
Targeting Pivotal Hallmarks of Cancer for Enhanced Therapeutic Strategies in Triple-Negative Breast Cancer Treatment—In Vitro, In Vivo and Clinical Trials Literature Review


Abstract
Simple Summary
Abstract
1. Introduction
1.1. Hallmarks of Cancer
1.2. Tripple-Negative Breast Cancer (TNBC)
1.3. Aim of This Study
2. Deregulating Cellular Metabolism and the Warburg Effect
2.1. Glycoconjugates as a Trojan Horse Method to Deliver Drugs to Malignant Cells
2.2. Shell-Shedable Micelles as Yet Another Drug Delivery System
2.3. Rg3 Liposomes Interacting with GLUT-1
3. HIF-1 at the Crossroads of Acidosis and Hypoxia
NDBT Inhibition as an Approach to Reduce Acidosis and Hypoxia in TNBC
4. Role of LOXL2 in Triggering Metastasis in TNBC
Evolution of LOX Enzyme Inhibitors
5. TILs-Key Contributors in TNBC’s Immune Evasion Strategy
5.1. CAR-T Therapy Generations
5.2. CAR-T Therapy Targets in TNBC
5.3. CAR-T Cell-Derived Exosomes as a Way to Reduce Toxicity on Healthy Cells
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pietras, K.; Ostman, A. Hallmarks of Cancer: Interactions with the Tumor Stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Karim, A.M.; Eun Kwon, J.; Ali, T.; Jang, J.; Ullah, I.; Lee, Y.G.; Park, D.W.; Park, J.; Jeang, J.W.; Kang, S.C. Triple-negative Breast Cancer: Epidemiology, Molecular Mechanisms, and Modern Vaccine-based Treatment Strategies. Biochem. Pharmacol. 2023, 212, 115545. [Google Scholar] [CrossRef] [PubMed]
- OECD. Krajowe Profile Dotyczące Nowotworów: Polska 2023; OECD: Paris, France, 2023. [Google Scholar] [CrossRef]
- Wilkinson, L.; Gathani, T. Understanding Breast Cancer as a Global Health Concern. Br. J. Radiol. 2022, 95, 20211033. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Geyer, F.C.; Reis-Filho, J.S. Histological Types of Breast Cancer: How Special Are They? Mol. Oncol. 2010, 4, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Bergin, A.R.T.; Loi, S. Triple-negative Breast Cancer: Recent Treatment Advances. F1000Research 2019, 8, 1342. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Aggarwal, R. An Overview of Triple-Negative Breast Cancer. Arch. Gynecol. Obstet. 2015, 293, 247–269. [Google Scholar] [CrossRef]
- Cetin, I.; Topcul, M. Triple Negative Breast Cancer. Asian Pac. J. Cancer Prev. 2014, 15, 2427–2431. [Google Scholar] [CrossRef]
- Nedeljković, M.; Damjanović, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef]
- Sukumar, J.; Gast, K.; Quiroga, D.; Lustberg, M.; Williams, N. Triple-negative Breast Cancer: Promising Prognostic Biomarkers Currently in Development. Expert Rev. Anticancer Ther. 2021, 21, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [PubMed]
- Borri, F.; Granaglia, A. Pathology of Triple Negative Breast Cancer. Semin. Cancer Biol. 2020, 72, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.A.; Oza, G.; Sharma, A.; Arriaga, L.G.; Hernández Hernández, J.M.; Rotello, V.M.; Ramirez, J.T. Triple-Negative Breast Cancer: A Review of Conventional and Advanced Therapeutic Strategies. Int. J. Environ. Res. Public Health 2020, 17, 2078. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.; Anselmi, K.F.; Kuroda, F.; Schwartz, J.C. Oncoplasty as the Standard of Care in Breast Cancer Surgery. Eur. Oncol. Haematol. 2014, 1, 43–47. [Google Scholar] [CrossRef]
- Aerts, L.; Christiaens, M.R.; Enzlin, P.; Neven, P.; Amant, F. Sexual Functioning in Women After Mastectomy Versus Breast Conserving Therapy for Early-Stage Breast Cancer: A Prospective Controlled Study. Breast 2014, 23, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Baranova, A.; Krasnoselskyi, M.; Starikov, V.; Kartashov, S.; Zhulkevych, I.; Vlasenko, V.; Oleshko, K.; Bilodid, O.; Sadchikova, M.; Vinnyk, Y. Triple-negative Breast Cancer: Current Treatment Strategies and Factors of Negative Prognosis. J. Med. Life 2022, 15, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Korde, L.A.; Somerfield, M.R.; Carey, L.A.; Crews, J.A.; Denduluri, N.; Hwang, E.S.; Khan, S.A.; Liobl, S.; Morris, E.A.; Perez, A.; et al. Neoadjuvant Chemotherapy, Endocrine Therapy, and Targeted Therapy for Breast Cancer: ASCO Guideline. J. Clin. Oncol. 2021, 39, 1485–1505. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, F.; Nuzzolese, I.; Ponzone, R. Neoadjuvant or Adjuvant Chemotherapy in Early Breast Cancer? Expert Opin. Pharmacother. 2020, 21, 1071–1082. [Google Scholar] [CrossRef]
- Kwapisz, D. Pembrolizumab and Atezolizumab in Triple-negative Breast Cancer. Cancer Immunol. Immunother. 2020, 70, 607–617. [Google Scholar] [CrossRef]
- Won, K.A.; Spruck, C. Triple-negative Breast Cancer Therapy: Current and Future Perspectives. Int. J. Oncol. 2020, 57, 1245–1261. [Google Scholar] [CrossRef]
- Emens, L.A.; Loi, S. Immunotherapy Approaches for Breast Cancer Patients in 2023. Cold Spring Harb. Perspect. Med. 2023, 13, a041332. [Google Scholar] [CrossRef]
- Slade, D. PARP And PARG Inhibitors in Cancer Treatment. Genes Dev. 2020, 34, 5–6. [Google Scholar] [CrossRef]
- Lu, B.; Natarajan, E.; Balaji Raghavendran, H.R.; Markandan, U.D. Molecular Classification, Treatment, and Genetic Biomarkers in Triple-Negative Breast Cancer: A Review. Technol. Cancer Res. Treat. 2023, 22, 15330338221145246. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Upadhyay, M.; Samal, J.; Kandpal, M.; Singh, O.V.; Vivekanandan, P. The Warburg effect: Insights from the past decade. Pharmacol. Ther. 2013, 137, 318–330. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Boese, A.C.; Kang, S. Mitochondrial metabolism-mediated redox regulation in cancer progression. Redox Biol. 2021, 42, 101870. [Google Scholar] [CrossRef]
- Mullapudi, S.S.; Mitra, D.; Li, M.; Kang, E.-T.; Chiong, E.; Neoh, K.G. Potentiating Anti-Cancer Chemotherapeutics and Antimicrobials via Sugar-Mediated Strategies. Mol. Syst. Des. Eng. 2020, 5, 772–791. [Google Scholar] [CrossRef]
- Baer, S.C.; Casaubon, L.; Younes, M. Expression of the human erythrocyte glucose transporter Glut1 in cutaneous neoplasia. J. Am. Acad. Dermatol. 1997, 37, 575–577. [Google Scholar] [CrossRef]
- Macheda, M.L.; Rogers, S.; Best, J.D. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2005, 202, 654–662. [Google Scholar] [CrossRef]
- Airley, R.E.; Mobasheri, A. Hypoxic regulation of glucose transport, anaerobic metabolism and angiogenesis in cancer: Novel pathways and targets for anticancer therapeutics. Chemotherapy 2007, 53, 233–256. [Google Scholar] [CrossRef]
- Carvalho, K.C.; Cunha, I.W.; Rocha, R.M.; Ayala, F.R.; Cajaíba, M.M.; Begnami, M.D.; Vilela, R.S.; Paiva, G.R.; Andrade, R.G.; Soares, F.A. GLUT1 Expression in Malignant Tumors and Its Use as an Immunodiagnostic Marker. Clinics 2011, 66, 965–972. [Google Scholar] [CrossRef]
- Barron, C.C.; Bilan, P.J.; Tsakiridis, T.; Tsiani, E. Facilitative Glucose Transporters: Implications for Cancer Detection, Prognosis and Treatment. Metabolism 2016, 65, 124–139. [Google Scholar] [CrossRef]
- Ben-Haim, S.; Ell, P. 18F-FDG PET and PET/CT in the Evaluation of Cancer Treatment Response. J. Nucl. Med. 2009, 50, 88–99. [Google Scholar] [CrossRef]
- Pastuch-Gawołek, G.; Szreder, J.; Domińska, M.; Pielok, M.; Cichy, P.; Grymel, M. A Small Sugar Molecule with Huge Potential in Targeted Cancer Therapy. Pharmaceutics 2023, 15, 913. [Google Scholar] [CrossRef]
- Liu, H.; Ertay, A.; Peng, P.; Li, J.; Liu, D.; Xiong, H.; Zou, Y.; Qiu, H.; Hancock, D.; Yuan, X.; et al. SGLT1 is required for the survival of triple-negative breast cancer cells via potentiation of EGFR activity. Mol. Oncol. 2019, 13, 1874–1886. [Google Scholar] [CrossRef]
- Lai, B.; Xiao, Y.; Pu, H.; Cao, Q.; Jing, H.; Liu, X. Overexpression of SGLT1 is correlated with tumor development and poor prognosis of ovarian carcinoma. Arch. Gynecol. Obstet. 2012, 285, 1455–1461. [Google Scholar] [CrossRef]
- Hanabata, Y.; Nakajima, Y.; Morita, K.; Kayamori, K.; Omura, K. Coexpression of SGLT1 and EGFR is associated with tumor differentiation in oral squamous cell carcinoma. Odontology 2012, 100, 156–163. [Google Scholar] [CrossRef]
- Casneuf, V.F.; Fonteyne, P.; Van Damme, N.; Demetter, P.; Pauwels, P.; De Hemptinne, B.; De Vos, M.; Van de Wiele, C.; Peeters, M. Expression of SGLT1, Bcl-2 and p53 in primary pancreatic cancer related to survival. Cancer Investig. 2008, 26, 852–859. [Google Scholar] [CrossRef]
- Cao, Y. Obesity protects cancer from drugs targeting blood vessels. Cell Metab. 2018, 27, 1163–1165. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Jin, X.; Wang, M.; Liu, D.; Luo, Q.; Tian, H.; Cai, L.; Meng, L.; Bi, R.; Wang, L.; et al. GLUT5-mediated fructose utilization drives lung cancer growth by stimulating fatty acid synthesis and AMPK/mTORC1 signaling. JCI Insight 2020, 5, e131596. [Google Scholar] [CrossRef]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system from concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Vellon, L.; Cuyàs, E.; Buxó, M.; Martin-Castillo, B.; Serra, D.; García, J.; Lupu, R.; Menendez, J.A. Clinical and therapeutic relevance of the metabolic oncogene fatty acid synthase in HER2+ breast cancer. Histol. Histopathol. 2017, 32, 687–698. [Google Scholar]
- Ogrodzinski, M.P.; Bernard, J.J.; Lunt, S.Y. Deciphering metabolic rewiring in breast cancer subtypes. Transl. Res. 2017, 189, 105–122. [Google Scholar] [CrossRef]
- Camarda, R.; Zhou, A.; Kohnz, R.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef]
- Pizer, E.S.; Thupari, J.; Han, W.F.; Pinn, M.L.; Chrest, F.J.; Frehywot, G.L.; Townsend, C.A.; Kuhajda, F.P. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res. 2000, 60, 213–218. [Google Scholar]
- Vergote, D.; Cren-Olive, C.; Chopin, V.; Toillon, R.-A.; Rolando, C.; Hondermarck, H.; Le Bourhis, X. (-)-Epigallocatechin (EGC) of green tea induces apoptosis of human breast cancer cells but not of their normal counterparts. Breast Cancer Res. Treat. 2002, 76, 195–201. [Google Scholar] [CrossRef]
- Yeh, C.W.; Chen, W.J.; Chiang, C.T.; Lin-Shiau, S.Y.; Lin, J.K. Suppression of fatty acid synthase in MCF-7 breast cancer cells by tea and tea polyphenols: A possible mechanism for their hypolipidemic effects. Pharmacogenom. J. 2003, 3, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Baliga, M.S.; Meleth, S.; Katiyar, S.K. Growth inhibitory and antimetastatic effect of green tea polyphenols on metastasis-specific mouse mammary carcinoma 4T1 cells in vitro and in vivo systems. Clin. Cancer Res. 2005, 11, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Puig, T.; Vázquez-Martín, A.; Relat, J.; Pétriz, J.; Menéndez, J.A.; Porta, R.; Colomer, R. Fatty acid metabolism in breast cancer cells: Differential inhibitory effects of epigallocatechin gallate (EGCG) and C75. Breast Cancer Res. Treat. 2007, 109, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Marín, V.; Burgos, V.; Pérez, R.; Maria, D.A.; Pardi, P.; Paz, C. The Potential Role of Epigallocatechin-3-Gallate (EGCG) in Breast Cancer Treatment. Int. J. Mol. Sci. 2023, 24, 10737. [Google Scholar] [CrossRef] [PubMed]
- El Ansari, R.; Craze, M.L.; Miligy, I.; Diez-Rodriguez, M.; Nolan, C.C.; Ellis, I.O.; Rakha, E.A.; Green, A.R. The amino acid transporter SLC7A5 confers a poor prognosis in the highly proliferative breast cancer subtypes and is a key therapeutic target in luminal B tumors. Breast Cancer Res. 2018, 20, 21. [Google Scholar] [CrossRef] [PubMed]
- Van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumor growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.D.; Lamichhane, S.; Lundgren, S.; Bofin, A.; Fjosne, H.; Giskeodegard, G.F.; Bathen, T.F. Metabolic characterization of triple negative breast cancer. BMC Cancer 2014, 14, 941. [Google Scholar] [CrossRef] [PubMed]
- Cassago, A.; Ferreira, A.P.; Ferreira, I.M.; Fornezari, C.; Gomes, E.R.; Greene, K.S.; Pereira, H.M.; Garratt, R.C.; Dias, S.M.; Ambrosio, A.L. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 1092–1097. [Google Scholar] [CrossRef]
- Matés, J.M.; Campos-Sandoval, J.A.; Márquez, J. Glutaminase isoenzymes in the metabolic therapy of cancer. Biochim. Biophys. Acta—Rev. Cancer 2018, 1870, 158–164. [Google Scholar] [CrossRef]
- McDermott, L.A.; Iyer, P.; Vernetti, L.; Rimer, S.; Sun, J.; Boby, M.; Yang, T.; Fioravanti, M.; O’Neill, J.; Wang, L.; et al. Design and evaluation of novel glutaminase inhibitors. Bioorg. Med. Chem. 2016, 24, 1819–1839. [Google Scholar] [CrossRef]
- Maeda, H.; Khatami, M. Analyses of Repeated Failures in Cancer Therapy for Solid Tumors: Poor Tumor-selective Drug Delivery, Low Therapeutic Efficacy and Unsustainable Costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Hyman, D.M.; Vasan, N.; Baselga, J. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar]
- Patel, T.K.; Adhikari, N.; Amin, S.A.; Biswas, S.; Jha, T.; Ghosh, B. Small Molecule Drug Conjugates (SMDCs): An Emerging Strategy for Anticancer Drug Design and Discovery. New J. Chem. 2021, 45, 5291–5321. [Google Scholar] [CrossRef]
- Molejon, M.I.; Weiz, G.; Breccia, J.D.; Vaccaro, M.I. Glycoconjugation: An Approach to Cancer Therapeutics. World J. Clin. Oncol. 2020, 11, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Yang, J.; Seeberger, P.H.; Yin, J. Glycoconjugates for glucose transporter-mediated cancer-specific targeting and treatment. Carbohydr. Res. 2020, 498, 108195. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, D. Glufosfamide: Can we improve the process of anticancer agent development? Expert Opin. Investig. Drugs 2012, 21, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Seker, H.; Bertram, B.; Burkle, A.; Kaina, B.; Pohl, J.; Koepsell, H.; Wiesser, M. Mechanistic aspects of the cytotoxic activity of glufosfamide, a new tumor therapeutic agent. Br. J. Cancer 2000, 82, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Meredith, A.M.; Dass, C.R. Increasing role of the cancer chemotherapeutic doxorubicin in cellular metabolism. J. Pharm. Pharmacol. 2016, 68, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- Hortobagyi, G.N. Anthracyclines in the treatment of cancer. An overview. Drugs 1997, 54, 1–7. [Google Scholar]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.; Sludden, J.; Griffin, M.J.; Cole, M.; Verrill, M.; Jamieson, D.; Boddy, A.V. Influence of Pharmacogenetics on Response and Toxicity in Breast Cancer Patients Treated with Doxorubicin and Cyclophosphamide. Br. J. Cancer 2010, 102, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.C.; Honrado, M.; Armada, A.; Viveiros, M.; Rueff, J.; Rodrigues, A.S. ABC Efflux Transporters and the Circuitry of MiRNAs: Kinetics of Expression in Cancer Drug Resistance. Int. J. Mol. Sci. 2020, 21, 2985. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.M.; Ni, J.D.; Song, D.; Ding, M.; Huang, J. Interplay between unfolded protein response and autophagy promotes tumor drug resistance. Oncol. Lett. 2015, 10, 1959–1969. [Google Scholar] [CrossRef] [PubMed]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Jiang, X.; Albright, C.F.; Graciani, N.; Yue, E.; Zhang, M.; Zhang, S.Y.; Bruckner, R.; Diamond, M.; Dowling, R.; et al. Discovery of matrix metalloproteases selective and activated peptide-doxorubicin prodrugs as anti-tumor agents. Bioorg. Med. Chem. Lett. 2010, 20, 853–856. [Google Scholar] [CrossRef] [PubMed]
- Stepankova, J.; Studenovsky, M.; Malina, J.; Kasparkova, J.; Liskova, B.; Novakova, O.; Ulbrich, K.; Brabec, V. DNA interactions of 2-pyrrolinodoxorubicin, a distinctively more potent daunosamine-modified analogue of doxorubicin. Biochem. Pharmacol. 2011, 82, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Simons, A.L.; Ahmad, I.M.; Mattson, D.M.; Dornfeld, K.J.; Spitz, D.R. 2-Deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007, 67, 3364–3370. [Google Scholar] [CrossRef] [PubMed]
- Mohanti, B.K.; Rath, G.K.; Anantha, N.; Kannan, V.; Das, B.S.; Chandramouli, B.A.; Banerjee, A.K.; Das, S.; Jena, A.; Ravichandran, R.; et al. Improving cancer radiotherapy with 2-deoxy-D-glucose: Phase I/II clinical trials on human cerebral gliomas. Int. J. Radiat. Oncol. Biol. Phys. 1996, 35, 103–111. [Google Scholar] [CrossRef]
- Maschek, G.; Savaraj, N.; Priebe, W.; Braunschweiger, P.; Hamilton, K.; Tidmarsh, G.F.; De Young, L.R.; Lampidis, T.J. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 2004, 64, 31–34. [Google Scholar] [CrossRef]
- Galanski, M.; Jakupec, M.; Keppler, B. Update of the preclinical situation of anticancer platinum complexes: Novel design strategies and innovative analytical approaches. Curr. Med. Chem. 2005, 12, 2075–2094. [Google Scholar] [CrossRef]
- Harper, B.W.; Krause-Heuer, A.M.; Grant, M.P.; Manohar, M.; Garbutcheon-Singh, K.B.; Aldrich-Wright, J.R. Advances in platinum chemotherapeutics. Chem.—Eur. J. 2010, 16, 7064–7077. [Google Scholar] [CrossRef] [PubMed]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [PubMed]
- Aldossary, S.A. Review on Pharmacology of Cisplatin: Clinical Use, Toxicity and Mechanism of Resistance of Cisplatin. Biomed. Pharmacol. J. 2019, 12, 7–15. [Google Scholar] [CrossRef]
- Kweekel, D.M.; Gelderblom, H.; Guchelaar, H.J. Pharmacology of oxaliplatin and the use of pharmacogenomics to individualize therapy. Cancer Treat. Rev. 2005, 31, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, S.A.; Ponte, F.; Sicilia, E.; Azzazy, H.M. Experimental and Computational Investigations of Carboplatin Supramolecular Complexes. ACS Omega 2020, 5, 31456–31466. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, S.A.; Brüßler, J.; Alawak, M.; El-Sayed, M.M.; Bakowsky, U.; Shoeib, T. Chemotherapy Based on Supramolecular Chemistry: A Promising Strategy in Cancer Therapy. Pharmaceutics 2019, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Comella, P.; Casaretti, R.; Sandomenico, C.; Avallone, A.; Franco, L. Role of oxaliplatin in the treatment of colorectal cancer. Ther. Clin. Risk Manag. 2009, 5, 229–238. [Google Scholar] [CrossRef]
- Jain, A.; Jain, S.K.; Ganesh, N.; Barve, J.; Beg, A.M. Design and development of ligand-appended polysaccharidic nanoparticles for the delivery of oxaliplatin in colorectal cancer. Nanomedicine 2010, 6, 179–190. [Google Scholar] [CrossRef]
- Labaye, J.; Sarret, D.; Duvic, C.; Herody, M.; Didelot, F.; Nedelec, G.; Noel, L.H. Renal toxicity of oxaliplatin. Nephrol. Dial. Transplant. 2005, 20, 1275–1276. [Google Scholar] [CrossRef]
- Bautista, M.A.; Stevens, W.T.; Chen, C.S.; Curtis, B.R.; Aster, R.H.; Hsueh, C.T. Hypersensitivity reaction and acute immune-mediated thrombocytopenia from oxaliplatin: Two case reports and a review of the literature. J. Hematol. Oncol. 2010, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.A.; Tripathi, R.; Mishra, B. Methotrexate: A detailed review on drug delivery and clinical aspects. Expert Opin. Drug Deliv. 2012, 9, 151–169. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.C.; McCormick, J.; Pui, C.; Buddington, R.K.; Harvey, R.D. Preventing and Managing Toxicities of High-Dose Methotrexate. Oncologist 2016, 21, 1471–1482. [Google Scholar] [CrossRef] [PubMed]
- Świerkot, J. Toxicity of low dose methotrexate in rheumatoid arthritis. Adv. Clin. Exp. Med. 2007, 16, 287–295. [Google Scholar]
- Visser, K.; Van der Heijde, D.M. Risk and management of liver toxicity during methotrexate treatment in rheumatoid and psoriatic arthritis: A systematic review of the literature. Clin. Exp. Rheumatol. 2009, 27, 1017–1025. [Google Scholar] [PubMed]
- Abolmaali, S.S.; Tamaddon, A.M.; Dinarvand, R. A review of therapeutic challenges and achievements of methotrexate delivery systems for treatment of cancer and rheumatoid arthritis. Cancer Chemother. Pharmacol. 2013, 71, 1115–1130. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.P. Accumulation of methotrexate in human tissues following high-dose methotrexate therapy. J. Pak. Med. Assoc. 1998, 48, 341–343. [Google Scholar] [PubMed]
- Sun, Q.; Zhou, Z.; Qiu, N.; Shen, Y. Rational Design of Cancer Nanomedicine: Nanoproperty Integration and Synchronization. Adv. Mater. 2017, 29, 1606628. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Luo, Z.; Wu, Y.; Li, Z.; Loh, X.J. Recent Progress in Polyhydroxyalkanoates-Based Copolymers for Biomedical Applications. Biotechnol. J. 2019, 14, e1900283. [Google Scholar] [CrossRef]
- Barouti, G.; Jaffredo, C.G.; Guillaume, S.M. Advances in drug delivery systems based on synthetic poly(hydroxybutyrate) (co)polymers. Prog. Polym. Sci. 2017, 73, 1–31. [Google Scholar] [CrossRef]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Deirram, N.; Zhang, C.; Kermaniyan, S.S.; Johnston, A.P.; Such, G.K. pH-Responsive Polymer Nanoparticles for Drug Delivery. Macromol. Rapid Commun. 2019, 40, e1800917. [Google Scholar] [CrossRef] [PubMed]
- Kanamala, M.; Wilson, W.R.; Yang, M.; Palmer, B.D.; Wu, Z. Mechanisms and biomaterials in pH-responsive tumor targeted drug delivery: A review. Biomaterials 2016, 85, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Chan, J.M.; Farokhzad, O.C. pH-Responsive Nanoparticles for Drug Delivery. Mol. Pharm. 2010, 7, 1913–1920. [Google Scholar] [CrossRef]
- Sonawane, S.J.; Kalhapure, R.S.; Govender, T. Hydrazone linkages in pH responsive drug delivery systems. Eur. J. Pharm. Sci. 2017, 99, 45–65. [Google Scholar] [CrossRef]
- Wang, Z.; Deng, X.; Ding, J.; Zhou, W.; Zheng, X.; Tang, G. Mechanisms of drug release in pH-sensitive micelles for tumor targeted drug delivery system: A review. Int. J. Pharm. 2018, 535, 253–260. [Google Scholar] [CrossRef]
- Abraham, S.; Guo, F.; Li, L.S.; Rader, C.; Liu, C.; Barbas, C.F.; Lerner, R.A.; Sinha, S.C. Synthesis of the next-generation therapeutic antibodies that combine cell targeting and antibody-catalyzed prodrug activation. Proc. Natl. Acad. Sci. USA 2007, 104, 5584–5589. [Google Scholar] [CrossRef]
- De Graaf, M.; Boven, E.; Scheeren, H.W.; Haisma, H.J.; Pinedo, H.M. Beta-glucuronidase-mediated drug release. Curr. Pharm. Des. 2002, 8, 1391–1403. [Google Scholar] [CrossRef]
- Wu, J.; Liu, Q.; Lee, R.J. A folate receptor-targeted liposomal formulation for paclitaxel. Int. J. Pharm. 2006, 316, 148–153. [Google Scholar] [CrossRef]
- Al-Mahayri, Z.N.; AlAhmad, M.M.; Ali, B.R. Current opinion on the pharmacogenomics of paclitaxel-induced toxicity. Expert Opin. Drug Metab. Toxicol. 2021, 17, 785–801. [Google Scholar] [CrossRef]
- Marupudi, N.I.; Han, J.E.; Li, K.W.; Renard, V.M.; Tyler, B.M.; Brem, H. Paclitaxel: A review of adverse toxicities and novel delivery strategies. Expert Opin. Drug Saf. 2007, 6, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Fu, Y.S.; Aziz, F.; Wang, X.Q.; Yan, Q.; Liu, J.W. Ginsenoside Rg3 inhibits melanoma cell proliferation through down-regulation of histone deacetylase 3 (HDAC3) and increase of p53 acetylation. PLoS ONE 2014, 9, e115401. [Google Scholar] [CrossRef]
- Sato, K.; Mochizuki, M.; Saiki, I.; Yoo, Y.C.; Samukawa, K.; Azuma, I. Inhibition of tumor angiogenesis and metastasis by a saponin of Panax ginseng, ginsenoside-Rb2. Biol. Pharm. Bull. 1994, 17, 635–639. [Google Scholar] [CrossRef]
- Liu, C.; Gong, Q.; Chen, T.; Lv, J.; Feng, Z.; Liu, P.; Deng, Z. Treatment with 20(S)-ginsenoside Rg3 reverses multidrug resistance in A549/DDP xenograft tumors. Oncol. Lett. 2018, 15, 4376–4382. [Google Scholar] [CrossRef]
- Chen, Z.; Wei, X.; Shen, L.; Zhu, H.; Zheng, X. 20(S)-ginsenoside-Rg3 reverses temozolomide resistance and restrains epithelial-mesenchymal transition progression in glioblastoma. Cancer Sci. 2019, 110, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Wang, D.; Liang, J.; Guo, Y.; Zhu, Y.; Xia, J.; Qin, J.; Zhan, H.; Wang, J. Novel ginsenoside-based multifunctional liposomal delivery system for combination therapy of gastric cancer. Theranostics 2019, 9, 4437–4449. [Google Scholar] [CrossRef]
- Zhu, Y.; Liang, J.; Gao, C.; Wang, A.; Xia, J.; Hong, C.; Zhong, Z.; Zuo, Z.; Kim, J.; Ren, H.; et al. Multifunctional ginsenoside Rg3-based liposomes for glioma targeting therapy. J. Control. Release 2021, 330, 641–657. [Google Scholar] [CrossRef] [PubMed]
- Giaccia, A.J.; Simon, M.C.; Johnson, R. The biology of hypoxia: The role of oxygen sensing in development, normal function, and disease. Genes Dev. 2004, 18, 2183–2194. [Google Scholar] [CrossRef]
- Mazzone, M.; Dettori, D.; Leite de Oliveira, R.; Loges, S.; Schmidt, T.; Jonckx, B.; Tian, Y.M.; Lanahan, A.A.; Pollard, P.; Ruiz de Almodovar, C.; et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 2009, 136, 839–851. [Google Scholar] [CrossRef]
- Darby, I.A.; Hewitson, T.D. Hypoxia in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Chang, S.H.; Shih, S.C.; Dvorak, A.M.; Dvorak, H.F. Heterogeneity of the tumor vasculature. Semin. Thromb. Hemost. 2010, 36, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Feki, A.; Irminger-Finger, I. Mutational spectrum of p53 mutations in primary breast and ovarian tumors. Crit. Rev. Oncol. Hematol. 2004, 52, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin. Cancer Biol. 2009, 19, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Smallbone, K.; Maini, P.K.; Rose, F.; Averill, J.; Nagle, R.B.; Worrall, L.; Gillies, R.J. Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br. J. Cancer 2007, 97, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.S.; Davies, A.J.; Halestrap, P. The plasma membrane lactate transporter MCT4, but not MCT1 is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 6, 472–482. [Google Scholar] [CrossRef]
- Bartosova, M.; Parkkila, S.; Pohlodek, K.; Karttunen, T.J.; Galbavy, S.; Mucha, V.; Harris, A.L.; Pastorek, J.; Pastoreková, S. Expression of carbonic anhydrase IX in breast is associated with malignant tissues and is related to overexpression of c-erbB2. J. Pathol. 2002, 197, 314–321. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Svastova, E.; Witarski, W.; Csaderova, L.; Kosik, I.; Skvarkova, L.; Hulikova, A.; Zatovicova, M.; Barathova, M.; Kopacek, J.; Pastorek, J.; et al. Carbonic anhydrase IX interacts with bicarbonate transporters in lamellipodia and increases cell migration via its catalytic domain. J. Biol. Chem. 2012, 287, 3392–3402. [Google Scholar] [CrossRef]
- Lee, S.; Axelsen, T.V.; Andersen, A.P.; Vahl, P.; Pedersen, S.F.; Boedtkjer, E. Disrupting Na+, HCO3−-cotransporter NBCn1 (Slc4a7) delays murine breast cancer development. Oncogene 2016, 35, 2112–2122. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, A.; Patiar, S.; Wigfield, S.; Li, J.L.; Ledaki, I.; Turley, H.; Leek, R.; Snell, C.; Gatter, K.; Sly, W.S.; et al. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin. Cancer Res. 2012, 18, 3100–3111. [Google Scholar] [CrossRef]
- Parks, S.K.; Pouyssegur, J. The Na(+)/HCO3(-) Co-Transporter SLC4A4 Plays a Role in Growth and Migration of Colon and Breast Cancer Cells. J. Cell. Physiol. 2015, 230, 1954–1963. [Google Scholar] [CrossRef]
- Shiraishi, T.; Verdone, J.E.; Huang, J.; Kahlert, U.D.; Hernandez, J.R.; Torga, G.; Zarif, J.C.; Epstein, T.; Gatenby, R.; McCartney, A.; et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 2015, 6, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.; Ford, H. Epithelial to mesenchymal transition in development of cancer. Future Oncol. 2009, 8, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Scimeca, M.; Antonacci, C.; Colombo, D.; Bonfiglio, R.; Buonomo, O.C.; Bonanno, E. Emerging prognostic markers related to mesenchymal characteristics of poorly differentiated breast cancers. Tumor Biol. 2016, 37, 5427–5435. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumors. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Liu, Q.; Guan, C.; Liu, C.; Li, H.; Wu, J.; Sun, C. Targeting hypoxia-inducible factor-1alpha: A new strategy for triple-negative breast cancer therapy. Biomed. Pharmacother. 2022, 156, 113861. [Google Scholar] [CrossRef]
- Pepple, A.L.; Guy, J.L.; McGinnis, R.; Felsted, A.E.; Song, B.; Hubbard, R.; Worlikar, T.; Garavaglia, H.; Dib, J.; Chao, H.; et al. Histotripsy focused ultrasound ablation induces immunological cell death in treated and distant untreated tumors. J. Immunother. Cancer 2020, 8, 730. [Google Scholar]
- Chang, J.C. Cancer stem cells: Role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine 2016, 95, S20–S25. [Google Scholar] [CrossRef] [PubMed]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef]
- Jiang, Z.S.; Sun, Y.Z.; Wang, S.M.; Ruan, J.S. Epithelial-mesenchymal transition: A potential regulator of ABC transporters in tumor progression. J. Cancer 2017, 8, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Smith-Mungo, L.I.; Kagan, H.M. Lysyl oxidase: Properties, regulation and multiple functions in biology. Matrix Biol. 1998, 16, 387–398. [Google Scholar] [CrossRef]
- Payne, S.L.; Hendrix, M.J.; Kirschmann, D.A. Paradoxical roles for lysyl oxidases in cancer—A prospect. J. Cell. Biochem. 2007, 101, 1338–1354. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Santamaría, P.G.; Moreno-Bueno, G. LOXL2 in epithelial cell plasticity and tumor progression. Future Oncol. 2012, 8, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Chang, J.; Cox, T.R.; Lang, G.; Bird, D.; Nicolau, M.; Evans, H.R.; Gartland, A.; Erler, J.T. LOXL2-mediated matrix remodeling in metastasis and mammary gland involution. Cancer Res. 2011, 71, 1561–1572. [Google Scholar] [CrossRef]
- Han, Y.; Lian, S.; Cui, X.; Meng, K.; Győrffy, B.; Jin, T.; Huang, D. Potential options for managing LOX+ER-breast cancer patients. Oncotarget 2016, 7, 32893–32901. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.; Cotic, C.; Pomp, V.; Fink, D.; Varga, Z. Overexpression of Lox in triple-negative breast cancer. Ann. Diagn. Pathol. 2018, 34, 98–102. [Google Scholar] [CrossRef]
- Ahn, S.G.; Kim, S.J.; Kim, C.; Jeong, J. Molecular Classification of Triple-Negative Breast Cancer. J. Breast Cancer 2016, 19, 223–230. [Google Scholar] [CrossRef]
- Ahn, S.G.; Dong, S.M.; Oshima, A.; Kim, W.H.; Lee, H.M.; Lee, S.A.; Kwon, S.H.; Lee, J.H.; Lee, J.M.; Jeong, J.; et al. LOXL2 expression is associated with invasiveness and negatively influences survival in breast cancer patients. Breast Cancer Res. Treat. 2013, 141, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Postovit, L.M.; Abbott, D.E.; Payne, S.L.; Wheaton, W.W.; Margaryan, N.V.; Sullivan, R.; Jansen, M.K.; Csiszar, K.; Hendrix, M.J.; Kirschmann, D.A. Hypoxia/reoxygenation: A dynamic regulator of lysyl oxidase-facilitated breast cancer migration. J. Cell. Biochem. 2008, 103, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Bignon, M.; Pichol-Thievend, C.; Hardouin, J.; Malbouyres, M.; Bréchot, N.; Nasciutti, L.; Barret, A.; Teillon, J.; Guillon, E.; Etienne, E.; et al. Lysyl oxidase-like protein-2 regulates sprouting angiogenesis and type IV collagen assembly in the endothelial basement membrane. Blood 2011, 118, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- De Jong, O.G.; Van Der Waals, L.M.; Kools, F.R.W.; Verhaar, M.C.; Van Balkom, B.W. Lysyl oxidase-like 2 is a regulator of angiogenesis through modulation of endothelial-to-mesenchymal transition. J. Cell. Physiol. 2018, 234, 10260–10269. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, S.; Tian, Y.; Ju, A.; Hou, Q.; Liu, J.; Fu, Y.; Luo, Y. Lysyl Oxidase-Like Protein 2 Promotes Tumor Lymphangiogenesis and Lymph Node Metastasis in Breast Cancer. Neoplasia 2019, 21, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Schilling, E.D.; Strong, F.M. Isolation, Structure and Synthesis of a Lathyrus Factor From L. Odoratus. J. Am. Chem. Soc. 1955, 77, 2843–2845. [Google Scholar] [CrossRef]
- Sherif, H.M. In search of a new therapeutic target for the treatment of genetically triggered thoracic aortic aneurysms and cardiovascular conditions: Insights from human and animal lathyrism. Interact. Cardiovasc. Thorac. Surg. 2010, 11, 271–276. [Google Scholar] [CrossRef]
- Bondareva, A.; Downey, C.M.; Ayres, F.; Liu, W.; Boyd, S.K.; Hallgrimsson, B.; Jirik, F.R. The Lysyl Oxidase Inhibitor, β-Aminopropionitrile, Diminishes the Metastatic Colonization Potential of Circulating Breast Cancer Cells. PLoS ONE 2009, 4, e5620. [Google Scholar] [CrossRef]
- Smithen, D.A.; Leung, L.M.H.; Challinor, M.; Lawrence, R.; Tang, H.; Niculescu-Duvaz, D.; Pearce, S.P.; McLeary, R.; Lopes, F.; Aljarah, M.; et al. 2-Aminomethylene-5-sulfonylthiazole Inhibitors of Lysyl Oxidase (LOX) and LOXL2 Show Significant Efficacy in Delaying Tumor Growth. J. Med. Chem. 2019, 63, 2308–2324. [Google Scholar] [CrossRef]
- Barry-Hamilton, V.; Spangler, R.; Marshall, D.; McCauley, S.; Rodriguez, H.M.; Oyasu, M.; Mikels, A.; Vaysberg, M.; Ghermazien, H.; Wai, C.; et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat. Med. 2010, 16, 1009–1017. [Google Scholar] [CrossRef]
- Chopra, V.; Sangarappillai, R.M.; Romero-Canelón, I.; Jones, A.M. Lysyl Oxidase Like-2 (LOXL2): An Emerging Oncology Target. Adv. Ther. 2020, 3, 2366–3987. [Google Scholar] [CrossRef]
- Schmelzer, C.E.; Heinz, A.; Troilo, H.; Lockhart-Cairns, M.P.; Jowitt, T.A.; Marchand, M.F.; Bidault, L.; Bignon, M.; Hedtke, T.; Barret, A.; et al. Lysyl oxidase-like 2 (LOXL2)-mediated cross-linking of tropoelastin. FASEB J. 2019, 33, 5468–5481. [Google Scholar] [CrossRef] [PubMed]
- Hollosi, P.; Yakushiji, J.K.; Fong, K.S.; Csiszar, K.; Fong, S.F. Lysyl oxidase-like 2 promotes migration in noninvasive breast cancer cells but not in normal breast epithelial cells. Int. J. Cancer 2009, 125, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Pharmaxis. Available online: https://www.pharmaxis.com.au/investor-centre/news/view/pharmaxis-releases-positive-results-of-phase-1-clinical-trial-for-second-loxl2-inhibitor-compound (accessed on 10 December 2023).
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Mamessier, E.; Sylvain, A.; Thibult, M.-L.; Houvenaeghel, G.; Jacquemier, J.; Castellano, R.; Gonçalves, A.; André, P.; Romagné, F.; Thibault, G. Human breast cancer cells enhance self-tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Investig. 2011, 121, 3609–3622. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.; Gray, R.J.; Demaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014, 32, 2959–2966. [Google Scholar] [CrossRef]
- Pruneri, G.; Gray, K.P.; Vingiani, A.; Viale, G.; Curigliano, G.; Criscitiello, C.; Láng, I.; Ruhstaller, T.; Gianni, L.; Goldhirsch, A.; et al. Tumor-infiltrating lymphocytes (TILs) are a powerful prognostic marker in patients with triple-negative breast cancer enrolled in the IBCSG phase III randomized clinical trial 22-00. Breast Cancer Res. Treat. 2016, 158, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Whitford, P.; George, W.D.; Campbell, A.M. Flow cytometric analysis of tumor infiltrating lymphocyte activation and tumor cell MHC class I and II expression in breast cancer patients. Cancer Lett. 1992, 61, 157–164. [Google Scholar] [CrossRef]
- Marra, A.; Viale, G.; Curigliano, G. Recent advances in triple negative breast cancer: The immunotherapy era. BMC Med. 2019, 17, 90. [Google Scholar] [CrossRef]
- Castle, J.C.; Kreiter, S.; Diekmann, J.J.; Löwer, M.; Van de Roemer, N.; De Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.D.; Warren, R.L.; Gibb, E.A.; Martin, S.D.; Spinelli, J.J.; Nelson, B.H.; Holt, R.A. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014, 24, 743–750. [Google Scholar] [CrossRef]
- Rajasagi, M.; Shukla, S.A.; Fritsch, E.F.; Keskin, D.B.; DeLuca, D.; Carmona, E.; Zhang, W.; Sougnez, C.; Cibulskis, K.; Sidney, J.; et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 2014, 124, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Dadmarz, R.; Sgagias, M.K.; Rosenberg, S.A.; Schwartzentruber, D.J. CD4+ T lymphocytes infiltrating human breast cancer recognise autologous tumor in an MHC-class-II restricted fashion. Cancer Immunol. Immunother. 1995, 40, 1–9. [Google Scholar] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: First clinical experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Cho, J.Y. Recent Advances in Allogeneic CAR-T Cells. Biomolecules 2020, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Tokarew, N.; Ogonek, J.; Endres, S.; Von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Xu, Z.; Zhuang, Y.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Hematological Malignancies. J. Cancer 2021, 12, 326–334. [Google Scholar] [CrossRef]
- Albinger, N.; Hartmann, J.; Ullrich, E. Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany. Gene Ther. 2021, 28, 513–527. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef] [PubMed]
- UPMC Hillman Cancer Center. Available online: https://hillman.upmc.com/mario-lemieux-center/treatment/car-t-cell-therapy/fda-approved-therapies (accessed on 22 March 2024).
- Abbott, R.C.; Cross, R.S.; Jenkins, M.R. Finding the Keys to the CAR: Identifying Novel Target Antigens for T Cell Redirection Immunotherapies. Int. J. Mol. Sci. 2020, 21, 515. [Google Scholar] [CrossRef] [PubMed]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef]
- Hsu, J.L.; Hung, M.C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Scanlan, M.J.; Gure, A.O.; Jungbluth, A.A.; Old, L.J.; Chen, Y.T. Cancer/testis antigens: An expanding family of targets for cancer immunotherapy. Immunol. Rev. 2002, 188, 22–32. [Google Scholar] [CrossRef]
- Dees, S.; Ganesan, R.; Singh, S.; Grewal, I.S. Emerging CAR-T Cell Therapy for the Treatment of Triple-Negative Breast Cancer. Mol. Cancer Ther. 2020, 19, 2409–2421. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H.; Liu, J.W.; Lu, C.; Wei, J.F. CAR-T Cell Therapy for Breast Cancer: From Basic Research to Clinical Application. Int. J. Biol. Sci. 2022, 18, 2609–2626. [Google Scholar] [CrossRef]
- Alewine, C.; Xiang, L.; Yamori, T.; Niederfellner, G.; Bosslet, K.; Pastan, I. Efficacy of RG7787, a next-generation mesothelin-targeted immunotoxin, against triple-negative breast and gastric cancers. Mol. Cancer Ther. 2014, 13, 2653–2661. [Google Scholar] [CrossRef]
- Zhang, B.L.; Qin, D.Y.; Mo, Z.M.; Li, Y.; Wei, W.; Wang, Y.S.; Wang, W.; Wei, Y.Q. Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors. Sci. China Life Sci. 2016, 59, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.; Kapoor, V.; Predina, J.; Powell, D.J., Jr.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [PubMed]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Bollard, C.M.; Rössig, C.; Calonge, M.J.; Huls, M.H.; Wagner, H.J.; Massague, J.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood 2002, 99, 3179–3187. [Google Scholar] [CrossRef]
- Nishio, N.; Dotti, G. Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Oncoimmunology. 2015, 4, e988098. [Google Scholar] [CrossRef]
- John, L.B.; Kershaw, M.H.; Darcy, P.K. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology 2013, 2, e26286. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, S.; Chow, K.K.; Mata, M.; Shaffer, D.R.; Song, X.T.; Wu, M.F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Lai, Y.; Li, J.; Qin, L.; Xu, Y.; Zhao, R.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology 2017, 6, e1284722. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, Q.; Zuo, T.; Wei, G.; Jiao, S. Combined antitumor effects of anti-EGFR variant III CAR-T cell therapy and PD-1 checkpoint blockade on glioblastoma in a mouse model. Cell. Immunol. 2020, 352, 104112. [Google Scholar] [CrossRef] [PubMed]
- De Toro, J.; Herschlik, L.; Waldner, C.; Mongini, C. Emerging roles of exosomes in normal and pathological conditions: New insights for diagnosis and therapeutic applications. Front. Immunol. 2015, 6, 203. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Andre, F.; Amigorena, S.; Soria, J.C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic cell-derived exosomes for cancer therapy. J. Clin. Investig. 2016, 126, 1224–1232. [Google Scholar] [CrossRef]
- Lu, J.; Wu, J.; Tian, J.; Wang, S. Role of T cell-derived exosomes in immunoregulation. Immunol. Res. 2018, 66, 313–322. [Google Scholar] [CrossRef]
- Wu, S.W.; Li, L.; Wang, Y.; Xiao, Z. CTL-Derived Exosomes Enhance the Activation of CTLs Stimulated by Low-Affinity Peptides. Front. Immunol. 2019, 10, 1274. [Google Scholar] [CrossRef]
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F.; et al. CAR exosomes derived from effector CAR-T cells have potent antitumor effects and low toxicity. Nat. Commun. 2019, 10, 4355. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Cui, S.; Li, S.; Du, C.; Tian, J.; Wan, S.; Qian, Z.; Gu, Y.; Chen, W.R.; Wang, G. Targeted Cancer Therapy with a 2-Deoxyglucose--Based Adriamycin Complex. Cancer Res. 2013, 73, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Lu, Y.; Gao, X.; Liu, R.; Zhang-Negrerie, D.; Shi, Y.; Wang, Y.; Wang, S.; Gao, Q. Highly water-soluble platinum(II) complexes as GLUT substrates for targeted therapy: Improved anticancer efficacy and transporter-mediated cytotoxic properties. Chem. Commun. 2013, 49, 2421. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, M.; Pastuch-Gawołek, G.; Makuch, S.; Wiśniewski, J.; Krenács, T.; Hamar, P.; Gamian, A.; Szeja, W.; Szkudlarek, D.; Krawczyk, M.; et al. In Vitro and In Vivo Efficacy of a Novel Glucose-Methotrexate Conjugate in Targeted Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 1748. [Google Scholar] [CrossRef] [PubMed]
- Domiński, A.; Domińska, M.; Skonieczna, M.; Pastuch-Gawołek, G.; Kurcok, P. Shell-Sheddable Micelles Based on Poly(ethylene glycol)-hydrazone-poly[R,S]-3-hydroxybutyrate Copolymer Loaded with 8-Hydroxyquinoline Glycoconjugates as a Dual Tumor-Targeting Drug Delivery System. Pharmaceutics 2022, 14, 290. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, A.; Zhang, S.; Kim, J.; Xia, J.; Zhang, F.; Wang, D.; Wang, Q.; Wang, J. Paclitaxel-loaded ginsenoside Rg3 liposomes for drug-resistant cancer therapy by dual targeting of the tumor microenvironment and cancer cells. J. Adv. Res. 2023, 49, 159–173. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, A.; Hulikova, A.; Ledaki, I.; Snell, C.; Singleton, D.; Steers, G.; Seden, P.; Jones, D.; Bridges, E.; Wigfield, S.; et al. Disrupting Hypoxia-Induced Bicarbonate Transport Acidifies Tumor Cells and Suppresses Tumor Growth. Cancer Res. 2016, 76, 3744–3755. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.P.; Bolland, H.; Vancauwenberghe, E.; Collier, P.; Ritchie, A.A.; Clarke, P.A.; Grabowska, A.M.; Harris, A.L.; McIntyre, A. Targeting hypoxia regulated sodium driven bicarbonate transporters reduces triple negative breast cancer metastasis. Neoplasia 2022, 25, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Lucas, M.C.; Leonte, L.E.; Garcia-Montolio, M.; Singh, L.B.; Findlay, A.D.; Deodhar, M.; Foot, J.S.; Jarolimek, W.; Timpson, P.; et al. Pre-clinical evaluation of small molecule LOXL2 inhibitors in breast cancer. Oncotarget 2017, 8, 26066–26078. [Google Scholar] [CrossRef]
- De Vita, A.; Liverani, C.; Molinaro, R.; Martinez, J.O.; Hartman, K.A.; Spadazzi, C.; Miserocchi, G.; Taraballi, F.; Evangelopoulos, M.; Pieri, F.; et al. Lysyl oxidase engineered lipid nanovesicles for the treatment of triple negative breast cancer. Sci. Rep. 2021, 11, 5107. [Google Scholar] [CrossRef]
- Nief, C.A.; Gonzales, A.; Chelales, E.; Agudogo, J.S.; Crouch, B.T.; Nair, S.K.; Ramanujam, N. Targeting Tumor Acidosis and Regulatory T Cells Unmasks Anti-Metastatic Potential of Local Tumor Ablation in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 8479. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Cao, X.; Cai, H.; Feng, P.; Chen, X.; Zhu, Y.; Yang, Y.; An, W.; Yang, Y.; Jie, J. The Exosomes Derived from CAR-T Cell Efficiently Target Mesothelin and Reduce Triple-Negative Breast Cancer Growth. Cell. Immunol. 2021, 360, 104262. [Google Scholar] [CrossRef] [PubMed]


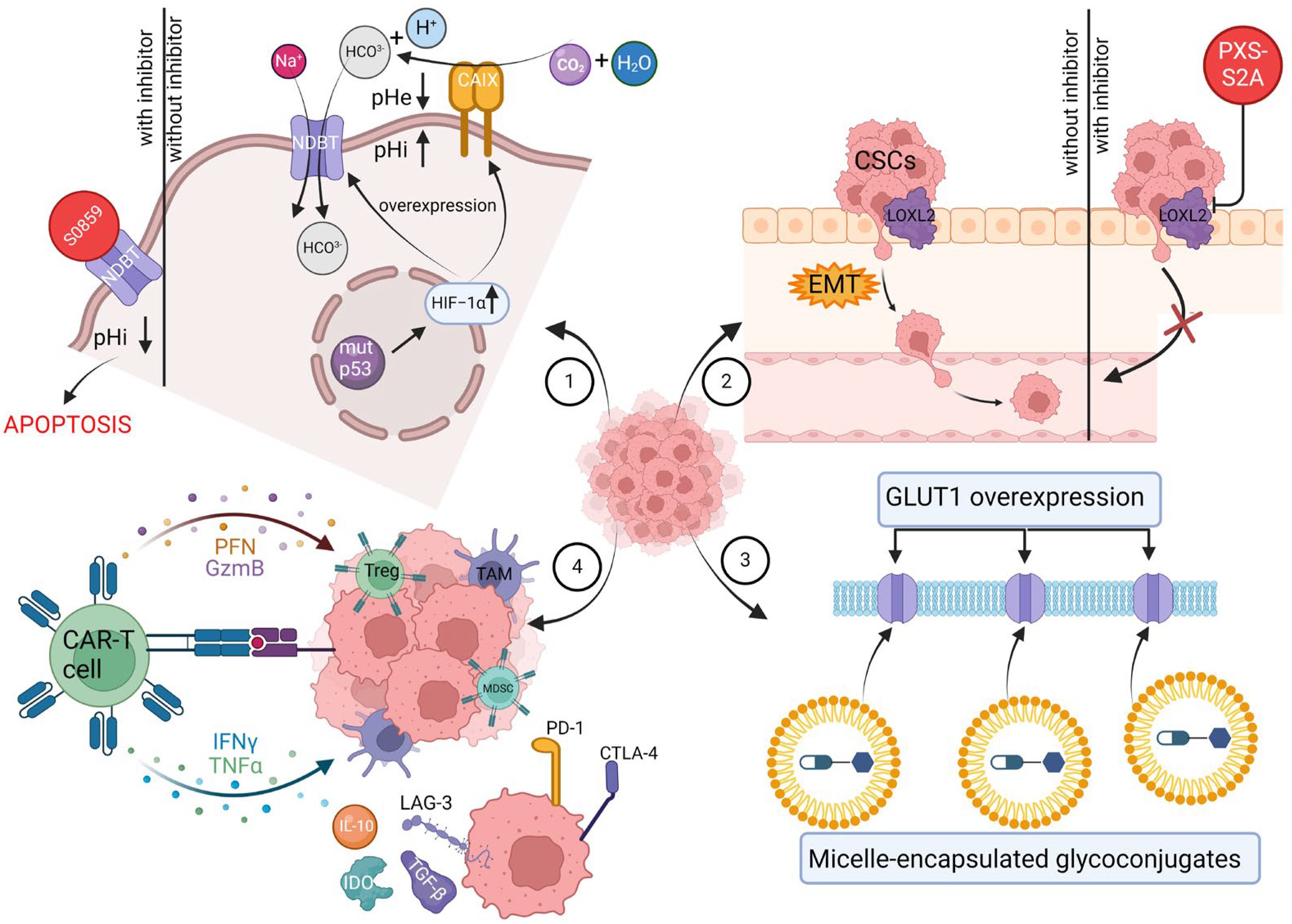{kind=link}
| Cancer Hallmark | Therapeutic Strategy | Model | Results | Reference |
|---|---|---|---|---|
| Deregulating cellular metabolism | Adriamycin-glycoconjugate targeting GLUT1 | In vitro MDA-MB-231 and MCF-7 cells | 2DG–SUC–ADM showed greater tumor-targeting ability and antitumor activity than free ADM. | [225] |
| Oxaliplatin-glycoconjugate targeting GLUT1 | In vitro MCF-7 cells | Fluro-substituted oxaliplatin-glycoconjugate exhibited the highest water-solubility among the three tested complexes. The test compound demonstrated a cytotoxicity level towards MCF7 cell lines that was twice as potent as the established clinical drug oxaliplatin. | [226] | |
| Methotrexate-glycoconjugate targeting GLUT1 | In vitro MCF-7 cells; in vivo 4T1 breast cancer mice | GLU–MTX was approximately 17-fold more preferentially accumulated in cancer cells compared to free MTX. GLU–MTX showed less cytotoxicity to healthy cells than MTX. GLU–MTX resulted in a substantial reduction of 74.4% in the growth of 4T1 allograft tumors on day 18, whereas MTX alone led to a much smaller inhibition of tumor growth, approximately 16.2%. | [227] | |
| Doxorubicin and 8-hydroxyquinoline glycoconjugate loaded into micelles | In vitro MCF-7 cells | Encapsulating drugs into micelles enhanced metabolic stability, improved tumor therapy selectivity, inhibited cancer cell proliferation more effectively, and induced apoptosis. | [228] | |
| Rg3 liposomes interacting with GLUT-1 | In vitro MCF-7 cells | The findings indicated that Rg3 improved the efficiency of liposome uptake and penetration into MCF-7 spheroids. | [229] | |
| Resisting cell death | Inhibiting NDBTs | In vitro MDA-MB-231 cells | NDBT inhibitor-S0859-reduced spheroid growth in MDA-MB231 cell lines by 48%. S0859 increased apoptosis in the core of MDA-MB-231 spheroids. | [230] |
| Activating invasion and metastasis | NDBT knockdown | In vitro MDA-MB-231 cells | NDBT knockdown significantly reduced pHi in MDA-MB-231 in normoxia and hypoxia, reducing spheroid growth. NDBT knockdown reduced both invasion and migration in normoxia and hypoxia. | [231] |
| LOXL2 inhibition | In vitro MDA-MB-231 cells; in vivo orthotopic MDA-MB-231 human breast cancers in mice | Both the dual inhibitor PXS-S1A and the LOXL2-specific inhibitor PXS-S2A suppressed cellular proliferation in a dose-dependent manner over an 8-day period in vitro. PXS-S1A exhibited an approximately 75% reduction in primary tumor volume, while PXS-S2B demonstrated a decrease of around 55% in tumor volume in vivo. | [232] | |
| Lipid nanovesicles targeting LOX | In vitro MDA-MB-231 cells; in vivo orthotopic MDA-MB-231 human breast cancers in mice | The delivery of EPI was significantly enhanced at both time intervals (6 and 48 h) when it was encapsulated within Lipo-EPI-LOX particles compared to free EPI and Lipo-EPI in vitro. Lipo-EPI-LOX displayed a substantial reduction in tumor growth compared to Lipo-EPI and free EPI in MDA-MB-231 murine xenografts over a 33-day period. Results demonstrated that Lipo-EPI-LOX treatment was the best tolerated therapy. | [233] | |
| Avoiding immune destruction | “BiCyclA” | In vivo 67NR, 4T1-Luc, E0771 breast cancer mice | While BiCyclA effectively cured most mice with 4T1-Luc, 67NR, and E0771 tumors, only mice bearing E0771 tumors fully resisted tumor rechallenge. | [234] |
| CAR-T cell-derived exosomes | In vitro MDA-MB-231 cells; in vivo orthotopic MDA-MB-231 human breast cancers in mice | The anti-MSLN CAR-T-cell exosomes demonstrated notably greater efficacy in killing cells in vitro compared to control exosomes when targeting MDA231-MSLN cell lines. ELISA results suggested that the anti-MSLN CAR-T-cell exosomes contained effector molecules, such as perforin and granzyme B, capable of directly targeting and killing TNBC cells. The results indicated that anti-MSLN CAR-T-cell exosome treatment effectively inhibited tumor growth in MDA231-MSLN xenograft models without causing toxicity at any dosage. | [235] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szulc, A.; Woźniak, M. Targeting Pivotal Hallmarks of Cancer for Enhanced Therapeutic Strategies in Triple-Negative Breast Cancer Treatment—In Vitro, In Vivo and Clinical Trials Literature Review. Cancers 2024, 16, 1483. https://doi.org/10.3390/cancers16081483
Szulc A, Woźniak M. Targeting Pivotal Hallmarks of Cancer for Enhanced Therapeutic Strategies in Triple-Negative Breast Cancer Treatment—In Vitro, In Vivo and Clinical Trials Literature Review. Cancers. 2024; 16(8):1483. https://doi.org/10.3390/cancers16081483
Chicago/Turabian StyleSzulc, Anna, and Marta Woźniak. 2024. "Targeting Pivotal Hallmarks of Cancer for Enhanced Therapeutic Strategies in Triple-Negative Breast Cancer Treatment—In Vitro, In Vivo and Clinical Trials Literature Review" Cancers 16, no. 8: 1483. https://doi.org/10.3390/cancers16081483
APA StyleSzulc, A., & Woźniak, M. (2024). Targeting Pivotal Hallmarks of Cancer for Enhanced Therapeutic Strategies in Triple-Negative Breast Cancer Treatment—In Vitro, In Vivo and Clinical Trials Literature Review. Cancers, 16(8), 1483. https://doi.org/10.3390/cancers16081483