
Reliable Flow-Cytometric Approach for Minimal Residual Disease Monitoring in Patients with B-Cell Precursor Acute Lymphoblastic Leukemia after CD19-Targeted Therapy
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Prerequisites
2.2. Protocol Testing: Patients and Samples
2.3. MRD Monitoring by MFC
2.4. Cytogenetics and Molecular Genetics (MRD Monitoring by FGT-PCR)
2.5. MRD Monitoring Using NGS of Specific IG/TR Gene Rearrangements
3. Results
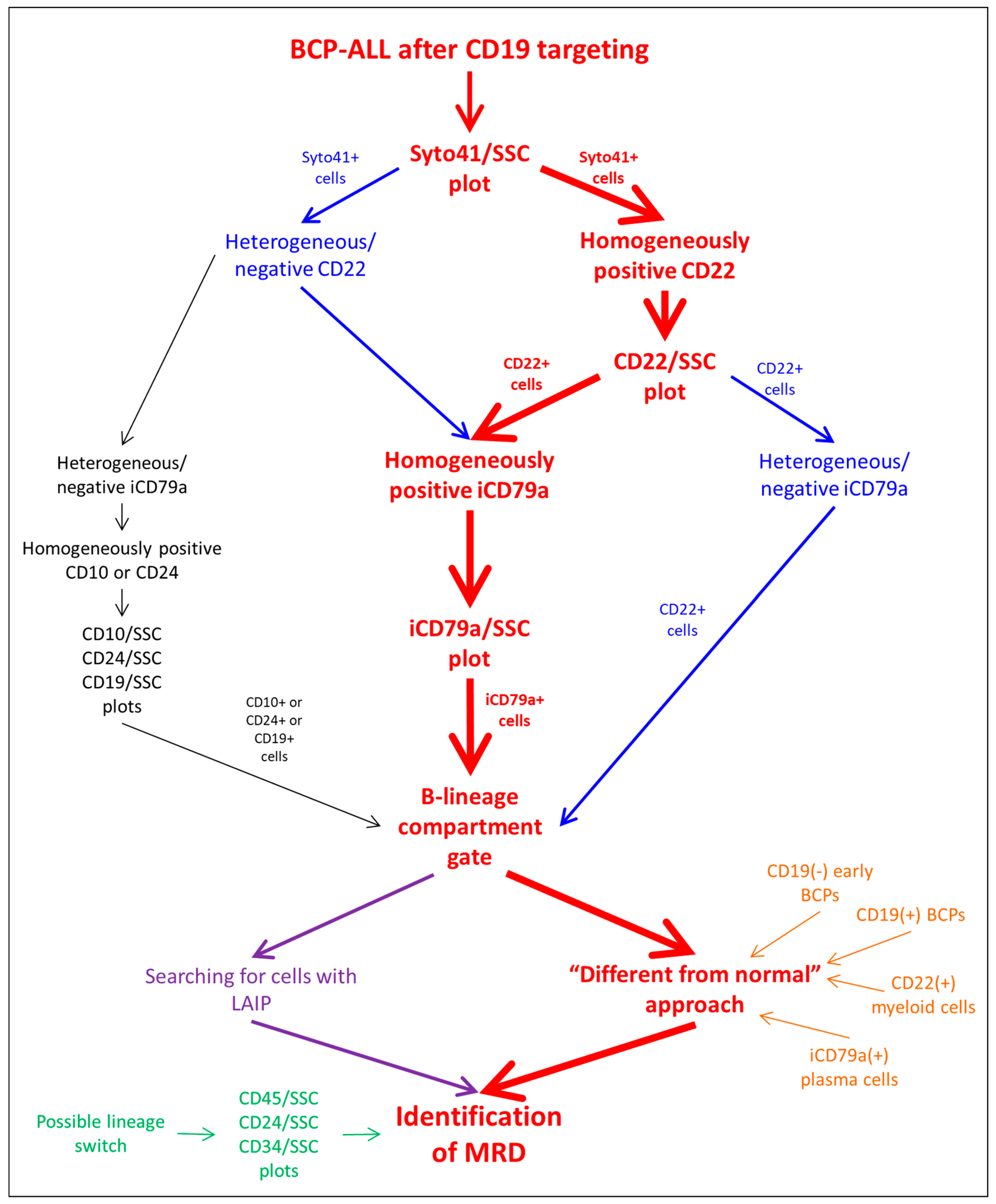
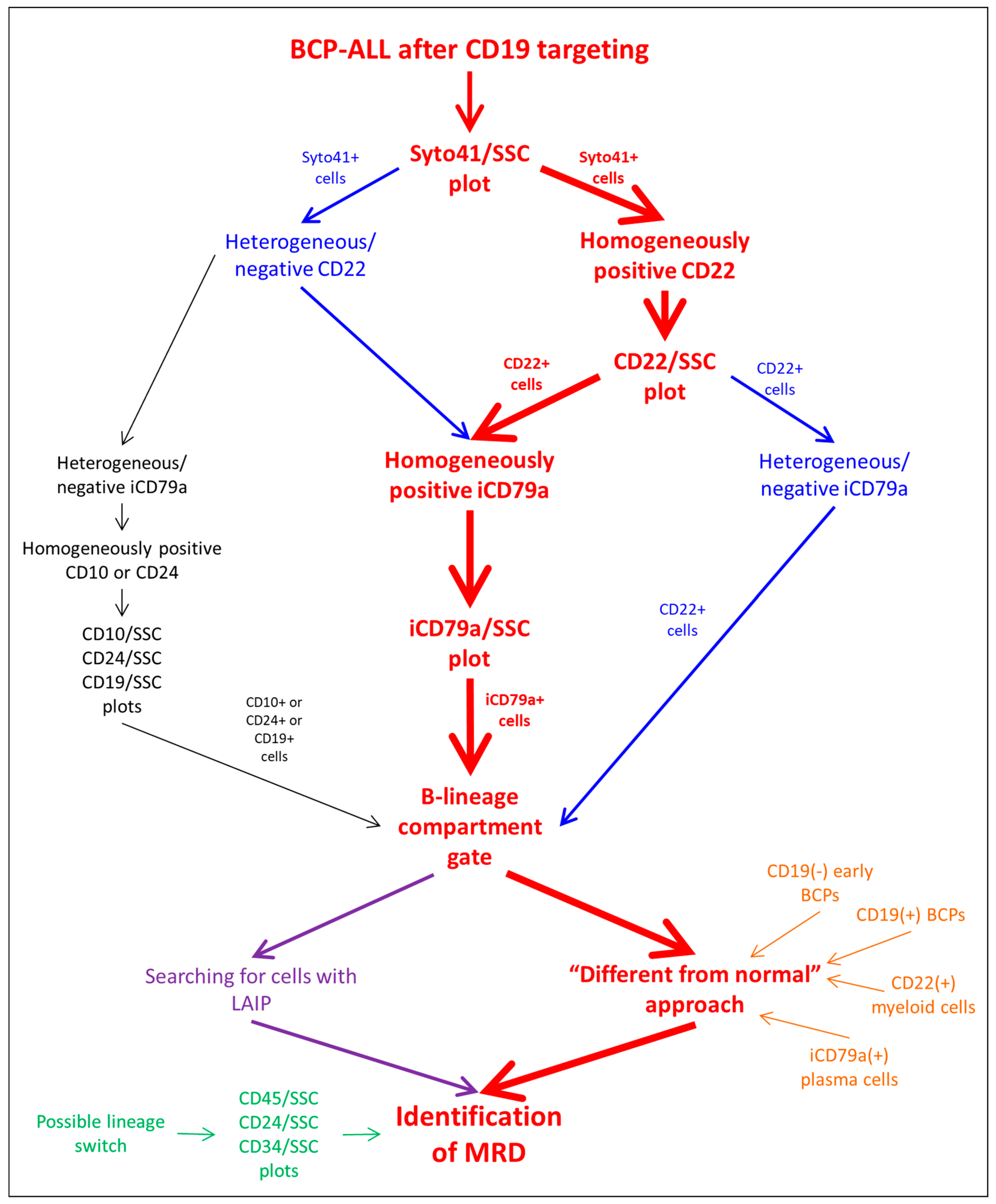
3.1. Algorithm Description
3.2. Protocol Testing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berry, D.A.; Zhou, S.; Higley, H.; Mukundan, L.; Fu, S.; Reaman, G.H.; Wood, B.L.; Kelloff, G.J.; Jessup, J.M.; Radich, J.P. Association of Minimal Residual Disease with Clinical Outcome in Pediatric and Adult Acute Lymphoblastic Leukemia: A Meta-analysis. JAMA Oncol. 2017, 3, e170580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wood, B.L. How do we measure MRD in ALL and how should measurements affect decisions. Re: Treatment and prognosis? Best Pract. Res. Clin. Haematol. 2017, 30, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wood, B.L. Monitoring minimal residual disease in acute leukemia: Technical challenges and interpretive complexities. Blood Rev. 2017, 31, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Gokbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Bruggemann, M.; Horst, H.A.; et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Keating, A.K.; Gossai, N.; Phillips, C.L.; Maloney, K.; Campbell, K.; Doan, A.; Bhojwani, D.; Burke, M.J.; Verneris, M.R. Reducing minimal residual disease with blinatumomab prior to HCT for pediatric patients with acute lymphoblastic leukemia. Blood Adv. 2019, 3, 1926–1929. [Google Scholar] [CrossRef] [Green Version]
- Queudeville, M.; Ebinger, M. Blinatumomab in Pediatric Acute Lymphoblastic Leukemia-From Salvage to First Line Therapy (A Systematic Review). J. Clin. Med. 2021, 10, 2544. [Google Scholar] [CrossRef]
- Dourthe, M.E.; Baruchel, A. CAR T-cells in acute lymphoblastic leukemia: Current results. Bull. Cancer 2021, 108, S40–S54. [Google Scholar] [CrossRef]
- Mejstrikova, E.; Klinger, M.; Markovic, A.; Zugmaier, G.; Locatelli, F. CD19 expression in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia pre- and post-treatment with blinatumomab. Pediatr. Blood Cancer 2021, 68, e29323. [Google Scholar] [CrossRef]
- Libert, D.; Yuan, C.M.; Masih, K.E.; Galera, P.; Salem, D.; Shalabi, H.; Yates, B.; Delbrook, C.; Shern, J.F.; Fry, T.J.; et al. Serial evaluation of CD19 surface expression in pediatric B-cell malignancies following CD19-targeted therapy. Leukemia 2020, 34, 3064–3069. [Google Scholar] [CrossRef]
- Bartram, J.; Patel, B.; Fielding, A.K. Monitoring MRD in ALL: Methodologies, technical aspects and optimal time points for measurement. Semin. Hematol. 2020, 57, 142–148. [Google Scholar] [CrossRef]
- Dworzak, M.N.; Gaipa, G.; Ratei, R.; Veltroni, M.; Schumich, A.; Maglia, O.; Karawajew, L.; Benetello, A.; Potschger, U.; Husak, Z.; et al. Standardization of flow cytometric minimal residual disease evaluation in acute lymphoblastic leukemia: Multicentric assessment is feasible. Cytom. Part B Clin. Cytom. 2008, 74, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Borowitz, M.J.; Wood, B.L.; Keeney, M.; Hedley, B.D. Measurable Residual Disease Detection in B-Acute Lymphoblastic Leukemia: The Children’s Oncology Group (COG) Method. Curr. Protoc. 2022, 2, e383. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [Green Version]
- Mo, G.; Wang, H.W.; Talleur, A.C.; Shahani, S.A.; Yates, B.; Shalabi, H.; Douvas, M.G.; Calvo, K.R.; Shern, J.F.; Chaganti, S.; et al. Diagnostic approach to the evaluation of myeloid malignancies following CAR T-cell therapy in B-cell acute lymphoblastic leukemia. J. Immunother. Cancer 2020, 8, e001563. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Miller, V.; McCullouch, V.; Dougherty, K.; Fromm, J.R.; Wood, B.L. A novel flow cytometric assay for detection of residual disease in patients with B-lymphoblastic leukemia/lymphoma post anti-CD19 therapy. Cytom. Part B Clin. Cytom. 2016, 94, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Fu, M.; Wang, A.; Wu, X.; Zhen, J.; Gong, M.; Zhang, X.; Yue, G.; Du, Q.; Zhao, W.; et al. Cytoplasmic CD79a is a promising biomarker for B lymphoblastic leukemia follow up post CD19 CAR-T therapy. Leuk. Lymphoma 2022, 63, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, M.W.C.; Buracchi, C.; Laqua, A.; Nierkens, S.; Sedek, L.; Flores-Montero, J.; Hofmans, M.; Sobral de Costa, E.; Novakova, M.; Mejstrikova, E.; et al. Flow cytometric minimal residual disease assessment in B-cell precursor acute lymphoblastic leukaemia patients treated with CD19-targeted therapies—A EuroFlow study. Br. J. Haematol. 2022, 197, 76–81. [Google Scholar] [CrossRef]
- Mikhailova, E.; Gluhanyuk, E.; Illarionova, O.; Zerkalenkova, E.; Kashpor, S.; Miakova, N.; Diakonova, Y.; Olshanskaya, Y.; Shelikhova, L.; Novichkova, G.; et al. Immunophenotypic changes of leukemic blasts in children with relapsed/refractory B-cell precursor acute lymphoblastic leukemia, who have been treated with Blinatumomab. Haematol. Ogica 2021, 106, 2009–2012. [Google Scholar] [CrossRef]
- Mikhailova, E.; Illarionova, O.; Shelikhova, L.; Zerkalenkova, E.; Molostova, O.; Olshanskaya, Y.; Novichkova, G.; Maschan, A.; Maschan, M.; Popov, A. Immunophenotypic changes in leukemic blasts in children with relapsed/refractory B-cell precursor acute lymphoblastic leukemia after treatment with CD19-directed chimeric antigen receptor (CAR)- expressing T cells. Haematol. Ogica 2022, 107, 970–974. [Google Scholar] [CrossRef]
- Mikhailova, E.; Itov, A.; Zerkalenkova, E.; Roumiantseva, J.; Olshanskaya, Y.; Karachunskiy, A.; Novichkova, G.; Maschan, M.; Popov, A. B-lineage antigens that are useful to substitute CD19 for minimal residual disease monitoring in B cell precursor acute lymphoblastic leukemia after CD19 targeting. Cytom. Part B Clin. Cytom. 2022, 102, 353–359. [Google Scholar] [CrossRef]
- Dworzak, M.N.; Fritsch, G.; Buchinger, P.; Fleischer, C.; Printz, D.; Zellner, A.; Schollhammer, A.; Steiner, G.; Ambros, P.F.; Gadner, H. Flow cytometric assessment of human MIC2 expression in bone marrow, thymus, and peripheral blood. Blood 1994, 83, 415–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhailova, E.; Semchenkova, A.; Illarionova, O.; Kashpor, S.; Brilliantova, V.; Zakharova, E.; Zerkalenkova, E.; Zangrando, A.; Bocharova, N.; Shelikhova, L.; et al. Relative expansion of CD19-negative very-early normal B-cell precursors in children with acute lymphoblastic leukaemia after CD19 targeting by blinatumomab and CAR-T cell therapy: Implications for flow cytometric detection of minimal residual disease. Br. J. Haematol. 2021, 193, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Mikhailova, E.; Roumiantseva, J.; Illarionova, O.; Lagoyko, S.; Miakova, N.; Zerkalenkova, E.; Zharikova, L.; Olshanskaya, Y.; Novichkova, G.; Maschan, M.; et al. Strong expansion of normal CD19-negative B-cell precursors after the use of blinatumomab in the first-line therapy of acute lymphoblastic leukaemia in children. Br. J. Haematol. 2022, 196, e6–e9. [Google Scholar] [CrossRef]
- Semchenkova, A.; Mikhailova, E.; Komkov, A.; Gaskova, M.; Abasov, R.; Matveev, E.; Kazanov, M.; Mamedov, I.; Shmitko, A.; Belova, V.; et al. Lineage Conversion in Pediatric B-Cell Precursor Acute Leukemia under Blinatumomab Therapy. Int. J. Mol. Sci. 2022, 23, 4019. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, S.; Obuchowski, N.A.; Lieber, M.L. Clinical evaluation of diagnostic tests. Am. J. Roentgenol. 2005, 184, 14–19. [Google Scholar] [CrossRef]
- Maurer-Granofszky, M.; Schumich, A.; Buldini, B.; Gaipa, G.; Kappelmayer, J.; Mejstrikova, E.; Karawajew, L.; Rossi, J.; Suzan, A.C.; Agriello, E.; et al. An Extensive Quality Control and Quality Assurance (QC/QA) Program Significantly Improves Inter-Laboratory Concordance Rates of Flow-Cytometric Minimal Residual Disease Assessment in Acute Lymphoblastic Leukemia: An I-BFM-FLOW-Network Report. Cancers 2021, 13, 6148. [Google Scholar] [CrossRef]
- Kalina, T.; Flores-Montero, J.; Lecrevisse, Q.; Pedreira, C.E.; van der Velden, V.H.; Novakova, M.; Mejstrikova, E.; Hrusak, O.; Bottcher, S.; Karsch, D.; et al. Quality assessment program for EuroFlow protocols: Summary results of four-year (2010–2013) quality assurance rounds. Cytom. Part A 2015, 87, 145–156. [Google Scholar] [CrossRef]
- Theunissen, P.; Mejstrikova, E.; Sedek, L.; van der Sluijs-Gelling, A.J.; Gaipa, G.; Bartels, M.; Sobral da Costa, E.; Kotrova, M.; Novakova, M.; Sonneveld, E.; et al. Standardized flow cytometry for highly sensitive MRD measurements in B-cell acute lymphoblastic leukemia. Blood 2017, 129, 347–357. [Google Scholar] [CrossRef]
- den Nijs, J.I.; Gonggrijp, H.S.; Augustinus, E.; Leeksma, C.H. Hot bands: A simple G-banding method for leukemic metaphases. Cancer Genet. Cytogenet. 1985, 15, 373–374. [Google Scholar] [CrossRef]
- ISCN-2020. ISCN, An International System for Human Cytogenomic Nomenclature; Karger Publishers: Basel, Switzerland, 2020. [Google Scholar]
- Gabert, J.; Beillard, E.; van der Velden, V.H.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cave, H.; et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe Against Cancer program. Leukemia 2003, 17, 2318–2357. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Lopes, B.A.; Caye-Eude, A.; Cave, H.; Arfeuille, C.; Cuccuini, W.; Sutton, R.; Venn, N.C.; Oh, S.H.; Tsaur, G.; et al. Human MLL/KMT2A gene exhibits a second breakpoint cluster region for recurrent MLL-USP2 fusions. Leukemia 2019, 33, 2306–2340. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.W.; van der Velden, V.H.; van Dongen, J.J. Efficient and easy detection of MLL-AF4, MLL-AF9 and MLL-ENL fusion gene transcripts by multiplex real-time quantitative RT-PCR in TaqMan and LightCycler. Leukemia 2005, 19, 2016–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beillard, E.; Pallisgaard, N.; van der Velden, V.H.; Bi, W.; Dee, R.; van der Schoot, E.; Delabesse, E.; Macintyre, E.; Gottardi, E.; Saglio, G.; et al. Evaluation of candidate control genes for diagnosis and residual disease detection in leukemic patients using ‘real-time’ quantitative reverse-transcriptase polymerase chain reaction (RQ-PCR)—A Europe against cancer program. Leukemia 2003, 17, 2474–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolotin, D.A.; Poslavsky, S.; Mitrophanov, I.; Shugay, M.; Mamedov, I.Z.; Putintseva, E.V.; Chudakov, D.M. MiXCR: Software for comprehensive adaptive immunity profiling. Nat. Methods 2015, 12, 380–381. [Google Scholar] [CrossRef]
- Komkov, A.; Miroshnichenkova, A.; Nugmanov, G.; Popov, A.; Pogorelyy, M.; Zapletalova, E.; Jelinkova, H.; Pospisilova, S.; Lebedev, Y.; Chudakov, D.; et al. High-throughput sequencing of T-cell receptor alpha chain clonal rearrangements at the DNA level in lymphoid malignancies. Br. J. Haematol. 2020, 188, 723–731. [Google Scholar] [CrossRef]
- Dube, S.; Qin, J.; Ramakrishnan, R. Mathematical analysis of copy number variation in a DNA sample using digital PCR on a nanofluidic device. PLoS ONE 2008, 3, e2876. [Google Scholar] [CrossRef] [Green Version]
- DiGiuseppe, J.A.; Wood, B.L. Applications of Flow Cytometric Immunophenotyping in the Diagnosis and Posttreatment Monitoring of B and T Lymphoblastic Leukemia/Lymphoma. Cytom. Part B Clin. Cytom. 2019, 96, 256–265. [Google Scholar] [CrossRef]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Bene, M.C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [Green Version]
- Toba, K.; Hanawa, H.; Fuse, I.; Sakaue, M.; Watanabe, K.; Uesugi, Y.; Higuchi, W.; Takahashi, M.; Aizawa, Y. Difference in CD22 molecules in human B cells and basophils. Exp. Hematol. 2002, 30, 205–211. [Google Scholar] [CrossRef]
- Han, K.; Kim, Y.; Lee, J.; Lim, J.; Lee, K.Y.; Kang, C.S.; Kim, W.I.; Kim, B.K.; Shim, S.I.; Kim, S.M. Human basophils express CD22 without expression of CD19. Cytometry 1999, 37, 178–183. [Google Scholar] [CrossRef]
- Reineks, E.Z.; Osei, E.S.; Rosenberg, A.; Auletta, J.; Meyerson, H.J. CD22 expression on blastic plasmacytoid dendritic cell neoplasms and reactivity of anti-CD22 antibodies to peripheral blood dendritic cells. Cytom. Part B Clin. Cytom. 2009, 76, 237–248. [Google Scholar] [CrossRef] [PubMed]
- De Zen, L.; Bicciato, S.; Te Kronnie, G.; Basso, G. Computational analysis of flow-cytometry antigen expression profiles in childhood acute lymphoblastic leukemia: An MLL/AF4 identification. Leukemia 2003, 17, 1557–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.N.; Stevenson, M.S.; Yuan, C.M.; Richards, K.; Delbrook, C.; Kreitman, R.J.; Pastan, I.; Wayne, A.S. Characterization of CD22 expression in acute lymphoblastic leukemia. Pediatr. Blood Cancer 2015, 62, 964–969. [Google Scholar] [CrossRef] [Green Version]
- Buechner, J.; Caruana, I.; Kunkele, A.; Rives, S.; Vettenranta, K.; Bader, P.; Peters, C.; Baruchel, A.; Calkoen, F.G. Chimeric Antigen Receptor T-Cell Therapy in Paediatric B-Cell Precursor Acute Lymphoblastic Leukaemia: Curative Treatment Option or Bridge to Transplant? Front. Pediatr. 2021, 9, 784024. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.A.; Ji, L.; Xu, X.; Devidas, M.; Hogan, L.E.; Borowitz, M.J.; Raetz, E.A.; Zugmaier, G.; Sharon, E.; Bernhardt, M.B.; et al. Effect of Postreinduction Therapy Consolidation with Blinatumomab vs Chemotherapy on Disease-Free Survival in Children, Adolescents, and Young Adults with First Relapse of B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 2021, 325, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Zugmaier, G.; Rizzari, C.; Morris, J.D.; Gruhn, B.; Klingebiel, T.; Parasole, R.; Linderkamp, C.; Flotho, C.; Petit, A.; et al. Effect of Blinatumomab vs Chemotherapy on Event-Free Survival Among Children with High-risk First-Relapse B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 2021, 325, 843–854. [Google Scholar] [CrossRef]
- Pulsipher, M.A.; Han, X.; Maude, S.L.; Laetsch, T.W.; Qayed, M.; Rives, S.; Boyer, M.W.; Hiramatsu, H.; Yanik, G.A.; Driscoll, T.; et al. Next-Generation Sequencing of Minimal Residual Disease for Predicting Relapse after Tisagenlecleucel in Children and Young Adults with Acute Lymphoblastic Leukemia. Blood Cancer Discov. 2022, 3, 66–81. [Google Scholar] [CrossRef]
- Abou Dalle, I.; Jabbour, E.; Short, N.J. Evaluation and management of measurable residual disease in acute lymphoblastic leukemia. Ther. Adv. Hematol. 2020, 11, 2040620720910023. [Google Scholar] [CrossRef]
- Cherian, S.; Stetler-Stevenson, M. Flow Cytometric Monitoring for Residual Disease in B Lymphoblastic Leukemia Post T Cell Engaging Targeted Therapies. Curr. Protoc. Cytom. 2018, 86, e44. [Google Scholar] [CrossRef]
- Reiter, M.; Diem, M.; Schumich, A.; Maurer-Granofszky, M.; Karawajew, L.; Rossi, J.G.; Ratei, R.; Groeneveld-Krentz, S.; Sajaroff, E.O.; Suhendra, S.; et al. Automated Flow Cytometric MRD Assessment in Childhood Acute B- Lymphoblastic Leukemia Using Supervised Machine Learning. Cytom. Part A 2019, 95, 966–975. [Google Scholar] [CrossRef]
- Bene, M.C.; Lacombe, F.; Porwit, A. Unsupervised flow cytometry analysis in hematological malignancies: A new paradigm. Int. J. Lab. Hematol. 2021, 43 (Suppl. S1), 54–64. [Google Scholar] [CrossRef] [PubMed]
- Bouriche, L.; Bernot, D.; Nivaggioni, V.; Arnoux, I.; Loosveld, M. Detection of Minimal Residual Disease in B Cell Acute Lymphoblastic Leukemia Using an Eight-Color Tube with Dried Antibody Reagents. Cytom. Part B Clin. Cytom. 2019, 96, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Semchenkova, A.; Brilliantova, V.; Shelikhova, L.; Zhogov, V.; Illarionova, O.; Mikhailova, E.; Raykina, E.; Skorobogatova, E.; Novichkova, G.; Maschan, A.; et al. Chimerism evaluation in measurable residual disease-suspected cells isolated by flow cell sorting as a reliable tool for measurable residual disease verification in acute leukemia patients after allogeneic hematopoietic stem cell transplantation. Cytom. Part B Clin. Cytom. 2021, 100, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Obro, N.F.; Ryder, L.P.; Madsen, H.O.; Andersen, M.K.; Lausen, B.; Hasle, H.; Schmiegelow, K.; Marquart, H.V. Identification of residual leukemic cells by flow cytometry in childhood B-cell precursor acute lymphoblastic leukemia: Verification of leukemic state by flow-sorting and molecular/cytogenetic methods. Haematol. Ogica 2012, 97, 137–141. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.J.; Coustan-Smith, E.; Kao, H.W.; Liu, H.C.; Chen, S.H.; Hsiao, C.C.; Yang, C.P.; Jaing, T.H.; Yeh, T.C.; Kuo, M.C.; et al. Concordance of two approaches in monitoring of minimal residual disease in B-precursor acute lymphoblastic leukemia: Fusion transcripts and leukemia-associated immunophenotypes. J. Formos. Med. Assoc. 2017, 116, 774–781. [Google Scholar] [CrossRef]
- Wood, B.; Wu, D.; Crossley, B.; Dai, Y.; Williamson, D.; Gawad, C.; Borowitz, M.J.; Devidas, M.; Maloney, K.W.; Larsen, E.; et al. Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood 2018, 131, 1350–1359. [Google Scholar] [CrossRef]
- Popov, A.; Tsaur, G.; Verzhbitskaya, T.; Riger, T.; Permikin, Z.; Demina, A.; Mikhailova, E.; Shorikov, E.; Arakaev, O.; Streneva, O.; et al. Comparison of minimal residual disease measurement by multicolour flow cytometry and PCR for fusion gene transcripts in infants with acute lymphoblastic leukaemia with KMT2A gene rearrangements. Br. J. Haematol. 2021. [Google Scholar] [CrossRef]
- Neale, G.A.; Coustan-Smith, E.; Stow, P.; Pan, Q.; Chen, X.; Pui, C.H.; Campana, D. Comparative analysis of flow cytometry and polymerase chain reaction for the detection of minimal residual disease in childhood acute lymphoblastic leukemia. Leukemia 2004, 18, 934–938. [Google Scholar] [CrossRef]
- Gaipa, G.; Cazzaniga, G.; Valsecchi, M.G.; Panzer-Grumayer, R.; Buldini, B.; Silvestri, D.; Karawajew, L.; Maglia, O.; Ratei, R.; Benetello, A.; et al. Time point-dependent concordance of flow cytometry and real-time quantitative polymerase chain reaction for minimal residual disease detection in childhood acute lymphoblastic leukemia. Haematol. Ogica 2012, 97, 1582–1593. [Google Scholar] [CrossRef]

{kind=link}
| Fluorochrome | FITC | PE | PE-CF594 | PerCP-Cy5.5 | PE-Cy7 | APC | A700 | APC-Cy7 | BV510 | BV768 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Antibody | CD58 | CD22 | CD10 | CD20 | CD19 | iCD79a | CD34 | CD45 | SYTO41 | CD38 | CD24 |
| Clone | 1C3 (AICD58.6) | S-HCL-1 | HI10a | L27 | SJ25C1 | HM47 | 8G12 | 2D1 | HIT2 | ML5 |
| B-Cell Gating Type | Normal BM Cell Populations |
|---|---|
| CD19-based | CD19(+) BCPs, plasma cells, mature B cells |
| CD22+iCD79a-based | CD19(+) BCPs, CD19(−) BCPs, mature B cells |
| CD22-based | CD19(+) BCPs, CD19(−) BCPs, mature B cells, basophils, plasmacytoid dendritic cells |
| iCD79a-based | CD19(+) BCPs, CD19(−) BCPs, plasma cells, mature B cells |
| CD10-based | CD19(+) BCPs, CD19(−) BCPs |
| CD24-based | CD19(+) BCPs, mature B cells |
| All samples | NGS-MRD | ||
| Positive | Negative | ||
| MFC-MRD | Positive | 46 | 3 |
| Negative | 55 | 234 | |
| Concordance: 82.8% | |||
| All samples | FGT-MRD | ||
| Positive | Negative | ||
| MFC-MRD | Positive | 12 | 3 |
| Negative | 10 | 103 | |
| Concordance: 89.8% | |||
| KMT2A-rearranged samples | NGS-MRD | ||
| Positive | Negative | ||
| MFC-MRD | Positive | 3 | 0 |
| Negative | 5 | 29 | |
| Concordance: 86.5% | |||
| KMT2A-rearranged samples | FGT-MRD | ||
| Positive | Negative | ||
| MFC-MRD | Positive | 3 | 0 |
| Negative | 1 | 50 | |
| Concordance: 98.1% | |||
| CD19(+) blasts after CAR-T therapy | NGS-MRD | ||
| Positive | Negative | ||
| MFC-MRD | Positive | 11 | 0 |
| Negative | 14 | 41 | |
| Concordance: 78.8% | |||
| CD19(+) blasts after CAR-T therapy | FGT-MRD | ||
| Positive | Negative | ||
| MFC-MRD | Positive | 1 | 2 |
| Negative | 1 | 27 | |
| Concordance: 90.3% | |||
| CD19(−) blasts after CAR-T therapy | NGS-MRD | ||
| Positive | Negative | ||
| MFC-MRD | Positive | 21 | 2 |
| Negative | 9 | 42 | |
| Concordance: 85.1% | |||
| CD19(−) blasts after CAR-T therapy | FGT-MRD | ||
| Positive | Negative | ||
| MFC-MRD | Positive | 4 | 1 |
| Negative | 2 | 19 | |
| Concordance: 88.5% | |||
| Less than 2 months after infusion | NGS-MRD | ||
| Positive | Negative | ||
| MFC-MRD | Positive | 4 | 1 |
| Negative | 17 | 45 | |
| Concordance: 73.1% | |||
| Less than 2 months after infusion | FGT-MRD | ||
| Positive | Negative | ||
| MFC-MRD | Positive | 2 | 0 |
| Negative | 1 | 30 | |
| Concordance: 97.0% | |||
| 2 months or more after infusion | NGS-MRD | ||
| Positive | Negative | ||
| MFC-MRD | Positive | 22 | 1 |
| Negative | 27 | 140 | |
| Concordance: 85.3% | |||
| 2 months or more after infusion | FGT-MRD | ||
| Positive | Negative | ||
| MFC-MRD | Positive | 3 | 2 |
| Negative | 6 | 51 | |
| Concordance: 87.1% | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhailova, E.; Illarionova, O.; Komkov, A.; Zerkalenkova, E.; Mamedov, I.; Shelikhova, L.; Olshanskaya, Y.; Miakova, N.; Novichkova, G.; Karachunskiy, A.; et al. Reliable Flow-Cytometric Approach for Minimal Residual Disease Monitoring in Patients with B-Cell Precursor Acute Lymphoblastic Leukemia after CD19-Targeted Therapy. Cancers 2022, 14, 5445. https://doi.org/10.3390/cancers14215445
Mikhailova E, Illarionova O, Komkov A, Zerkalenkova E, Mamedov I, Shelikhova L, Olshanskaya Y, Miakova N, Novichkova G, Karachunskiy A, et al. Reliable Flow-Cytometric Approach for Minimal Residual Disease Monitoring in Patients with B-Cell Precursor Acute Lymphoblastic Leukemia after CD19-Targeted Therapy. Cancers. 2022; 14(21):5445. https://doi.org/10.3390/cancers14215445
Chicago/Turabian StyleMikhailova, Ekaterina, Olga Illarionova, Alexander Komkov, Elena Zerkalenkova, Ilgar Mamedov, Larisa Shelikhova, Yulia Olshanskaya, Natalia Miakova, Galina Novichkova, Alexander Karachunskiy, and et al. 2022. "Reliable Flow-Cytometric Approach for Minimal Residual Disease Monitoring in Patients with B-Cell Precursor Acute Lymphoblastic Leukemia after CD19-Targeted Therapy" Cancers 14, no. 21: 5445. https://doi.org/10.3390/cancers14215445
APA StyleMikhailova, E., Illarionova, O., Komkov, A., Zerkalenkova, E., Mamedov, I., Shelikhova, L., Olshanskaya, Y., Miakova, N., Novichkova, G., Karachunskiy, A., Maschan, M., & Popov, A. (2022). Reliable Flow-Cytometric Approach for Minimal Residual Disease Monitoring in Patients with B-Cell Precursor Acute Lymphoblastic Leukemia after CD19-Targeted Therapy. Cancers, 14(21), 5445. https://doi.org/10.3390/cancers14215445