An Overview of the Photocatalytic Water Splitting over Suspended Particles


Abstract
1. Introduction
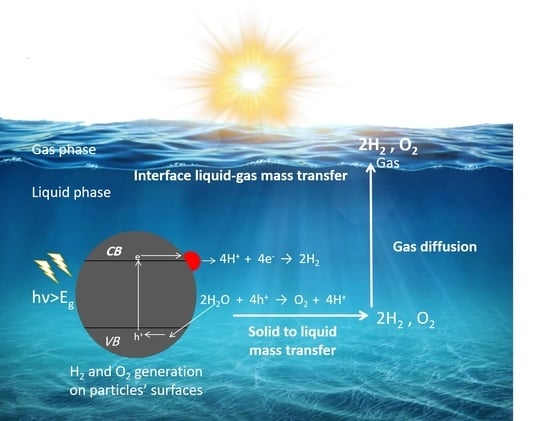
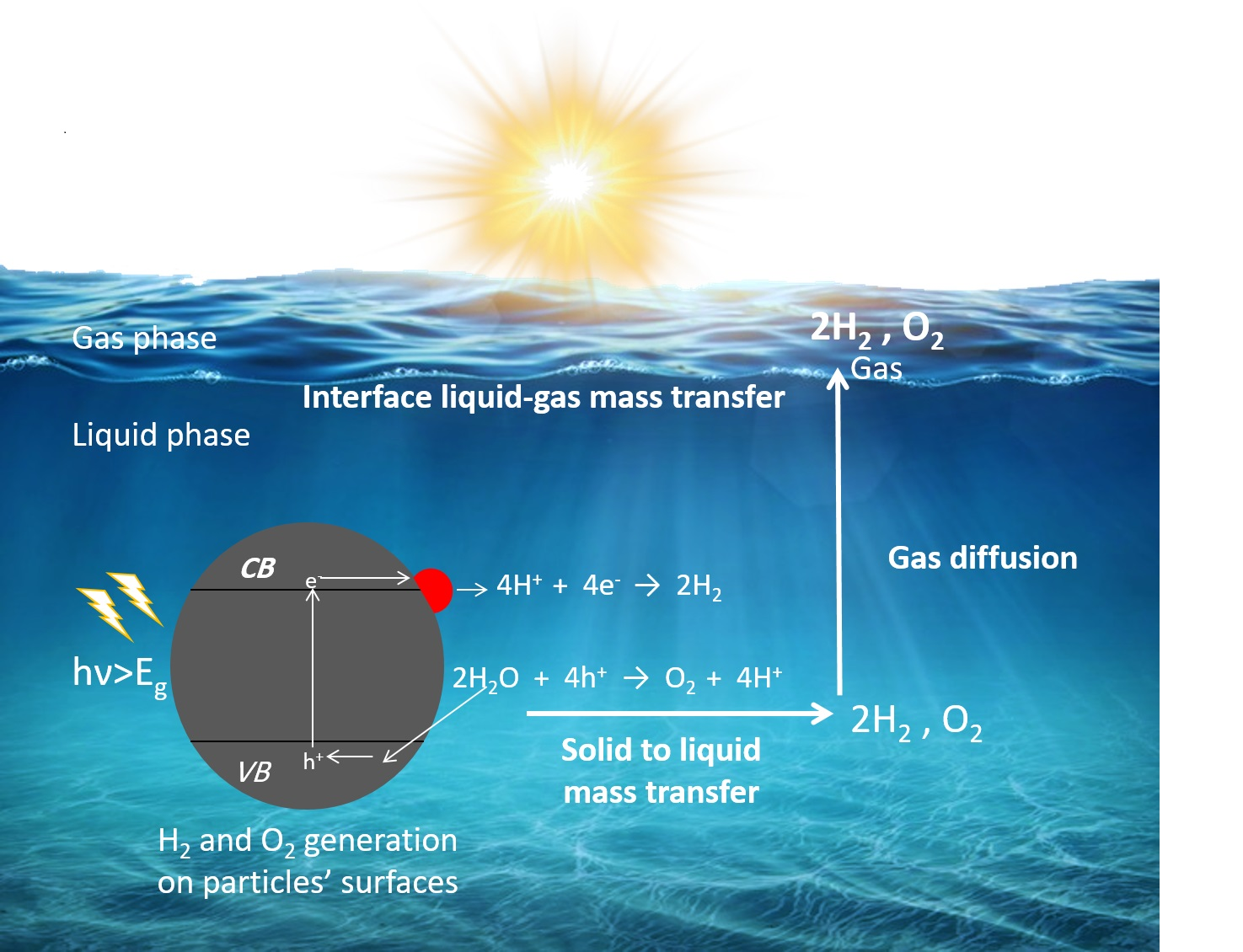
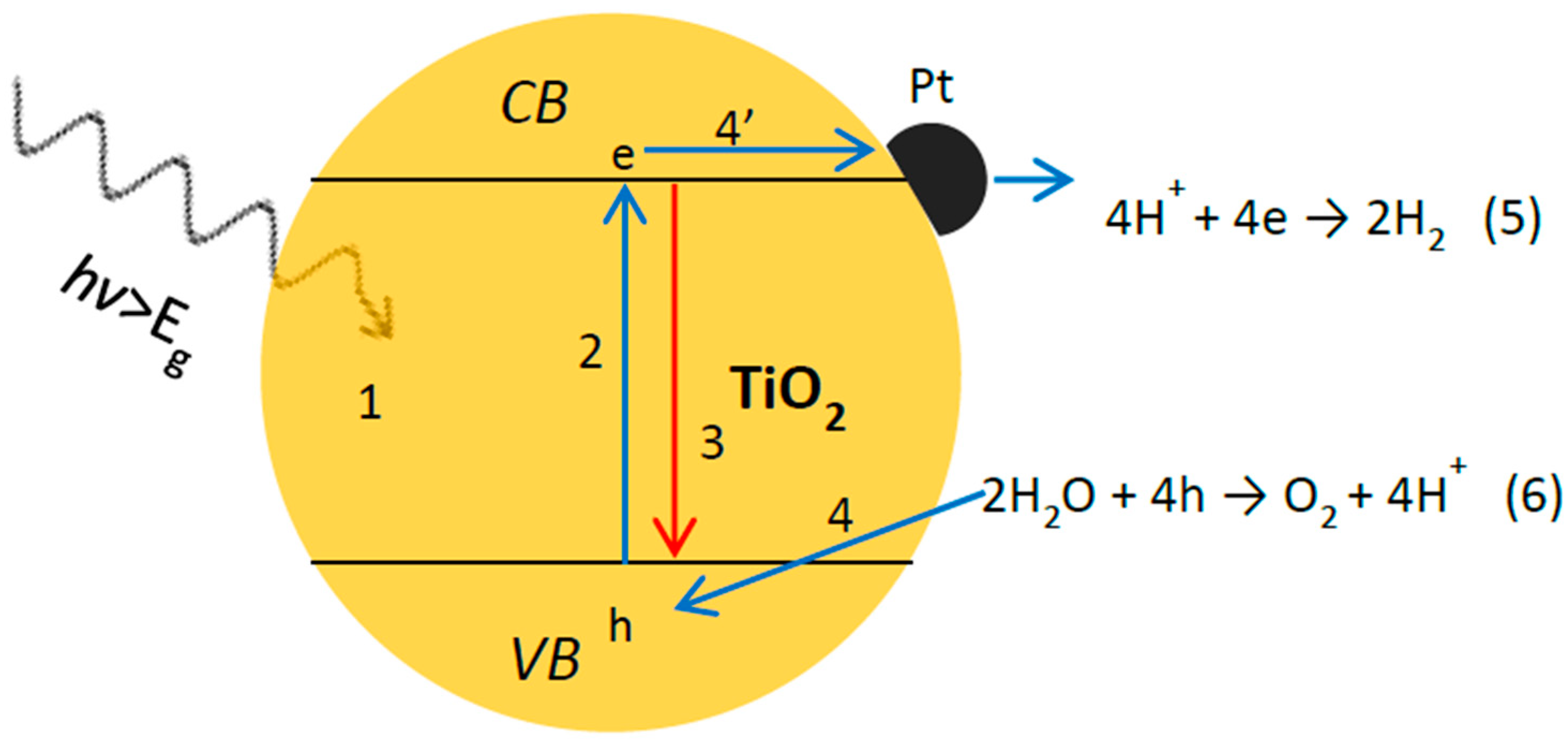
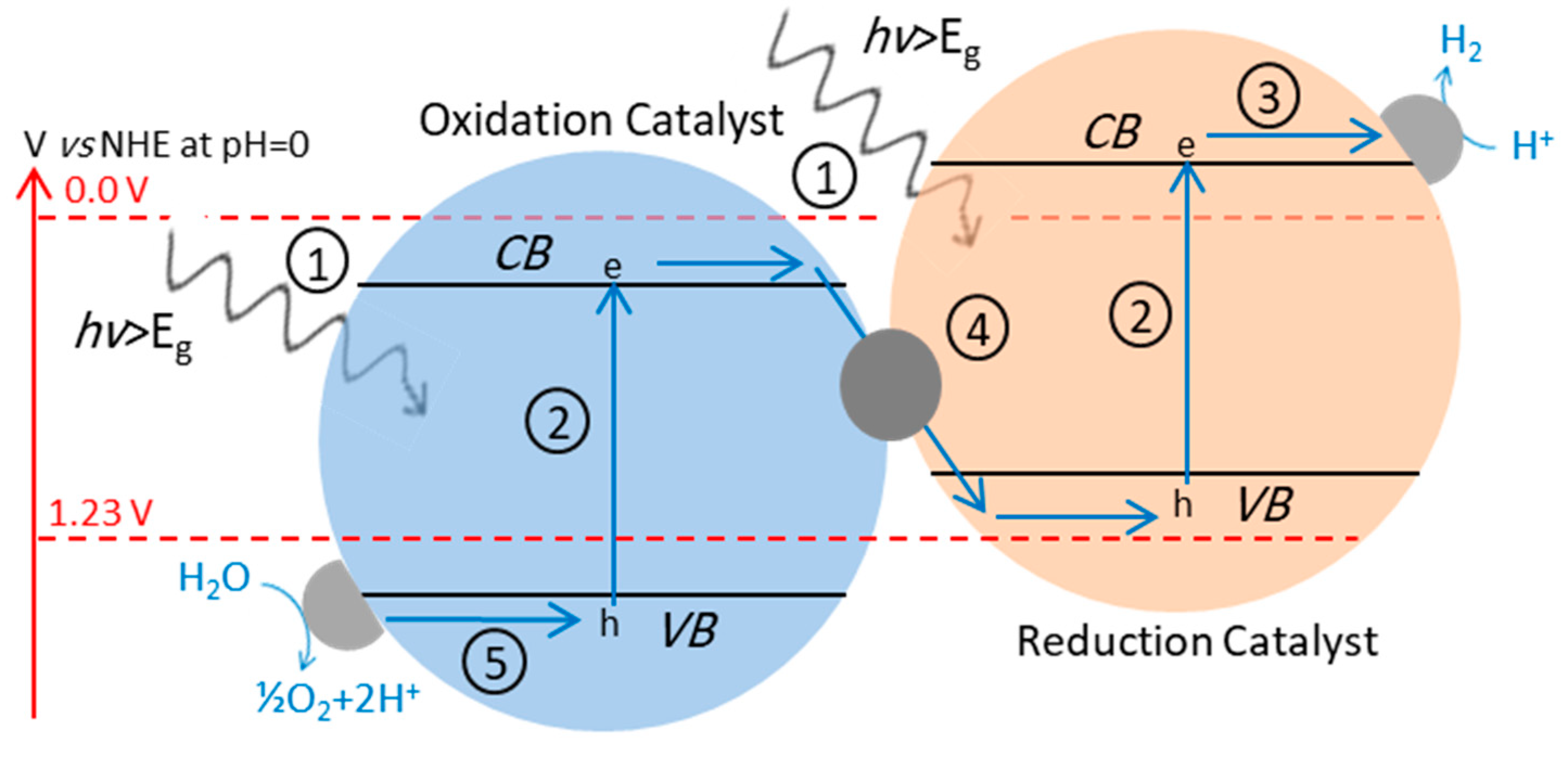
2. Fundamental Processes in Photocatalytic Overall Water Splitting
3. Design and Synthesis of Particulate Photocatalytic Systems
4. Improving Light Absorption
4.1. UV Light Photocatalysts
4.2. Visible Light Photocatalysts
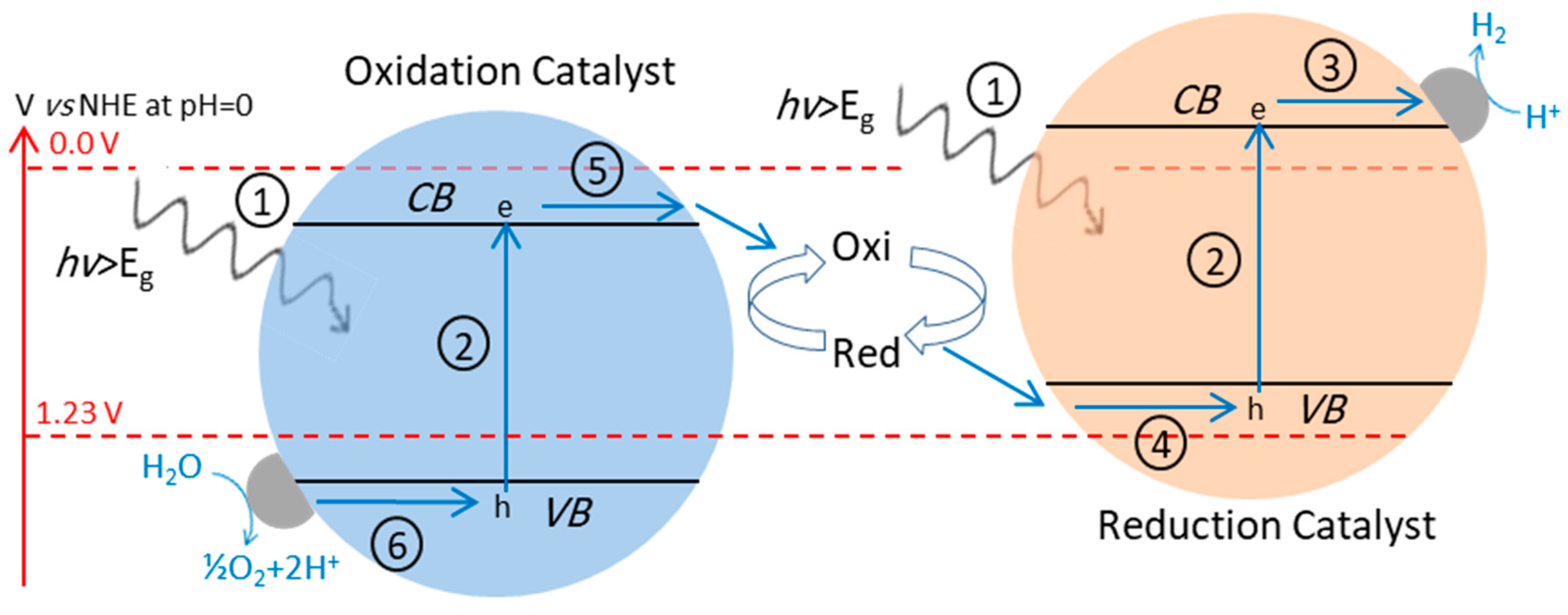
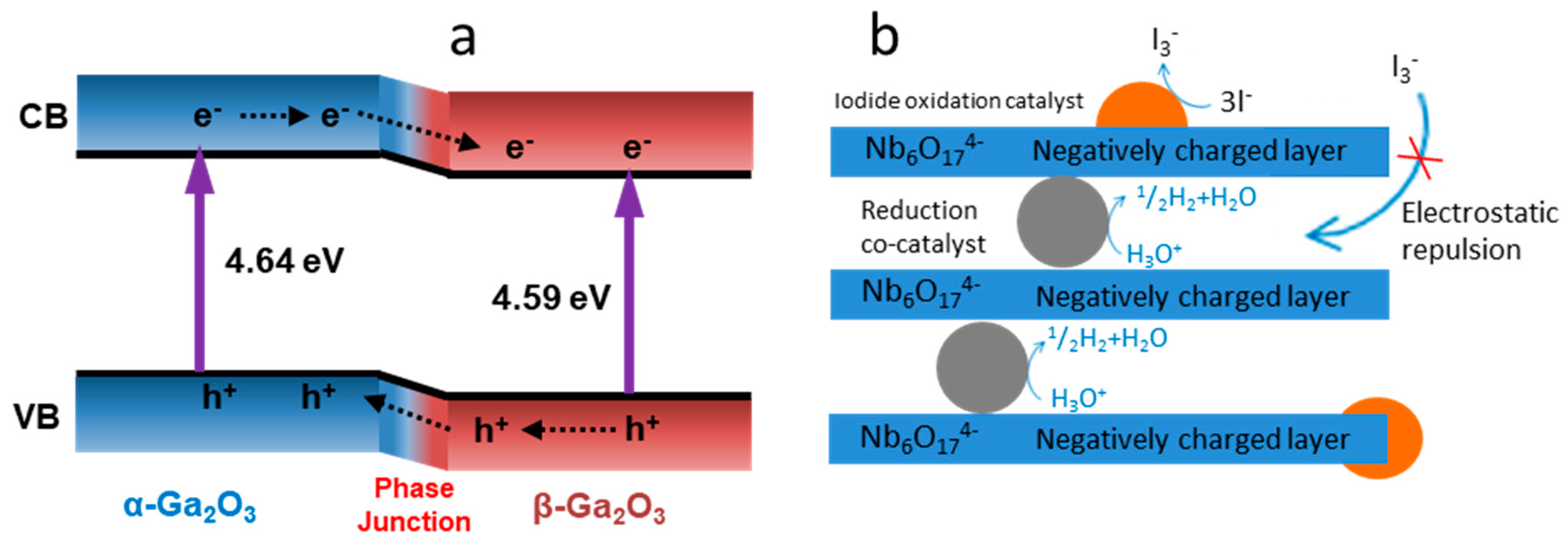
4.3. Z-Scheme: A Two-Step Approach
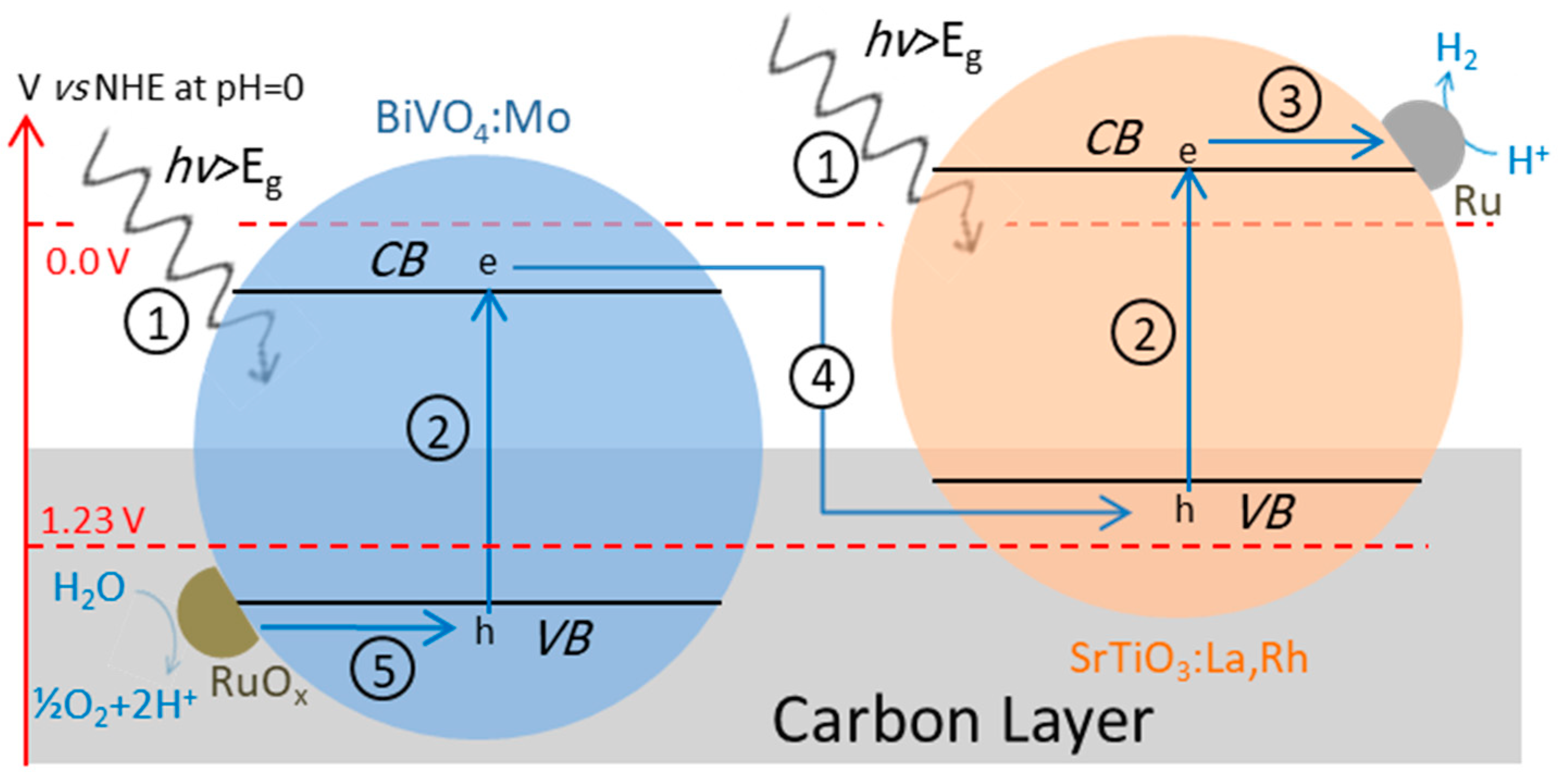
4.3.1. Z-Scheme with Aqueous Redox Mediator

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H2 Photocatalyst (wt. % Unless Indicated) | O2 Photocatalyst (wt.%) | Mediator | H2 Rate (mmolh−1) | O2 Rate (mmolh−1) | AQY (%) | Ref. |
|---|---|---|---|---|---|---|
| Pt/SrTiO3:Rh | BiVO4 | Fe3+/Fe2+ | 15 | 7.2 | 0.4 at 420 nm | [110] |
| Pt/SrTiO3:Cr/Ta (Pt = 0.3, Cr, Ta = 4.0 mol% each) | PtOx/WO3 (Pt = 0.5) | IO3−/I− | 16 | 8 | 1 at 420 nm | [111] |
| Pt/TaON (Pt = 0.3) | PtOx/WO3 (Pt = 0.5) | IO3−/I−_ | 16.5 | 8 | 0.5 at 420 nm | [112] |
| Pt/ZrO2/TaON (Pt = 1.0, Zn/Ta = 0.1) | PtOx/WO3 (Pt = 0.5) | IO3−/I−_ | 52 | 27 | 6.3 at 420 nm | [109] |
| Ru/SrTiO3:Rh (Ru = 1.0, Sr:Ti:Rh = 1.1:0.98:0.02) | PtOx/WO3 (Pt = 0.3) | Fe3+/Fe2+ | 88 | 44 | 4.2 at 420 nm | [40] |
| Pt/MgTa2O6−xNy/TaON (Pt = 0.4, Mg/Ta = 0.2) | PtOx/WO3 (Pt = 0.45) | IO3−/I− | 108 | 55 | 6.8 at 420 nm | [110] |
4.3.2. Z-Scheme with Solid-State Electron Mediator
5. Improving Efficiency
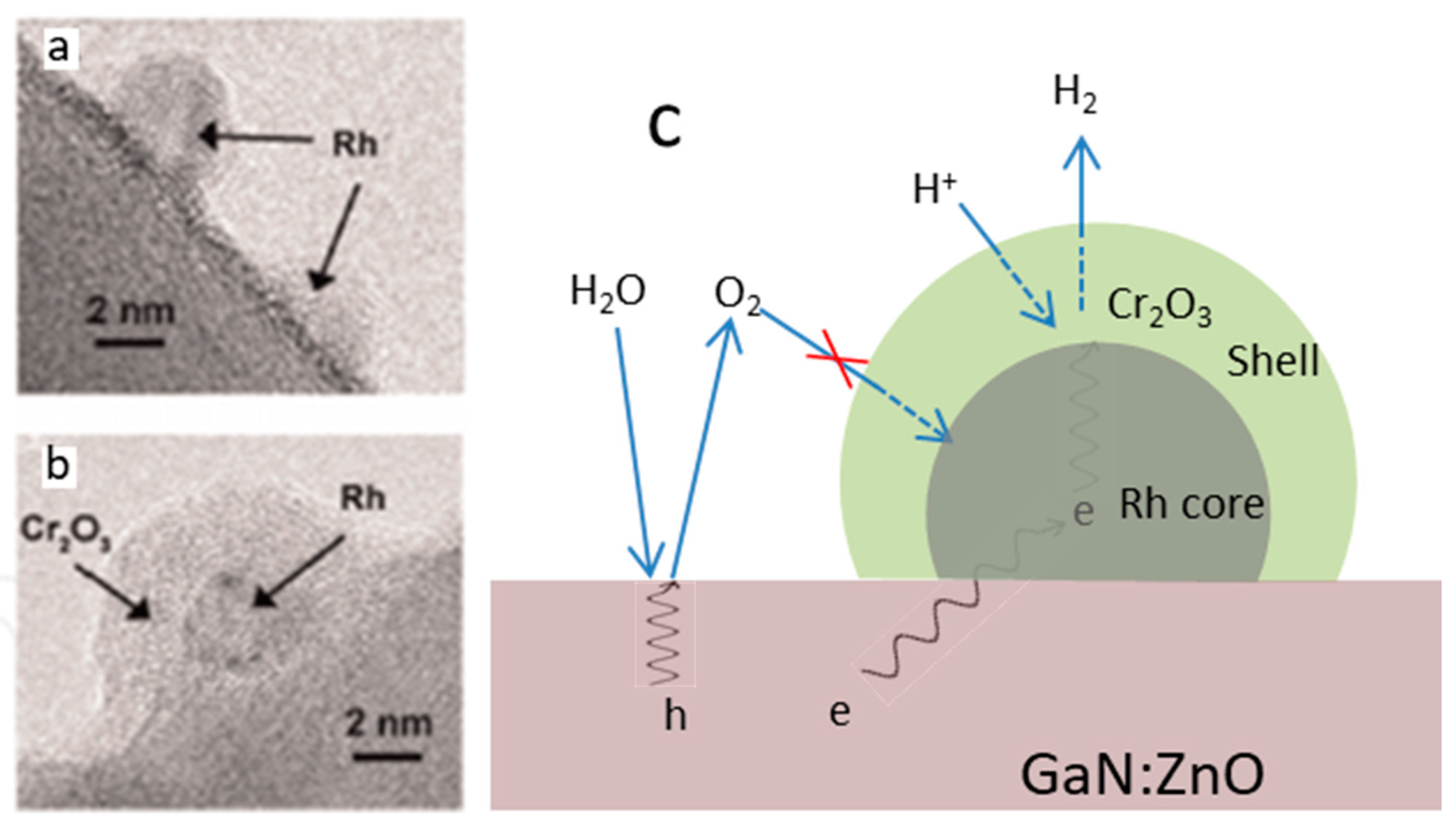
5.1. Charge Recombination
5.2. Back-Reaction (2 H2+O2 → 2 H2O)
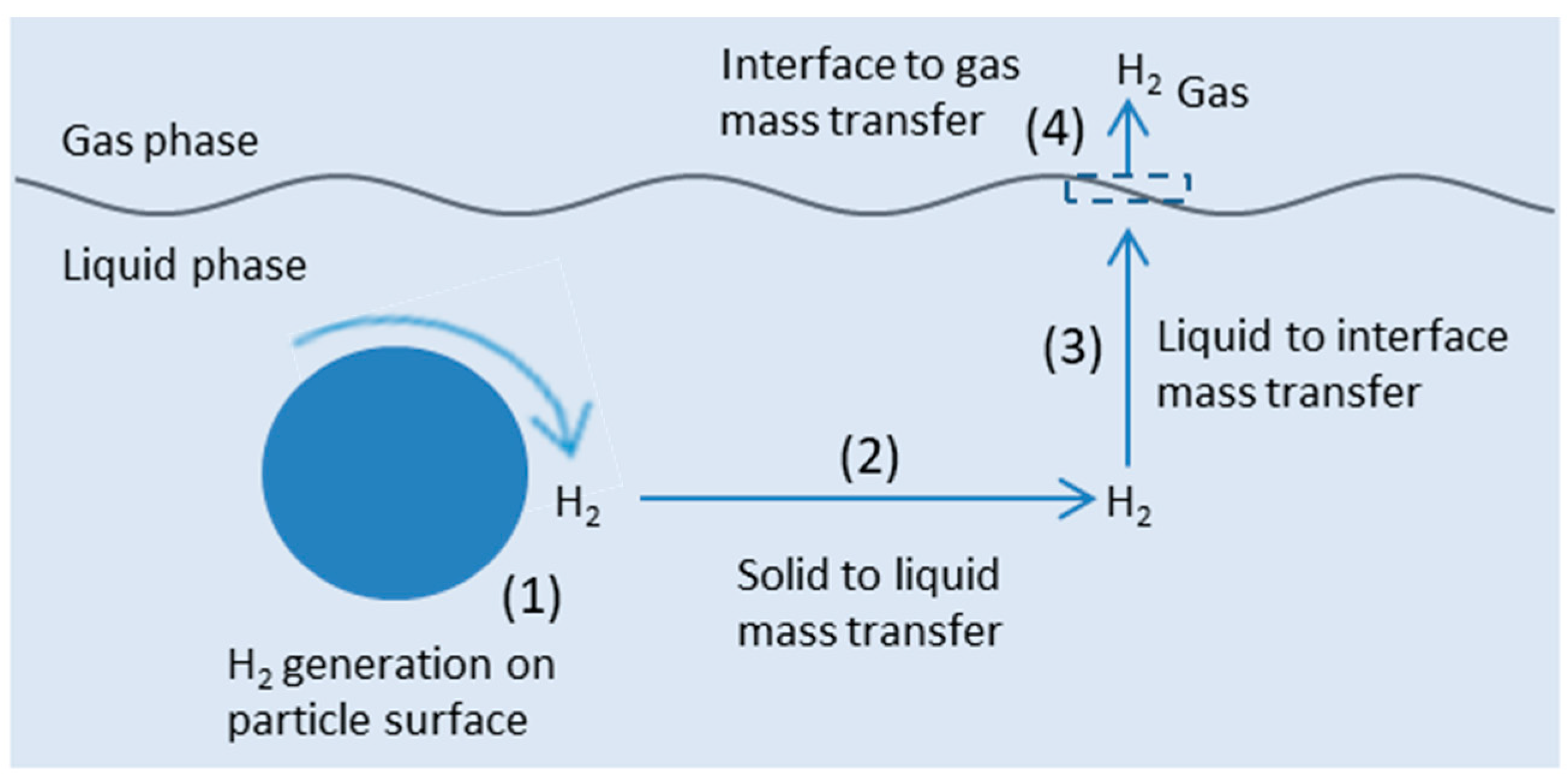
5.3. Mass Transfer Limitations
6. More Recent Overall Water Splitting Systems
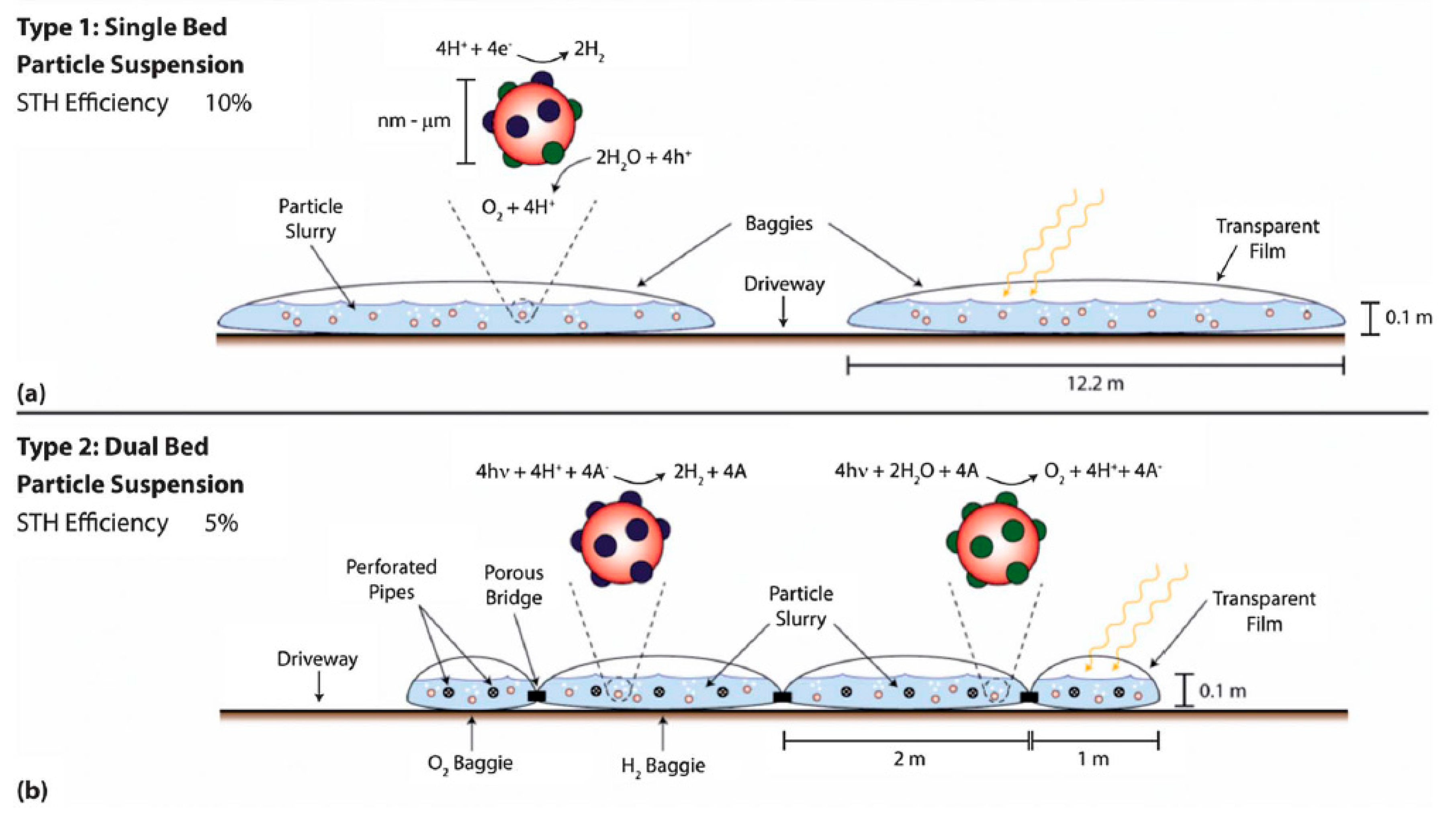
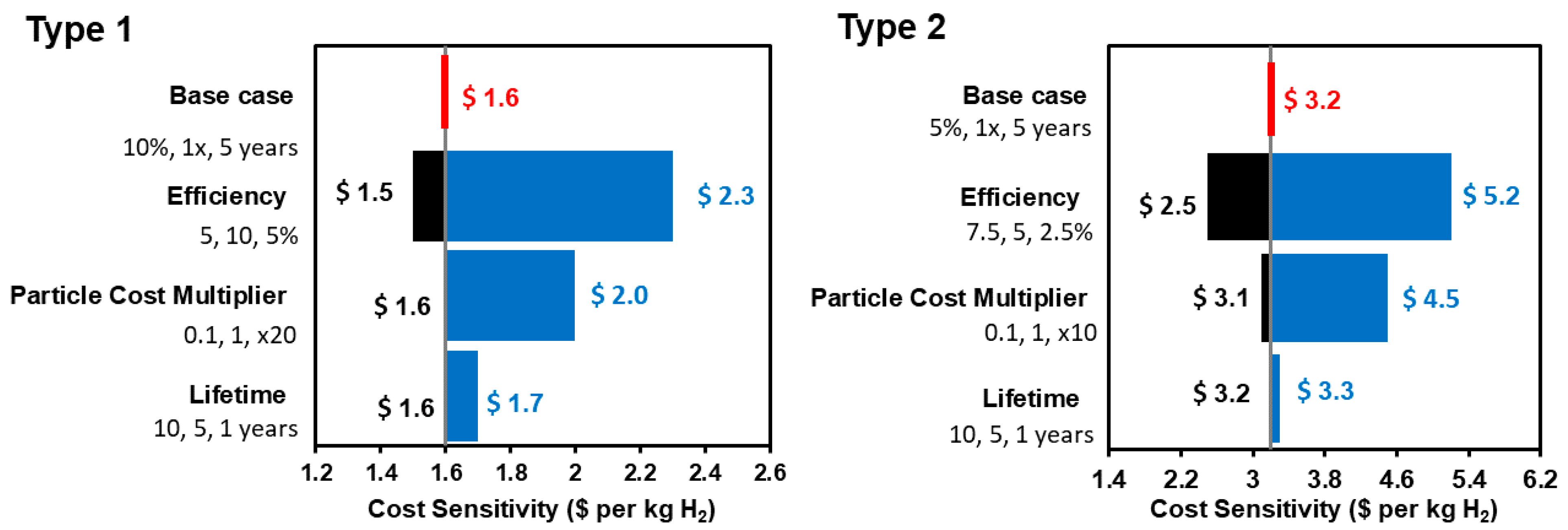
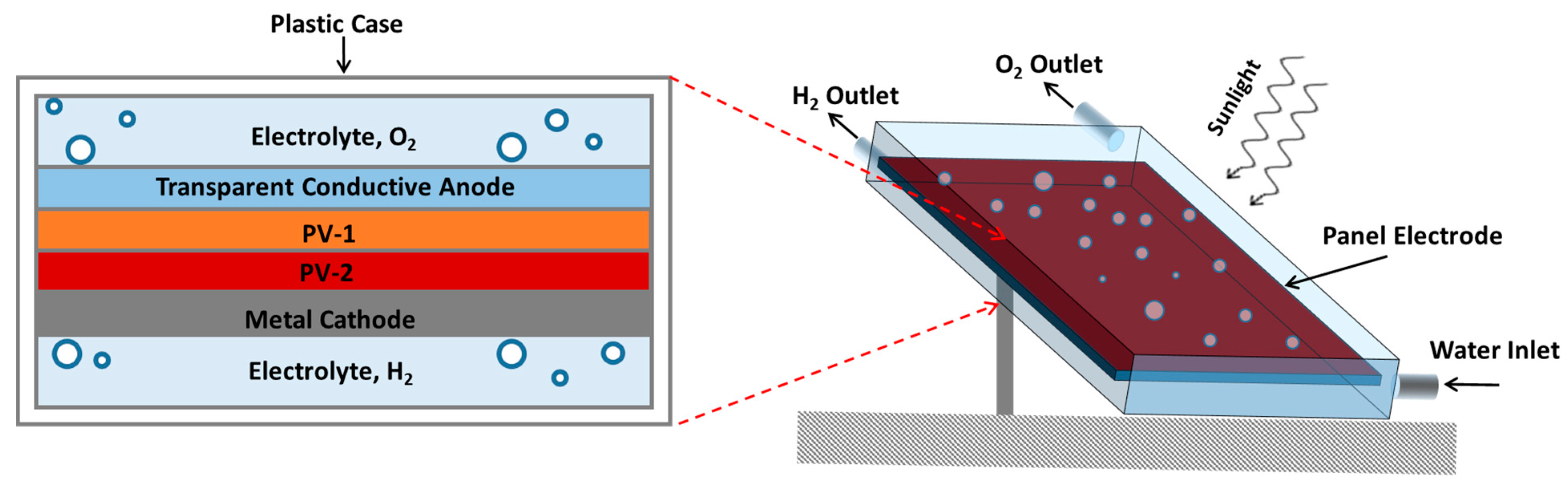
7. Reactor Design and Cost of Hydrogen
8. Conclusions and Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Siegmund, P.; Abermann, J.; Baddour, O.; Canadell, P.; Cazenave, A.; Derksen, C.; Garreau, A.; Howell, S.; Huss, H.; Isensee, K.; et al. World Meteorological Organization, The Global Climate in 2015–2019. In The Global Climate in 2015–2019: Climate Change Accelerates; WMO Publication Board: Geneva, Switzerland, 2019. [Google Scholar]
- Morton, O. A New Day Dawning? Silicon Valley Sunrise; Nature Publishing Group: London, UK, 2006.
- Barber, J. Photosynthetic energy conversion: Natural and artificial. Chem. Soc. Rev. 2009, 38, 185–196. [Google Scholar] [CrossRef]
- Lewis, N.S. Toward Cost-Effective Solar Energy Use. Science 2007, 315, 798–801. [Google Scholar] [CrossRef]
- Grätzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Erisman, J.W.; Sutton, M.A.; Galloway, J.; Klimont, Z.; Winiwarter, W. How a century of ammonia synthesis changed the world. Nat. Geosci. 2008, 1, 636–639. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Aasberg-Petersen, K.; Dybkjær, I.; Ovesen, C.; Schjødt, N.; Sehested, J.; Thomsen, S. Natural gas to synthesis gas–catalysts and catalytic processes. J. Nat. Gas Sci. Eng. 2011, 3, 423–459. [Google Scholar] [CrossRef]
- Nadeem, M.A.; Idriss, H. Effect of pH, temperature, and low light flux on the performance (16% STH) of coupled triple junction solar cell to water electrolysis. J. Power Sources 2020, 459, 228074. [Google Scholar] [CrossRef]
- Bashir, S.M.; Nadeem, M.A.; Al-Oufi, M.; Al-Hakami, M.; Isimjan, T.T.; Idriss, H. Sixteen Percent Solar-to-Hydrogen Efficiency Using a Power-Matched Alkaline Electrolyzer and a High Concentrated Solar Cell: Effect of Operating Parameters. ACS Omega 2020, 5, 10510–10518. [Google Scholar] [CrossRef] [PubMed]
- James, B.D.; Baum, G.N.; Perez, J.; Baum, K.N.J.D.r. Technoeconomic Analysis of Photoelectrochemical (PEC) Hydrogen Production. Office of Energy Efficiency and Renewable Energy: Arlington, Virginia, 2009. [Google Scholar]
- Shaner, M.R.; Atwater, H.A.; Lewis, N.S.; McFarland, E.W.J.E.; Science, E. A comparative technoeconomic analysis of renewable hydrogen production using solar energy. Energy Environ. Sci. 2016, 9, 2354–2371. [Google Scholar] [CrossRef]
- Pinaud, B.A.; Benck, J.D.; Seitz, L.C.; Forman, A.J.; Chen, Z.; Deutsch, T.G.; James, B.D.; Baum, K.N.; Baum, G.N.; Ardo, S.; et al. Technical and economic feasibility of centralized facilities for solar hydrogen production via photocatalysis and photoelectrochemistry. Energy Environ. Sci. 2013, 6, 1983–2002. [Google Scholar] [CrossRef]
- Fabian, D.M.; Hu, S.; Singh, N.; Houle, F.A.; Hisatomi, T.; Domen, K.; Osterloh, F.E.; Ardo, S. Particle suspension reactors and materials for solar-driven water splitting. Energy Environ. Sci. 2015, 8, 2825–2850. [Google Scholar] [CrossRef]
- Goto, Y.; Hisatomi, T.; Wang, Q.; Higashi, T.; Ishikiriyama, K.; Maeda, T.; Sakata, Y.; Okunaka, S.; Tokudome, H.; Katayama, M.; et al. A Particulate Photocatalyst Water-Splitting Panel for Large-Scale Solar Hydrogen Generation. Joule 2018, 2, 509–520. [Google Scholar] [CrossRef]
- Wang, Q.; Hisatomi, T.; Suzuki, Y.; Pan, Z.; Seo, J.; Katayama, M.; Minegishi, T.; Nishiyama, H.; Takata, T.; Seki, K.; et al. Particulate Photocatalyst Sheets Based on Carbon Conductor Layer for Efficient Z-Scheme Pure-Water Splitting at Ambient Pressure. J. Am. Chem. Soc. 2017, 139, 1675–1683. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, Y.; Hisatomi, T.; Nakabayashi, M.; Shibata, N.; Kubota, J.; Domen, K. Z-scheme water splitting using particulate semiconductors immobilized onto metal layers for efficient electron relay. J. Catal. 2015, 328, 308–315. [Google Scholar] [CrossRef]
- Pan, Z.; Hisatomi, T.; Wang, Q.; Chen, S.; Iwase, A.; Nakabayashi, M.; Shibata, N.; Takata, T.; Katayama, M.; Minegishi, T.; et al. Photoreduced Graphene Oxide as a Conductive Binder to Improve the Water Splitting Activity of Photocatalyst Sheets. Adv. Funct. Mater. 2016, 26, 7011–7019. [Google Scholar] [CrossRef]
- Minegishi, T.; Nishimura, N.; Kubota, J.; Domen, K. Photoelectrochemical properties of LaTiO2N electrodes prepared by particle transfer for sunlight-driven water splitting. Chem. Sci. 2013, 4, 1120–1124. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Wagner, F.; Somorjai, G. Photocatalytic hydrogen production from water on Pt-free SrTiO3 in alkali hydroxide solutions. Nature 1980, 285, 559–560. [Google Scholar] [CrossRef]
- Yamaguti, K.; Sato, S. Photolysis of water over metallized powdered titanium dioxide. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1985, 81, 1237–1246. [Google Scholar] [CrossRef]
- Kudo, A.; Domen, K.; Maruya, K.-i.; Onishi, T. Photocatalytic activities of TiO2 loaded with NiO. Chem. Phys. Lett. 1987, 133, 517–519. [Google Scholar] [CrossRef]
- Sayama, K.; Arakawa, H. Effect of carbonate salt addition on the photocatalytic decomposition of liquid water over Pt–TiO2 catalyst. J. Chem. Soc. Faraday Trans. 1997, 93, 1647–1654. [Google Scholar] [CrossRef]
- Tabata, S.; Nishida, H.; Masaki, Y.; Tabata, K. Stoichiometric photocatalytic decomposition of pure water in Pt/TiO2 aqueous suspension system. Catal. Lett. 1995, 34, 245–249. [Google Scholar] [CrossRef]
- Moon, S.-C.; Mametsuka, H.; Suzuki, E.; Anpo, M. Stoichiometric decomposition of pure water over Pt-loaded Ti/B binary oxide under UV-irradiation. Chem. Lett. 1998, 27, 117–118. [Google Scholar] [CrossRef]
- Sayama, K.; Arakawa, H. Effect of Na2CO3 addition on photocatalytic decomposition of liquid water over various semiconductor catalysis. J. Photochem. Photobiol. A Chem. 1994, 77, 243–247. [Google Scholar] [CrossRef]
- Bard, A.J. Photoelectrochemistry. Science 1980, 207, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, M.A.; Al-Oufi, M.; Wahab, A.K.; Anjum, D.; Idriss, H. Hydrogen Production on Ag-Pd/TiO2 Bimetallic Catalysts: Is there a Combined Effect of Surface Plasmon Resonance with Schottky Mechanism on the Photo-Catalytic Activity? ChemistrySelect 2017, 2, 2754–2762. [Google Scholar] [CrossRef]
- Khan, M.A.; Al-Oufi, M.; Toseef, A.; Nadeem, M.A.; Idriss, H. Comparing the Reaction Rates of Plasmonic (Gold) and Non-Plasmonic (Palladium) Metal Particles in Photocatalytic Hydrogen Production. Catal. Lett. 2018, 148, 1–10. [Google Scholar] [CrossRef]
- Nadeem, A.; Muir, J.M.; Waterhouse, G.W.; Idriss, H. Hydrogen Photo-Production from Ethanol on TiO2: A Surface Science and Catalysis Study; SPIE Bellingham: Washington, DC, USA, 2011; Volume 8109. [Google Scholar]
- Wahab, A.K.; Nadeem, M.A.; Idriss, H. Hydrogen Production During Ethylene Glycol Photoreactions Over Ag-Pd/TiO2 at Different Partial Pressures of Oxygen. Front. Chem. 2019, 7. [Google Scholar] [CrossRef]
- Nadeem, A.; Waterhouse, G.I.; Metson, J.; Idriss, H. Hydrogen Production by Photoreaction of Ethanol Over Au/TiO2 Anatase: The Effect of TiO2 Particle Size; SPIE Bellingham: Washington, DC, USA, 2010; Volume 7770. [Google Scholar]
- Hussain, E.; Majeed, I.; Nadeem, M.A.; Iqbal, A.; Chen, Y.; Choucair, M.; Jin, R.; Nadeem, M.A. Remarkable effect of BaO on photocatalytic H2 evolution from water splitting via TiO2(P25) supported palladium nanoparticles. J. Environ. Chem. Eng. 2019, 7, 102729. [Google Scholar] [CrossRef]
- Domen, K.; Naito, S.; Soma, M.; Onishi, T.; Tamaru, K. Photocatalytic decomposition of water vapour on an NiO–SrTiO3 catalyst. J. Chem. Soc. Chem. Commun. 1980, 12, 543–544. [Google Scholar] [CrossRef]
- Domen, K.; Kudo, A.; Onishi, T.; Kosugi, N.; Kuroda, H. Photocatalytic decomposition of water into H2 and O2 over NiO-SrTiO3 powder. 1. Structure of the catalyst. J. Phys. Chem. 1986, 90, 292–295. [Google Scholar] [CrossRef]
- Pan, C.; Takata, T.; Nakabayashi, M.; Matsumoto, T.; Shibata, N.; Ikuhara, Y.; Domen, K. A complex perovskite type oxynitride: The first photocatalyst for water splitting operable at up to 600 nm. Angew. Chem. Int. Ed. 2015, 54, 2955–2959. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Bai, L.; Hisatomi, T.; Maeda, K.; Domen, K. Photocatalytic water splitting using modified GaN:ZnO solid solution under visible light: Long-time operation and regeneration of activity. J. Am. Chem. Soc. 2012, 134, 8254–8259. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Teramura, K.; Takata, T.; Hara, M.; Saito, N.; Toda, K.; Inoue, Y.; Kobayashi, H.; Domen, K. Overall Water Splitting on (Ga1-xZnx)(N1-xOx) Solid Solution Photocatalyst: Relationship between Physical Properties and Photocatalytic Activity. J. Phys. Chem. B 2005, 109, 20504–20510. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sasaki, Y.; Shirakura, N.; Kudo, A. Synthesis of highly active rhodium-doped SrTiO3 powders in Z-scheme systems for visible-light-driven photocatalytic overall water splitting. J. Mater. Chem. A 2013, 1, 12327–12333. [Google Scholar] [CrossRef]
- Kudo, A. Z-scheme photocatalyst systems for water splitting under visible light irradiation. MRS Bull. 2011, 36, 32–38. [Google Scholar] [CrossRef]
- Fujito, H.; Kunioku, H.; Kato, D.; Suzuki, H.; Higashi, M.; Kageyama, H.; Abe, R. Layered Perovskite Oxychloride Bi4NbO8Cl: A Stable Visible Light Responsive Photocatalyst for Water Splitting. J. Am. Chem. Soc. 2016, 138, 2082–2085. [Google Scholar] [CrossRef]
- Tabata, M.; Maeda, K.; Higashi, M.; Lu, D.; Takata, T.; Abe, R.; Domen, K. Modified Ta3N5 Powder as a Photocatalyst for O2 Evolution in a Two-Step Water Splitting System with an Iodate/Iodide Shuttle Redox Mediator under Visible Light. Langmuir 2010, 26, 9161–9165. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, J.; Wang, L.; Han, M.; Zhang, M.; Wang, H.; Huang, H.; Liu, Y.; Kang, Z. Carbon dots as solid-state electron mediator for BiVO4/CDs/CdS Z-scheme photocatalyst working under visible light. Appl. Catal. B Environ. 2017, 206, 501–509. [Google Scholar] [CrossRef]
- Srinivasan, N.; Sakai, E.; Miyauchi, M. Balanced Excitation between Two Semiconductors in Bulk Heterojunction Z-Scheme System for Overall Water Splitting. ACS Catal. 2016, 6, 2197–2200. [Google Scholar] [CrossRef]
- Kobayashi, R.; Takashima, T.; Tanigawa, S.; Takeuchi, S.; Ohtani, B.; Irie, H. A heterojunction photocatalyst composed of zinc rhodium oxide, single crystal-derived bismuth vanadium oxide, and silver for overall pure-water splitting under visible light up to 740 nm. Phys. Chem. Chem. Phys. 2016, 18, 27754–27760. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hisatomi, T.; Ma, S.S.K.; Li, Y.; Domen, K. Core/Shell Structured La- and Rh-Codoped SrTiO3 as a Hydrogen Evolution Photocatalyst in Z-Scheme Overall Water Splitting under Visible Light Irradiation. Chem. Mater. 2014, 26, 4144–4150. [Google Scholar] [CrossRef]
- Iwase, A.; Ng, Y.H.; Ishiguro, Y.; Kudo, A.; Amal, R. Reduced Graphene Oxide as a Solid-State Electron Mediator in Z-Scheme Photocatalytic Water Splitting under Visible Light. J. Am. Chem. Soc. 2011, 133, 11054–11057. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hwang, D.W.; Kim, H.G.; Bae, S.W.; Lee, J.S.; Li, W.; Oh, S.H. Highly efficient overall water splitting through optimization of preparation and operation conditions of layered perovskite photocatalysts. Top. Catal. 2005, 35, 295–303. [Google Scholar] [CrossRef]
- Asai, R.; Nemoto, H.; Jia, Q.; Saito, K.; Iwase, A.; Kudo, A. A visible light responsive rhodium and antimony-codoped SrTiO3 powdered photocatalyst loaded with an IrO2 cocatalyst for solar water splitting. Chem. Commun. 2014, 50, 2543–2546. [Google Scholar] [CrossRef]
- Kato, H.; Kudo, A. Water splitting into H2 and O2 on alkali tantalate photocatalysts ATaO3 (A = Li, Na, and K). J. Phys. Chem. B 2001, 105, 4285–4292. [Google Scholar] [CrossRef]
- Jo, W.J.; Kang, H.J.; Kong, K.-J.; Lee, Y.S.; Park, H.; Lee, Y.; Buonassisi, T.; Gleason, K.K.; Lee, J.S. Phase transition-induced band edge engineering of BiVO4 to split pure water under visible light. Proc. Natl. Acad. Sci. USA 2015, 112, 13774–13778. [Google Scholar] [CrossRef]
- Liu, H.; Yuan, J.; Shangguan, W.; Teraoka, Y. Visible-light-responding BiYWO6 solid solution for stoichiometric photocatalytic water splitting. J. Phys. Chem. C 2008, 112, 8521–8523. [Google Scholar] [CrossRef]
- Chiang, T.H.; Lyu, H.; Hisatomi, T.; Goto, Y.; Takata, T.; Katayama, M.; Minegishi, T.; Domen, K. Efficient photocatalytic water splitting using al-doped SrTiO3 coloaded with molybdenum oxide and rhodium–chromium oxide. ACS Catal. 2018, 8, 2782–2788. [Google Scholar] [CrossRef]
- Liao, L.; Zhang, Q.; Su, Z.; Zhao, Z.; Wang, Y.; Li, Y.; Lu, X.; Wei, D.; Feng, G.; Yu, Q.; et al. Efficient solar water-splitting using a nanocrystalline CoO photocatalyst. Nat. Nanotechnol. 2014, 9, 69–73. [Google Scholar] [CrossRef]
- Sakata, Y.; Matsuda, Y.; Yanagida, T.; Hirata, K.; Imamura, H.; Teramura, K. Effect of metal ion addition in a Ni supported Ga2O3 photocatalyst on the photocatalytic overall splitting of H2O. Catal. Lett. 2008, 125, 22–26. [Google Scholar] [CrossRef]
- Sakata, Y.; Hayashi, T.; Yasunaga, R.; Yanaga, N.; Imamura, H. Remarkably high apparent quantum yield of the overall photocatalytic H2O splitting achieved by utilizing Zn ion added Ga2O3 prepared using dilute CaCl2 solution. Chem. Commun. 2015, 51, 12935–12938. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.; Saito, N.; Yamada, Y.; Maeda, K.; Takata, T.; Kondo, J.N.; Hara, M.; Kobayashi, H.; Domen, K.; Inoue, Y. RuO2-Loaded β-Ge3N4 as a Non-Oxide Photocatalyst for Overall Water Splitting. J. Am. Chem. Soc. 2005, 127, 4150–4151. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Takata, T.; Domen, K. Overall water splitting on the transition-metal oxynitride photocatalyst LaMg1/3Ta2/3O2N over a large portion of the visible-light spectrum. Chem. A Eur. J. 2016, 22, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Lu, D.; Domen, K. Direct Water Splitting into Hydrogen and Oxygen under Visible Light by using Modified TaON Photocatalysts with d0 Electronic Configuration. Chem. A Eur. J. 2013, 19, 4986–4991. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Teramura, K.; Domen, K. Effect of post-calcination on photocatalytic activity of (Ga1-xZnx)(N1−xOx) solid solution for overall water splitting under visible light. J. Catal. 2008, 254, 198–204. [Google Scholar] [CrossRef]
- Kibria, M.G.; Nguyen, H.P.; Cui, K.; Zhao, S.; Liu, D.; Guo, H.; Trudeau, M.L.; Paradis, S.; Hakima, A.-R.; Mi, Z. One-step overall water splitting under visible light using multiband InGaN/GaN nanowire heterostructures. ACS Nano 2013, 7, 7886–7893. [Google Scholar] [CrossRef]
- Xu, J.; Pan, C.; Takata, T.; Domen, K. Photocatalytic overall water splitting on the perovskite-type transition metal oxynitride CaTaO2N under visible light irradiation. Chem. Commun. 2015, 51, 7191–7194. [Google Scholar] [CrossRef]
- Pan, C.; Takata, T.; Kumamoto, K.; Ma, S.S.K.; Ueda, K.; Minegishi, T.; Nakabayashi, M.; Matsumoto, T.; Shibata, N.; Ikuhara, Y. Band engineering of perovskite-type transition metal oxynitrides for photocatalytic overall water splitting. J. Mater. Chem. A 2016, 4, 4544–4552. [Google Scholar] [CrossRef]
- Zhang, G.; Lan, Z.-A.; Lin, L.; Lin, S.; Wang, X. Overall water splitting by Pt/g-C3N4 photocatalysts without using sacrificial agents. Chem. Sci. 2016, 7, 3062–3066. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Liu, N.; Han, Y.; Zhang, X.; Huang, H.; Lifshitz, Y.; Lee, S.-T.; Zhong, J.; Kang, Z. Metal-free efficient photocatalyst for stable visible water splitting via a two-electron pathway. Science 2015, 347, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Takata, T.; Domen, K. Particulate photocatalysts for overall water splitting. Nat. Rev. Mater. 2017, 2, 17050. [Google Scholar] [CrossRef]
- Kato, H.; Asakura, K.; Kudo, A. Highly efficient water splitting into H2 and O2 over lanthanum-doped NaTaO3 photocatalysts with high crystallinity and surface nanostructure. J. Am. Chem. Soc. 2003, 125, 3082–3089. [Google Scholar] [CrossRef] [PubMed]
- Ham, Y.; Hisatomi, T.; Goto, Y.; Moriya, Y.; Sakata, Y.; Yamakata, A.; Kubota, J.; Domen, K. Flux-mediated doping of SrTiO3 photocatalysts for efficient overall water splitting. J. Mater. Chem. A 2016, 4, 3027–3033. [Google Scholar] [CrossRef]
- Ikarashi, K.; Sato, J.; Kobayashi, H.; Saito, N.; Nishiyama, H.; Inoue, Y. Photocatalysis for water decomposition by RuO2-dispersed ZnGa2O4 with d10 configuration. J. Phys. Chem. B 2002, 106, 9048–9053. [Google Scholar] [CrossRef]
- Sato, J.; Saito, N.; Nishiyama, H.; Inoue, Y. New photocatalyst group for water decomposition of RuO2-loaded p-block metal (In, Sn, and Sb) oxides with d10 configuration. J. Phys. Chem. B 2001, 105, 6061–6063. [Google Scholar] [CrossRef]
- Inoue, Y. Photocatalytic water splitting by RuO2-loaded metal oxides and nitrides with d0-and d10-related electronic configurations. Energy Environ. Sci. 2009, 2, 364–386. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef]
- Ajmal, A.; Majeed, I.; Malik, R.N.; Idriss, H.; Nadeem, M.A. Principles and mechanisms of photocatalytic dye degradation on TiO2 based photocatalysts: A comparative overview. RSC Adv. 2014, 4, 37003–37026. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 photocatalysis: Mechanisms and materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef]
- Wang, M.; Li, Z.; Wu, Y.; Ma, J.; Lu, G. Inhibition of hydrogen and oxygen reverse recombination reaction over Pt/TiO2 by F− ions and its impact on the photocatalytic hydrogen formation. J. Catal. 2017, 353, 162–170. [Google Scholar] [CrossRef]
- Kitano, M.; Matsuoka, M.; Ueshima, M.; Anpo, M. Recent developments in titanium oxide-based photocatalysts. Appl. Catal. A Gen. 2007, 325, 1–14. [Google Scholar] [CrossRef]
- Gaya, U.I.; Abdullah, A.H. Heterogeneous photocatalytic degradation of organic contaminants over titanium dioxide: A review of fundamentals, progress and problems. J. Photochem. Photobiol. C Photochem. Rev. 2008, 9, 1–12. [Google Scholar] [CrossRef]
- Maeda, K.; Domen, K. New non-oxide photocatalysts designed for overall water splitting under visible light. J. Phys. Chem. C 2007, 111, 7851–7861. [Google Scholar] [CrossRef]
- Kamata, K.; Maeda, K.; Lu, D.; Kako, Y.; Domen, K. Synthesis and photocatalytic activity of gallium–zinc–indium mixed oxynitride for hydrogen and oxygen evolution under visible light. Chem. Phys. Lett. 2009, 470, 90–94. [Google Scholar] [CrossRef]
- Inoue, Y.; Niiyama, T.; Asai, Y.; Sato, K. Stable photocatalytic activity of BaTi4O9 combined with ruthenium oxide for decomposition of water. J. Chem. Soc. Chem. Commun. 1992, 7, 579–580. [Google Scholar] [CrossRef]
- Iwase, A.; Kato, H.; Kudo, A. Nanosized Au particles as an efficient cocatalyst for photocatalytic overall water splitting. Catal. Lett. 2006, 108, 7–10. [Google Scholar] [CrossRef]
- Murdoch, M.; Waterhouse, G.; Nadeem, M.; Metson, J.; Keane, M.; Howe, R.; Llorca, J.; Idriss, H. The effect of gold loading and particle size on photocatalytic hydrogen production from ethanol over Au/TiO2 nanoparticles. Nat. Chem. 2011, 3, 489–492. [Google Scholar] [CrossRef]
- Majeed, I.; Nadeem, M.A.; Hussain, E.; Waterhouse, G.I.N.; Badshah, A.; Iqbal, A.; Nadeem, M.A.; Idriss, H. On the Synergism between Cu and Ni for Photocatalytic Hydrogen Production and their Potential as Substitutes of Noble Metals. ChemCatChem 2016, 8, 3146–3155. [Google Scholar] [CrossRef]
- Li, R.; Han, H.; Zhang, F.; Wang, D.; Li, C. Highly efficient photocatalysts constructed by rational assembly of dual-cocatalysts separately on different facets of BiVO4. Energy Environ. Sci. 2014, 7, 1369–1376. [Google Scholar] [CrossRef]
- Alsabban, M.M.; Yang, X.; Wahyudi, W.; Fu, J.-H.; Hedhili, M.N.; Ming, J.; Yang, C.-W.; Nadeem, M.A.; Idriss, H.; Lai, Z.; et al. Design and Mechanistic Study of Highly Durable Carbon-Coated Cobalt Diphosphide Core–Shell Nanostructure Electrocatalysts for the Efficient and Stable Oxygen Evolution Reaction. ACS Appl. Mater. Interfaces 2019, 11, 20752–20761. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ji, Z.; Zou, W.; Gu, X.; Deng, Y.; Gao, F.; Tang, C.; Dong, L. In situ loading transition metal oxide clusters on TiO2 nanosheets as co-catalysts for exceptional high photoactivity. ACS Catal. 2013, 3, 2052–2061. [Google Scholar] [CrossRef]
- Maeda, K.; Wang, X.; Nishihara, Y.; Lu, D.; Antonietti, M.; Domen, K. Photocatalytic activities of graphitic carbon nitride powder for water reduction and oxidation under visible light. J. Phys. Chem. C 2009, 113, 4940–4947. [Google Scholar] [CrossRef]
- Youngblood, W.J.; Lee, S.-H.A.; Kobayashi, Y.; Hernandez-Pagan, E.A.; Hoertz, P.G.; Moore, T.A.; Moore, A.L.; Gust, D.; Mallouk, T.E. Photoassisted overall water splitting in a visible light-absorbing dye-sensitized photoelectrochemical cell. J. Am. Chem. Soc. 2009, 131, 926–927. [Google Scholar] [CrossRef]
- Kim, H.; Hwang, D.; Kim, Y.; Lee, J. Highly donor-doped (110) layered perovskite materials as novel photocatalysts for overall water splitting. J. Chem. Soc. Chem. Commun. 1999, 12, 1077–1078. [Google Scholar] [CrossRef]
- Sayama, K.; Arakawa, H.; Domen, K. Photocatalytic water splitting on nickel intercalated A4TaxNb6-xO17 (A = K, Rb). Catal. Today 1996, 28, 175–182. [Google Scholar] [CrossRef]
- Laurent-Applegate, L.; Roques, S. Biological Actions of Infrared Radiation. In Sensing, Signaling and Cell Adaptation; Storey, K.B., Storey, J.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2002; pp. 233–242. [Google Scholar]
- Maeda, K.; Takata, T.; Hara, M.; Saito, N.; Inoue, Y.; Kobayashi, H.; Domen, K. GaN:ZnO Solid Solution as a Photocatalyst for Visible-Light-Driven Overall Water Splitting. J. Am. Chem. Soc. 2005, 127, 8286–8287. [Google Scholar] [CrossRef]
- Wang, Z.; Inoue, Y.; Hisatomi, T.; Ishikawa, R.; Wang, Q.; Takata, T.; Chen, S.; Shibata, N.; Ikuhara, Y.; Domen, K. Overall water splitting by Ta3N5 nanorod single crystals grown on the edges of KTaO3 particles. Nat. Catal. 2018, 1, 756–763. [Google Scholar] [CrossRef]
- Murthy, D.H.K.; Matsuzaki, H.; Wang, Z.; Suzuki, Y.; Hisatomi, T.; Seki, K.; Inoue, Y.; Domen, K.; Furube, A. Origin of the overall water splitting activity of Ta3N5 revealed by ultrafast transient absorption spectroscopy. Chem. Sci. 2019, 10, 5353–5362. [Google Scholar] [CrossRef]
- Kudo, A. Recent progress in the development of visible light-driven powdered photocatalysts for water splitting. Int. J. Hydrog. Energy 2007, 32, 2673–2678. [Google Scholar] [CrossRef]
- Konta, R.; Ishii, T.; Kato, H.; Kudo, A. Photocatalytic Activities of Noble Metal Ion Doped SrTiO3 under Visible Light Irradiation. J. Phys. Chem. B 2004, 108, 8992–8995. [Google Scholar] [CrossRef]
- Park, K.-W.; Kolpak, A.M. Understanding photocatalytic overall water splitting on CoO nanoparticles: Effects of facets, surface stoichiometry, and the CoO/water interface. J. Catal. 2018, 365, 115–124. [Google Scholar] [CrossRef]
- Wang, X.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J.M.; Domen, K.; Antonietti, M. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 2009, 8, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.J.; Reardon, P.J.T.; Moniz, S.J.A.; Tang, J. Visible Light-Driven Pure Water Splitting by a Nature-Inspired Organic Semiconductor-Based System. J. Am. Chem. Soc. 2014, 136, 12568–12571. [Google Scholar] [CrossRef] [PubMed]
- Ng, B.J.; Putri, L.K.; Kong, X.Y.; Teh, Y.W.; Pasbakhsh, P.; Chai, S.P. Z-Scheme Photocatalytic Systems for Solar Water Splitting. Adv. Sci. (Weinh. Baden-Wurtt. Ger.) 2020, 7, 1903171. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Sayama, K.; Domen, K.; Arakawa, H. A new type of water splitting system composed of two different TiO2 photocatalysts (anatase, rutile) and a IO3−/I− shuttle redox mediator. Chem. Phys. Lett. 2001, 344, 339–344. [Google Scholar] [CrossRef]
- Ryu, A. Development of a New System for Photocatalytic Water Splitting into H2 and O2 under Visible Light Irradiation. Bull. Chem. Soc. Jpn. 2011, 84, 1000–1030. [Google Scholar] [CrossRef]
- Ma, G.; Chen, S.; Kuang, Y.; Akiyama, S.; Hisatomi, T.; Nakabayashi, M.; Shibata, N.; Katayama, M.; Minegishi, T.; Domen, K. Visible Light-Driven Z-Scheme Water Splitting Using Oxysulfide H2 Evolution Photocatalysts. J. Phys. Chem. Lett. 2016, 7, 3892–3896. [Google Scholar] [CrossRef]
- Qi, Y.; Chen, S.; Li, M.; Ding, Q.; Li, Z.; Cui, J.; Dong, B.; Zhang, F.; Li, C. Achievement of visible-light-driven Z-scheme overall water splitting using barium-modified Ta3N5 as a H2-evolving photocatalyst. Chem. Sci. 2017, 8, 437–443. [Google Scholar] [CrossRef]
- Kato, T.; Hakari, Y.; Ikeda, S.; Jia, Q.; Iwase, A.; Kudo, A. Utilization of Metal Sulfide Material of (CuGa)1–xZn2xS2 Solid Solution with Visible Light Response in Photocatalytic and Photoelectrochemical Solar Water Splitting Systems. J. Phys. Chem. Lett. 2015, 6, 1042–1047. [Google Scholar] [CrossRef]
- Chen, S.; Qi, Y.; Hisatomi, T.; Ding, Q.; Asai, T.; Li, Z.; Ma, S.S.K.; Zhang, F.; Domen, K.; Li, C. Efficient Visible-Light-Driven Z-Scheme Overall Water Splitting Using a MgTa2O6−xNy /TaON Heterostructure Photocatalyst for H2 Evolution. Angew. Chem. Int. Ed. 2015, 54, 8498–8501. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, J.; Li, C.; Tian, W. Achieving solar overall water splitting with hybrid photosystems of photosystem II and artificial photocatalysts. Nat. Commun. 2014, 5, 4647. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Higashi, M.; Lu, D.; Abe, R.; Domen, K. Efficient nonsacrificial water splitting through two-step photoexcitation by visible light using a modified oxynitride as a hydrogen evolution photocatalyst. J. Am. Chem. Soc. 2010, 132, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Hori, M.; Konta, R.; Shimodaira, Y.; Kudo, A. Construction of Z-scheme type heterogeneous photocatalysis systems for water splitting into H2 and O2 under visible light irradiation. Chem. Lett. 2004, 33, 1348–1349. [Google Scholar] [CrossRef]
- Abe, R.; Sayama, K.; Sugihara, H. Development of new photocatalytic water splitting into H2 and O2 using two different semiconductor photocatalysts and a shuttle redox mediator IO3-/I. J. Phys. Chem. B 2005, 109, 16052–16061. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Higashi, M.; Domen, K. Overall Water Splitting under Visible Light through a Two-Step Photoexcitation between TaON and WO3 in the Presence of an Iodate–Iodide Shuttle Redox Mediator. ChemSusChem 2011, 4, 228–237. [Google Scholar] [CrossRef]
- Tsuji, K.; Tomita, O.; Higashi, M.; Abe, R. Manganese-Substituted Polyoxometalate as an Effective Shuttle Redox Mediator in Z-Scheme Water Splitting under Visible Light. ChemSusChem 2016, 9, 2201–2208. [Google Scholar] [CrossRef]
- Sasaki, Y.; Kato, H.; Kudo, A. [Co(bpy)3]3+/2+ and [Co(phen)3]3+/2+ Electron Mediators for Overall Water Splitting under Sunlight Irradiation Using Z-Scheme Photocatalyst System. J. Am. Chem. Soc. 2013, 135, 5441–5449. [Google Scholar] [CrossRef]
- Zhao, W.; Maeda, K.; Zhang, F.; Hisatomi, T.; Domen, K. Effect of post-treatments on the photocatalytic activity of Sm2Ti2S2O5 for the hydrogen evolution reaction. Phys. Chem. Chem. Phys. 2014, 16, 12051–12056. [Google Scholar] [CrossRef]
- Sasaki, Y.; Nemoto, H.; Saito, K.; Kudo, A. Solar Water Splitting Using Powdered Photocatalysts Driven by Z-Schematic Interparticle Electron Transfer without an Electron Mediator. J. Phys. Chem. C 2009, 113, 17536–17542. [Google Scholar] [CrossRef]
- Tada, H.; Mitsui, T.; Kiyonaga, T.; Akita, T.; Tanaka, K. All-solid-state Z-scheme in CdS–Au–TiO2 three-component nanojunction system. Nat. Mater. 2006, 5, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Jeong, E.D.; Borse, P.H.; Jeon, S.; Yong, K.; Lee, J.S.; Li, W.; Oh, S.H. Photocatalytic Ohmic layered nanocomposite for efficient utilization of visible light photons. Appl. Phys. Lett. 2006, 89, 064103. [Google Scholar] [CrossRef]
- Bai, S.; Jiang, J.; Zhang, Q.; Xiong, Y. Steering charge kinetics in photocatalysis: Intersection of materials syntheses, characterization techniques and theoretical simulations. Chem. Soc. Rev. 2015, 44, 2893–2939. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Song, M.; Wang, H.; Zhang, X.; Sui, N.; Zhang, Q.; Colvin, V.L.; Yu, W.W. Latest progress in constructing solid-state Z scheme photocatalysts for water splitting. Nanoscale 2019, 11, 11071–11082. [Google Scholar] [CrossRef] [PubMed]
- Hisatomi, T.; Takanabe, K.; Domen, K. Photocatalytic Water-Splitting Reaction from Catalytic and Kinetic Perspectives. Catal. Lett. 2015, 145, 95–108. [Google Scholar] [CrossRef]
- Tang, J.; Durrant, J.R.; Klug, D.R. Mechanism of Photocatalytic Water Splitting in TiO2. Reaction of Water with Photoholes, Importance of Charge Carrier Dynamics, and Evidence for Four-Hole Chemistry. J. Am. Chem. Soc. 2008, 130, 13885–13891. [Google Scholar] [CrossRef]
- Scott, M.; Nadeem, A.M.; Waterhouse, G.I.W.; Idriss, H. Hydrogen Production from Ethanol. Comparing Thermal Catalytic Reactions to Photo-catalytic Reactions. MRS Proc. 2011, 1326, mrss11-1326-f1307-1307. [Google Scholar] [CrossRef]
- Leytner, S.; Hupp, J.T. Evaluation of the energetics of electron trap states at the nanocrystalline titanium dioxide/aqueous solution interface via time-resolved photoacoustic spectroscopy. Chem. Phys. Lett. 2000, 330, 231–236. [Google Scholar] [CrossRef]
- Majeed, I.; Nadeem, M.A.; Al-Oufi, M.; Nadeem, M.A.; Waterhouse, G.I.N.; Badshah, A.; Metson, J.B.; Idriss, H. On the role of metal particle size and surface coverage for photo-catalytic hydrogen production: A case study of the Au/CdS system. Appl. Catal. B Environ. 2016, 182, 266–276. [Google Scholar] [CrossRef]
- Nadeem, M.A.; Murdoch, M.; Waterhouse, G.I.N.; Metson, J.B.; Keane, M.A.; Llorca, J.; Idriss, H. Photoreaction of ethanol on Au/TiO2 anatase: Comparing the micro to nanoparticle size activities of the support for hydrogen production. J. Photochem. Photobiol. A Chem. 2010, 216, 250–255. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Q.; Li, M.; Shen, S.; Wang, X.; Wang, Y.; Feng, Z.; Shi, J.; Han, H.; Li, C. Photocatalytic overall water splitting promoted by an α–β phase junction on Ga2O3. Angew. Chem. Int. Ed. 2012, 51, 13089–13092. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Peng, Y.-K.; Hu, L.; Zheng, J.; Prabhakaran, D.; Wu, S.; Puchtler, T.J.; Li, M.; Wong, K.-Y.; Taylor, R.A.; et al. Photocatalytic water splitting by N-TiO2 on MgO (111) with exceptional quantum efficiencies at elevated temperatures. Nat. Commun. 2019, 10, 4421. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Ren, B.; Chang, S.; Mi, W.; He, J.; Wang, W. Achieving effective control of the photocatalytic performance for CoFe2O4/MoS2 heterojunction via exerting external magnetic fields. Mater. Lett. 2020, 260, 126979. [Google Scholar] [CrossRef]
- Gao, W.; Lu, J.; Zhang, S.; Zhang, X.; Wang, Z.; Qin, W.; Wang, J.; Zhou, W.; Liu, H.; Sang, Y. Suppressing Photoinduced Charge Recombination via the Lorentz Force in a Photocatalytic System. Adv. Sci. 2019, 6, 1901244. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pei, Q.; Wang, R.; Zhou, Y.; Zhang, Z.; Cao, Q.; Wang, D.; Mi, W.; Du, Y. Enhanced Photocatalytic Performance through Magnetic Field Boosting Carrier Transport. ACS Nano 2018, 12, 3351–3359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yates, J.T. Band Bending in Semiconductors: Chemical and Physical Consequences at Surfaces and Interfaces. Chem. Rev. 2012, 112, 5520–5551. [Google Scholar] [CrossRef]
- Yoshida, Y.; Matsuoka, M.; Moon, S.; Mametsuka, H.; Suzuki, E.; Anpo, M. Photocatalytic decomposition of liquid-water on the Pt-loaded TiO2 catalysts: Effects of the oxidation states of Pt species on the photocatalytic reactivity and the rate of the back reaction. Res. Chem. Intermed. 2000, 26, 567–574. [Google Scholar] [CrossRef]
- Sanap, K.K.; Varma, S.; Dalavi, D.; Patil, P.; Waghmode, S.; Bharadwaj, S. Variation in noble metal morphology and its impact on functioning of hydrogen mitigation catalyst. Int. J. Hydrog. Energy 2011, 36, 10455–10467. [Google Scholar] [CrossRef]
- Berto, T.F.; Sanwald, K.E.; Byers, J.P.; Browning, N.D.; Gutiérrez, O.Y.; Lercher, J.A. Enabling overall water splitting on photocatalysts by CO-covered noble metal co-catalysts. J. Phys. Chem. Lett. 2016, 7, 4358–4362. [Google Scholar] [CrossRef]
- Dionigi, F.; Vesborg, P.C.; Pedersen, T.; Hansen, O.; Dahl, S.; Xiong, A.; Maeda, K.; Domen, K.; Chorkendorff, I. Suppression of the water splitting back reaction on GaN:ZnO photocatalysts loaded with core/shell cocatalysts, investigated using a μ-reactor. J. Catal. 2012, 292, 26–31. [Google Scholar] [CrossRef]
- Yoshida, M.; Takanabe, K.; Maeda, K.; Ishikawa, A.; Kubota, J.; Sakata, Y.; Ikezawa, Y.; Domen, K. Role and function of noble-metal/Cr-layer core/shell structure cocatalysts for photocatalytic overall water splitting studied by model electrodes. J. Phys. Chem. C 2009, 113, 10151–10157. [Google Scholar] [CrossRef]
- Takata, T.; Pan, C.; Nakabayashi, M.; Shibata, N.; Domen, K. Fabrication of a core–shell-type photocatalyst via photodeposition of group IV and V transition metal oxyhydroxides: An effective surface modification method for overall water splitting. J. Am. Chem. Soc. 2015, 137, 9627–9634. [Google Scholar] [CrossRef] [PubMed]
- Abe, R.; Sayama, K.; Arakawa, H. Significant effect of iodide addition on water splitting into H2 and O2 over Pt-loaded TiO2 photocatalyst: Suppression of backward reaction. Chem. Phys. Lett. 2003, 371, 360–364. [Google Scholar] [CrossRef]
- Nadeem, M.; Idriss, H. Photo-thermal reactions of ethanol over Ag/TiO2 catalysts. The role of silver plasmon resonance in the reaction kinetics. Chem. Commun. 2018, 54, 5197–5200. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hisatomi, T.; Jia, Q.; Tokudome, H.; Zhong, M.; Wang, C.; Pan, Z.; Takata, T.; Nakabayashi, M.; Shibata, N.; et al. Scalable water splitting on particulate photocatalyst sheets with a solar-to-hydrogen energy conversion efficiency exceeding 1%. Nat. Mater. 2016, 15, 611–615. [Google Scholar] [CrossRef] [PubMed]
- Standard Test Methods for Measurement of Electrical Performance and Spectral Response of Nonconcentrator Multijunction Photovoltaic Cells and Modules. In E2236-10(2019); ASTM International: West Conshohocken, PA, USA, 2019.
- Ipek, B.; Uner, D. Artificial photosynthesis from a chemical engineering perspective. In Artificial Photosynthesis; IntechOpen: London, UK, 2012; pp. 13–36. [Google Scholar]
- Skillen, N.; Adams, M.; McCullagh, C.; Ryu, S.Y.; Fina, F.; Hoffmann, M.R.; Irvine, J.T.S.; Robertson, P.K.J. application of a novel fluidised photo reactor under UV–Visible and natural solar irradiation in the photocatalytic generation of hydrogen. Chem. Eng. J. 2016, 286, 610–621. [Google Scholar] [CrossRef][Green Version]
- Reilly, K.; Wilkinson, D.P.; Taghipour, F. Photocatalytic water splitting in a fluidized bed system: Computational modeling and experimental studies. Appl. Energy 2018, 222, 423–436. [Google Scholar] [CrossRef]
- Chen, D.; Li, F.; Ray, A.K. Effect of mass transfer and catalyst layer thickness on photocatalytic reaction. AIChE J. 2000, 46, 1034–1045. [Google Scholar] [CrossRef]
- Wang, Q.; Hisatomi, T.; Katayama, M.; Takata, T.; Minegishi, T.; Kudo, A.; Yamada, T.; Domen, K. Particulate photocatalyst sheets for Z-scheme water splitting: Advantages over powder suspension and photoelectrochemical systems and future challenges. Faraday Discuss. 2017, 197, 491–504. [Google Scholar] [CrossRef]
- Paramelle, D.; Sadovoy, A.; Gorelik, S.; Free, P.; Hobley, J.; Fernig, D.G. A rapid method to estimate the concentration of citrate capped silver nanoparticles from UV-visible light spectra. Analyst 2014, 139, 4855–4861. [Google Scholar] [CrossRef]
- Huang, X.; El-Sayed, M.A. Gold nanoparticles: Optical properties and implementations in cancer diagnosis and photothermal therapy. J. Adv. Res. 2010, 1, 13–28. [Google Scholar] [CrossRef]
- Khan, M.A.; Sinatra, L.; Oufi, M.; Bakr, O.M.; Idriss, H. Evidence of Plasmonic Induced Photocatalytic Hydrogen Production on Pd/TiO2 Upon Deposition on Thin Films of Gold. Catal. Lett. 2017, 147, 811–820. [Google Scholar] [CrossRef]
- Majeed, I.; Nadeem, M.A.; Hussain, E.; Badshah, A.; Gilani, R.; Nadeem, M.A. Effect of deposition method on metal loading and photocatalytic activity of Au/CdS for hydrogen production in water electrolyte mixture. Int. J. Hydrog. Energy 2017, 42, 3006–3018. [Google Scholar] [CrossRef]
- Mubeen, S.; Lee, J.; Singh, N.; Krämer, S.; Stucky, G.D.; Moskovits, M. An autonomous photosynthetic device in which all charge carriers derive from surface plasmons. Nat. Nanotechnol. 2013, 8, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Wolff, C.M.; Frischmann, P.D.; Schulze, M.; Bohn, B.J.; Wein, R.; Livadas, P.; Carlson, M.T.; Jäckel, F.; Feldmann, J.; Würthner, F.; et al. All-in-one visible-light-driven water splitting by combining nanoparticulate and molecular co-catalysts on CdS nanorods. Nat. Energy 2018, 3, 862–869. [Google Scholar] [CrossRef]
- Lyu, H.; Hisatomi, T.; Goto, Y.; Yoshida, M.; Higashi, T.; Katayama, M.; Takata, T.; Minegishi, T.; Nishiyama, H.; Yamada, T.; et al. An Al-doped SrTiO3 photocatalyst maintaining sunlight-driven overall water splitting activity for over 1000 h of constant illumination. Chem. Sci. 2019, 10, 3196–3201. [Google Scholar] [CrossRef]
- Takata, T.; Jiang, J.; Sakata, Y.; Nakabayashi, M.; Shibata, N.; Nandal, V.; Seki, K.; Hisatomi, T.; Domen, K. Photocatalytic water splitting with a quantum efficiency of almost unity. Nature 2020, 581, 411–414. [Google Scholar] [CrossRef]
- Alsayegh, S.; Johnson, J.; Ohs, B.; Lohaus, J.; Wessling, M.J.I.J.o.H.E. Systematic optimization of H2 recovery from water splitting process using membranes and N2 diluent. Int. J. Hydrog. Energy 2017, 42, 6000–6011. [Google Scholar] [CrossRef]
- Alsayegh, S.; Johnson, J.; Wei, X.; Ohs, B.; Lohaus, J.; Wessling, M. CO2 aided H2 recovery from water splitting processes. Int. J. Hydrog. Energy 2017, 42, 21793–21805. [Google Scholar] [CrossRef]
- Kibria, M.G.; Chowdhury, F.A.; Zhao, S.; AlOtaibi, B.; Trudeau, M.L.; Guo, H.; Mi, Z. Visible light-driven efficient overall water splitting using p-type metal-nitride nanowire arrays. Nat. Commun. 2015, 6, 6797. [Google Scholar] [CrossRef]
- Takata, T.; Pan, C.; Domen, K. Design and Development of Oxynitride Photocatalysts for Overall Water Splitting under Visible Light Irradiation. ChemElectroChem 2016, 3, 31–37. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, C.; Dong, H.; Shen, J.-R.; Dau, H.; Zhao, J. A synthetic Mn4Ca-cluster mimicking the oxygen-evolving center of photosynthesis. Science 2015, 348, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Kanan, M.W.; Nocera, D.G. In Situ Formation of an Oxygen-Evolving Catalyst in Neutral Water Containing Phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Teramura, K.; Hosokawa, S.; Kominami, H.; Tanaka, T. Visible light-induced water splitting in an aqueous suspension of a plasmonic Au/TiO2 photocatalyst with metal co-catalysts. Chem. Sci. 2017, 8, 2574–2580. [Google Scholar] [CrossRef]
- Zhu, J.; Fan, F.; Chen, R.; An, H.; Feng, Z.; Li, C. Direct Imaging of Highly Anisotropic Photogenerated Charge Separations on Different Facets of a Single BiVO4 Photocatalyst. Angew. Chem. Int. Ed. 2015, 54, 9111–9114. [Google Scholar] [CrossRef]










| Photocatalysts | Preparation Method | Examples | Cocatalyst Loading | Examples | ||||
|---|---|---|---|---|---|---|---|---|
| Metal oxides | → | Molten salt (Flux) Solid state reactions Ammonia precipitation | → | SrTiO3:Al, SrTiO3:Rh,Sb, La2Ti2O7:Ba, NaTaO3, Ga2O3:Zn, BiYWO6, Bi1−xInxV1−xMoxO4 | → | Impregnation Photodeposition | → | NiOx, CoOx, IrO2, RuO2, Rh2−yCryO3 |
| Calcination under controlled atmosphere | → | MxOy | ||||||
| Metal (oxy) nitrides | → | Thermal nitridation of metal oxides using NH3 | → | Ge3N4, TaON:ZrO2, (Zn0.18Ga0.82)(N0.82O0.18), LaMg1/3Ta2/3O2N, CaTaO2N, Ta3N5, LaScxTa1−xO1−2xN2−2x, GaN:Mg/InGaN:Mg | → | Impregnation Photodeposition | → | RuO2, Rh2−yCryO3 |
| Metal-free photocatalysts | → | Thermal polymerization Electrochemical | → | g-C3N4, C-dot/g-C3N4 | ||||
| Semiconductor | Metal/Metal Oxide (wt.%) | Eg (eV) | H2 Rate (mmolh−1) | O2 Rate (mmolh−1) | AQY (%) | Ref. |
|---|---|---|---|---|---|---|
| La2Ti2O7:Ba(8.0 mol %) | Ni (2.0) | 3.2 | 5 | 2.5 | 50 (not given) | [49] |
| SrTiO3:Al(0.1 mol %) | RhxCryO3 (Rh = 0.1, Cr = 0.1) | 3.2 | 1.4 | 0.7 | 56 at 365 nm | [15,54] |
| SrTiO3:Al(0.1 mol %) | MoOy/RhCrOx (Mo = 0.03, Rh = 0.1, Cr = 0.1) | 3.2 | 1.8 | 0.9 | 69 at 365 nm | [54] |
| SrTiO3:Rh,Sb(0.5 & 2.0 wt.%) | IrO2 (3.0) | 3.2 | 4.4 | 1.9 | 0.1 at 420 nm | [50] |
| NaTaO3 | NiO (0.05) | 4.0 | 3.4 | 1.6 | 20 at 270 nm | [51] |
| Ga2O3:Zn(1.0 mol %) | Ni (1.0) | 4.4 | 4.1 | 2.2 | 20 at 270 nm | [56] |
| Ga2O3:Zn(3.0 mol %) | RhxCryO3 (Rh = 0.5, Cr = 1.5) | 4.4 | 3.2 | 1.6 | 71 at 254 nm | [57] |
| Ge3N4 | RuO2 (1.0) | 3.8 | 0.2 | 0.1 | 9 at 300 nm | [58] |
| Semiconductor | Metal Oxide (wt.% Unless Indicated) | Eg (eV) | H2 Rate (µmolh−1) | O2 Rate (µmolh−1) | AQY (%) | Ref. |
|---|---|---|---|---|---|---|
| Bi1−xInxV1−xMoxO4 | RuO2(3.0) | 2.5 | 17 | 7.8 | 3.2 at 420 nm | [52] |
| BiYWO6 | RuO2(1.0) | 2.7 | 4.1 | 1.8 | 0.17 at 420 nm | [53] |
| LaMg1/3Ta2/3O2N | RhCrOx (Rh = 0.5 Cr = 0.5) | - | 22 | 11 | 0.18 at 440 nm | [59] |
| TaON:ZrO2 (Zr/Ta = 0.1) | RuOx/Cr2O3/IrO2 (Ru = 3.0, Cr = 2.5, Ir/Ta = 0.04) | 2.5 | 6.7 × 10−3 | 2.3 × 10−3 | <0.1 at 420 nm | [60] |
| CoO | - | 2.6 | 1785 | 848 | 5% (STH) | [55] |
| (Zn0.18Ga0.82) (N0.82O0.18) | Rh2−yCryO3 (Rh = 2.5, Cr = 2.0) | 2.64 | 927 | 460 | 5.9 at 420 nm | [61] |
| GaN:Mg/InGaN (grown using MBE) | Rh/Cr2O3 (Not applicable) | 2.22 | 38 | 21 | 12.3 at 400 nm | [62] |
| CaTaO2N | RhCrOy (Rh = 0.5, Cr = 0.5) | 2.43 | 14 × 10−2 | 7 × 10−2 | 0.003 at 440 nm | [63] |
| LaSc0.5Ta0.5O2N | RhCrOy (Rh = 0.5, Cr = 0.5) | 2.1 | 2.4 × 10−3 | 1.2 × 10−3 | - | [64] |
| Ta3N5/KTaO3 (Ta3N5 = 1.4 wt.%) | Rh/Cr2O3 (Rh = 0.002, Cr = 0.004) | 2.1 | 6 × 10−3 | 3 × 10−3 | 0.25 at 400 nm | [94] |
| g-C3N4 | CoOx (1.0) | 2.8 | 8.5 | 3.5 | 0.3 at 405 nm | [65] |
| C3N4/C-dots | - | 2.74 | 46 | - | 16 at 420 nm | [66] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nadeem, M.A.; Khan, M.A.; Ziani, A.A.; Idriss, H. An Overview of the Photocatalytic Water Splitting over Suspended Particles. Catalysts 2021, 11, 60. https://doi.org/10.3390/catal11010060
Nadeem MA, Khan MA, Ziani AA, Idriss H. An Overview of the Photocatalytic Water Splitting over Suspended Particles. Catalysts. 2021; 11(1):60. https://doi.org/10.3390/catal11010060
Chicago/Turabian StyleNadeem, Muhammad Amtiaz, Mohd Adnan Khan, Ahmed Abdeslam Ziani, and Hicham Idriss. 2021. "An Overview of the Photocatalytic Water Splitting over Suspended Particles" Catalysts 11, no. 1: 60. https://doi.org/10.3390/catal11010060
APA StyleNadeem, M. A., Khan, M. A., Ziani, A. A., & Idriss, H. (2021). An Overview of the Photocatalytic Water Splitting over Suspended Particles. Catalysts, 11(1), 60. https://doi.org/10.3390/catal11010060