
Novel Cellular Therapies for Hepatocellular Carcinoma


Abstract
Simple Summary
Abstract
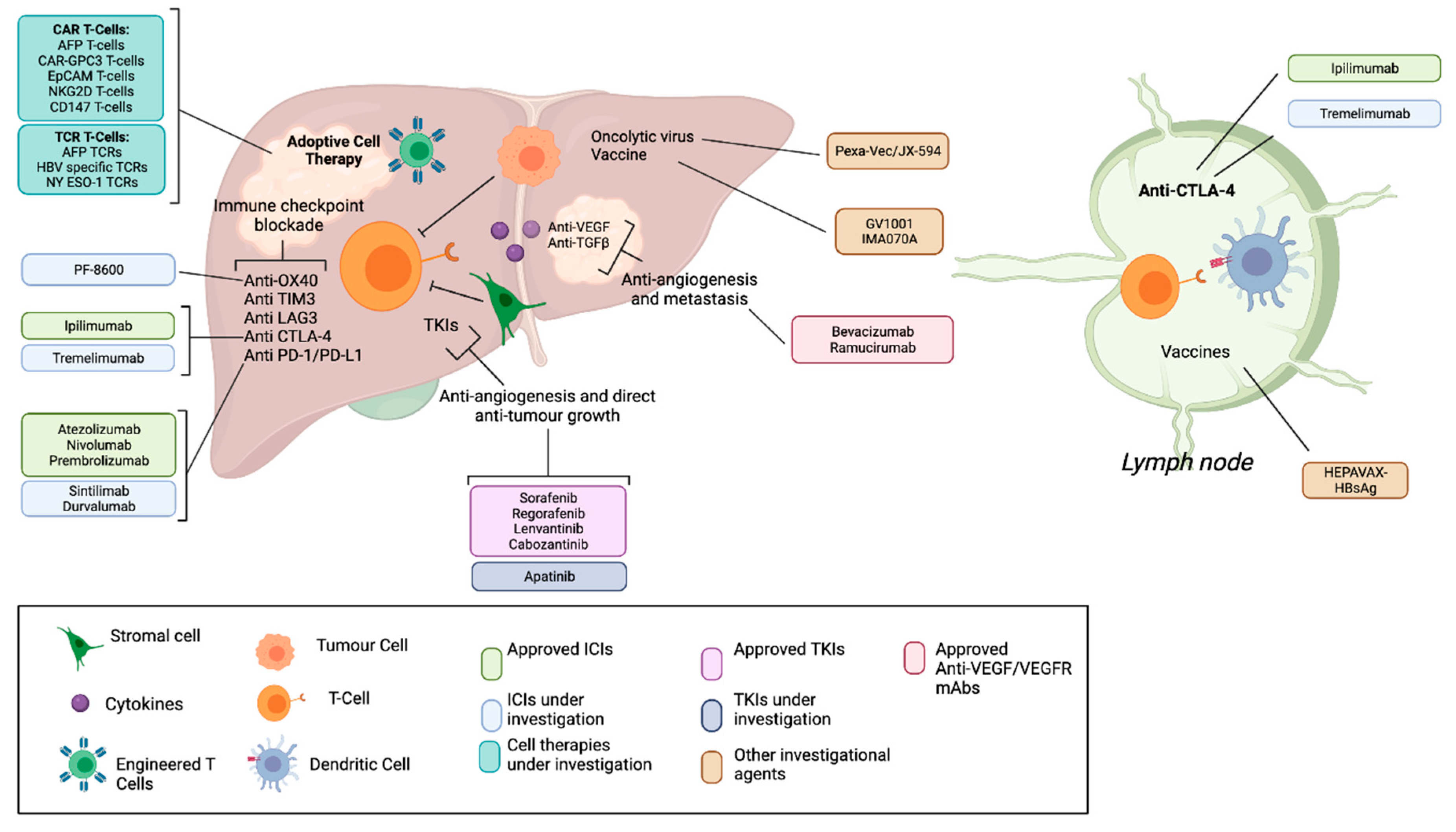
1. Introduction
2. Non-Gene Modified Adoptive Cell Therapies
2.1. Cytokine Induced Killer (CIK) Cells
2.1.1. CIK Biology and Background
2.1.2. CIKs for HCC
2.1.3. Combinatorial Approaches: CIKs + TACE
2.1.4. Combinatorial Approaches: CIKs + Dendritic cells (DCs)
2.1.5. Future Directions for CIK Therapy
2.2. Tumour Infiltrating Lymphocytes (TILs)
2.2.1. TIL Biology and Background
2.2.2. TILs for HCC
3. Gene Modified Adoptive Cell Therapies
3.1. Chimeric Antigen Receptor T-Cells (CAR-T)
3.1.1. CAR-T Biology, Background, Targets
3.1.2. Glypican-3 (GPC3) Biology
3.1.3. Glypican-3 (GPC3) Targeting CAR-T (Preclinical)
3.1.4. Glypican-3 (GPC3) Targeting CAR-T (Clinical)
3.1.5. Alpha Fetoprotein (AFP) Biology
3.1.6. Alpha Fetoprotein (AFP) Targeting CAR-T (Preclinical)
3.1.7. Alpha Fetoprotein (AFP) Targeting CAR-T (Clinical)
3.1.8. c-MET Biology and Background
3.1.9. c-MET Targeting CAR-T (Preclinical)
3.1.10. Mucin 1 Glycoprotein 1 (MUC1) Biology and Background
3.1.11. MUC1 Targeting CAR-T (Preclinical)
3.1.12. MUC1 Targeting CAR-T (Clinical)
3.1.13. NK Group 2 Member D Ligand (NKG2DL) Biology and Background
3.1.14. NKG2DL Targeting CAR-T (Preclinical)
3.1.15. NKG2DL Targeting CAR-T (Clinical)
3.1.16. Other Potential CAR-T Targets for HCC
3.2. T-Cell Receptor Transduced T Cells (TCR-T)
3.2.1. TCR-T Biology, Background, Targets
3.2.2. AFP TCR-T Biology and Background
3.2.3. AFP TCR-T (Clinical)
3.2.4. Viral Associated Peptides as TCR-T Targets
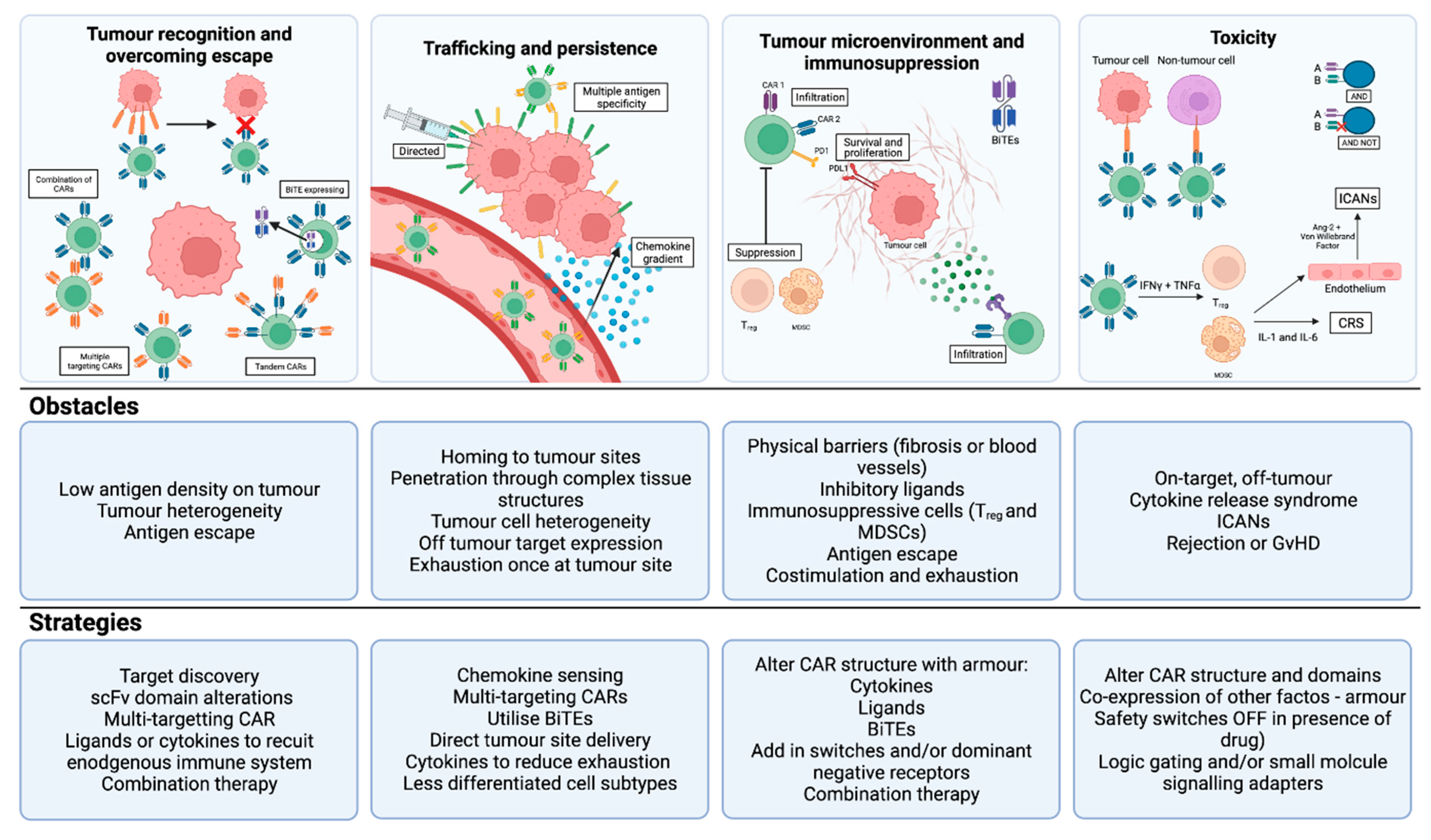
4. What Is the Future of T cell therapy for HCC?
4.1. CAR-T: Enhancing Tumour Recognition, Overcoming Escape and Reducing Toxicity
4.2. CAR-T: Enhancing Trafficking and Persistence
4.3. CAR-T: Overcoming the Immunosuppressive Tumour Microenvironment
4.4. TCR-T: Enhancing Tumour Recognition and Reducing Off-Tumour Toxicity
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Di Bisceglie, A.M.; Rustgi, V.K.; Hoofnagle, J.H.; Dusheiko, G.M.; Lotze, M.T. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Forgues, M.; Ye, Q.-H.; Jia, H.-L.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.-X.; Tang, Z.-Y.; et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef]
- Unitt, E.; Marshall, A.; Gelson, W.; Rushbrook, S.M.; Davies, S.; Vowler, S.L.; Morris, L.S.; Coleman, N.; Alexander, G.J. Tumour lymphocytic infiltrate and recurrence of hepatocellular carcinoma following liver transplantation. J. Hepatol. 2006, 45, 246–253. [Google Scholar] [CrossRef]
- Stroehl, Y.W.; Letzen, B.S.; Van Breugel, J.M.M.; Geschwind, J.-F.; Chapiro, J. Intra-arterial therapies for liver cancer: Assessing tumor response. Expert Rev. Anticancer Ther. 2017, 17, 119–127. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; Bruix, J. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 13, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H., 3rd; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Martinelli, E.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.; Meyer, T.; Nault, J.-C.; Neumann, U.; Ricke, J.; et al. Updated treatment recommendations for hepatocellular carcinoma (HCC) from the ESMO Clinical Practice Guidelines. Ann. Oncol. 2021, 32, 801–805. [Google Scholar] [CrossRef]
- Kelley, R.K.; Sangro, B.; Harris, W.; Ikeda, M.; Okusaka, T.; Kang, Y.-K.; Qin, S.; Tai, D.W.-M.; Lim, H.Y.; Yau, T.; et al. Safety, Efficacy, and Pharmacodynamics of Tremelimumab Plus Durvalumab for Patients With Unresectable Hepatocellular Carcinoma: Randomized Expansion of a Phase I/II Study. J. Clin. Oncol. 2021, 39, 2991–3001. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Sangro, B.; Harris, W.P.; Ikeda, M.; Okusaka, T.; Kang, Y.-K.; Qin, S.; Tai, W.M.D.; Lim, H.Y.; Yau, T.; et al. Efficacy, tolerability, and biologic activity of a novel regimen of tremelimumab (T) in combination with durvalumab (D) for patients (pts) with advanced hepatocellular carcinoma (aHCC). J. Clin. Oncol. 2020, 38, 4508. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Hsu, C. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Zou, W. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012, 56, 1342–1351. [Google Scholar] [CrossRef]
- Du, W.; Yang, M.; Turner, A.; Xu, C.; Ferris, R.L.; Huang, J.; Kane, L.P.; Lu, B. TIM-3 as a Target for Cancer Immunotherapy and Mechanisms of Action. Int. J. Mol. Sci. 2017, 18, 645. [Google Scholar] [CrossRef]
- Zhou, G.; Sprengers, D.; Boor, P.P.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; de Jonge, J.; Gaspersz, M.; et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology 2017, 153, 1107–1119. [Google Scholar] [CrossRef]
- Sangro, B.; Chan, S.L.; Meyer, T.; Reig, M.; El-Khoueiry, A.; Galle, P.R. Diagnosis and management of toxicities of immune checkpoint inhibitors in hepatocellular carcinoma. J. Hepatol. 2020, 72, 320–341. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.H.; Negrin, R.S. A novel population of expanded human CD3+CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency. J. Immunol. 1994, 153, 1687–1696. [Google Scholar] [PubMed]
- Alvarnas, J.C.; Linn, Y.-C.; Hope, E.G.; Negrin, R.S. Expansion of cytotoxic CD3+ CD56+ cells from peripheral blood progenitor cells of patients undergoing autologous hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2001, 7, 216–222. [Google Scholar] [CrossRef]
- Nishimura, R.; Baker, J.; Beilhack, A.; Zeiser, R.; Olson, J.A.; Sega, E.I.; Karimi, M.; Negrin, R.S. In vivo trafficking and survival of cytokine-induced killer cells resulting in minimal GVHD with retention of antitumor activity. Blood 2008, 112, 2563–2574. [Google Scholar] [CrossRef]
- Schmidt-Wolf, I.G.H.; Finke, S.; Trojaneck, B.; Denkena, A.; Lefterova, P.; Schwella, N.; Heuft, H.-G.; Prange, G.; Korte, M.; Takeya, M.; et al. Phase I clinical study applying autologous immunological effector cells transfected with the interleukin-2 gene in patients with metastatic renal cancer, colorectal cancer and lymphoma. Br. J. Cancer 1999, 81, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Linn, Y.C.; Lau, L.C.; Hui, K. Generation of cytokine-induced killer cells from leukaemic samples with in vitro cytotoxicity against autologous and allogeneic leukaemic blasts. Br. J. Haematol. 2002, 116, 78–86. [Google Scholar] [CrossRef]
- Li, Y.-C.; Zhao, L.; Wu, J.-P.; Qu, C.-X.; Song, Q.-K.; Wang, R.-B. Cytokine-induced killer cell infusion combined with conventional treatments produced better prognosis for hepatocellular carcinoma patients with barcelona clinic liver cancer B or earlier stage: A systematic review and meta-analysis. Cytotherapy 2016, 18, 1525–1531. [Google Scholar] [CrossRef]
- Cao, J.; Kong, F.-H.; Liu, X.; Wang, X.-B. Immunotherapy with dendritic cells and cytokine-induced killer cells for hepatocellular carcinoma: A meta-analysis. World J. Gastroenterol. 2019, 25, 3649–3663. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, B.; Tang, Z.-R.; Lei, Z.-Y.; Wang, H.-F.; Feng, Y.-Y.; Fan, Z.-P.; Xu, D.-P.; Wang, F.-S. Autologous cytokine-induced killer cell therapy in clinical trial phase I is safe in patients with primary hepatocellular carcinoma. World J. Gastroenterol. 2004, 10, 1146–1151. [Google Scholar] [CrossRef]
- Olioso, P.; Giancola, R.; Di Riti, M.; Contento, A.; Accorsi, P.; Iacone, A. Immunotherapy with cytokine induced killer cells in solid and hematopoietic tumours: A pilot clinical trial. Hematol. Oncol. 2009, 27, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.-Z.; Lin, H.-L.; Chen, Q.; Ye, Y.-B.; Chen, Q.-Z.; Chen, M.-S. Efficacy of transcatheter arterial chemoembolization combined with cytokine-induced killer cell therapy on hepatocellular carcinoma: A comparative study. Chin. J. Cancer 2010, 29, 172–177. [Google Scholar] [CrossRef]
- Zhao, M.; Wu, P.-H.; Zeng, Y.-X.; Xia, J.-C.; Zhang, F.-J.; Xian, L.-J.; Zhang, Y.-P.; Zhou, K.; Fan, W.-J.; Zhang, L.; et al. Cytokine-induced killer cell fusion to lower recurrence of hepatocellular carcinoma after transcatheter arterial chemoembolization sequentially combined with radiofrequency ablation: A randomized trial. Zhonghua Yi Xue Za Zhi 2006, 86, 1823–1828. [Google Scholar]
- Chen, F.; Yang, M.; Song, Q.; Wu, J.; Wang, X.; Zhou, X.; Yuan, Y.; Song, Y.; Jiang, N.; Zhao, Y.; et al. Enhanced antitumor effects and improved immune status of dendritic cell and cytokine-induced killer cell infusion in advanced cancer patients. Mol. Clin. Oncol. 2017, 7, 903–910. [Google Scholar] [CrossRef]
- Mosińska, P.; Gabryelska, A.; Zasada, M.; Fichna, J. Dual Functional Capability of Dendritic Cells—Cytokine-Induced Killer Cells in Improving Side Effects of Colorectal Cancer Therapy. Front. Pharmacol. 2017, 8, 126. [Google Scholar] [CrossRef]
- Zhang, D.; He, J.-T. [In vitro cytotox icity effects of cocultured DC-C IK cells combined with sorafenib against hepa to cellular carcinoma]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi = Chin. J. Cell. Mol. Immunol. 2011, 27, 664–667. [Google Scholar]
- Yu, X.; Xia, W.; Zhang, T.; Wang, H.; Xie, Y.; Yang, J.; Miao, J. Enhanced cytotoxicity of IL-24 gene-modified dendritic cells co-cultured with cytokine-induced killer cells to hepatocellular carcinoma cells. Int. J. Hematol. 2010, 92, 276–282. [Google Scholar] [CrossRef]
- Besser, M.J.; Shapira-Frommer, R.; Itzhaki, O.; Treves, A.J.; Zippel, D.B.; Levy, D.; Kubi, A.; Shoshani, N.; Zikich, D.; Ohayon, Y.; et al. Adoptive Transfer of Tumor-Infiltrating Lymphocytes in Patients with Metastatic Melanoma: Intent-to-Treat Analysis and Efficacy after Failure to Prior Immunotherapies. Clin. Cancer Res. 2013, 19, 4792–4800. [Google Scholar] [CrossRef]
- Wu, R.; Forget, M.A.; Chacon, J.; Bernatchez, C.; Haymaker, C.; Chen, J.Q.; Radvanyi, L. Adoptive T-Cell Therapy Using Autologous Tumor-Infiltrating Lymphocytes for Metastatic Melanoma: Current Status and Future Outlook. Cancer J. 2012, 18, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Gao, F.; Xie, K.; Xiang, Q.; Qin, Y.; Chen, P.; Wan, H.; Deng, Y.; Huang, J.; Wu, H. The density of tumor-infiltrating lymphocytes and prognosis in resectable hepatocellular carcinoma: A two-phase study. Aging 2021, 13, 9665–9678. [Google Scholar] [CrossRef]
- Ge, Z.; Zhou, G.; Carrascosa, L.C.; Gausvik, E.; Boor, P.P.; Noordam, L.; Doukas, M.; Polak, W.G.; Terkivatan, T.; Pan, Q.; et al. TIGIT and PD1 Co-blockade Restores ex vivo Functions of Human Tumor-Infiltrating CD8+ T Cells in Hepatocellular Carcinoma. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 443–464. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The promise and potential pitfalls of chimeric antigen receptors. Curr. Opin. Immunol. 2009, 21, 215–223. [Google Scholar] [CrossRef]
- Davenport, A.J.; Jenkins, M.R.; Cross, R.S.; Yong, C.S.; Prince, H.M.; Ritchie, D.S.; Trapani, J.; Kershaw, M.; Darcy, P.K.; Neeson, P.J. CAR-T Cells Inflict Sequential Killing of Multiple Tumor Target Cells. Cancer Immunol. Res. 2015, 3, 483–494. [Google Scholar] [CrossRef]
- Gong, M.C.; Latouche, J.-B.; Krause, A.; Heston, W.D.; Bander, N.H.; Sadelain, M. Cancer Patient T Cells Genetically Targeted to Prostate-Specific Membrane Antigen Specifically Lyse Prostate Cancer Cells and Release Cytokines in Response to Prostate-Specific Membrane Antigen. Neoplasia 1999, 1, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Brocker, T.; Karjalainen, K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J. Exp. Med. 1995, 181, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Fesnak, A.; June, C.H.; Levine, A.D.F.C.H.J.B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Dudley, M.E. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr. Opin. Immunol. 2009, 21, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Ghorashian, S.; Pule, M.; Amrolia, P. CD19 chimeric antigen receptor T cell therapy for haematological malignancies. Br. J. Haematol. 2015, 169, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R.; Weir, A.N. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar] [PubMed]
- Zhang, H.; Merchant, M.S.; Chua, K.S.; Khanna, C.; Helman, L.J.; Telford, B.; Ward, Y.; Summers, J.; Toretsky, J.A.; Thomas, E.K.; et al. Tumor Expression of 4-1BB Ligand Sustains Tumor Lytic T Cells. Cancer Biol. Ther. 2003, 2, 579–586. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roddie, C.; Dias, J.; O’Reilly, M.A.; Abbasian, M.; Cadinanos-Garai, A.; Vispute, K.; Bosshard-Carter, L.; Mitsikakou, M.; Mehra, V.; Roddy, H.; et al. Durable Responses and Low Toxicity After Fast Off-Rate CD19 Chimeric Antigen Receptor-T Therapy in Adults With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2021, 39, 3352–3363. [Google Scholar] [CrossRef] [PubMed]
- Nakatsura, T.; Yoshitake, Y.; Senju, S.; Monji, M.; Komori, H.; Motomura, Y.; Hosaka, S.; Beppu, T.; Ishiko, T.; Kamohara, H.; et al. Glypican-3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem. Biophys. Res. Commun. 2003, 306, 16–25. [Google Scholar] [CrossRef]
- Zhou, F.; Shang, W.; Yu, X.; Tian, J. Glypican-3: A promising biomarker for hepatocellular carcinoma diagnosis and treatment. Med. Res. Rev. 2018, 38, 741–767. [Google Scholar] [CrossRef]
- Capurro, M.; Wanless, I.R.; Sherman, M.; Deboer, G.; Shi, W.; Miyoshi, E.; Filmus, J. Glypican-3: A novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology 2003, 125, 89–97. [Google Scholar] [CrossRef]
- Capurro, M.; Martin, T.; Shi, W.; Filmus, J. Glypican-3 binds to frizzled and plays a direct role in the stimulation of canonical Wnt signaling. J. Cell Sci. 2014, 127, 1565–1575. [Google Scholar] [CrossRef]
- Stadlmann, S.; Gueth, U.; Baumhoer, D.; Moch, H.; Terracciano, L.; Singer, G. Glypican-3 Expression in Primary and Recurrent Ovarian Carcinomas. Int. J. Gynecol. Pathol. 2007, 26, 341–344. [Google Scholar] [CrossRef]
- Aviel-Ronen, S.; Lau, S.K.; Pintilie, M.; Lau, D.; Liu, N.; Tsao, M.; Jothy, S. Glypican-3 is overexpressed in lung squamous cell carcinoma, but not in adenocarcinoma. Mod. Pathol. 2008, 21, 817–825. [Google Scholar] [CrossRef]
- Li, W.; Guo, L.; Rathi, P.; Marinova, E.; Gao, X.; Wu, M.-F.; Liu, H.; Dotti, G.; Gottschalk, S.; Metelitsa, L.S.; et al. Redirecting T Cells to Glypican-3 with 4-1BB Zeta Chimeric Antigen Receptors Results in Th1 Polarization and Potent Antitumor Activity. Hum. Gene Ther. 2017, 28, 437–448. [Google Scholar] [CrossRef]
- Phung, Y.; Gao, W.; Man, Y.-G.; Nagata, S.; Ho, M. High-affinity monoclonal antibodies to cell surface tumor antigen glypican-3 generated through a combination of peptide immunization and flow cytometry screening. mAbs 2012, 4, 592–599. [Google Scholar] [CrossRef]
- Yamauchi, N.; Watanabe, A.; Hishinuma, M.; Ohashi, K.-I.; Midorikawa, Y.; Morishita, Y.; Niki, T.; Shibahara, J.; Mori, M.; Makuuchi, M.; et al. The glypican 3 oncofetal protein is a promising diagnostic marker for hepatocellular carcinoma. Mod. Pathol. 2005, 18, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Montalbano, M.; Rastellini, C.; McGuire, J.T.; Prajapati, J.; Shirafkan, A.; Vento, R.; Cicalese, L. Role of Glypican-3 in the growth, migration and invasion of primary hepatocytes isolated from patients with hepatocellular carcinoma. Cell. Oncol. 2017, 41, 169–184. [Google Scholar] [CrossRef]
- Gao, H.; Li, K.; Tu, H.; Pan, X.; Jiang, H.; Shi, B.; Kong, J.; Wang, H.; Yang, S.; Gu, J.; et al. Development of T Cells Redirected to Glypican-3 for the Treatment of Hepatocellular Carcinoma. Clin. Cancer Res. 2014, 20, 6418–6428. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T Cells Suppress the Growth of Tumor Cells in Patient-Derived Xenografts of Hepatocellular Carcinoma. Front. Immunol. 2017, 7, 690. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Devaud, C.P.; Duong, C.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 Antibody Therapy Potently Enhances the Eradication of Established Tumors by Gene-Modified T Cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Wang, L.-C.; Dolfi, D.V.; Wilson, C.B.; Ranganathan, R.; Sun, J.; Kapoor, V.; Scholler, J.; Puré, E.; Milone, M.C.; et al. Multifactorial T-cell Hypofunction That Is Reversible Can Limit the Efficacy of Chimeric Antigen Receptor–Transduced Human T cells in Solid Tumors. Clin. Cancer Res. 2014, 20, 4262–4273. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Melenhorst, J.J. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef]
- Roddie, C.; O’Reilly, M.A.; Marzolini, M.A.V.; Wood, L.; Pinto, J.D.A.; Abbasian, M.; Vispute, K.; Bosshard, L.; Lowdell, M.W.; Wheeler, G.; et al. Automated Manufacture of Matched Donor-Derived Allogeneic CD19 CAR T-Cells for Relapsed/Refractory B-ALL Following Allogeneic Stem Cell Transplantation: Toxicity, Efficacy and the Important Role of Lymphodepletion. Blood 2019, 134, 776. [Google Scholar] [CrossRef]
- Steffin, D.H.M.; Batra, S.A.; Rathi, P.; Guo, L.; Li, W.; Courtney, A.N.; Metelitsa, L.S.; Heczey, A. A phase I clinical trial using armored GPC3 CAR T cells for children with relapsed/refractory liver tumors. J. Clin. Oncol. 2019, 37, TPS2647. [Google Scholar] [CrossRef]
- Sun, L.; Gao, F.; Gao, Z.; Ao, L.; Li, N.; Ma, S.; Jia, M.; Lu, P.; Sun, B.; Ho, M.; et al. Shed antigen-induced blocking effect on CAR-T cells targeting Glypican-3 in Hepatocellular Carcinoma. J. Immunother. Cancer 2021, 9, e001875. [Google Scholar] [CrossRef] [PubMed]
- Docta, R.Y.; Ferronha, T.; Sanderson, J.P.; Weissensteiner, T.; Pope, G.R.; Bennett, A.D.; Gerry, A.B. Tuning T-Cell Receptor Affinity to Optimize Clinical Risk-Benefit When Targeting Alpha-Fetoprotein-Positive Liver Cancer. Hepatol. Baltim. Md. 2019, 69, 2061–2075. [Google Scholar] [CrossRef]
- Sideras, K.; Bots, S.J.; Biermann, K.; Sprengers, D.; Polak, W.G.; Ijzermans, J.N.M.; de Man, R.A.; Pan, Q.; Sleijfer, S.; Bruno, M.J.; et al. Tumour antigen expression in hepatocellular carcinoma in a low-endemic western area. Br. J. Cancer 2015, 112, 1911–1920. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; He, T.; Cui, H.; Wang, Y.; Huang, C.; Han, F. Effects of AFP gene silencing on apoptosis and proliferation of a hepatocellular carcinoma cell line. Discov. Med. 2012, 14, 115–124. [Google Scholar] [PubMed]
- Liu, H.; Xu, Y.; Xiang, J.; Long, L.; Green, S.; Yang, Z.; Zimdahl, B.; Lu, J.; Cheng, N.; Horan, L.; et al. Targeting Alpha-Fetoprotein (AFP)–MHC Complex with CAR T-Cell Therapy for Liver Cancer. Clin. Cancer Res. 2017, 23, 478–488. [Google Scholar] [CrossRef]
- Ishikawa, T.; Factor, V.M.; Marquardt, J.U.; Raggi, C.; Seo, D.; Kitade, M.; Thorgeirsson, S.S. Hepatocyte growth factor/c-met signaling is required for stem-cell-mediated liver regeneration in mice. Hepatology 2012, 55, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Boccaccio, C.; Comoglio, P. Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat. Rev. Cancer 2006, 6, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Rimassa, L.; Assenat, E.; Peck-Radosavljevic, M.; Pracht, M.; Zagonel, V.; Mathurin, P.; Caremoli, E.R.; Porta, C.; Daniele, B.; Bolondi, L.; et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): A final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 2018, 19, 682–693. [Google Scholar] [CrossRef]
- Jiang, W.; Li, T.; Guo, J.; Wang, J.; Jia, L.; Shi, X.; Yang, T.; Jiao, R.; Wei, X.; Feng, Z.; et al. Bispecific c-Met/PD-L1 CAR-T Cells Have Enhanced Therapeutic Effects on Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 546586. [Google Scholar] [CrossRef] [PubMed]
- Kotera, Y.; Fontenot, J.D.; Pecher, G.; Metzgar, R.S.; Finn, O.J. Humoral immunity against a tandem repeat epitope of human mucin MUC-1 in sera from breast, pancreatic, and colon cancer patients. Cancer Res. 1994, 54, 2856–2860. [Google Scholar] [PubMed]
- Kufe, D.W. Mucins in cancer: Function, prognosis and therapy. Nat. Cancer 2009, 9, 874–885. [Google Scholar] [CrossRef]
- Wang, J.; Liu, G.; Li, Q.; Wang, F.; Xie, F.; Zhai, R.; Guo, Y.; Chen, T.; Zhang, N.; Ni, W.; et al. Mucin1 promotes the migration and invasion of hepatocellular carcinoma cells via JNK-mediated phosphorylation of Smad2 at the C-terminal and linker regions. Oncotarget 2015, 6, 19264–19278. [Google Scholar] [CrossRef]
- Yuan, S.-F.; Li, K.-Z.; Wang, L.; Dou, K.-F.; Yan, Z.; Han, W.; Zhang, Y.-Q. Expression of MUC1 and its significance in hepatocellular and cholangiocarcinoma tissue. World J. Gastroenterol. WJG 2005, 11, 4661–4666. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdanifar, M.; Roy, L.D.; Whilding, L.M.; Gavrill, A.; Maher, J.; Mukherjee, P. CAR T Cells Targeting the Tumor MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth. Front. Immunol. 2019, 10, 1149. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-D.; Wang, Z.; Gong, R.-Z.; Li, L.-F.; Wu, H.-P.; Jin, H.-J.; Qian, Q.-J. Specific cytotoxicity of MUC1 chimeric antigen receptor-engineered Jurkat T cells against hepatocellular carcinoma. Acad. J. Second. Mil. Med Univ. 2014, 35, 1177. [Google Scholar] [CrossRef]
- Mei, Z.; Zhang, K.; Lam, A.K.; Huang, J.; Qiu, F.; Qiao, B.; Zhang, Y. MUC1 as a target for CAR-T therapy in head and neck squamous cell carinoma. Cancer Med. 2019, 9, 640–652. [Google Scholar] [CrossRef]
- Sun, B.; Yang, N.; Dai, H.; Liu, X.; Jia, R.; Cui, X.; Li, W.; Cai, C.; Xu, J.; Zhao, X. Eradication of Hepatocellular Carcinoma by NKG2D-Based CAR-T Cells. Cancer Immunol. Res. 2019, 7, 1813–1823. [Google Scholar] [CrossRef]
- Hue, S.; Mention, J.-J.; Monteiro, R.; Zhang, S.; Cellier, C.; Schmitz, J.; Verkarre, V.; Fodil, N.; Bahram, S.; Cerf-Bensussan, N.; et al. A Direct Role for NKG2D/MICA Interaction in Villous Atrophy during Celiac Disease. Immunity 2004, 21, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 100–112. [Google Scholar] [CrossRef]
- Zerbini, A.; Pilli, M.; Soliani, P.; Ziegler, S.; Pelosi, G.; Orlandini, A.; Cavallo, C.; Uggeri, J.; Scandroglio, R.; Crafa, P.; et al. Ex vivo characterization of tumor-derived melanoma antigen encoding gene-specific CD8+cells in patients with hepatocellular carcinoma. J. Hepatol. 2004, 40, 102–109. [Google Scholar] [CrossRef]
- Korangy, F.; Ormandy, L.A.; Bleck, J.S.; Klempnauer, J.; Wilkens, L.; Manns, M.P.; Greten, T.F. Spontaneous Tumor-Specific Humoral and Cellular Immune Responses to NY-ESO-1 in Hepatocellular Carcinoma. Clin. Cancer Res. 2004, 10, 4332–4341. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, E.; Nakamoto, Y.; Marukawa, Y.; Arai, K.; Yamashita, T.; Tsuji, H.; Kuzushima, K.; Takiguchi, M.; Kaneko, S. Cytotoxic T cell responses to human telomerase reverse transcriptase in patients with hepatocellular carcinoma. Hepatology 2006, 43, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Vasanthakumar, S.; Sasikala, P.; Padma, M.; Balachandar, V.; Venkatesh, B.; Ganesan, S. EpCAM as a novel therapeutic target for hepatocellular carcinoma. J. Oncol. Sci. 2017, 3, 71–76. [Google Scholar]
- Li, Y.; Xu, J.; Chen, L.; Zhong, W.D.; Zhang, Z.; Mi, L.; Zhang, Y.; Liao, C.G.; Bian, H.J.; Jiang, J.L.; et al. HAb18G (CD147), a cancer-associated biomarker and its role in cancer detection. Histopathology 2009, 54, 677–687. [Google Scholar] [CrossRef]
- Wu, J.; Lu, M.; Li, Y.; Shang, Y.-K.; Wang, S.-J.; Meng, Y.; Wang, Z.; Li, Z.-S.; Chen, H.; Chen, Z.-N.; et al. Regulation of a TGF-β1-CD147 self-sustaining network in the differentiation plasticity of hepatocellular carcinoma cells. Oncogene 2016, 35, 5468–5479. [Google Scholar] [CrossRef]
- Zhang, R.-Y.; Wei, D.; Liu, Z.-K.; Yong, Y.-L.; Wei, W.; Zhang, Z.-Y.; Lv, J.-J.; Zhang, Z.; Chen, Z.-N.; Bian, H. Doxycycline Inducible Chimeric Antigen Receptor T Cells Targeting CD147 for Hepatocellular Carcinoma Therapy. Front. Cell Dev. Biol. 2019, 7, 233. [Google Scholar] [CrossRef]
- Tahara, K.; Mori, M.; Sadanaga, N.; Sakamoto, Y.; Kitano, S.; Makuuchi, M. Expression of the MAGE gene family in human hepatocellular carcinoma. Cancer 1999, 85, 1234–1240. [Google Scholar] [CrossRef]
- Weon, J.; Potts, P.R. The MAGE protein family and cancer. Curr. Opin. Cell Biol. 2015, 37, 1–8. [Google Scholar] [CrossRef]
- Wang, H.; Chen, D.; Wang, R.; Quan, W.; Xia, D.; Mei, J.; Liu, C. NY-ESO-1 expression in solid tumors predicts prognosis: A systematic review and meta-analysis. Medicine 2019, 98, e17990. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Nouso, K.; Noguchi, Y.; Higashi, T.; Ono, T.; Jungbluth, A.; Chen, Y.-T.; Old, L.J.; Nakayama, E.; Shiratori, Y. Expression and immunogenicity of NY-ESO-1 in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2006, 21, 1281–1285. [Google Scholar] [CrossRef]
- Bjorkman, P.J.; Saper, M.A.; Samraoui, B.; Bennett, W.S.; Strominger, J.L.; Wiley, D.C. Structure of the human class I histocompatibility antigen, HLA-A2. Nat. Cell Biol. 1987, 329, 506–512. [Google Scholar] [CrossRef]
- Huang, J.; Brameshuber, M.; Zeng, X.; Xie, J.; Li, Q.-J.; Chien, Y.-H.; Valitutti, S.; Davis, M.M. A Single Peptide-Major Histocompatibility Complex Ligand Triggers Digital Cytokine Secretion in CD4+ T Cells. Immunity 2013, 39, 846–857. [Google Scholar] [CrossRef] [PubMed]
- González-Galarza, F.F.; Takeshita, L.Y.; Santos, E.J.; Kempson, F.; Maia, M.H.T.; Da Silva, A.L.S.; Silva, A.L.T.E.; Ghattaoraya, G.; Alfirevic, A.; Jones, A.; et al. Allele frequency net 2015 update: New features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 2015, 43, D784–D788. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, X.; Zhou, W.-L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.-D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct. Target. Ther. 2019, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ghorashian, S.; Veliça, P.; Chua, I.; McNicol, A.-M.; Carpenter, B.; Holler, A.; Nicholson, E.; Ahmadi, M.; Zech, M.; Xue, S.-A.; et al. CD8 T Cell Tolerance to a Tumor-Associated Self-Antigen Is Reversed by CD4 T Cells Engineered to Express the Same T Cell Receptor. J. Immunol. 2014, 194, 1080–1089. [Google Scholar] [CrossRef]
- Goyal, L.; Frigault, M.; Meyer, T.; Feun, L.G.; Bruix, J.; El-Khoueiry, A.; Finn, R.S. Abstract 3183: Initial safety of AFP SPEAR T-cells in patients with advanced hepatocellular carcinoma. Cancer Res. 2019, 79 (Suppl. 13), 3183. [Google Scholar]
- Butterfield, L.H.; Ribas, A.; Meng, W.S.; Dissette, V.B.; Amarnani, S.; Vu, H.T.; Seja, E.; Todd, K.; Glaspy, J.A.; McBride, W.H.; et al. T-cell responses to HLA-A*0201 immunodominant peptides derived from alpha-fetoprotein in patients with hepatocellular cancer. Clin. Cancer Res. 2003, 9, 5902–5908. [Google Scholar] [PubMed]
- Adaptimmune Updates Data from its Phase 1 Trial for Liver Cancer at ILCA Showing Clinical Benefit [Internet]. Adaptimmune Therapeutics plc. Available online: https://www.adaptimmune.com/investors-and-media/news-media/press-releases/detail/196/adaptimmune-updates-data-from-its-phase-1-trial-for-liver (accessed on 10 September 2021).
- Wang, S.-H.; Yeh, S.-H.; Chen, P.-J. Unique Features of Hepatitis B Virus-Related Hepatocellular Carcinoma in Pathogenesis and Clinical Significance. Cancers 2021, 13, 2454. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W.; Brunetto, M.; Gehring, A.; Xue, S.-A.; Schurich, A.; Khakpoor, A.; Zhan, H.; Ciccorossi, P.; Gilmour, K.; Cavallone, D.; et al. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J. Hepatol. 2015, 62, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Gehring, A.; Xue, S.-A.; Ho, Z.Z.; Teoh, D.; Ruedl, C.; Chia, A.; Koh, S.; Lim, S.G.; Maini, M.; Stauss, H.; et al. Engineering virus-specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. J. Hepatol. 2011, 55, 103–110. [Google Scholar] [CrossRef]
- Lai, Q.; Avolio, A.; Lerut, J.; Singh, G.; Chan, S.C.; Berloco, P.B.; Tisone, G.; Agnes, S.; Chok, K.S.H.; Sharr, W.; et al. Recurrence of hepatocellular cancer after liver transplantation: The role of primary resection and salvage transplantation in East and West. J. Hepatol. 2012, 57, 974–979. [Google Scholar] [CrossRef]
- Faria, L.C.; Gigou, M.; Roque–Afonso, A.M.; Sebagh, M.; Roche, B.; Fallot, G.; Ferrari, T.C.D.A.; Guettier, C.; Dussaix, E.; Castaing, D.; et al. Hepatocellular Carcinoma Is Associated With an Increased Risk of Hepatitis B Virus Recurrence After Liver Transplantation. Gastroenterol. 2008, 134, 1890–1899. [Google Scholar] [CrossRef]
- Li, T.; Jiang, W.; Feng, Z.Q. The research advances of CAR-T cell therapy in solid tumor. J. Med. Postgrad. 2019, 886–890. [Google Scholar] [CrossRef]
- Tseng, H.-C.; Xiong, W.; Badeti, S.; Yang, Y.; Ma, M.; Liu, T.; Ramos, C.A.; Dotti, G.; Fritzky, L.; Jiang, J.-G.; et al. Efficacy of anti-CD147 chimeric antigen receptors targeting hepatocellular carcinoma. Nat. Commun. 2020, 11, 4810. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef]
- Brandt, L.J.B.; Barnkob, M.B.; Michaels, Y.S.; Heiselberg, J.; Barington, T. Emerging Approaches for Regulation and Control of CAR T Cells: A Mini Review. Front. Immunol. 2020, 11, 326. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef]
- Zou, B.; Liu, X.; Zhang, B.; Gong, Y.; Cai, C.; Li, P.; Chen, J.; Xing, S.; Chen, J.; Peng, S.; et al. The Expression of FAP in Hepatocellular Carcinoma Cells is Induced by Hypoxia and Correlates with Poor Clinical Outcomes. J. Cancer 2018, 9, 3278–3286. [Google Scholar] [CrossRef]
- Baglieri, J.; Brenner, D.A.; Kisseleva, T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1723. [Google Scholar] [CrossRef]
- Katz, S.C.; Burga, R.A.; McCormack, E.; Wang, L.J.; Mooring, W.; Point, G.R.; Khare, P.D.; Thorn, M.; Ma, Q.; Stainken, B.F.; et al. Phase I Hepatic Immunotherapy for Metastases Study of Intra-Arterial Chimeric Antigen Receptor–Modified T-cell Therapy for CEA+ Liver Metastases. Clin. Cancer Res. 2015, 21, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Vilgelm, A.E.; Richmond, A. Chemokines Modulate Immune Surveillance in Tumorigenesis, Metastasis, and Response to Immunotherapy. Front. Immunol. 2019, 10, 333. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.C.; Sadelain, M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell–mediated rejection of systemic lymphoma in immunodeficient mice. Blood 2010, 115, 3508–3519. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.-F.; Bulsara, S.; et al. Glypican-3–Specific CAR T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity against Hepatocellular Carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef]
- Hou, A.J.; Chang, Z.L.; Lorenzini, M.H.; Zah, E.; Chen, Y.Y. TGF-β-responsive CAR-T cells promote anti-tumor immune function. Bioeng. Transl. Med. 2018, 3, 75–86. [Google Scholar] [CrossRef]
- Spear, P.; Barber, A.; Rynda-Apple, A.; Sentman, C.L. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-γ and GM-CSF. J. Immunol. 2012, 188, 6389–6398. [Google Scholar] [CrossRef]
- Hosseinkhani, N.; Derakhshani, A.; Kooshkaki, O.; Shadbad, M.A.; Hajiasgharzadeh, K.; Baghbanzadeh, A.; Safarpour, H.; Mokhtarzadeh, A.; Brunetti, O.; Yue, S.; et al. Immune Checkpoints and CAR-T Cells: The Pioneers in Future Cancer Therapies? Int. J. Mol. Sci. 2020, 21, 8305. [Google Scholar] [CrossRef]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Zheng, Z.; Robbins, P.F.; Tran, E.; Prickett, T.D.; Gartner, J.J.; Li, Y.F.; Ray, S.; Franco, Z.; Bliskovsky, V.; et al. An Efficient Single-Cell RNA-Seq Approach to Identify Neoantigen-Specific T Cell Receptors. Mol. Ther. 2018, 26, 379–389. [Google Scholar] [CrossRef]
- Ye, B.; Smerin, D.; Gao, Q.; Kang, C.; Xiong, X. High-throughput sequencing of the immune repertoire in oncology: Applications for clinical diagnosis, monitoring, and immunotherapies. Cancer Lett. 2018, 416, 42–56. [Google Scholar] [CrossRef]
- Obenaus, M.; Leitão, C.; Leisegang, M.; Chen, X.; Gavvovidis, I.; Van Der Bruggen, P.; Uckert, W.; Schendel, D.J.; Blankenstein, T. Identification of human T-cell receptors with optimal affinity to cancer antigens using antigen-negative humanized mice. Nat. Biotechnol. 2015, 33, 402–407. [Google Scholar] [CrossRef]
- Tan, M.P.; Gerry, A.B.; Brewer, J.E.; Melchiori, L.; Bridgeman, J.S.; Bennett, A.D.; Pumphrey, N.J.; Jakobsen, B.K.; Price, D.; Ladell, K.; et al. T cell receptor binding affinity governs the functional profile of cancer-specific CD8 + T cells. Clin. Exp. Immunol. 2015, 180, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Kohrt, H.; Caux, C.; Levy, R. Intratumoral Immunization: A New Paradigm for Cancer Therapy. Clin. Cancer Res. 2014, 20, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.T.; Kranz, D.M. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol. Sci. 2016, 37, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Brenner, M.K. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Study Number | Phase | Intervention | CAR-T Type | Population under Study | Inclusion/Exclusion Criteria | End Points + Expected Toxicities | Target Sample Size (n) | Status |
|---|---|---|---|---|---|---|---|---|
| NCT03993743 | I | CD147 targeted CAR T | 3rd Generation doxycycline inducible system. Autologous | Advanced HCC | Untreatable by surgery or local therapy or has postoperative progressions, has failed at least one line of standard systemic chemotherapy | DLT and MTD of CD-147 CAR-T hepatic artery infusions Activity of CAR-T cell hepatic artery infusions CAR-T detection in extrahepatic sites | 34 | Recruiting |
| NCT04121273 | I | GPC3 targeted CAR T | 2nd Generation Autologous | Advanced HCC | Cancer has progressed after treatment, or cannot receive standard of care. | Dose limiting toxicity such as fever or jaundice Radiological evaluation of tumour size Peripheral tumour marker Peripheral CAR-T detection by flow cytometry | 20 | Recruiting |
| NCT02905188 | I | GPC3 targeted CAR T | 2nd Generation Autologous Lymphodepletion regimen (Cytoxan and Fludarabine) | Previously treated HCC | Relapsed or refractory to treatment that has metastasized or cannot receive other standard lines of therapy | Dose limiting toxicity such as any grade 5 event, grade 2 or 4 allergic reaction, both haematologica and non haematologic of grade 4 that fails to return to grade 2 within 72 hurs. Grade 3 or 4 CRS. CR/PR. CAR-T persistence by PCR. | 14 | Recruiting |
| NCT03198546 | I | GPC3 T2 targeted CAR T | 3rd/4th Generation GPC3 and/or TGFb CAR with/without IL7/CCL19 and/or scfv against PD1/CTLA4/Tigit Autologous | Advanced HCC | Advanced disease and not eligible for alternative therapy | Dose limiting toxicity that is irreversible or life threatening; haematologic or non-haematologic grade 3–5. CR/PR Persistence measured by PCR | 30 | Recruiting |
| NCT03884751 | I | GPC3 targeted CAR T | 2nd Generation Autologous | Advanced HCC | Patients must not be eligible for surgery or have progressive disease after standard therapies. | Dose limiting toxicity and maximum tolerated dose Pharmacokinetics including CAR-T expansion and persistence measured by PCR | 15 | Recruiting |
| NCT03941626 | I/II | EGFRviii/DR5 targeted CAR-T/TCR-T | 2nd Generation Autologous Lymphodepletion regimen (Cyclophosphamide and fludarabine) | Basket trial including HCC | Multi tumour type but for liver patients must be untreatable by surgery or postoperative recurrence or no effective treatment | Adverse events and clinical response measured by change in tumour volume by CT and MRI | 50 | Recruiting |
| NCT03013712 | I | EpCAM targeted CAR T | 2nd Generation Autologous | Basket trial including HCC | Multi tumour type but for liver patients must be untreatable by surgery or postoperative recurrence or no effective treatment | Toxicity profile and antitumour efficacy Persistence of CAR-T cells in the blood measured by flow cytometry | 60 | Recruiting |
| NCT03980288 | I | GPC3 Targeted CAR T | 4th Generation Autologous Lymphodepletion regimen (Cyclophosphamide and fludarabine) | Advanced HCC | Advanced hepatocellular carcinoma and refractory or intolerant to current standard systemic treatment | Dose limiting toxicity and maximum tolerated dose CAR-T expansion measured by PCR | 36 | Recruiting |
| NCT04506983 | I | GPC3-CAR T Cell | 2nd Generation Autologous | Advanced HCC | Advanced disease BCLC B/C | Percentage of any adverse events Overall remission rate Proliferation of CAR T Cells | 12 | Not yet recruiting |
| NCT04550663 | I | NKG2D CAR-T (KD-025) | Autologous | Basket (including HCC) | For those with advanced disease that is not eligible for other treatment. NKG2DL+ | Maximum tolerated dose Adverse events occurrence monitoring Objective remission rate | 10 | Not yet recruiting |
| NCT02395250 | I | GPC3 CAR T cell | 2nd Generation Autologous | Previously treated HCC | Untreatable by surgery or postoperative recurrence with no effective treatment | Any adverse events incidence as a result of CAR-T cells | 13 | Completed |
| NCT02723942 | I | GPC3 CAR T cell | No generation mentioned Autologous | Previously treated HCC | Untreatable by surgery or postoperative recurrence with no effective treatment | Effect of CAR on tumour through reduction in tumour burden, assessment via CT or PET. Safety profile related issues such as fever and jaundice. | N/A | Withdrawn |
| NCT04270461 | I | NKG2D CAR T cell | 2nd Generation Autologous | Basket NKG2DL+(including HCC) | Patients with relapsed/refractory disease | OS and safety profile Delivery via hepatic portal artery | N/A | Withdrawn |
| NCT04093648 | I | GPC3 CAR T with IL21 and 15 (TEGAR) | 4th Generation Autologous Lymphodepletion (Cytoxan and Fludarabine) | Preciously treated HCC | Untreatable by surgery or postoperative recurrence with no effective treatment | DLT including neurotoxicity and CRS Response rate (partial or complete) | N/A | Withdrawn—incorporated into another study |
| NCT03349255 | I | ET1402L1-CAR T cell | 2nd Generation Autologous | Previously treated liver cancer AFP+ | No available curative therapeutic options and a poor overall prognosis. | DLT and toxicity such as fever, CRS, neutropenia Response rate CAR-T cell engraftment | 3 | Terminated |
| Study Number | Phase | Intervention | Source of Cells | Population under Study | Inclusion/ Exclusion Criteria | Trial End Points | Target Sample Size (n) | Status |
|---|---|---|---|---|---|---|---|---|
| NCT03132792 | I | AFPC332 T Cells | Autologous | HLA-A02+ AFP+ HCC | Relapsed or refractory disease | DLTs and AEs Time intervals between infusion and response | 45 | Recruiting |
| NCT04368182 | I | C-TCR055 (AFP TCR) | Autologous | HLA-A02+ AFP+ HCC | Unresectable disease, relapsed or refractory | Safety through adverse event monitoring and ORR | 3 | Recruiting |
| NCT03971747 | I | C-TCR055 (AFP TCR) | Autologous | HLA-A02+ AFP+ HCC | Unresectable disease, relapsed or refractory | Safety through adverse event monitoring and ORR | 9 | Recruiting |
| NCT03634683 | I/II | LioCyx | Autologous | HLA Class I HBV HCC | Recurrent HBV related HCC post transplantation | Safety by reporting of adverse events, ORR and quality of life measurements | 72 | Not yet recruiting |
| NCT04502082 | I/II | ET140203 T cells (ARYA-1) | Autologous | HLA-A02+ AFP+ HCC | Advanced disease | Incidence and severity of adverse events | 50 | Recruiting |
| NCT03888859 | I | ET1402L1-ARTEMISTM T cells | Autologous | HLA-A02+ AFP+ HCC | Advanced disease where patients have no available curable therapeutics | DLTs Administration route assessment; IV/intrahepatic/intratumoural | 12 | Completed |
| NCT03965546 | I | ET140202 AFP T cell combination with wither TAE or Sorafenib | Autologous | HLA-A02+ AFP+ HCC | Advanced disease where patients have no available curative therapeutics | Adverse events frequency and T cell expansion | 27 | Recruiting |
| NCT03899415 | I | HBV specific TCR redirected T Cell | Autologous | HLA Class I HBV HCC | Advanced HBV related HCC post hepatectomy or radiofrequency ablation | Incidence of adverse events and ORR | 10 | Recruiting |
| NCT03132792 | I | AFPᶜ³³²T cells | Autologous | HLA-A02+ AFP+ HCC | Advanced, relapsed or refractory disease | DLTs and adverse event incidence and any response rate | 45 | Recruiting |
| NCT01967823 | II | Anti-NY ESO-1 mTCR (murine TCR) | Autologous | HLA-A*0201 Basket (including HCC) | Advanced disease NY-ESO-1 positive tumours | Clinical response (CR/PR) and TCR expansion by PCR | 11 | Completed |
| NCT03441100 | I | IMA202–101 TCR T cells targeting MAGE-1 | Autologous | Basket (including HCC) HLA restricted | Advanced or metastatic tumour MAGE-1 + tumour | Adverse event incidence and persistence of T Cells | 15 | Recruiting |
| Cell Therapy | Antigen(s) Recognised | MHC Restricted | Advantages | Disadvantages |
|---|---|---|---|---|
| TCR-T TCR-engineered T Cells | Peptide/MHC, intracellular targets possible | Yes |
|
|
| CAR-T Chimeric antigen receptor T-cells | Cell surface antigens | No |
|
|
| CIKs Cytokine induced killer cells. | Not antigen specific | No |
|
|
| TILs Tumour infiltrating lymphocytes | Multiple tumour associated antigens | Yes |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roddy, H.; Meyer, T.; Roddie, C. Novel Cellular Therapies for Hepatocellular Carcinoma. Cancers 2022, 14, 504. https://doi.org/10.3390/cancers14030504
Roddy H, Meyer T, Roddie C. Novel Cellular Therapies for Hepatocellular Carcinoma. Cancers. 2022; 14(3):504. https://doi.org/10.3390/cancers14030504
Chicago/Turabian StyleRoddy, Harriet, Tim Meyer, and Claire Roddie. 2022. "Novel Cellular Therapies for Hepatocellular Carcinoma" Cancers 14, no. 3: 504. https://doi.org/10.3390/cancers14030504
APA StyleRoddy, H., Meyer, T., & Roddie, C. (2022). Novel Cellular Therapies for Hepatocellular Carcinoma. Cancers, 14(3), 504. https://doi.org/10.3390/cancers14030504