Abstract
In this work, molybdena-promoted Li/MgO is studied as a catalyst for the oxidative conversion of n-hexane. The structure of the catalysts is investigated with X-ray Diffraction (XRD) and Raman spectroscopy. The MoO3/Li/MgO catalyst contains three types of molybdena-containing species, the presence of which depend on molybdena loading. At low Mo/Li ratios (i) isolated dispersed [MoO4]2− anionic species are observed. At high Mo/Li ratios, the formation of crystalline lithium molybdate phases such as (ii) monomeric Li2MoO4 and tentatively (iii) polymeric Li2Mo4O13 are concluded. The presence of these lithium molybdates diminishes the formation of Li2CO3 in the catalyst. Subsequently, the catalyst maintains high surface area and stability with time-on-stream during oxidative conversion. Molybdena loading as low as 0.5 wt % is sufficient to induce these improvements, maintaining the non-redox characteristics of the catalyst, whereas higher loadings enhance deep oxidation and oxidative dehydrogenation reactions. Promoting a Li/MgO catalyst with 0.5 wt % MoO3 is thus efficient for selective conversion of n-hexane to alkenes, giving alkene yield up to 24% as well as good stability.
1. Introduction
Catalytic oxidative conversion of alkanes to alkenes has gained interest over the years for on-purpose alkene production. The main challenge is that alkenes are highly reactive and susceptible to further oxidation in the presence of gaseous oxygen via adsorption on the catalyst surface. Therefore, to maintain high alkene yields in oxidative conditions, the use of basic catalysts is essential.
Li/MgO is a promising catalyst for the oxidative conversion (dehydrogenation/cracking) of lower alkanes to alkenes [1,2,3,4,5,6,7,8,9,10,11,12]. This catalyst has no formal redox character, i.e., Li+ and Mg2+ are not susceptible to oxidation state changes during the above reactions, and together with its inherent strong Brønsted basicity, it minimizes the re-adsorption and sequential combustion of formed alkenes [1,2,3,4,5,6,7,8]. Thus, the catalyst results in high selectivity to alkenes, which is highly desirable in the oxidative conversion of alkanes. This makes Li/MgO a better catalyst for oxidative reactions compared to acidic or redox-type catalysts, such as alumina or vanadia. It has been established through the work of Lunsford [13,14,15,16,17,18] on the oxidative coupling of methane that [Li+O−]-type defect sites are responsible for catalytic activity. The nucleophilic [O−] site is a strong hydrogen abstractor and initiates alkane activation via homolytic scission of the C-H bond in the alkane forming a radical. However, contradicting this, more recently Schlögl and co-workers [19,20] concluded, supported by quantum chemical calculations, that [Li+O−] is not the active site. Calculations on cluster models illustrated that both Li/MgO and MgO possess the same nature of active sites; i.e., low coordinated Mg2+O2− sites (Mg2+LCO2−LC) at steps and corners [19,20,21,22]. Thus, promotion with lithium does not introduce new active sites, but enhances the concentration of defect sites in MgO. In agreement, previous work from our lab suggests that lithium cations (Li+) and oxygen vacancies tend to segregate at steps and corners at the MgO surface, increasing the number of low coordinated Mg2+O2− sites (Mg2+LCO2−LC), hence enhancing catalyst activity and selectivity [4,19,20,23]. The mechanism for oxidative conversion reaction over Li/MgO is analogous to that of the oxidative coupling of methane suggested by Schlögl and co-workers; i.e., heterolytic addition of the C–H bond of the alkane on the Mg2+O2− pair in MgO, leading to a surface OH group and an alkyl radical [19,20]. The formed radical then undergoes a complex set of reactions in the gas phase in the presence of oxygen forming alkenes and alkanes, as well as combustion products, like H2O and COx [1,2,3,4,5,6,7,8,9,10,11,24].
Recently, we reported [6,7] on the oxidative cracking of n-hexane over the Li/MgO catalyst. Our goal was to achieve higher selectivity to alkenes and lower combustion as compared to redox catalysts, which were previously attempted for the oxidative conversion of alkanes [25]. Indeed, Li/MgO resulted in lower combustion selectivity compared to catalysts containing oxides of facile redox properties, e.g., V2O5/MgO [25]. The low oxidation activity of the Li/MgO catalyst resulted in lower n-hexane conversions at the typical reaction temperatures (500–600 °C) studied [6]. Furthermore, Li/MgO catalysts deactivated during time-on-stream, which was attributed to the reaction of CO2 with [Li+O−] catalytic sites and Li2CO3 formation [3,18,26]. Lunsford and co-workers confirmed that the presence of CO2 increases the activation energy for methyl radical generation during the oxidative coupling of methane [18]. The same effect was reported by our group for the oxidative cracking of propane [2,3]. The product CO2 interacts with [Li+O−] sites, forming an intermediate Li+CO3−, which via further reaction with another [Li+O−] site is converted into the more stable Li2CO3 [18].
In order to improve both the activity and stability of Li/MgO, we promoted the catalyst with low amounts of oxides with redox properties [6]. Our goal was to add slight redox activity to the catalyst to enhance C–H bond scission and eventually n-hexane conversions. Of the different transition metal oxides attempted, molybdena-promoted Li/MgO showed the best alkene yields. Significantly, the presence of molybdena also prevented deactivation and catalyst stability was restored [6,7].
Supported molybdena catalysts have been often studied for the oxidative dehydrogenation of light alkanes, e.g., ethane, propane, and butane [27,28,29,30,31,32,33,34,35,36,37,38]. It has been suggested [31,32] that oxidation of the C–H bond in the alkane proceeds via a Mars-van-Krevelen redox mechanism with the participation of molybdena lattice oxygen, followed by re-oxidation with gas-phase oxygen.
Various molybdenum oxide systems have been reported, e.g., supported on MgO, ZrO2, Al2O3, TiO2, and SiO2 [32,35,36,39,40,41,42,43]. The presence of molybdena influences the physiochemical properties of the oxide support. In general, molybdena content, state of molybdena species on the surface of the support, and calcination temperature influence both the textural and acidic properties of the support. For example, in alumina-supported molybdena, in addition to Lewis acid sites, Brønsted acid sites are detected, the concentration of which depends on molybdena loading and calcination temperature [44]. In zirconia-supported molybdena, the increase in acidity with an increase in molybdena loading is correlated to the formation of Mo–O–Zr surface species as precursors for crystalline Zr(MoO4)2 [45]. Moreover, molybdena enhances the surface area of zirconia through stabilizing the tetragonal zirconia phase [46,47].
Generally, the performance of molybdena-based catalysts is related to the extent of crystallinity and chemical structure of the oxidic molybdena species on the support, e.g., free MoO3, monomeric MoO42− or polymeric Mo6O192−, Mo7O24−6 units [32,35,41]. Hence, structure-performance correlations for supported molybdena catalysts have been of continuous interest. Magnesium oxide-supported molybdena (MoO3/MgO), in particular, is reported to be an efficient and selective catalyst for the oxidative dehydrogenation of C3–C4 alkanes to the corresponding alkenes [33,34]. The activity of the MoO3/MgO for alkane activation depends on the molybdena loading [37,38]. In the oxidative dehydrogenation of propane over a MgMoOx catalyst, for example, Yoon et al. [37,38] reported that optimal molybdena loadings are necessary to maintain high propane conversions. Mg0.95MoOx crystallites with a slight excess of molybdenum (Mg/Mo = 0.9–1.0) showed the highest activity for propane conversion (22% conversion at 515 °C). Above these optimal loadings oxidation activity became significant. The high oxidation activity in these catalysts is due to the facile redox properties of molybdena, where the cation easily undergoes a change in the oxidation state (e.g., Mo6+ to Mo4+) [25].
In relation to the chemical structure of the molybdates on magnesium oxide, extensive literature has been published on the topic [39,40,41,42,43]. Bare and co-workers [41,42] reported that, in the case of MoO3/MgO catalysts, the structure of molybdena species depends on the molybdenum coverage of the support. For sub-monolayer coverages, dispersed species are observed. These consist of highly distorted octahedral molybdena species, e.g., MoO6, at low molybdenum loadings and regular octahedrally-coordinated polymolybdate species, e.g., [Mo7O24]6−, at high molybdenum loadings [41]. For coverages exceeding the monolayer, crystalline magnesium molybdate (MgMoO4), in which molybdenum is tetrahedrally coordinated, is observed as the dominant species. Raman spectroscopic studies [39,40] showed that for magnesium oxide-supported catalysts, surface molybdena species are sensitive to hydration. Upon exposure to water, octahedrally-coordinated molybdena species transform to tetrahedrally-coordinated MoO42− species.
In this work, we present a detailed study on the role of molybdena in improving the performance of Li/MgO. A detailed characterization of the MoO3/Li/MgO catalysts is presented in order to identify the chemical structure of the different molybdena species (MoOx) present on surface of Li/MgO. Our objective is to determine the influence of different molybdena species on (i) n-hexane conversion, (ii) alkene versus combustion selectivity, and (iii) catalyst stability. This is expected to help establish guidelines for developing an optimal catalyst for the oxidative conversion of n-hexane.
2. Results
2.1. Catalytic Tests
Figure 1 presents n-hexane conversions within 6 h of reaction time-on-stream. The Li/MgO catalyst suffered fast deactivation during the first hour of reaction. The 0.5MoO3/Li/MgO catalyst resulted in similar initial n-hexane conversions to Li/MgO, however it exhibited better stability, hence significantly higher hexane conversions after 6 h time-on-stream. Both the 3.6MoO3/Li/MgO and 7.1MoO3/Li/MgO catalysts were very stable, but exhibited lower initial n-hexane conversions than the unpromoted Li/MgO.
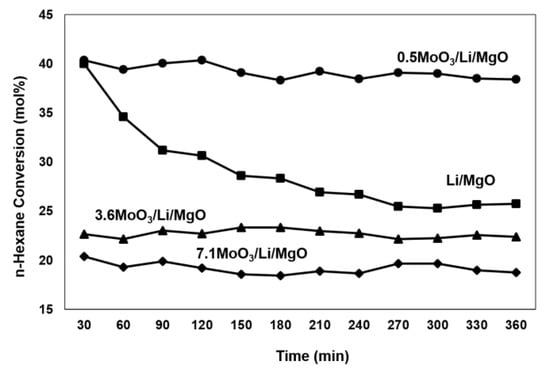
Figure 1.
n-Hexane conversion versus time. Reaction conditions: 100 mL/min, 10% n-hexane, 8% oxygen and balance helium, T = 575 °C. WHSV = 154 h−1.
More specifically, Figure 2 presents n-hexane conversions as a function of Mo/Li ratio after 6 h of time-on-stream. The catalyst promoted with 0.5wt % molybdena (Mo/Li = 0.03) resulted in the highest activity, presenting significant improvement in activity compared to undoped Li/MgO.
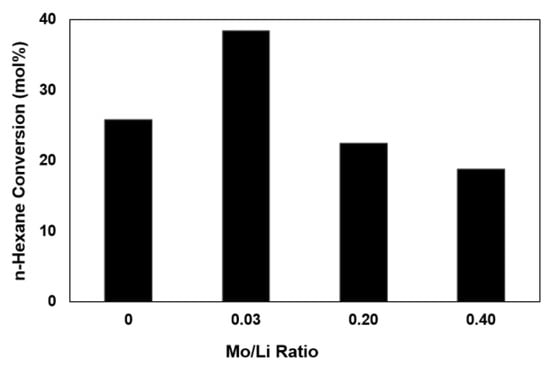
Figure 2.
n-Hexane conversion versus Mo/Li after 6 h of time-on-stream. Reaction conditions: 100 mL/min, 10% n-hexane, 8% oxygen and balance helium, T = 575 °C. WHSV = 154 h−1.
The influence of varying loadings of molybdena on the performance of Li/MgO during the oxidative conversion of n-hexane at 575 °C is shown in Table 1. Initial conversions and corresponding product selectivities after 30 min time-on-stream are reported. It is important to note that product selectivities remained constant with time. 0.5MoO3/Li/MgO showed similar conversion and selectivity to products as Li/MgO, however catalysts with higher molybdena loadings resulted in lower conversion and more combustion products.

Table 1.
Performance of various MoO3/Li/MgO catalysts for the oxidative conversion of n-hexane. Reaction conditions: 100 mL/min, 10% n-hexane, 8% oxygen and balance helium, T = 575 °C. WHSV = 154 h−1.
Moreover, in Figure 3 we compare selectivities to products at the same n-hexane conversion of ca. 10 mol %. The same n-hexane conversions were achieved through changing contact time with the catalyst. The 0.5MoO3/Li/MgO catalyst showed a similar selectivity pattern to that of Li/MgO, while both the 3.6MoO3/Li/MgO and 7.1MoO3/Li/MgO catalysts resulted in the formation of more combustion products. Significant formation of C6 alkene (hexene) was observed in the case of both the 3.6MoO3/Li/MgO and 7.1MoO3/Li/MgO catalysts. Oxygen conversion was almost same (~35 mol %) over both the Li/MgO and 0.5MoO3/Li/MgO catalysts, while it increased to 43 mol % and 53 mol % over 3.6MoO3/Li/MgO and 7.1MoO3/Li/MgO, respectively.
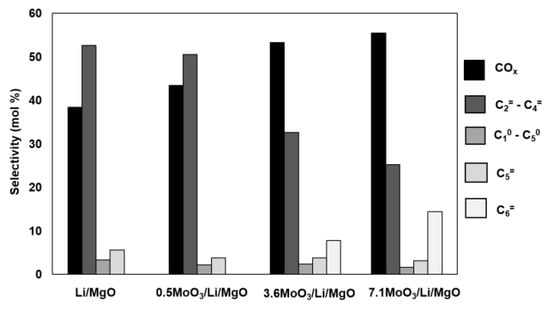
Figure 3.
Selectivity to products based on carbon at an n-hexane conversion of 10 mol %. Oxygen conversions = 35 mol % (Li/MgO), 36 mol % (0.5MoO3/Li/MgO), 43 mol % (3.6MoO3/Li/MgO), and 53 mol % (7.1MoO3/Li/MgO). Reaction conditions: 100 mL/min total flow, 10% n-hexane, 8% oxygen and balance helium, T = 575 °C. WHSV = 154–385 h−1.
Figure 4 shows the selectivities to C2–C4 alkenes as a function of n-hexane conversion. These results clearly indicate that the 0.5MoO3/Li/MgO catalyst preserved the characteristics of the unpromoted catalyst and maintained the high alkene selectivity, even with increasing n-hexane conversion. However, both the 3.6MoO3/Li/MgO and 7.1MoO3/Li/MgO catalysts exhibited typical performance of redox catalysts, i.e., decreasing selectivity to alkenes with increasing conversion.
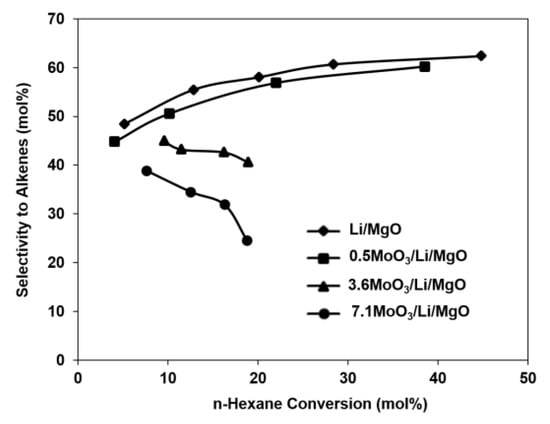
Figure 4.
Selectivity to C2–C4 alkenes as function of n-hexane conversion. Reaction conditions: 100 mL/min, 10% n-hexane, 8% oxygen and balance helium, T = 575 °C, WHSV = 5–154 h−1.
2.2. Surface Area and XRD
The characteristics of the studied catalysts are presented in Table 2. It was observed that Li/MgO suffered a dramatic decrease in surface area upon calcination at 600 °C (15 m2/g), while in the presence of molybdena, high surface areas (70–82 m2/g) were retained. In the case of MgO, the loss in surface area upon calcination at 600 °C was less severe. Nevertheless, promotion with ≥3.3 wt % of MoO3 resulted in even higher surface areas than the unpromoted sample calcined at 600 °C (148 m2/g). The MoOx surface coverage (θ) of molybdena-promoted catalysts shown in Table 2 were calculated based on the surface area of each catalyst and using 22 Å2 as the mean surface area occupied by one Mo6+ oxide unit (MoO3) [5]. Values indicated sub-monolayer coverage for all samples studied.
Table 2.
Characteristics of the catalysts.
Figure 5 presents the XRD patterns of the catalysts. All catalysts exhibited characteristic peaks for crystalline MgO. No XRD peaks corresponding to crystalline MgMoO4 or MgMo2O7 phases were observed, which would be expected at 2theta values between 20–300 [38]. The absence of such peaks confirms that molybdena phases were finely dispersed and no crystalline phases were observed. Our results are in good agreement with literature findings; in supported molybdenum oxide systems with sub-monolayer coverages the presence of dispersed molybdena anionic species such as MoO42-, Mo6O192−, and Mo7O246− are commonly reported [32,35,36,41].
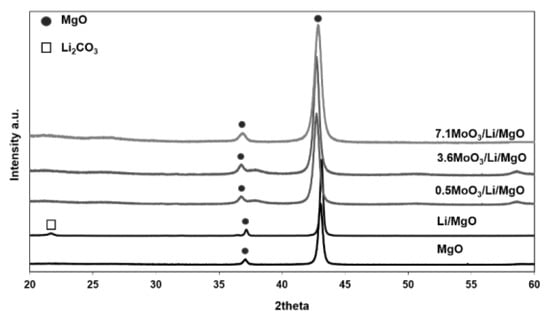
Figure 5.
XRD patterns of Li/MgO and MoO3/Li/MgO catalysts.
In addition, no peaks corresponding to any crystalline Li phases, e.g., Li2CO3, were observed in any molybdena-promoted catalysts. The observed shift in the MgO peak positions in the MoOx-containing samples indicates the formation of solid solutions between MoOx and Li/MgO. The observed shift was stronger in the 0.5MoO3/Li/MgO and 3.6MoO3/Li/MgO samples than in 7.1MoO3/Li/MgO, suggesting that the formation of lithium molybdates is dominant in the latter. This was further investigated by characterization of the catalysts with Raman spectroscopy.
Figure 6 presents the XRD pattern of an equimolar mixture of Li2CO3 and (NH4)2MoO4 calcined at conditions similar to that during catalyst preparation (600 °C for 5 h). In the same figure, results are compared to the XRD measurements of the Li2CO3, Li2MoO4, and Li2Mo4O13 reference compounds and show the characteristic peaks of Li2MoO4 and Li2Mo4O13.
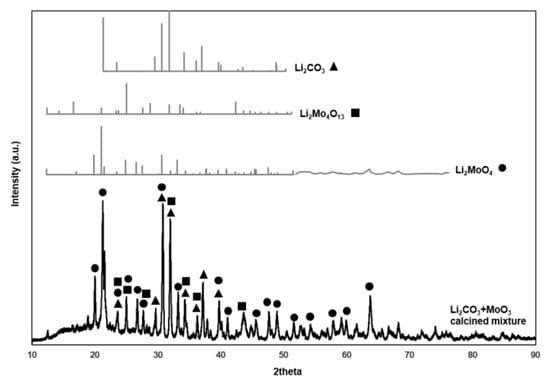
Figure 6.
XRD pattern of an equimolar mixture of Li2CO3 and (NH4)2MoO4 calcined at 600 °C, in comparison with the XRD of the Li2CO3, Li2MoO4, and Li2Mo4O13 reference compounds.
2.3. Temperature Programmed Desorption (TPD)
Figure 7 presents results of TPD experiments for both fresh (a) and used (b) catalysts. The TPD of Li/MgO exhibited a broad CO2 desorption peak with a maximum at 860 °C, which is typical for Li2CO3 [18]. The presence of Li2CO3 (formed with CO2 from ambient) is an inherent property of Li/MgO [4,26]. The TPD of the molybdena-promoted catalysts exhibited similar Li2CO3 peaks, however with lower intensity, decreasing with increasing molybdena loading. This confirms the gradual decrease in amounts of Li2CO3 in Li/MgO with increasing molybdena loading.
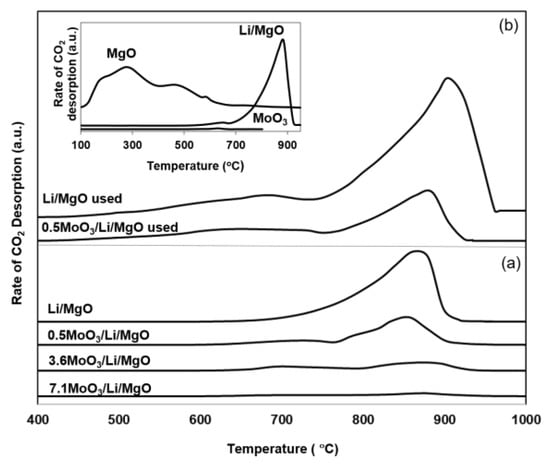
Figure 7.
Temperature programmed desorption for (a) fresh Li/MgO and MoO3/Li/MgO catalysts pretreated at 600 °C and (b) used Li/MgO and 0.5MoO3/Li/MgO catalysts (signals are normalized to the BET surface area). For comparison TPD of MgO, fresh Li/MgO and MoO3 are presented.
Similarly, the CO2 desorption peak observed for the used 0.5MoO3/Li/MgO is significantly smaller than that of used Li/MgO. The broad desorption peak observed between 600 and 700 °C in the used catalysts is attributed to the desorption of weakly adsorbed CO2 on [Li+O−] sites forming [Li+CO3−], as illustrated earlier by Lunsford [18].
2.4. Raman Spectra
All MoO3/Li/MgO catalysts were characterized by Raman spectroscopy (Figure 8b). For better assignment of Raman bands, MgO promoted with comparable molybdena loadings was prepared and characterized (Figure 8a). At first, we assign the Raman bands of MoO3/MgO. Figure 8a presents the Raman spectra of MoO3/MgO samples. The Raman band at 800 cm−1 was characteristic in all samples and also appeared in the Raman spectra of Li/MgO in (Figure 8b). This band was not observed in any of the Raman spectra of MgO reported in literature [48]. Thus, the presence of such a band in our catalysts should relate to the sol–gel synthesized MgO support. This band was used as an internal standard when comparing intensities of Raman bands corresponding to the presence of molybdena species.
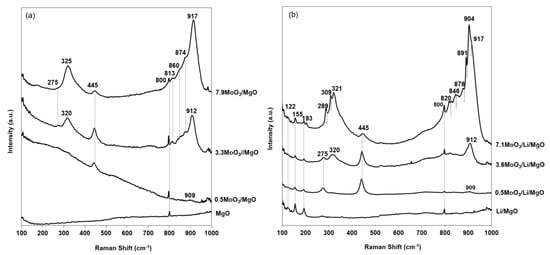
Figure 8.
Raman spectra of (a) molybdenum oxide supported on MgO and (b) molybdenum oxide supported on Li/MgO as function of loading of molybdena.
Bare et al. studied the surface chemistry of magnesium oxide-supported molybdenum oxide [41]. They reported that the structure of the molybdate species on MgO support after calcination at 600 °C in dry air depends on the molybdenum loadings. Below 3.3 wt %, isolated distorted octahedrally-coordinated molybdate species are present, which upon exposure to water saturated air transform to tetrahedrally-coordinated species. Above 6.7 wt % molybdenum, octahedrally-coordinated polymolybdate species are formed. Only above 20 wt % molybdenum is bulk magnesium molybdate formed from the reaction of MoO3 with MgO.
The Raman spectra of our MoO3/MgO catalysts show close resemblance to those of MoO3/MgO calcined at 600 °C and exposed to water-saturated air, reported by Bare et al. (see Table 3) [41]. The Raman band 445 cm−1 appearing in all MoO3/MgO samples is a characteristic for the lattice vibration and Mg–O stretching vibration in Mg(OH)2 [41]. The 0.5MoO3/MgO exhibited a Raman band at 908 cm−1, which, with increasing the molybdena amount, increased in intensity and shifted to 912 cm−1and 917 cm−1 for 3.3MoO3/MgO and 7.9MoO3/MgO, respectively. Thus, in agreement with Bare and co-workers [41], we assigned the Raman bands at 912 cm−1, 874 cm−1, and 320 cm−1 in the 3.3MoO3/MgO and the bands at 917 cm−1, 874 cm−1, and 325 cm−1 in the 7.9MoO3/MgO to the symmetric, antisymmetric stretching, and bending modes, respectively, of terminal Mo–Ot in tetrahedrally-coordinated isolated MoO42− species. These assignments are summarized in Table 3.
Table 3.
Raman bands of MoO3/MgO catalysts.
Usually in MoO3/MgO systems, a Raman band at 220 cm−1 is characteristic of heptamolybdates [41,49]. The absence of this band in all the spectra (Figure 8a,b) indicates the absence of well-defined polymolybdate species in our systems. In the Raman spectra of 4.5–8.9 wt % molybdenum supported on MgO, calcined at 600 °C in dry air, Bare and co-workers [41] attributed Raman bands at 807 cm−1 and 860 cm−1 to Mo–O–Mg vibrations. The Raman band in our case at 813 cm−1 and the shoulder at 860 cm−1 in both 3.3MoO3/MgO and 7.9MoO3/MgO (Figure 8a) is therefore probably due to Mo–O–Mg vibration of Mo–O–Mg surface species.
Now, we study the Raman spectra of the molybdena-promoted Li/MgO catalysts (Figure 8b). The Raman bands 122 cm−1, 155 cm−1, 193 cm−1 appearing in Li/MgO are characteristic for lattice vibrations in Li2CO3 [50,51]. Raman spectra of the molybdenum-containing catalysts showed significantly lower amounts of Li2CO3. These results are consistent with results obtained via temperature-programmed desorption of CO2 (Figure 7a), and confirm the role of molybdena in lowering the amount of Li2CO3 in Li/MgO.
Upon the promotion of Li/MgO with 0.5 wt % MoO3, Raman bands at 275 cm−1, 445 cm−1, and 909 cm−1 were observed. The Raman bands at 275 cm−1 and 445 cm−1 correspond, similar to in MoO3/MgO, to the lattice vibration and Mg–O stretching vibration in Mg(OH)2, respectively [41]. Generally, the Raman spectra of MoO3/Li/MgO catalysts showed close resemblance to those of the MoO3/MgO samples. The 0.5MoO3/Li/MgO catalyst exhibited a very weak Raman band at 909 cm−1, the latter increased in intensity and shifted to 912 cm−1 and 917cm−1 (shoulder) for the 3.3MoO3/MgO and 7.9MoO3/MgO catalysts, respectively. Therefore, similar to in MoO3/MgO, we assigned the Raman band 909cm−1 in the 0.5MoO3/Li/MgO catalyst, the bands at 320 cm−1 and 920cm−1 in the 3.6MoO3/Li/MgO catalyst and the 321cm−1, 878cm−1, and 917cm−1 shoulder bands in the 7.1MoO3/Li/MgO catalyst to the stretching modes of terminal Mo–Ot in tetrahedrally-coordinated hydrated MoO42− species. No free MoO3 phases were detected in any MoO3/Li/MgO catalyst.
The catalyst with the highest MoO3 loading, 7.1MoO3/Li/MgO, however, showed additional Raman bands at 289 cm−1, 309 cm−1, 820 cm−1, 846 cm−1, 878 cm−1, 891 cm−1, and 904 cm−1 (Figure 8b). These were absent in the corresponding 7.9MoO3/MgO catalyst. To investigate the nature of these phases we subtracted the Raman spectrum of the 7.9MoO3/MgO catalyst from the corresponding spectrum of the 7.3MoO3/Li/MgO catalyst. Figure 9 shows the difference spectrum. The majority of the bands appearing in the difference spectrum of the 7.1MoO3/Li/MgO catalyst (309 cm−1, 321 cm−1, 358 cm−1, 820 cm−1, 846 cm−1, 878 cm−1, 904 cm−1) match those of Li2MoO4. The bands at 904 cm−1, 820–878 cm−1, and 309–358 cm−1 correspond to the symmetric, antisymmetric stretching, and bending modes of the terminal Mo–Ot bond in Li2MoO4, respectively [52]. Thus, in the case of the 7.1MoO3/Li/MgO catalyst, part of the molybdenum is present as tetrahedrally-coordinated in a Li2MoO4 phase.
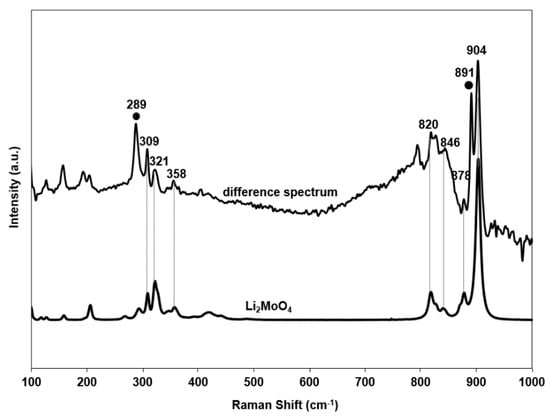
Figure 9.
Raman spectrum of Li2MoO4 and the subtracted spectrum of 7.1MoO3/Li/MgO–7.9MoO3/MgO.
Additionally, the difference spectrum shows two sharp Raman bands at 289 cm−1 and 891 cm−1. The quasi-binary phase diagram of Li2O and MoO3 reported in literature [53] shows that in MoO3-rich conditions (MoO3/Li2O > 0.5) at T > 600 °C, the formation of Li2Mo4O13 is favored. We also observed the formation of such species by XRD (Figure 6). We tentatively assign the Raman bands at 289 cm−1 and 891 cm−1 to the bending and symmetric stretching modes of the terminal Mo–Ot bond in a well-defined Li2Mo4O13 phase, while other Raman bands expected for Li2Mo4O13 (160–420 cm−1, 420–746 cm−1, and 845–991 cm−1) [54] were not clearly detected. We hence suggest in catalysts with 7.1 wt % MoO3 the possible presence of Li2Mo4O13 polymolybdate species. Li2Mo4O13 is reported in literature [55] to have a regular derivative structure of V6O13, in which Mo is octahedrally-coordinated. Table 4 summarizes the Raman bands and corresponding molybdena phases in molybdena-promoted Li/MgO.
Table 4.
Raman bands of MoO3/Li/MgO catalysts.
In conclusion, the molybdena-promoted Li/MgO catalyst contains three types of molybdate species, the formation of which depends on the molybdena loading. At low Mo/Li ratios, in both the 0.5MoO3/Li/MgO and 3.6MoO3/Li/MgO catalysts, isolated dispersed [MoO4]2− anionic species were detected. At high Mo/Li ratios, in the 7.1MoO3/Li/MgO catalyst, the formation of crystalline lithium molybdate phases such as (i) monomeric Li2MoO4, and tentatively, (ii) polymeric Li2Mo4O13 were detected. These lithium molybdate phases were present at low concentrations, below the detection limit of XRD and their formation became significant with increasing Mo/Li atomic ratio.
3. Discussion
The results presented above indicate that the promotion of Li/MgO with sub monolayer coverages of molybdena introduces structural changes, which influence both physical properties and the performance of the catalyst in the oxidative conversion of n-hexane. XRD and Raman spectra confirmed the presence of well-dispersed molybdena species in addition to lithium molybdates at high Mo/Li ratios, influencing catalyst performance, as discussed below.
3.1. Textural Properties and Stability of the Catalyst
The presence of Li2CO3, as confirmed by TPD results (Figure 7a) and previously reported [2,3,18], is an inherent property of Li/MgO. Trionfetti et al. [4] reported that only ~40% of the lithium incorporates into the MgO lattice as [Li+O−] sites during the sol–gel preparation of Li/MgO, whereas the rest stays as Li2O which forms Li2CO3 via reaction with ambient CO2. The presence of Li2CO3 leads to detrimental changes in surface area upon exposure of the catalyst to high temperatures, as observed from the BET results in Table 2. Molybdena-promoted Li/MgO catalysts, however, exhibit less presence of Li2CO3, clearly observed from the TPD and Raman results, and consequently maintain a higher surface area even after exposure to high temperature treatment (Table 2). Hence, we conclude that the presence of molybdena reduces the amount of Li2CO3 in Li/MgO.
We suggest that during catalyst calcination at 600 °C, MoO3, formed as a result of the decomposition of ammonium molybdate, reacts with the Li2CO3 present in Li/MgO, forming lithium molybdate mixed oxide phases. Indeed, the XRD pattern (Figure 6) of an equimolar mixture of Li2CO3 and (NH4)2MoO4 calcined at similar conditions during catalyst preparation (600 °C for 5 h) resulted in characteristic peaks of Li2MoO4 and Li2Mo4O13. The absence of characteristic peaks of MoO3 confirms the reactivity of MoO3 with Li2CO3 in the catalyst, leading to the formation of lithium molybdate phases [47]. In our catalysts, these molybdates, as detected by Raman in Figure 8b, were present as dispersed species at low Mo/Li ratios (0.5MoO3/Li/MgO and 3.6MoO3/Li/MgO) and well-defined crystalline Li2MoO4 and tentatively Li2Mo4O13 at high Mo/Li ratios.
In addition to Li2CO3, inherently present in the catalyst, Li2CO3 is formed during oxidative reaction from the reaction of the product CO2 with [Li+O−] sites of Li/MgO. The formation of Li2CO3 in Li/MgO during the catalytic reaction was confirmed by the TPD of the used catalyst in Figure 7b. Li2CO3 sinters the catalyst at reaction temperatures [56], and consequently, this leads to degradation of defect sites and loss of catalytic activity.
The promotion of Li/MgO with molybdena improves the stability of the catalyst during oxidative conversion; all MoO3/Li/MgO catalysts maintained stability within 6 h of reaction time-on-stream (Figure 1). The stability of all molybdena-promoted catalysts is attributed to the presence of the various aforementioned molybdate phases, which increase the surface acidity of Li/MgO. This in turn plays a positive role in minimizing the extent of CO2 adsorption on [Li+O−] sites of Li/MgO and the subsequent formation of Li2CO3. The TPD of the used 0.5MoO3/Li/MgO catalyst (Figure 7b), for example, confirmed that the formation of Li2CO3 was clearly partly suppressed. With less Li2CO3 formation during catalytic reaction, the catalyst preserves its high surface area, and hence catalytic activity.
3.2. Activity and Selectivity
The 0.5MoO3/Li/MgO catalyst exhibited similar initial n-hexane conversion to Li/MgO, while catalysts with higher molybdena loading (3.6 and 7.1MoO3/Li/MgO) exhibited even lower n-hexane conversions than Li/MgO (Figure 1). Increase in molybdena loading led to a clear increase in selectivity to COx, accompanied with a significant decrease in selectivity to C2–C4 alkenes. In addition, a noticeable formation of hexene in the case of both 3.6 and 7.1 MoO3/Li/MgO catalysts was observed (Figure 3).
The product selectivity patterns observed over the molybdena-doped catalysts in Figure 3 suggest that three competing parallel reaction pathways exist: (1) surface-initiated gas-phase oxidative conversion of n-hexane, (2) oxidative dehydrogenation (ODH) of n-hexane, and (3) surface-enhanced deep oxidation.
Hereby, we briefly describe, based on previous literature findings, the mechanisms of the above-mentioned three reaction pathways and the significance of their occurrence at various Mo/Li ratios.
At low Mo/Li ratios, surface-initiated oxidative conversion of n-hexane was significant, leading to high selectivity to light alkenes. At these conditions dispersed molybena species were present, which did not contribute to catalytic activity. The conversion of n-hexane was initiated via heterolytic C–H bond scission and hexyl radical formation on the O2− of low coordinated Mg2+LCO2−LC sites, in analogy to the suggestion by Sclögl and co-workers [19,20]. It is well established in the oxidative conversion of alkanes over Li/MgO that alkane activation proceeds via surface radical initiation [2,15,16,17,18,19,20]. The alkyl radical formed would undergo a series of gas-phase reactions in the presence of molecular oxygen, leading to the observed product composition for Li/MgO and 0.5MoO3/Li/MgO catalysts in Figure 3.
Adversely, high Mo/Li ratios led to the formation of crystalline Li2MoO4 and Li2Mo4O13 phases which enhanced the oxidative dehydrogenation of n-hexane to hexene and deep oxidation reactions. The ODH reaction occurs through subsequent hydrogen abstraction from the n-hexane via O2− of MoOx [25,31,32]. This, in agreement with literature [38], was most significant in catalyst with 7.1 wt % MoO3, in which the presence of crystalline Li2MoO4 and Li2Mo4O13 were detected by Raman. In studies by Vrieland and Murchison [38] on the oxidative dehydrogenation of butane to butadiene over MoO3/MgO, maximum activity was attributed to presence of crystalline MgMoO4.
Deep oxidation reactions occur via adsorption and stabilization of intermediate carbocations/radicals on O2− of MoOx and their consequent combustion to COx in the presence of molecular oxygen. Both ODH and deep oxidation reactions explain the altered product composition observed for both 3.6MoO3/Li/MgO and 7.1MoO3/Li/MgO catalysts in Figure 3.
In the ODH of light alkanes, ethane and propane, it is well established that reactions on molybdena follow the Mars-van-Krevelen redox mechanism; i.e., molybdenum is reduced via loss of lattice oxygen and is then re-oxidized via molecular oxygen [25,31,32]. Consequently, in our catalysts with high molybdena loadings, excessive redox reactions resulted in complete consumption of molecular oxygen, hence limiting the n-hexane conversion route via molecular oxygen and inhibiting the extent of gas-phase radical chain reactions. Therefore, although C–H bond scission from n-hexane and ODH activity increased with the increase in molybdena loading, overall n-hexane conversions remained lower than in Li/MgO and 0.5MoO3/Li/MgO catalysts.
The performance of 0.5MoO3/Li/MgO mimicked that of Li/MgO, with similar initial activity as well as selectivity (Figure 4); moreover, the catalyst preserved activity with time-on-stream. The optimal Mo/Li ratio (Figure 2) is thus essential in order to, on one hand, maintain the non-redox nature of Li/MgO, and hence the desired high selectivity to alkenes (Figure 3 and Figure 4), and on the other hand, to stabilize the catalyst activity (Figure 1).
4. Materials and Methods
4.1. Materials
Mg(OCH3)2 (6–8 wt%) solution in methanol (Sigma-Aldrich, Darmstadt, Germany), methanol (Merck, Darmstadt, Germany), and LiNO3 (assay ≥ 99.99%, Sigma-Aldrich, Darmstadt, Germany) were used for preparation of MgO and Li/MgO catalysts. Ammonium molybdate (99.98%, Sigma-Aldrich, Darmstadt, Germany) was used as the MoO3 precursor. Pure n-hexane (GC assay ≥ 99.0%, Fluka, Honeywell, USA) was used for catalytic experiments. The reference compounds Li2MoO4 (assay ≥ 99.99%, Sigma-Aldrich, Darmstadt, Germany), Li2CO3 (assay ≥ 99.0%, Sigma-Aldrich, Darmstadt, Germany), MoO3 (assay 99.99%, Sigma-Aldrich, Darmstadt, Germany), and (NH4)6Mo7O24 (assay 99.98%, Sigma-Aldrich, Darmstadt, Germany) were used as received.
4.2. Catalyst Preparation
The MgO and Li/MgO catalysts used in this study were prepared according to the method described in detail earlier [4,7]. A methanol solution containing Mg(OCH3)2 (0.4 M) was mixed at room temperature with another methanol solution containing water (0.8 M) to form a sol. For Li/MgO, the required amount of LiNO3 was added to the solution to obtain ~1 wt % Li. The solution was allowed to stay for gelation for 24 h. The gel formed was dried at 50 °C in vacuum for 7 h, and calcined at 600 °C in a flowing air of 50 mL/min for 1 h with a heating rate of 5 °C/min. Modified MoO3/Li/MgO catalysts were prepared by wet impregnation of the sol–gel synthesized Li/MgO (calcined at 500 °C) using an aqueous solution of the ammonium molybdate. These were then dried at 50 °C in vacuum for 7 h and calcined at 600 °C in a flowing air of 50 mL/min for 5 h with a heating rate of 5 °C/min. Similarly, MoO3/MgO catalysts were prepared with wet impregnation of the sol–gel synthesized MgO. Molybdena-promoted samples are denoted as xMoO3/Li/MgO and xMoO3/MgO, where x is wt % of MoO3. Table 1 presents the list of the catalysts prepared.
4.3. Catalyst Characterization
The Brunauer–Emmett–Teller (BET) surface area of the catalyst was determined with nitrogen physisorption using a Micro-metrics Tristar instrument (Micro-metrics, USA).
X-ray Diffraction (XRD) patterns were recorded with a Philips PW 1830 diffractometer (Philips, Netherlands) using Cu Kα radiation, λ = 0.1544 nm. Spectra were registered in the 2θ range of 35 to 50 with step size of 0.01 and integration time of 1 s per step. Elemental composition of the catalysts was determined with atomic absorption spectroscopy (AAS). Li content in all the catalysts was 0.86 wt %. Mo loadings were determined with X-ray fluorescence spectroscopy (XRF), using Phillips PW 1480 spectrometer (Philips, Netherlands).
Temperature programmed desorption (TPD) experiments were performed to decompose the Li2CO3 inherently present in both fresh and used catalysts. A total of 100 mg of the catalyst was pretreated in O2/He at 600 °C for one hour to decompose any MgCO3 present. After cooling the catalyst to 100 °C in helium, TPD was conducted from 100 °C to 950 °C, with an increment of 10 °C/min, with helium flow of 10 mL/min as a carrier gas.
Raman spectral measurements were conducted with a SENTERRA instrument (Bruker Optics, Netherlands) equipped with a cooled charge-coupled device (CCD) detector (−60 °C). The samples were excited with a 785 nm red laser of 100 mW power. Spectra were recorded at room temperature from 100 to 1000 cm−1, at a resolution of 3 cm−1 and a 5 min integration time.
4.4. Catalytic Tests
The catalytic tests were carried out at atmospheric pressure and isothermal conditions in a fixed-bed reactor [6]. An alumina tube reactor of 4 mm internal diameter was used. Powder catalyst was pressed, crushed, and sieved to particle size of 0.4–0.6 mm before use. A total of 10–100 mg of catalyst sample (to obtain varying contact times) was diluted in quartz particles and placed in the isothermal region of the furnace (1 cm). An alumina rod of 3 mm internal diameter was placed right below the catalytic bed to reduce the post catalytic volume in order to minimize homogenous gas-phase reactions. A Chromel-Alumel thermocouple inside a quartz tube was inserted above the catalytic bed to record reaction temperature. The temperature of the furnace was controlled by a second thermocouple placed outside the reactor tube within the isothermal zone of the tubular furnace.
Reactions were studied at 575 °C. Feed (100 mL/min) consisted of 10 mol % of n-hexane vapor, 8 mol % of oxygen, and balance helium. Similar n-hexane conversions were achieved by varying the weight hourly space velocity (WHSV). Before each catalytic test, the catalysts were pretreated at 625 °C in 50% O2/He (60 mL/min) for 1 h. For analysis of the product, samples of outlet gas stream were injected into two micro gas chromatographies (micro GCs) (Varian CP4900, Netherlands) every 30 min during a period of 6 h. The first micro GC was a quad system consisting of four channels for the separation of O2, N2, CH4 CO, CO2, H2O, C2–C4 hydrocarbons (alkanes and alkenes). The second micro GC was a dual system consisting of two channels for the separation of He, H2, and C6–C8 hydrocarbons (alkanes and alkenes).
n-Hexane conversions were calculated on carbon mol basis; i.e., (C6in moles − C6out moles) / C6in moles × 100%. The carbon balance closed between 100% and 105%. Selectivity to individual products was also calculated based on the number of moles of carbon contained in the products, divided by the total number of moles of carbon in the product mixture excluding unconverted feed; i.e., (niCi / ∑ niCi) × 100%
5. Conclusions
Doping of Li/MgO catalyst with molybdena improves the performance in oxidative conversion of n-hexane. Promotion of Li/MgO with molybdena loading as low as 0.5wt % is sufficient to bring considerable improvements in the stability of the catalyst. Moreover, the catalyst exhibits similar activity for n-hexane conversion and selectivity to alkenes as the unpromoted Li/MgO.
MoO3/Li/MgO catalysts contain three types of molybdena-containing phases, the formation of which depends on MoOx loading. At low Mo/Li ratios, isolated dispersed [MoO4]2− anionic species are observed. At high Mo/Li ratios, the formation of crystalline lithium molybdate phases such as (i) monomeric Li2MoO4, and (ii) polymeric Li2Mo4O13 are concluded. The presence of these lithium molybdates diminishes the formation of Li2CO3 in the catalyst. Subsequently, the catalyst maintains high surface area and stability with time-on-stream during oxidative conversion.
Author Contributions
Conceptualization, C.B. and L.L.; data curation, C.B.; writing—original draft preparation, C.B.; writing—review and editing, L.L.; supervision, L.L.; funding acquisition, L.L. and C.B. All authors have read and agreed to the published version of the manuscript.
Funding
The authors gratefully thank ASPECT program in the Netherlands (project number 053.62.011) and the University Research Board at the American University of Beirut (award number: 103371, project number 23960) for financial support.
Acknowledgments
The authors gratefully thank and acknowledge the significant scientific contribution of K. Seshan in the presented work. The authors also acknowledge B. Geerdink and K. Altena-Schildkamp for technical support, L. Vrielink for XRF measurements, G. Meima (Dow Benelux) for AAS measurements, and Arjan van Zijp (Bruker Optics) for part of the Raman measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Leveles, L.; Fuchs, S.; Seshan, K.; Lercher, J.A.; Lefferts, L. Oxidative conversion of light alkanes to olefins over alkali promoted oxide catalysts. Appl. Catal. A 2002, 227, 287–297. [Google Scholar] [CrossRef]
- Leveles, L.; Seshan, K.; Lercher, J.A.; Lefferts, L. Oxidative conversion of propane over lithium-promoted magnesia catalyst II. Active site characterization and hydrocarbon activation. J. Catal. 2003, 218, 307–314. [Google Scholar] [CrossRef]
- Leveles, L.; Seshan, K.; Lercher, J.A.; Lefferts, L. Oxidative conversion of propane over lithium-promoted magnesia catalyst I. Kinetics and mechanism. J. Catal. 2003, 218, 296–306. [Google Scholar] [CrossRef]
- Trionfetti, C.; Babich, I.V.; Seshan, K.; Lefferts, L. Formation of high surface area Li/MgO—Efficient catalyst for the oxidative dehydrogenation/cracking of propane. Appl. Catal. A 2006, 310, 105–113. [Google Scholar] [CrossRef]
- Trionfetti, C.; Babich, I.V.; Seshan, K.; Lefferts, L. Presence of Lithium Ions in MgO Lattice: Surface Characterization by Infrared Spectroscopy and Reactivity towards Oxidative Conversion of Propane. Langmuir 2008, 24, 8220–8228. [Google Scholar] [CrossRef]
- Boyadjian, C.; Lefferts, L.; Seshan, K. Catalytic oxidative cracking of hexane as a route to olefins. Appl. Catal. A 2010, 372, 167–174. [Google Scholar] [CrossRef]
- Boyadjian, C.; van der Veer, B.; Babich, I.V.; Lefferts, L.; Seshan, K. Catalytic oxidative cracking as a route to olefins: Oxidative conversion of hexane over MoO3-Li/MgO. Catal. Today 2010, 157, 345–350. [Google Scholar] [CrossRef]
- Gaab, S.; Find, J.; Grasselli, R.K.; Lercher, J.A. Oxidative ethane activation over oxide supported molten alkali metal chloride catalysts. Stud. Surf. Sci. Catal. 2004, 147, 673–678. [Google Scholar]
- Lin, C.-H.; Campbell, K.D.; Wang, J.-X.; Lunsford, J.H. Oxidative dimerizatlon of methane over Lanthanum Oxide. J. Phys. Chem. 1986, 90, 534–537. [Google Scholar] [CrossRef]
- Xu, M.; Lunsford, J.H. Oxidative dehydrogenation of propane. React. Kin. Catal. Lett. 1996, 57, 3–11. [Google Scholar] [CrossRef]
- Lunsford, J.H.; Qiu, P.; Rosynek, M.P.; Xu, Z. Catalytic conversion of methane and ethylene to propylene. J. Phys. Chem. 1998, 102, 167–173. [Google Scholar] [CrossRef]
- Morales, E.; Lunsford, J.H. Oxidative dehydrogenation of ethane over a lithium-promoted magnesium oxide catalyst. J. Catal. 1989, 118, 255–265. [Google Scholar] [CrossRef]
- Ito, T.; Wang, J.-X.; Lin, C.-H.; Lunsford, J.H. Oxidative dimerization of methane over a lithium-promoted magnesium oxide catalyst. J. Am. Chem. Soc. 1985, 107, 5062–5068. [Google Scholar] [CrossRef]
- Wang, J.-X.; Lunsford, J.H. Characterization of [Li+O−] centers in lithium-doped MgO catalysts. J. Phys. Chem. 1986, 90, 5883–5887. [Google Scholar] [CrossRef]
- Shi, C.; Hatano, M.; Lunsford, J.H. A kinetic model for the oxidative coupling of methane over Li+/MgO Catalysts. Catal. Today 1992, 13, 191–199. [Google Scholar] [CrossRef]
- Lunsford, J.H. The role of surface-generated gas-phase radicals in catalysis. Langmuir 1989, 5, 12–16. [Google Scholar] [CrossRef]
- Shi, C.; Xu, M.; Rosynek, M.P.; Lunsford, J.H. Origin of kinetic isotope effects during the oxidative coupling of methane over a Li+/MgO catalyst. J. Phys. Chem. 1993, 97, 216–222. [Google Scholar] [CrossRef]
- Xu, M.; Shi, C.; Yang, X.; Rosynek, M.P.; Lunsford, J.H. Effect of carbon dioxide on the activation energy for methyl radical generatlon over Li/MgO catalysts. J. Phys. Chem. 1992, 96, 6395–6398. [Google Scholar] [CrossRef]
- Zavyalova, U.; Geske, M.; Horn, R.; Weinberg, G.; Frandsen, W.; Schuster, M.; Schlögl, R. Morphology and microstructure of Li/MgO catalysts for the oxidative coupling of methane. ChemCatChem 2011, 3, 949–959. [Google Scholar] [CrossRef]
- Kwapien, K.; Paier, J.; Sauer, J.; Geske, M.; Zavyalova, U.; Horn, R.; Schwach, P.; Trunschke, A.; Schlögl, R. Sites for methane activation on lithium-doped magnesium oxide surfaces. Angew. Chem. Int. Ed. 2014, 53, 8774–8778. [Google Scholar] [CrossRef]
- Schwach, P.; Frandsen, W.; Willinger, M.-G.; Schlögl, R.; Trunschke, A. Structure sensitivity of the oxidative activation of methane over MgO model catalysts: I. Kinetic study. J. Catal. 2015, 329, 560–573. [Google Scholar] [CrossRef]
- Schwach, P.; Hamilton, N.; Eichelbaum, M.; Thum, L.; Lunkenbein, T.; Schlögl, R.; Trunschke, A. Structure sensitivity of the oxidative activation of methane over MgO model catalysts: II. Nature of active sites and reaction mechanism. J. Catal. 2015, 329, 574–587. [Google Scholar] [CrossRef]
- Berger, T.; Schuh, J.; Sterrer, M.; Diwald, O.; Knözinger, E. Lithium ion induced surface reactivity changes on MgO nanoparticles. J. Catal. 2007, 247, 61–67. [Google Scholar] [CrossRef]
- Sinev, M.Y. Free radicals in catalytic oxidation of light alkanes: Kinetic and thermochemical aspects. J. Catal. 2003, 216, 468–476. [Google Scholar] [CrossRef]
- Cavani, F.; Trifiro, F. The oxidative dehydrogenation of ethane and propane as an alternative way for the production of light olefins. Catal. Today 1995, 24, 307–313. [Google Scholar] [CrossRef]
- Smyrl, N.R.; Fuller, E.L., Jr.; Powell, G.L. Monitoring the heterogeneous reaction of LiH and LiOH with H2O and CO2 by diffuse reflectance infrared fourier transform spectroscopy. Appl. Spectrosc. 1983, 37, 38–44. [Google Scholar] [CrossRef]
- Abello, M.C.; Gomez, M.F.; Ferretti, O. Mo/γ-Al2O3 catalysts for the oxidative dehydrogenation of propane: Effect of Mo loading. Appl. Catal. A 2001, 207, 421–431. [Google Scholar] [CrossRef]
- Abello, M.C.; Gomez, M.F.; Cadus, L.E. Selective oxidation of propane on MgO/γ-Al2O3-supported molybdenum catalyst: Influence of promoters. Catal. Lett. 1998, 53, 185–192. [Google Scholar] [CrossRef]
- Cadus, L.E.; Abello, M.C.; Gomez, M.F.; Rivarola, J.B. Oxidative dehydrogenation of propane over molybdenum supported on MgO−γ-Al2O3. Ind. Eng. Chem. Res. 1996, 35, 14–18. [Google Scholar] [CrossRef]
- Ueda, W.; Lee, K.H.; Yoon, Y.-S.; Moro-oka, Y. Selective oxidative dehydrogenation of propane over surface molybdenum-enriched MgMoO4 catalyst. Catal. Today 1998, 44, 199–203. [Google Scholar] [CrossRef]
- Heracleous, E.; Machli, M.; Lemonidou, A.A.; Vasalos, I.A. Oxidative dehydrogenation of ethane and propane over vanadia and molybdena supported catalysts. J. Mol. Catal A Chem. 2005, 232, 29–39. [Google Scholar] [CrossRef]
- Tsilomelekis, G.; Christodoulakis, A.; Boghosian, S. Support effects on structure and activity of molybdenum oxide catalysts for the oxidative dehydrogenation of ethane. Catal. Today 2007, 127, 139–147. [Google Scholar] [CrossRef]
- Dejoz, A.; Lopez Nieto, J.M.; Marquez, F.; Vazquez, M.I. The role of molybdenum in Mo-doped V–Mg–O catalysts during the oxidative dehydrogenation of n-butane. Appl. Catal. A 1999, 180, 83–94. [Google Scholar] [CrossRef]
- Pless, J.D.; Bardin, B.B.; Kim, H.-S.; Ko, D.; Smith, M.T.; Hammond, R.R.; Stair, P.C.; Poeppelmeier, K.R. Catalytic oxidative dehydrogenation of propane over Mg–V/Mo oxides. J. Catal. 2004, 223, 419–431. [Google Scholar] [CrossRef]
- Christodoulakis, A.; Heracleous, E.; Lemonidou, A.A.; Boghosian, S. An operando Raman study of structure and reactivity of alumina-supported molybdenum oxide catalysts for the oxidative dehydrogenation of ethane. J. Catal. 2006, 242, 16–25. [Google Scholar] [CrossRef]
- Christodoulakis, A.; Boghosian, S. Molecular structure and activity of molybdena catalysts supported on zirconia for ethane oxidative dehydrogenation studied by operando Raman spectroscopy. J. Catal. 2008, 260, 178–187. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Ueda, W.; Moro-oka, Y. Oxidative dehydrogenation of propane over magnesium molybdate catalysts. Catal. Lett. 1995, 35, 57–64. [Google Scholar] [CrossRef]
- Vrieland, G.E.; Murchison, C.B. Anaerobic oxidation of butane to butadiene over magnesium molybdate catalysts. I. Magnesia supported catalysts. Appl. Catal. A 1996, 134, 101–121. [Google Scholar] [CrossRef]
- Kim, D.S.; Segawa, K.; Soeya, T.; Wachs, I.E. Surface structures of supported molybdenum oxide catalysts under ambient conditions. J. Catal. 1992, 136, 539–553. [Google Scholar] [CrossRef]
- Vuurman, M.A.; Wachs, I.E. In situ Raman spectroscopy of alumina-supported metal oxide catalysts. J. Phys. Chem. 1992, 96, 5008–5016. [Google Scholar] [CrossRef]
- Chang, S.-C.; Leugers, M.A.; Bare, S. Surface chemlstry of magnesium oxide-supported molybdenum oxide: An in situ Raman spectroscoplc study. J. Phys. Chem. 1992, 96, 10358–10365. [Google Scholar] [CrossRef]
- Bare, S.R.; Mitchell, G.E.; Maj, J.J.; Vrieland, G.E.; Gland, J.L. Local site symmetry of dispersed molybdenum oxide catalysts: XANES at the Mo L2,3-Edges. J. Phys. Chem. 1993, 97, 6048–6053. [Google Scholar] [CrossRef]
- Bare, S.R. Surface structure of highly dispersed MoO3 on MgO using in situ Mo L3-Edge XANES. Langmuir 1998, 14, 1500–1504. [Google Scholar] [CrossRef]
- Tomoyuki, K.; Okazaki, S.; Shishido, T.; Teramura, K.; Tanaka, T. Brønsted acid generation of alumina-supported molybdenum oxide calcined at high temperatures: Characterization by acid-catalyzed reactions and spectroscopic methods. J. Mol. Catal. A Chem. 2013, 371, 21–28. [Google Scholar]
- El-Sharkawy, E.A.; Khder, A.S.; Ahmed, A.I. Structural characterization and catalytic activity of molybdenum oxide supported zirconia catalysts. Microporous Mesoporous Mater. 2007, 102, 128–137. [Google Scholar] [CrossRef]
- Valigi, M.; Cimino, A.; Cordischi, D.; De Rossi, C.; Ferraris, G.; Gazzoli, D.; Indovina, V.; Occhiuzzi, M. Molybdenum (VI) interaction with zirconia surface and its influence on the crystallization and sintering. Solid State Ion. 1993, 63–65, 136–142. [Google Scholar] [CrossRef]
- Afanasiev, P.; Geantet, C.; Breysse, M. Preparation of high surface area Mo/ZrO2 catalysts by a molten salt method: Aplication to Hydrosulfurization. J. Catal. 1995, 153, 17–24. [Google Scholar] [CrossRef]
- Ulla, A.A.; Spretz, R.; Lombardo, E.; Daniell, W.; Knözinger, H. Catalytic combustion of methane on Co/MgO: Characterisation of active cobalt sites. Appl. Catal. B 2001, 29, 217–229. [Google Scholar] [CrossRef]
- Payen, E.; Grimblot, J.; Kasztelan, S. Study of oxidic and reduced alumina-supported molybdate and heptamolybdate species by in situ laser Raman spectroscopy. J. Phys. Chem. 1987, 91, 6642–6648. [Google Scholar] [CrossRef]
- Brooker, M.H.; Wang, J. Raman and infrared studies of lithium and cesium carbonates. Spectrochim. Acta A 1992, 48, 999–1008. [Google Scholar] [CrossRef]
- Li, G.; Li, H.; Mo, Y.; Chen, L.; Huang, X. Further identification to the SEI film on Ag electrode in lithium batteries by surface enhanced Raman scattering (SERS). J. Power Sources 2002, 104, 190–194. [Google Scholar] [CrossRef]
- Erdoheyli, A.; Fodor, K.; Nemeth, R.; Hancz, A.; Oszko, A. Partial oxidation of methane on silica-supported different alkali metal molybdates. J. Catal. 2001, 199, 328–337. [Google Scholar] [CrossRef]
- Moser, M.; Klimm, D.; Ganschow, S.; Kwasniewski, A.; Jacobs, K. Re-determination of the pseudobinary system Li2O—MoO3. Cryst. Res. Technol. 2008, 43, 350–354. [Google Scholar] [CrossRef]
- Wan, S.; Zhang, B.; Yao, Y.; Zheng, G.; Zhang, S.; You, J. Raman and density functional theory studies of Li2Mo4O13 structures in crystalline and molten states. Inorg. Chem. 2017, 56, 14129–14134. [Google Scholar] [CrossRef] [PubMed]
- Gatehouse, B.M.; Miskin, B.K. Structural studies in the Li2MoO4−MoO3 system: Part 2. The high-temperature form of lithium tetramolybdate, H-Li2Mo4O13. J. Solid State Chem. 1975, 15, 274–282. [Google Scholar] [CrossRef]
- Perrichon, V.; Durupty, M.C. Thermal stability of alkali metals deposited on oxide supports and their influence on the surface area of the support. Appl. Catal. 1988, 42, 217–227. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).