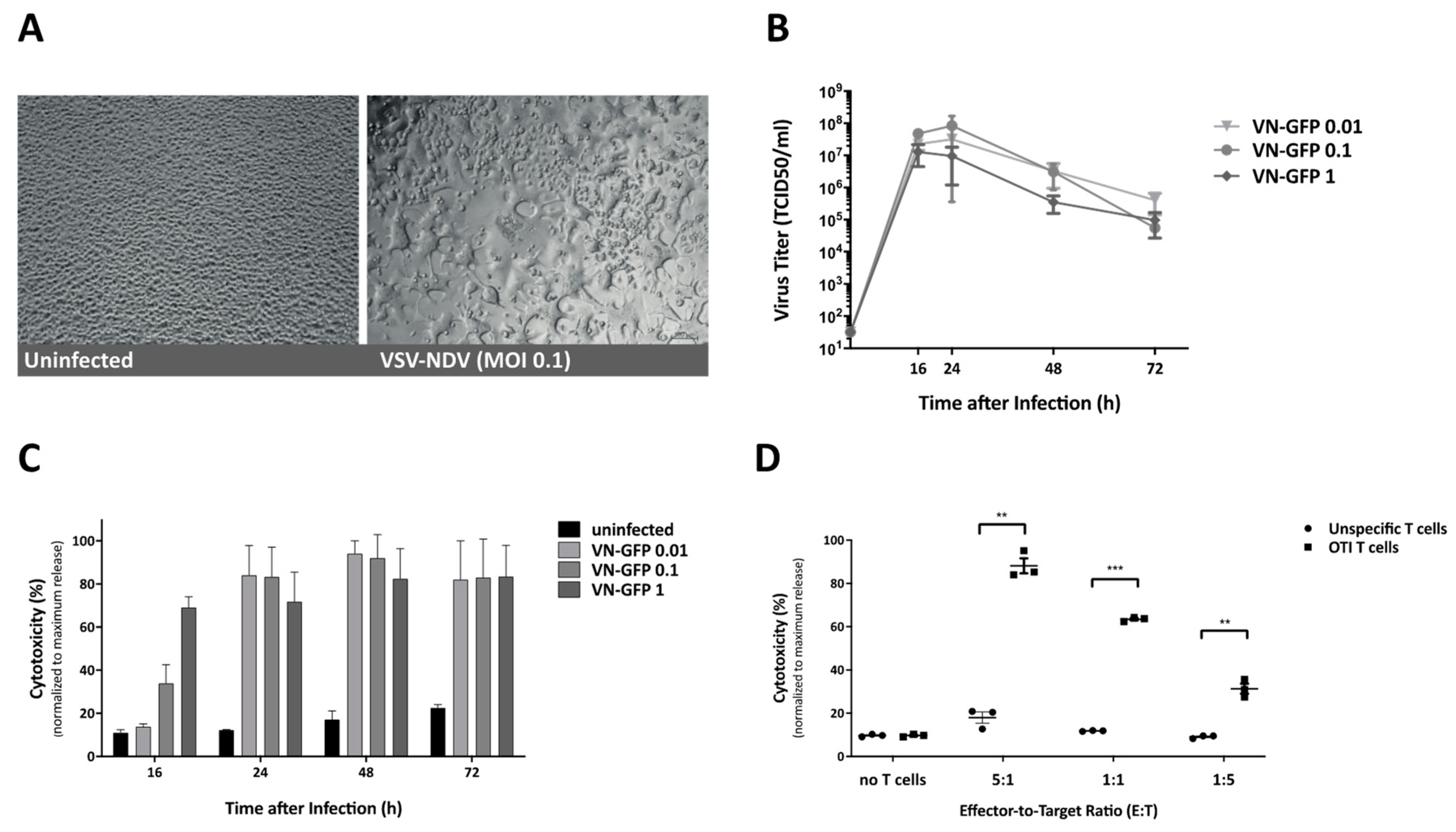
The growth curve analysis of virus replication was determined in B16 cells. The cells were seeded at 70–80% confluency the day before infection. For infection, the cells were incubated with either rVSV-NDV-GFP at the indicated MOI or PBS as negative control for 1 h at 37 °C in PBS with Ca2+ and Mg2+. The cells were then washed 3x to remove remaining free virus particles, and the normal cell culture medium was added. A sample was immediately collected and used to determine baseline virus titers directly after infection and washing (0 h), and additional aliquots were collected at 16, 24, 48 and 72 h after infection for determination of virus titers via a 50% tissue culture infective dose (TCID50) assay in AGE1.CR.pIX cells.
2.3. Cytotoxicity Assay
Cytotoxicity was determined based on the release of lactate dehydrogenase (LDH) in cell culture supernatant. LDH was measured with the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega, Madison, WI, USA), according to the manufacturer’s protocol. Absorption at 450 nm wavelength was detected with an absorbance microplate reader (Tecan, Austria). To obtain a maximum release control for each time-point, additional uninfected wells were treated with the supplied lysis buffer and incubated for 30 min, prior to collection of the supernatant. All experimental results were normalized to the maximum release control and performed in triplicate.
2.4. Microscopic Analysis
For visualization of the infection, representative images (bright field and fluorescence) were captured at 200× magnification using an Axiovert 40CFL microscope (Zeiss, Oberkochen, Germany) with an AxioCam ICm1 camera (Zeiss) attached to the microscope.
2.5. Co-Culture Experiments
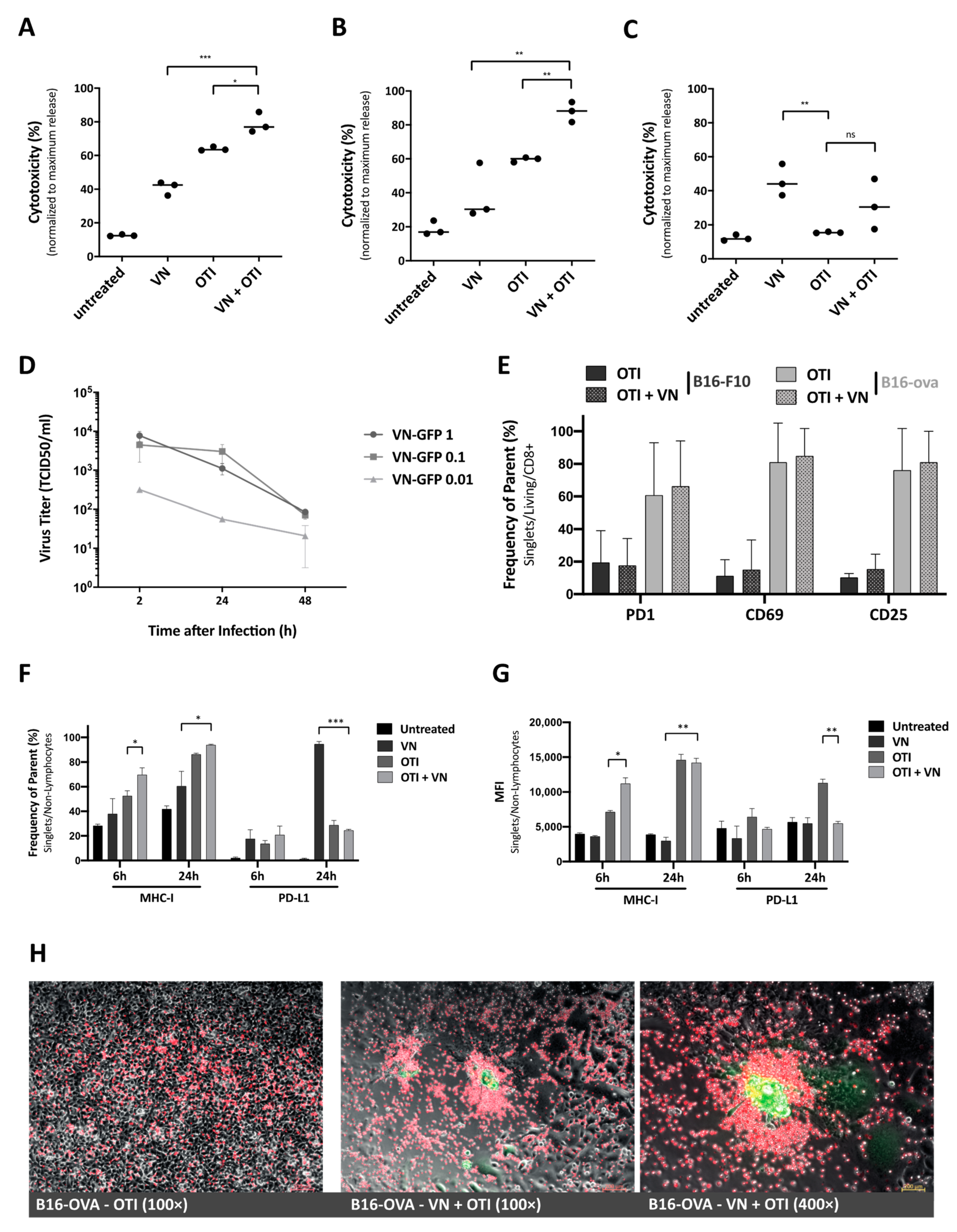
The cells were pre-infected at the indicated MOI and incubated for up to 24 h, depending on the cell line susceptibility, before the addition of T cells. Supernatants were collected 16 or 24 h after the addition of OTI T cells to determine cytotoxicity via LDH analysis, as described previously. T cells and tumor cells harvested from these co-culture experiments were used to examine activation and tumor cell response by flow cytometry. For photomicroscopy of co-cultured cells, B16-OVA cells were seeded and infected with rVSV-NDV-GFP (MOI 0.1) for 16 h. T cells were labeled with CellTrace Far Red Cell Proliferation Kit (Thermo Fisher Scientific, Waltham, MA, USA) and added to the co-culture. Representative fluorescence images were captured after 4 h.
2.6. Flow Cytometry
Flow cytometric measurements were performed using the CytoFLEX S platform (Beckman Coulter, Brea, CA, USA). If not stated otherwise, samples were incubated for 25 min with FcR blocking reagent (Miltenyi Biotech, Bergisch Gladbach, Germany), washed in PBS, incubated for 1 h at room temperature with Viobility Fixable Dye, CD8, CD4 (Miltenyi Biotech, Bergisch Gladbach, Germany), PD1, IFNγ, and TNFα (BD Biosciences, Franklin Lakes, NJ, USA) staining antibodies or SIINFEKL(OVA)-/ (VSV-NP)-specific tetramers (MBL International, Woburn, MA), washed again and processed according to the manufacturers’ protocols. The compensation was performed based on staining results from UltraComp beads (Thermo Fisher Scientific, Waltham, MA, USA). The results were analyzed using Flow Jo (Ashland, OR, USA) software.
2.7. Animal Experiments
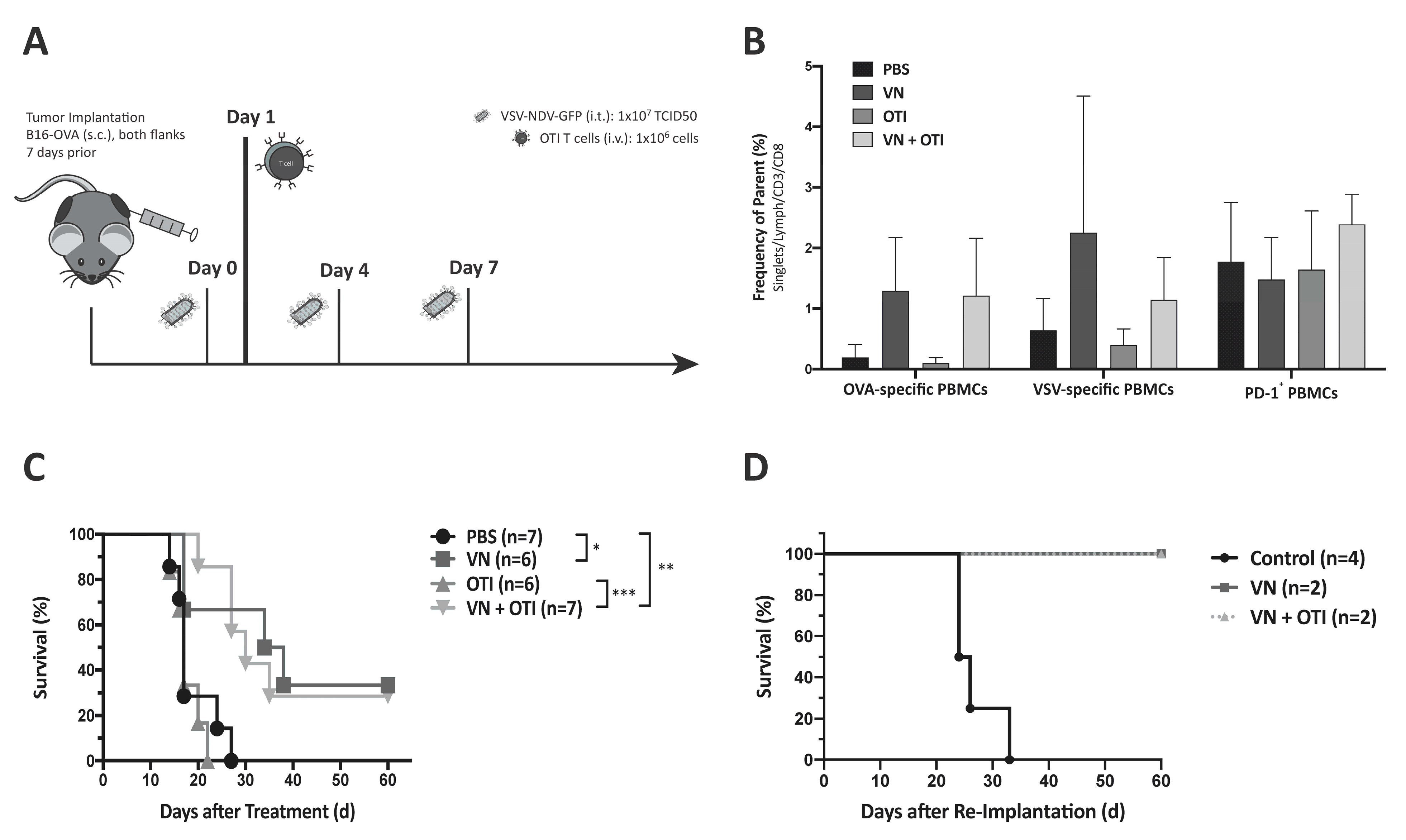
All animal experiments were performed in accordance with protocols approved by the institute’s Center for Preclinical Research and the regional government commission for animal protection (Regierung von Oberbayern, Munich, Germany). For survival analysis, six-week-old female C57BL/6J mice (Janvier Labs, Le Genest-Saint-Isle, France) were shaved and then injected with 2.4 × 10
5 (right flank) and 1.2 × 10
5 (left flank) B16-OVA cells subcutaneously in contralateral flanks. One week after tumor implantation (with an approximate tumor size of 20–50 mm
3), the treatment was initiated. Intratumoral injections were only administered into the right tumor (injected tumor) in order to allow the observation of abscopal effects in the left tumor (uninjected tumor). The treatment schedules for the experiments are provided in the Results section or in the
Supplemental Figures. Tumor width and length were measured regularly with a caliper, and the volume was calculated according to the modified ellipsoid formula: Tumor volume = 1/2(length × width
2) [
14]. Blood was collected on day 8 and 16 for flow cytometric analysis. The mice were monitored and euthanized at humane endpoints or at the latest when their tumor reached a diameter of 15 mm. Survival times with respect to the first injection of treatment were plotted in a Kaplan–Meier survival curve, and median survival times were calculated. Long-term surviving mice, together with age- and sex-matched control mice, were (re)implanted with 1.2 × 10
5 B16-OVA cells to investigate memory immunity against the tumor.
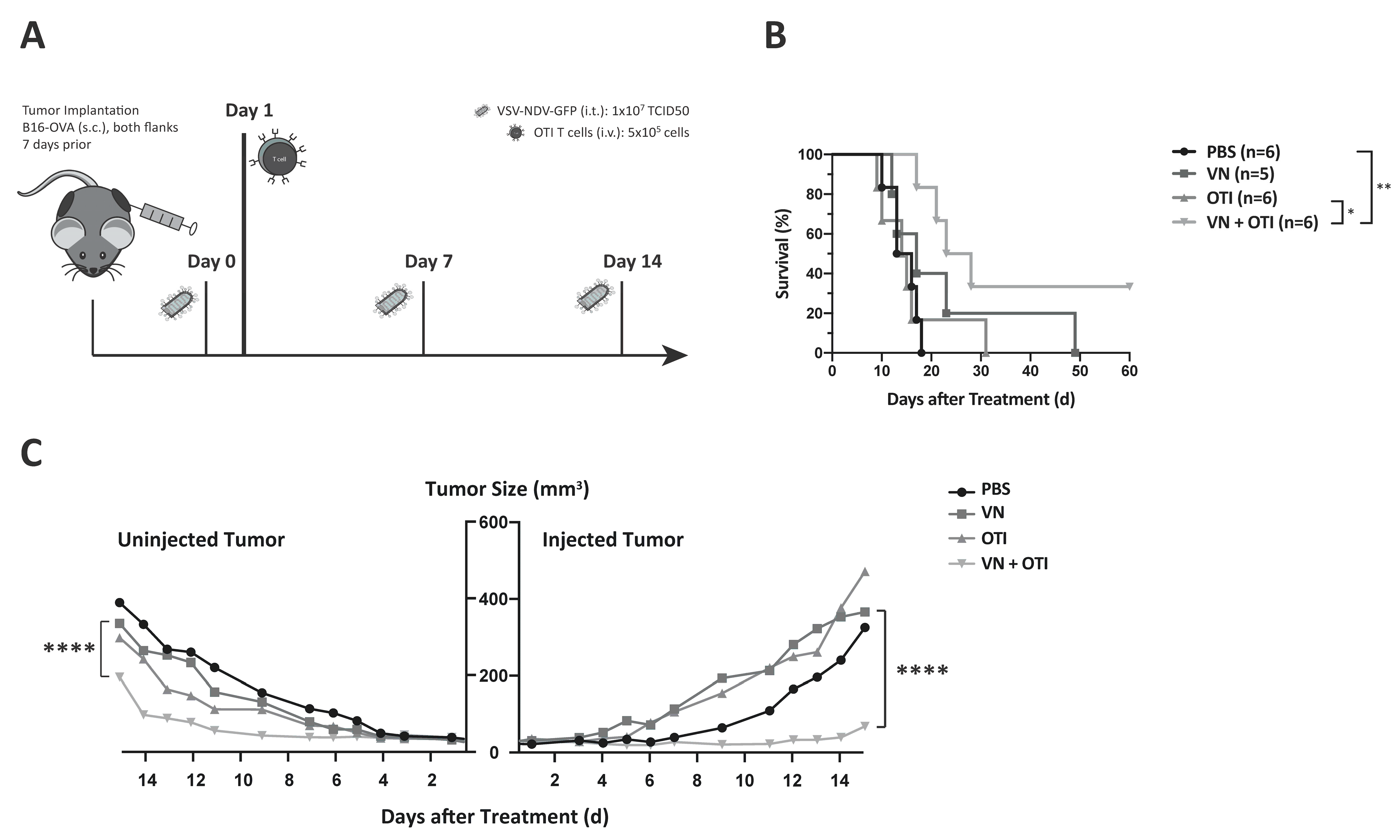
For kinetics experiments, six-week-old female C57BL/6J mice were shaved and then injected with 2.4 × 105 B16-OVA cells subcutaneously in both flanks. One week after tumor implantation, the treatment was initiated. The mice received virus (1 × 107 TCID50) or PBS on indicated days and, depending on the treatment group, an OTI T cell injection (1 × 106) by tail vein on day 1. Tumors and blood were harvested at specified time points after treatment. Tumors were divided in order to provide tissue for flow cytometry and immunohistochemistry (IHC). For flow cytometry, the tumor piece was mashed through a 40 µm filter after a 30-min incubation in Liberase TM (Roche, Basel, Switzerland) at a concentration of 20 µg/mL, and a single cell suspension was used for flow cytometry staining of infiltrating antigen-specific T cells. Alternatively, immune cells were concentrated via gradient centrifugation (20 min, 1025× g) in LymphSep (Biowest, Nuaillé, France) and used for flow cytometry staining. The measurement time was limited to 60 s in order to normalize each cell count to the size of the tumor before euthanization and correlate immune cell infiltration with tumor size. Blood was collected in EDTA-microvettes (Sarstedt Inc., Newton, NC, USA) and centrifuged (10 min, 1000× g). The resulting plasma was used to analyze the systemic cytokine profile after treatment with a bead-based cytokine array (Mouse Inflammation Panel, LEGENDplex, BioLegend, San Diege, CA, USA). Harvested spleens were mashed through a 40 µm filter and incubated with red blood cell lysis buffer for 2 min. After washing with mTCM and a second filtration step, the single cell suspension of splenocytes was frozen in 10% DMSO in FCS at −80 °C and thawn for the peptide activation assay.
2.8. Peptide Activation Assay
Frozen splenocytes collected from treated tumor-bearing mice at day 2, 5, and 8 after the first treatment were thawn and incubated in mTCM for 5 h at 37 °C. For the peptide activation assay, 2 × 106 splenocytes were added to a 96-well plate prepared with peptide solution at a final concentration of 1 μg/mL OVA or TRP2 peptide. Brefeldin A was added to each well after a 1-h stimulation. After incubation for 15 h at 37 °C, the cells were transferred to a V-bottom 96-well plate and stained for extracellular CD4 and CD8, as well as intracellular IFNγ and TNFα after fixation and washing using BD Cytox and Perm/Wash Buffer. Staining was analyzed by flow cytometry.
2.9. Histology and Immunohistochemistry
For immunohistochemistry (IHC), tumor pieces were fixed in 4% paraformaldehyde overnight, followed by dehydration and embedding in paraffin. Then, 2 µm-thin slices were subjected to immunohistochemical staining using a rabbit monoclonal antibody against CD3 (DCS, Hamburg, Germany) or CD8 (Dianova, Hamburg, Germany) on a Bond RX automated staining instrument (Leica, Biosystems, Nußloch, Germany). An analysis of pathological changes and confirmation of positive immunohistochemical reaction were performed by a certified pathologist who was blinded to the treatment groups of the specimens.
2.10. Statistical Analysis
The data were plotted and analyzed using GraphPad Prism 7.0 (GraphPad Software, San Diego, CA, USA). Means and standard error of the mean (SEM) were plotted when applicable. Individual data points were compared for statistical significance using an unpaired Student’s t-test, and p-values of less than 0.05 were considered to be statistically significant (* p < 0.05, ** p < 0.005, *** p < 0.001, **** p < 0.0001). Survival data was plotted in Kaplan–Meier curves, and statistical significance calculated by log-rank test.
4. Discussion
With the recent approval of CAR T cell therapies targeting CD19 [
21], as well as numerous other T cell therapies currently under clinical investigation, the concept of adoptive T cell therapy for cancer is at the forefront of the immune-oncology field. However, despite the promising responses to these therapies in specific subsets of patients, a weakness of this approach is that it relies on the identification of suitable tumor-associated antigens (TAAs) and neoantigens to target. Due to intratumoral heterogeneity and the process of cancer immune-editing, strategies that target a single antigen can result in the selection for tumor cells which do not express the targeted antigen, leading to escape variants causing relapse [
22]. Furthermore, since each tumor has its own distinct gene signature, targeted therapies often require expensive and time-consuming molecular screening of tumor biopsies and subsequent production of personalized treatments. Therefore, the concept of combining ACT approaches with oncolytic virus therapy has emerged as a rational strategy to, not only broaden the scope of the response by using the virus to kill cells that are not targeted by the ACT, but also to potentially enhance the infiltration and expansion of adoptively transferred cells through pro-inflammatory signaling mediated by the virus infection. This approach therefore has the potential to drive multiple mechanisms leading to synergistic tumor debulking and long-term systemic protection against the tumor.
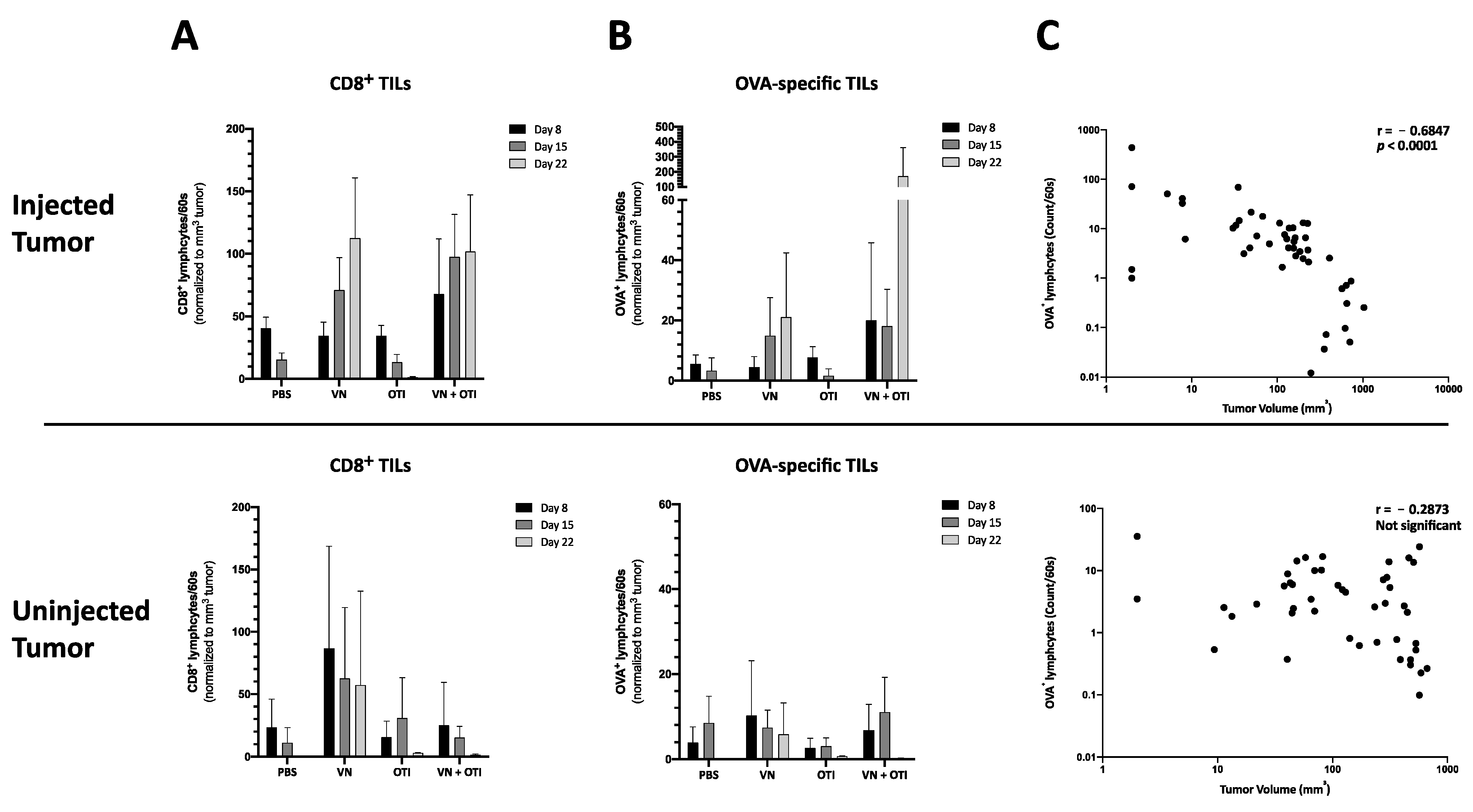
In the study reported here, we combined a highly immune-stimulatory oncolytic virus candidate with adoptive TCR T cell therapy in a model mouse system for melanoma, in order to demonstrate proof-of-concept and a preliminary mechanism to support the further development of the approach. Our results emphasize that the combination of oncolytic virus and antigen-specific T cells can work together to beneficially modulate the tumor microenvironment, including the induction of MHC-I expression on tumor cells (
Figure 2F,G), which is essential for an efficient elimination by antigen-specific T cells [
23]. Synergy was demonstrated, not only through the complementary mechanisms of viral-mediated oncolysis and T cell effector functions, but also through the important finding that T cells seem to be specifically recruited to areas of VSV-NDV infection (
Figure 2H), potentially enhancing the contribution of the adoptively transferred T cells in the combination therapy, as shown previously [
24,
25]. Although the mechanism remains unclear, this finding is supported by the observation of an influx of CD8
+ tumor infiltrating lymphocytes in response to combination therapy in vivo (
Figure 4A and
Figure 6A). OVA-specific infiltration was significantly increased compared to OTI monotherapy in both the injected and uninjected tumor at early timepoints (
Figure 4A, upper panel). An improved infiltration of adoptively transferred T cells compared to VSV-NDV monotherapy could not be confirmed after a single virus injection, but data suggest an infiltration of adoptively transferred T cells and an accelerated expansion of OVA-specific T cells after multiple virus injections compared to VSV-NDV monotherapy. This could not be observed in uninjected tumors indicating a required threshold of infection to attract the adoptively transferred population into the tumor. Research on transgene-expressing oncolytic viruses in combination therapy suggests that this effect could be further improved by engineering VSV-NDV to express cytokines, chemokines or other T cell engagers, such as BiTEs. A TNFα- and IL-12-expressing adenovirus has been shown to improve ACT in different combination approaches [
26,
27,
28]. Similarly, a vaccinia virus expressing the chemokine CXCL11 or IL15Rα enhanced the anti-tumor activity of CAR T cells in solid tumors [
29,
30].
The finding of virus-specific T cells in the uninjected tumor (
Figure 4A, upper panel) was unexpected and warrants further investigation. Since we failed to detect infectious virus in contralateral uninjected tumors in previous experiments (data not shown), we suspect that the presence of virus-specific T cells in the uninjected tumor is a reflection of the systemic accumulation of these cells at the investigated time-point, rather than being an indication of a specific intratumoral infiltration in response to virus spread to the uninjected tumor. This hypothesis will be explored in follow-up studies.
The in vivo studies shown here indicate that adoptively transferred tumor-specific TCR T cells lead to only negligible tumor responses, when administered as a monotherapy (
Figure 3C and
Figure 5B). This was likely due to the relatively low number of T cells injected, as well as challenges of effective T cell engraftment, both of which are relevant limitations that are faced in clinical application. Lymphodepletion plays a key role in improving engraftment of transferred T cells. Cole et al. showed that the anti-tumor effects of VSV combined with OTI T cells were further improved in lymphopenic hosts [
31]. In contrast, not only was the hybrid VSV-NDV virus extremely effective in delaying tumor growth in the injected tumor, but it also led to impressive abscopal effects in contralateral uninjected tumors, when injected frequently over the course of one week. By reducing the injection frequency to a more clinically applicable dosing scheme, we could then appreciate the power of the combination therapy, which led to a survival prolongation and significant delay in tumor growth of injected and distant tumors, with more than 30% of treated mice achieving long-term remission.
5. Conclusions
Our results demonstrate the robustness of the combination treatment approach when applied in a clinically relevant dosing scheme and without prior lymphodepletion. This is promising, in that this treatment would be available to a broader patient population, without an aggressive myeloablative radiation protocol [
32]. Furthermore, the effectiveness of combination therapy in conditions where monotherapy showed no effect, could suggest a better chance of treatment response, despite unknown variables of virus and T cell kinetics in the patient.
The first clinical trials combining oncolytic adenovirus and ACT are on the way to demonstrate the clinical benefit of this approach and its relevance in patient treatment (NCT03740256, NCT04217473). Despite promising preclinical data [
26], the clinical outcome will depend on numerous factors, including the immunogenicity of the virus and its potency in T cell recruitment, as well as the efficiency of the adoptive T cell therapy. The full potential of ACT and the immune response induced by the combination therapy may be limited by prevailing suppressive signaling pathways, immune checkpoints or secondary escape mechanisms. This calls for a third partner in the combination approach, i.e., immune checkpoint inhibitors. On their own, checkpoint inhibitors already stand out as extremely promising treatment options for several tumor indications [
33], but their systemic administration is still limited by the onset of severe adverse events [
34], and response rates are dependent on many pre-existing features within the tumor, such as a high mutational load [
35,
36]. Oncolytic viruses can upregulate the local expression of immune checkpoints at the tumor site and create changes in the tumor microenvironment to sensitize the tumor to immune checkpoint blockade [
37]. A triple combination approach, using adenovirus expressing PD-L1 antibody with CAR T cells, demonstrated therapeutic improvements in preclinical experiments [
38,
39,
40]. Of course, the complexity and high costs of combination treatment approaches in their transfer to the clinic is greatly increased by the addition of more therapeutic agents, but it seems that a curative outcome may depend on targeting multiple mechanisms [
22]. We predict that as we rapidly gain a new understanding of the biology of tumor pathogenesis, new rationally designed combination approaches will emerge. Due to the multi-mechanistic nature of oncolytic VSV-NDV, as well as its ability to optimally synergize with other immunotherapeutic agents, as shown here, it represents an attractive candidate for combinatorial approaches in immune-oncology.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





