
Dissecting the Functional Role of the TRIM8 Protein on Cancer Pathogenesis

 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
1. Introduction
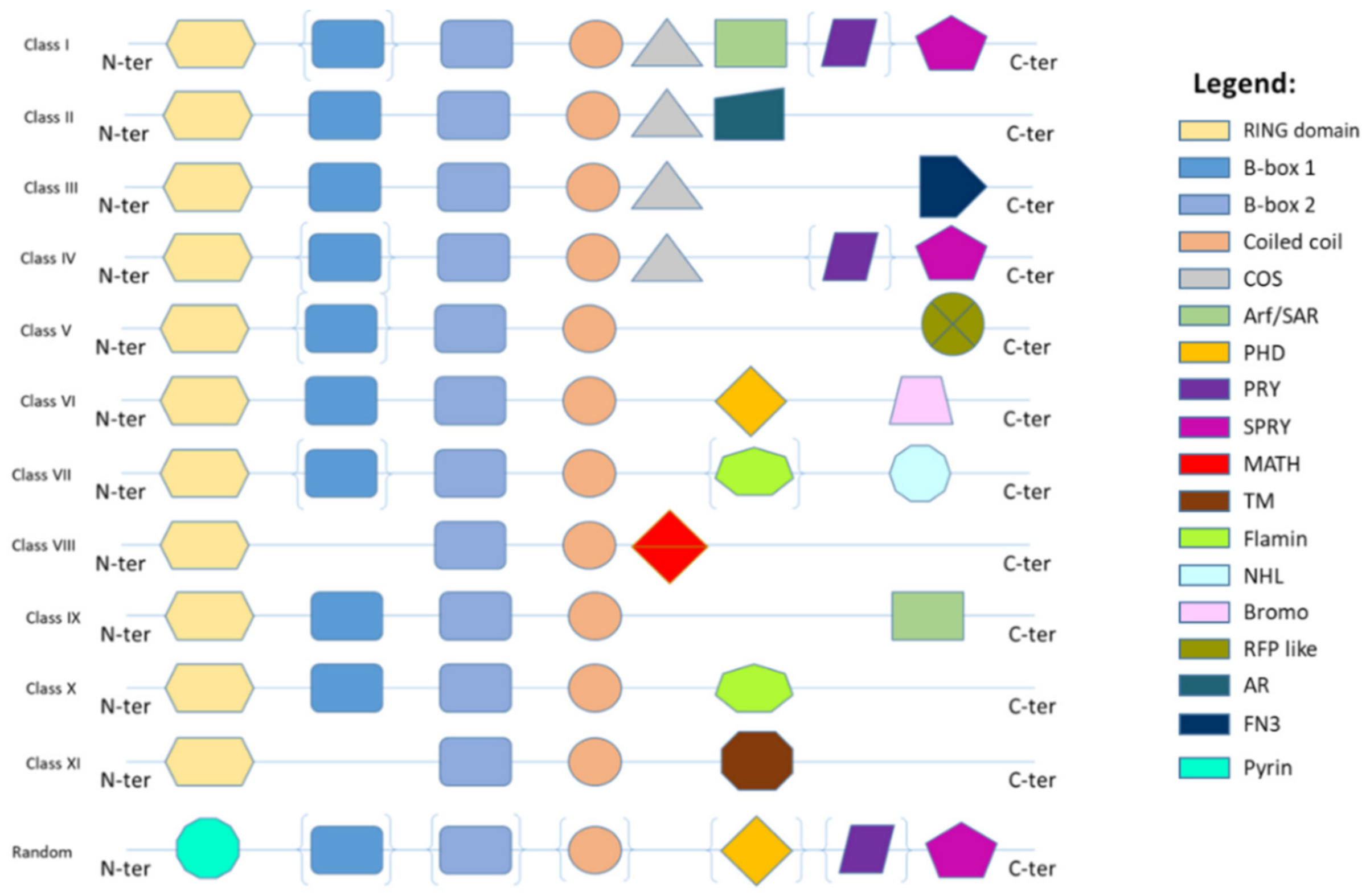
2. TRIM Proteins Biological Functions
- Degradation of toxic protein aggregates [39];
3. TRIM Proteins and Cancer Pathogenesis
- Chromosomal translocation [45]. It could generate a fusion protein without activity or with a different activity that could dysregulate some signaling pathways, leading to the generation of some tumor shapes. An example is a translocation between the TRIM19 gene (PML) on chromosome 15 and the retinoic acid receptor α (RARa) gene on chromosome 17. This translocation leads to the formation of a fusion protein that represses acid signaling retinoic and is associated with Acute Promyelocytic Leukemia [46]. Such similar examples are the following: TRIM24, TRIM27 and TRIM33 were found in translocations with the RET gene and are involved in papillary thyroid cancer, lymphoma and non-small cell lung carcinoma, respectively. Similarly, TRIM24 was found translocated with the BRAF gene in melanoma and lung cancer and with the FGFR1 gene in myeloproliferative syndrome [43];
- Modulation of the activity and stability of p53. TRIM11, TRIM13, TRIM21, TRIM24, TRIM25, TRIM28, TRIM29, TRIM31, TRIM32, TRIM39 and TRIM59 can ubiquitinate the p53 protein, a fundamental macromolecule in cell development whose purpose is to promote genomic stability and induce cell cycle arrest and apoptosis if extensive DNA damage is found in the cell. The ubiquitination of this protein leads to its direct degradation or to its sequestration in the cytoplasm: since it can no longer penetrate the nucleus, the ubiquitinated protein is no longer able to detect any damage to the DNA; consequently, the cell replicates itself by transmitting the same error in the nucleic acid sequence, resulting in the possible onset of tumor forms [47];
- Regulation of pathways to cancer stemness, including STAT signaling, AKT signaling, NANOGSox2-Oct-3/4 networks. Specifically, through these pathways, TRIM28 is involved in breast cancer, TRIM24 in glioblastoma and colorectal cancer, TRIMs 14 in gastric cancer and TRIM16 has been associated as a negative regulator of stemness in breast and ovarian cancer cells [43].
4. TRIM8
5. TRIM8 and Cancer Pathogenesis
6. TRIM8 as Tumor Suppressor
- By inducing the TP53 tumor suppressor activity through a positive feedback loop formation.
- Restoring TP53 functions by blunting N-MYC activity in chemo-resistant tumors.
- Quenching the DNp63a oncogenic activity by forming a negative feedback loop.
7. TRIM 8 as Oncogenic Protein
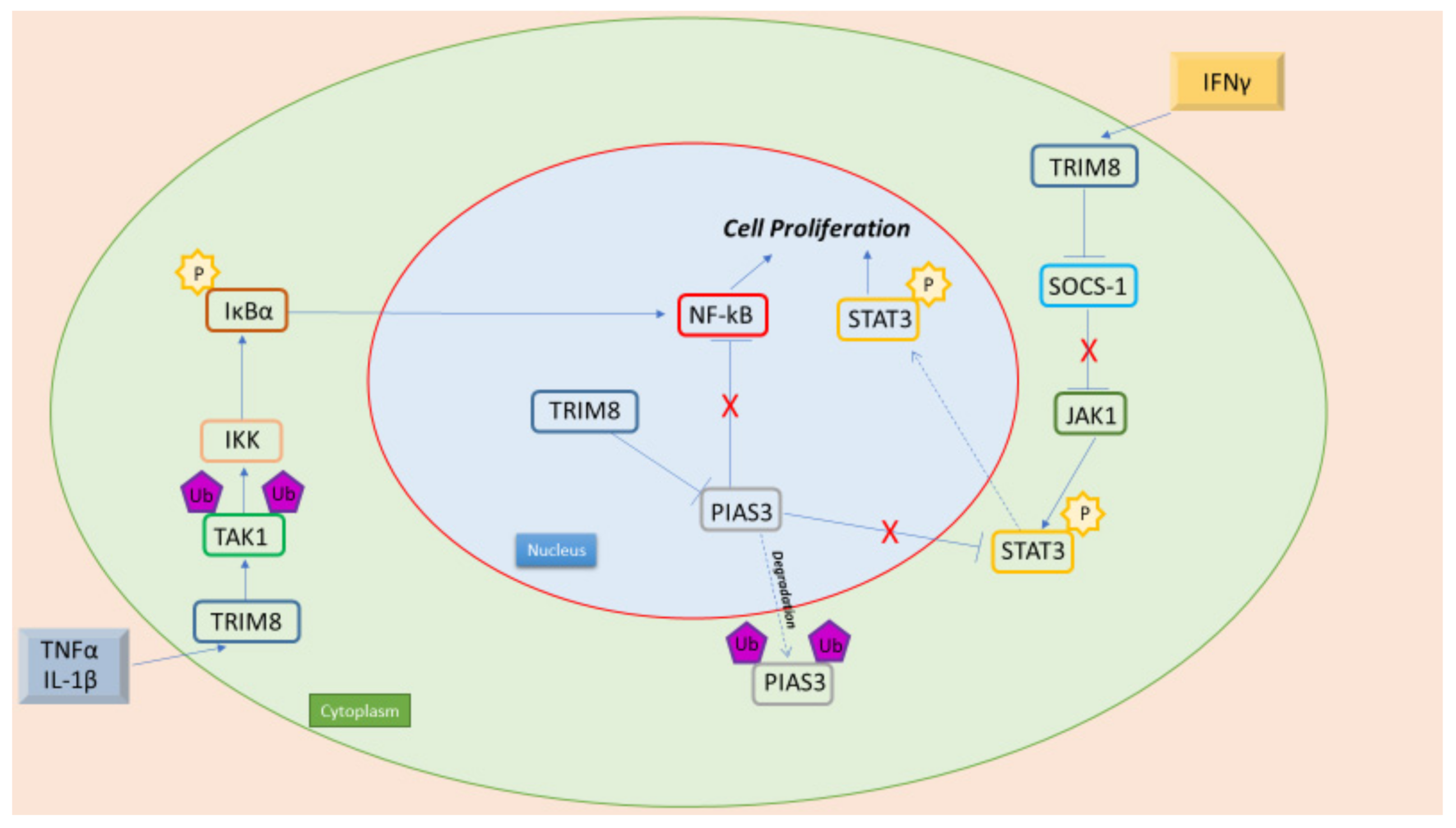
7.1. TRIM8 and NF-kB
7.2. TRIM8 and STAT3
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vincent, S.R.; Kwasnicka, D.A.; Fretier, P. A novel RING finger-B box-coiled-coil protein, GERP. Biochem. Biophys. Res. Commun. 2000, 279, 482–486. [Google Scholar] [CrossRef]
- Gushchina, L.V.; Kwiatkowski, T.A.; Bhattacharya, S.; Weisleder, N.L. Conserved structural and functional aspects of the tripartite motif gene family point towards therapeutic applications in multiple diseases. Pharmacol. Ther. 2018, 185, 12–25. [Google Scholar] [CrossRef]
- Short, K.M.; Cox, T.C. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J. Biol. Chem. 2006, 281, 8970–8980. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wu, H.; Wu, W.; Zhuo, W.; Liu, W.; Zhang, Y.; Cheng, M.; Chen, Y.G.; Gao, N.; Yu, H.; et al. Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 2014, 24, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Meroni, G. Genomics and evolution of the TRIM gene family. Adv. Exp. Med. Biol. 2012, 770, 1–9. [Google Scholar] [CrossRef]
- Sardiello, M.; Cairo, S.; Fontanella, B.; Ballabio, A.; Meroni, G. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol. Biol. 2008, 8, 225. [Google Scholar] [CrossRef] [Green Version]
- Han, K.; Lou, D.I.; Sawyer, S.L. Identification of a genomic reservoir for new TRIM genes in primate genomes. PLoS Genet. 2011, 7, e1002388. [Google Scholar] [CrossRef] [Green Version]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Marzano, F.; Guerrini, L.; Pesole, G.; Sbisà, E.; Tullo, A. Emerging Roles of TRIM8 in Health and Disease. Cells 2021, 10, 561. [Google Scholar] [CrossRef]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef] [Green Version]
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. Bioessays 2005, 27, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villán, E.; García-Sastre, A.; Gack, M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012, 8, e1003059. [Google Scholar] [CrossRef] [PubMed]
- Uchil, P.D.; Quinlan, B.D.; Chan, W.T.; Luna, J.M.; Mothes, W. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 2008, 4, e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchil, P.D.; Hinz, A.; Siegel, S.; Coenen-Stass, A.; Pertel, T.; Luban, J.; Mothes, W. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 2013, 87, 257–272. [Google Scholar] [CrossRef] [Green Version]
- Venuto, S.; Monteonofrio, L.; Cozzolino, F.; Monti, M.; Appolloni, I.; Mazza, T.; Canetti, D.; Giambra, V.; Panelli, P.; Fusco, C.; et al. TRIM8 interacts with KIF11 and KIFC1 and controls bipolar spindle formation and chromosomal stability. Cancer Lett. 2020, 473, 98–106. [Google Scholar] [CrossRef]
- Toniato, E.; Chen, X.P.; Losman, J.; Flati, V.; Donahue, L.; Rothman, P. TRIM8/GERP RING finger protein interacts with SOCS-1. J. Biol. Chem. 2002, 277, 37315–37322. [Google Scholar] [CrossRef] [Green Version]
- Dang, X.; He, B.; Ning, Q.; Liu, Y.; Chang, Y.; Chen, M. Suppression of TRIM8 by microRNA-182-5p restricts tumor necrosis factor-α-induced proliferation and migration of airway smooth muscle cells through inactivation of NF-Κb. Int. Immunopharmacol. 2020, 83, 106475. [Google Scholar] [CrossRef]
- McNab, F.W.; Rajsbaum, R.; Stoye, J.P.; O’Garra, A. Tripartite-motif proteins and innate immune regulation. Curr. Opin. Immunol. 2011, 23, 46–56. [Google Scholar] [CrossRef]
- Sato, T.; Okumura, F.; Kano, S.; Kondo, T.; Ariga, T.; Hatakeyama, S. TRIM32 promotes neural differentiation through retinoic acid receptor-mediated transcription. J. Cell Sci. 2011, 124 Pt 20, 3492–3502. [Google Scholar] [CrossRef] [Green Version]
- Schwamborn, J.C.; Berezikov, E.; Knoblich, J.A. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 2009, 136, 913–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, F.; Mukhopadhyay, R.; Ovaa, H.; Mulder, M. Targeting TRIM Proteins: A Quest towards Drugging an Emerging Protein Class. Chembiochem 2021, 22, 2011–2031. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 2019, 8, e42426. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Zhang, D.E. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem. 2006, 281, 3989–3994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Bhaduri, U.; Merla, G. Rise of TRIM8: A Molecule of Duality. Mol. Ther. Nucleic Acids 2020, 22, 434–444. [Google Scholar] [CrossRef]
- Iwai, K. Discovery of linear ubiquitination, a crucial regulator for immune signaling and cell death. FEBS J. 2021, 288, 1060–1069. [Google Scholar] [CrossRef]
- Brazee, P.; Dada, L.A.; Sznajder, J.I. Role of Linear Ubiquitination in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 761–768. [Google Scholar] [CrossRef] [Green Version]
- Elton, L.; Carpentier, I.; Verhelst, K.; Staal, J.; Beyaert, R. The multifaceted role of the E3 ubiquitin ligase HOIL-1: Beyond linear ubiquitination. Immunol. Rev. 2015, 266, 208–221. [Google Scholar] [CrossRef]
- Bell, J.L.; Malyukova, A.; Holien, J.K.; Koach, J.; Parker, M.W.; Kavallaris, M.; Marshall, G.M.; Cheung, B.B. TRIM16 acts as an E3 ubiquitin ligase and can heterodimerize with other TRIM family members. PLoS ONE 2012, 7, e37470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, T.G.; Möller, A.; Sirma, H.; Zentgraf, H.; Taya, Y.; Dröge, W.; Will, H.; Schmitz, M.L. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 2002, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Dikic, I. Atypical ubiquitin chains: New molecular signals. ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008, 9, 536–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010, 141, 315–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Xia, H. TRIM Proteins in Inflammation: From Expression to Emerging Regulatory Mechanisms. Inflammation 2021, 44, 811–820. [Google Scholar] [CrossRef]
- Venuto, S.; Castellana, S.; Monti, M.; Appolloni, I.; Fusilli, C.; Fusco, C.; Pucci, P.; Malatesta, P.; Mazza, T.; Merla, G.; et al. TRIM8-driven transcriptomic profile of neural stem cells identified glioma-related nodal genes and pathways. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 491–501. [Google Scholar] [CrossRef]
- Venuto, S.; Merla, G. E3 Ubiquitin Ligase TRIM Proteins, Cell Cycle and Mitosis. Cells 2019, 8, 510. [Google Scholar] [CrossRef] [Green Version]
- Roy, M.; Tomar, D.; Singh, K.; Lakshmi, S.; Prajapati, P.; Bhatelia, K.; Gohel, D.; Singh, R. TRIM8 regulated autophagy modulates the level of cleaved Caspase-3 subunit to inhibit genotoxic stress induced cell death. Cell. Signal. 2018, 48, 1–12. [Google Scholar] [CrossRef]
- Maarifi, G.; Smith, N.; Maillet, S.; Moncorgé, O.; Chamontin, C.; Edouard, J.; Sohm, F.; Blanchet, F.P.; Herbeuval, J.P.; Lutfalla, G.; et al. TRIM8 is required for virus-induced IFN response in human plasmacytoid dendritic cells. Sci. Adv. 2019, 5, eaax3511. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Zhang, Y.L.; Liu, L.N. Inhibition of TRIM8 restrains ischaemia-reperfusion-mediated cerebral injury by regulation of NF-κB activation associated inflammation and apoptosis. Exp. Cell Res. 2020, 388, 111818. [Google Scholar] [CrossRef]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Hatakeyama, S. TRIM proteins and diseases. J. Biochem. 2017, 161, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Mandell, M.A.; Saha, B.; Thompson, T.A. The Tripartite Nexus: Autophagy, Cancer, and Tripartite Motif-Containing Protein Family Members. Front. Pharmacol. 2020, 11, 308. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Shim, H.S.; Kenudson, M.; Zheng, Z.; Liebers, M.; Cha, Y.J.; Hoang Ho, Q.; Onozato, M.; Phi Le, L.; Heist, R.S.; Iafrate, A.J. Unique Genetic and Survival Characteristics of Invasive Mucinous Adenocarcinoma of the Lung. J. Thorac. Oncol. 2015, 10, 1156–1162. [Google Scholar] [CrossRef] [Green Version]
- Cambiaghi, V.; Giuliani, V.; Lombardi, S.; Marinelli, C.; Toffalorio, F.; Pelicci, P.G. TRIM proteins in cancer. Adv. Exp. Med. Biol. 2012, 770, 77–91. [Google Scholar] [CrossRef]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026120. [Google Scholar] [CrossRef]
- Jaworska, A.M.; Wlodarczyk, N.A.; Mackiewicz, A.; Czerwinska, P. The role of TRIM family proteins in the regulation of cancer stem cell self-renewal. Stem. Cells 2020, 38, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem. Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef]
- Czerwińska, P.; Shah, P.K.; Tomczak, K.; Klimczak, M.; Mazurek, S.; Sozańska, B.; Biecek, P.; Korski, K.; Filas, V.; Mackiewicz, A.; et al. TRIM28 multi-domain protein regulates cancer stem cell population in breast tumor development. Oncotarget 2017, 8, 863–882. [Google Scholar] [CrossRef] [Green Version]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef] [PubMed]
- Tse, J.C.; Kalluri, R. Mechanisms of metastasis: Epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J. Cell. Biochem. 2007, 101, 816–829. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Rienzo, M.; Romagnoli, A.; Antonioli, M.; Piacentini, M.; Fimia, G.M. TRIM proteins in autophagy: Selective sensors in cell damage and innate immune responses. Cell Death Differ. 2020, 27, 887–902. [Google Scholar] [CrossRef]
- Guo, L.; Dong, W.; Fu, X.; Lin, J.; Dong, Z.; Tan, X.; Zhang, T. Tripartite Motif 8 (TRIM8) Positively Regulates Pro-inflammatory Responses in Pseudomonas aeruginosa-Induced Keratitis Through Promoting K63-Linked Polyubiquitination of TAK1 Protein. Inflammation 2017, 40, 454–463. [Google Scholar] [CrossRef]
- Ye, W.; Hu, M.M.; Lei, C.Q.; Zhou, Q.; Lin, H.; Sun, M.S.; Shu, H.B. TRIM8 Negatively Regulates TLR3/4-Mediated Innate Immune Response by Blocking TRIF-TBK1 Interaction. J. Immunol. 2017, 199, 1856–1864. [Google Scholar] [CrossRef]
- Li, Q.; Yan, J.; Mao, A.P.; Li, C.; Ran, Y.; Shu, H.B.; Wang, Y.Y. Tripartite motif 8 (TRIM8) modulates TNFα- and IL-1β-triggered NF-κB activation by targeting TAK1 for K63-linked polyubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 19341–19346. [Google Scholar] [CrossRef] [Green Version]
- Caratozzolo, M.F.; Marzano, F.; Abbrescia, D.I.; Mastropasqua, F.; Petruzzella, V.; Calabrò, V.; Pesole, G.; Sbisà, E.; Guerrini, L.; Tullo, A. TRIM8 Blunts the Pro-proliferative Action of ΔNp63α in a p53 Wild-Type Background. Front. Oncol. 2019, 9, 1154. [Google Scholar] [CrossRef] [Green Version]
- Liau, N.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Commun. 2018, 9, 1558. [Google Scholar] [CrossRef]
- Micale, L.; Fusco, C.; Fontana, A.; Barbano, R.; Augello, B.; De Nittis, P.; Copetti, M.; Pellico, M.T.; Mandriani, B.; Cocciadiferro, D.; et al. TRIM8 downregulation in glioma affects cell proliferation and it is associated with patients survival. BMC Cancer 2015, 15, 470. [Google Scholar] [CrossRef] [Green Version]
- Tian, Z.; Tang, J.; Liao, X.; Gong, Y.; Yang, Q.; Wu, Y.; Wu, G. TRIM8 inhibits breast cancer proliferation by regulating estrogen signaling. Am. J. Cancer Res. 2020, 10, 3440–3457. [Google Scholar] [PubMed]
- Marzano, F.; Caratozzolo, M.F.; Pesole, G.; Sbisà, E.; Tullo, A. TRIM Proteins in Colorectal Cancer: TRIM8 as a Promising Therapeutic Target in Chemo Resistance. Biomedicines 2021, 9, 241. [Google Scholar] [CrossRef] [PubMed]
- Caratozzolo, M.F.; Valletti, A.; Gigante, M.; Aiello, I.; Mastropasqua, F.; Marzano, F.; Ditonno, P.; Carrieri, G.; Simonnet, H.; D’Erchia, A.M.; et al. TRIM8 anti-proliferative action against chemo-resistant renal cell carcinoma. Oncotarget 2014, 5, 7446–7457. [Google Scholar] [CrossRef] [PubMed]
- Caratozzolo, M.F.; Micale, L.; Turturo, M.G.; Cornacchia, S.; Fusco, C.; Marzano, F.; Augello, B.; D’Erchia, A.M.; Guerrini, L.; Pesole, G.; et al. TRIM8 modulates p53 activity to dictate cell cycle arrest. Cell Cycle 2012, 11, 511–523. [Google Scholar] [CrossRef]
- Mastropasqua, F.; Marzano, F.; Valletti, A.; Aiello, I.; Di Tullio, G.; Morgano, A.; Liuni, S.; Ranieri, E.; Guerrini, L.; Gasparre, G.; et al. TRIM8 restores p53 tumor suppressor function by blunting N-MYC activity in chemo-resistant tumors. Mol. Cancer 2017, 16, 67. [Google Scholar] [CrossRef] [Green Version]
- De Iuliis, V.; Marino, A.; Caruso, M.; Capodifoglio, S.; Flati, V.; Marynuk, A.; Marricareda, V.; Ursi, S.; Lanuti, P.; Talora, C.; et al. Autophagy processes are dependent on EGF receptor signaling. Oncotarget 2018, 9, 30289–30303. [Google Scholar] [CrossRef]
- Zhang, C.; Mukherjee, S.; Tucker-Burden, C.; Ross, J.L.; Chau, M.J.; Kong, J.; Brat, D.J. TRIM8 regulates stemness in glioblastoma through PIAS3-STAT3. Mol. Oncol. 2017, 11, 280–294. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, J.E.; De Iuliis, V.; Avolio, F.; Liberatoscioli, E.; Pulcini, R.; Di Francesco, S.; Pennelli, A.; Martinotti, S.; Toniato, E. Dissecting the Functional Role of the TRIM8 Protein on Cancer Pathogenesis. Cancers 2022, 14, 2309. https://doi.org/10.3390/cancers14092309
Esposito JE, De Iuliis V, Avolio F, Liberatoscioli E, Pulcini R, Di Francesco S, Pennelli A, Martinotti S, Toniato E. Dissecting the Functional Role of the TRIM8 Protein on Cancer Pathogenesis. Cancers. 2022; 14(9):2309. https://doi.org/10.3390/cancers14092309
Chicago/Turabian StyleEsposito, Jessica Elisabetta, Vincenzo De Iuliis, Francesco Avolio, Eliana Liberatoscioli, Riccardo Pulcini, Simona Di Francesco, Alfonso Pennelli, Stefano Martinotti, and Elena Toniato. 2022. "Dissecting the Functional Role of the TRIM8 Protein on Cancer Pathogenesis" Cancers 14, no. 9: 2309. https://doi.org/10.3390/cancers14092309