Abstract
The utilisation of fossil fuels has resulted in the continuous increase in anthropogenic carbon dioxide (CO2) emissions and has led to significant environmental impacts. To this end, the catalytic hydrogenation of captured CO2 into value-added C1 chemicals has attracted great attention. In this case, significant research efforts have been directed towards the development of heterogeneous catalysts. Owing to the unique properties and functionalities of hydridic hydrogen (H−), metal hydrides have shown great promise in hydrogen-involved catalytic processes. This is attributed to their enhanced hydrogen (H2) absorption-desorption reversibility and newly developed active sites. Nevertheless, their application in the activation and hydrogenation of CO2 has been overlooked. In this review paper, we provide an overview of recent advances in catalytic CO2 hydrogenation using metal hydride-based materials. Firstly, the reaction mechanisms of CO2 hydrogenation toward different C1 products (CO, CH4, CH3OH and HCOOH) are introduced to better understand their application trend. Thereafter, we highlight the challenges of developing robust hydride catalysts with different components and structures that enable tuning of their catalytic activity and selectivity. A brief introduction of the CO2 hydrogenation over typical homogeneous metal hydrides complexes is also presented. Lastly, conclusion, future outlook and perspectives are discussed.
1. Introduction
Human-induced activities have resulted in increased greenhouse gas (GHG) emissions, particularly CO2, which have led to the accelerated rate of global warming beyond the pre-industrial stage [1]. The continuous releases of CO2 emissions have a significant effect on the earth’s natural carbon cycle. The increased concentration of CO2 in the atmosphere results in great environmental impacts such as global warming, rises in sea level, ocean acidification and climate change [2]. The transformation of CO2 by using renewable green hydrogen into sustainable and value-added C1 chemicals such as methanol (CH3OH), formic acid (HCOOH) and methane (CH4) can be an ideal solution to mitigate the emissions of CO2, depleting fossil resources, and maintaining energy security. Due to its strong thermodynamic stability and kinetic inertness, the non-polar covalent CO2 has a very low conversion efficiency and relatively high water solubility (1.45 g L−1) at standard temperature and pressure [3]. This has led to extreme difficulty in CO2 activation and conversion to value-added hydrocarbons [4]. The catalytic hydrogenation of greenhouse CO2 to C1 chemicals is a promising route to store energy within chemical bonds and/or convert it into green, renewable chemicals [5].
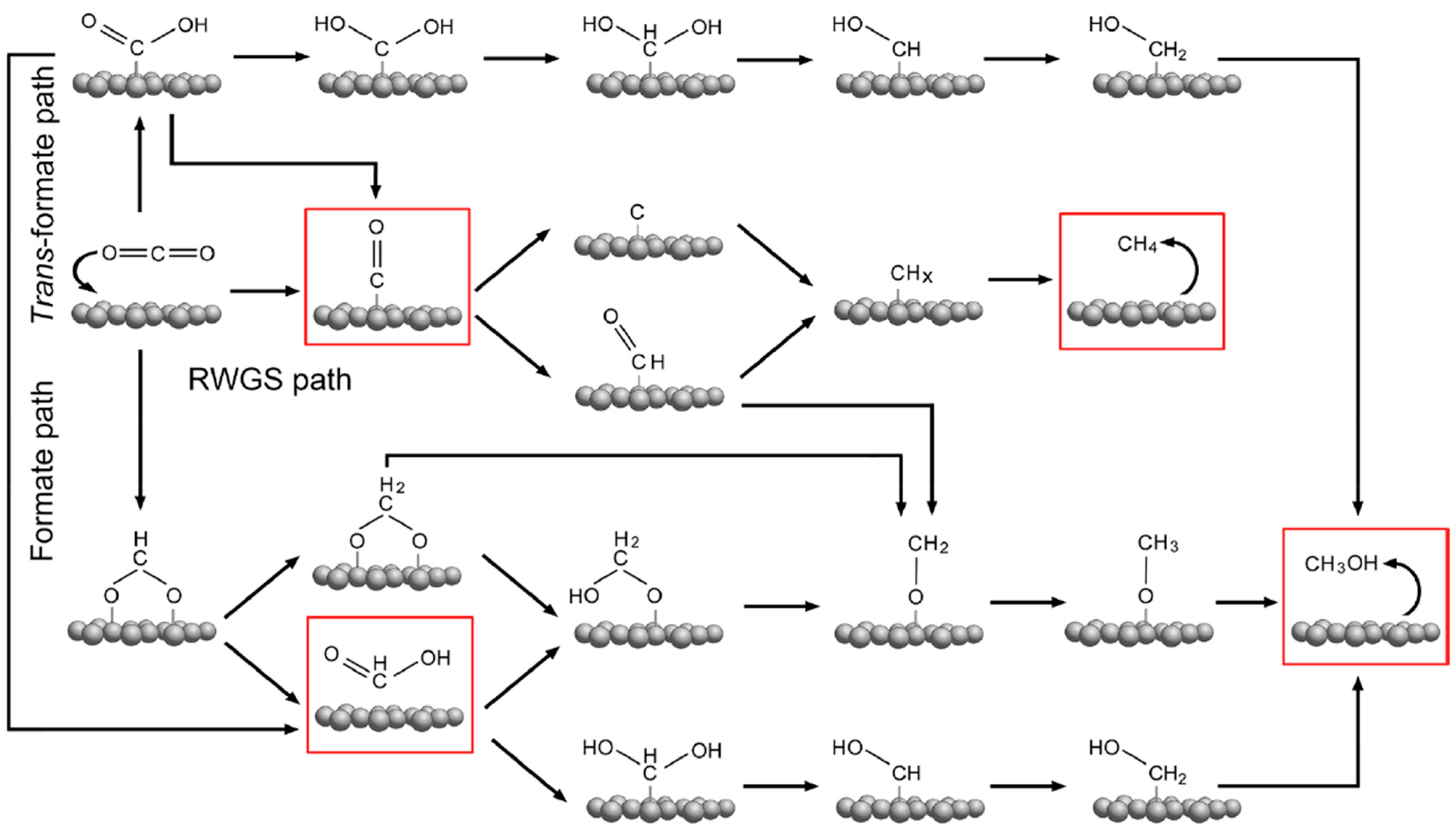
Catalysts play an important role in promoting the activation and conversion of CO2 and dissociation of hydrogen (H2), and thus yielding desired products, particularly C1 chemicals. This is based on the recent studies reported using different catalysts [6,7,8]. The C1 chemicals find applications in various fields, such as transport, textile, agriculture, pharmaceutical, petroleum, and the food industry, and in particular, as feedstock for the manufacturing of pharmaceuticals, plastics, leathers, dyes, rubbers etc. (Figure 1) [9,10,11,12]. They also play an important role as a potential clean energy carrier and storage, and substitute fuels in fuel cells, ships, and vehicles [13]. In heterogeneous catalysis, the reaction mechanism can be influenced by the different exposure of catalyst surfaces, defects, or electronic structures [14,15]. In recent years, in situ experimental and advanced theoretical studies have given advances in the identification and presentation of mechanisms in the CO2 hydrogenation reactions into C1 chemicals. The simplified reaction pathways for CO2 hydrogenation to different C1 products, i.e., methane, formic acid, carbon monoxide and methanol have been summarised in Figure 2 [16]. On the other hand, the reaction equations and thermodynamic data of CO2 hydrogenation to produce different C1 chemicals are described by Equations 1 to 5 [15,17,18,19,20,21,22].
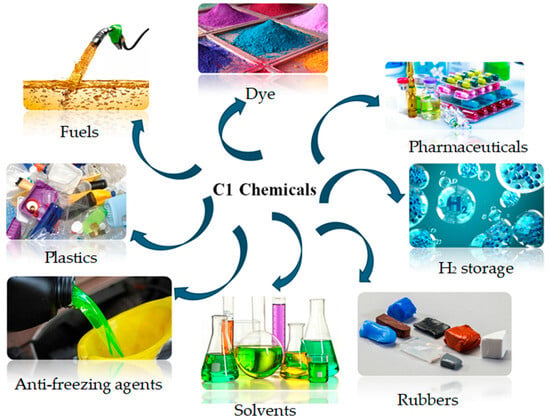
Figure 1.
Schematic diagram for the applications of C1 chemicals.
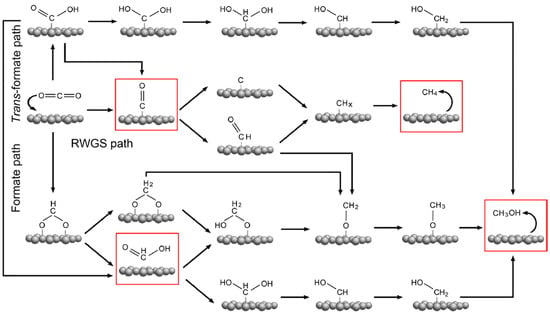
Figure 2.
Reaction mechanisms of CO2 hydrogenation to C1chemicals: methane, carbon monoxide, formic acid and methanol. Note: the C1 chemicals are in a highlighted red box. Adapted from Wang et al. [16].
CO2(g) + 4 H2(g)  CH4(g) + 2 H2O(g) ΔG298K = −113.5 kJ/mol
CH4(g) + 2 H2O(g) ΔG298K = −113.5 kJ/mol
ΔH298K = −164.7 kJ/mol
CH4(g) + 2 H2O(g) ΔG298K = −113.5 kJ/molΔH298K = −164.7 kJ/mol
CO2(g) + H2(g) CO(g) + H2O(g) ΔG298K = +28.6 kJ/mol
ΔH298K = +41.2 kJ/mol
CO(g) + H2O(g) ΔG298K = +28.6 kJ/molΔH298K = +41.2 kJ/mol
CO2(g) + H2(g) HCOOH(l) ∆G298K = +32.9 KJ/mol
∆H298K = −31.2 KJ/mol
HCOOH(l) ∆G298K = +32.9 KJ/mol∆H298K = −31.2 KJ/mol
CO2(g) + H2(g) + NH3(aq) HCOO−(aq) + NH4+(aq) ∆G298K = −9.5 KJ/mol
∆H298K = −83.4 KJ/mol
HCOO−(aq) + NH4+(aq) ∆G298K = −9.5 KJ/mol∆H298K = −83.4 KJ/mol
CO2(g) + 3 H2(g) CH3OH(l) + H2O(g) ΔG298K = 3.5 kJ/mol
∆H298K = −49.5 KJ/mol
CH3OH(l) + H2O(g) ΔG298K = 3.5 kJ/mol∆H298K = −49.5 KJ/mol
During the catalytic process on a solid catalyst surface, the CO2 molecules undergo adsorption, dissociation, and bond cleavage. Since the direct dissociation of CO2 into intermediate species such as CO and O atoms [23,24] is one of the key steps in the hydrogenation reaction into C1 chemicals such as methane and methanol [16], it is important to select a functional catalyst with a surface that can adsorb CO and O adequately. The catalyst must also promote the hydrogenation of the dissociated intermediate species into the desired products. The surface should not have a strong binding force to avoid surface poisoning and further prevent the adsorption of CO2 [25,26]. For example, the Cu(110) surface exhibited the most activity towards CO2 dissociation into CO and atomic O. However, the adsorbed atomic oxygen was found to poison and block the surface, leading to the inhibition of further CO2 adsorption [25]. In contrast, by increasing the catalytic reaction temperature and applying a molar ratio of CO2:H2 of 1:1, the atomic O adsorbed at the surface of Pt(111) favoured the reverse water gas shift (RWGS) reaction due to the reduction in the dissociated O2 [26]. Metal hydrides are starting to emerge and showing significant improvements in the hydrogen-involved catalytic process such as ammonia synthesis, alkyne hydrogenation and carbon dioxide methanation. In the next chapter we will highlight the properties of metal hydrides and their applications in hydrogenation reactions.
2. Metal Hydrides
2.1. Basic Properties of Metal Hydrides
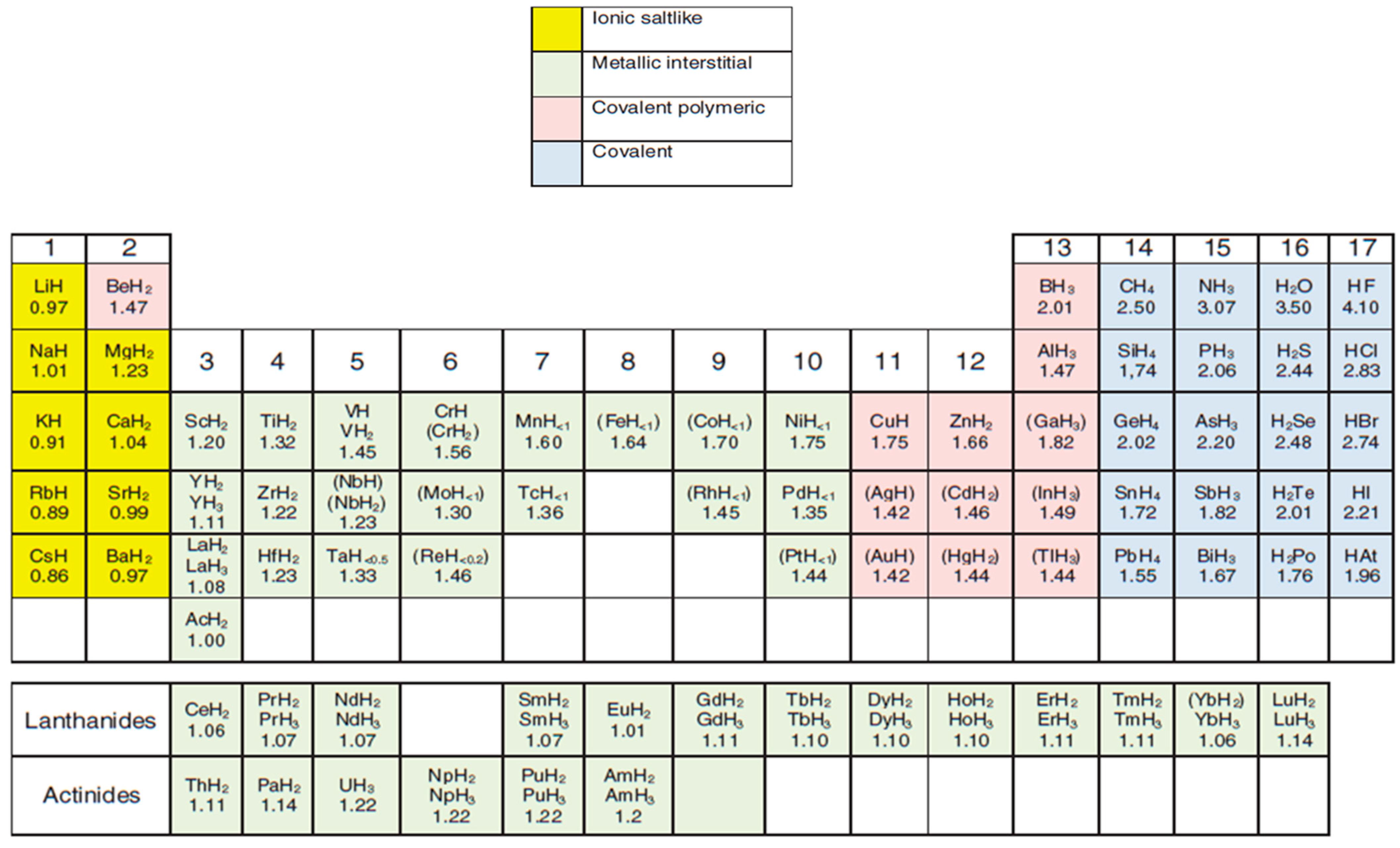
Metal hydrides are compounds that consist of metal cations and hydride anions. When pressurised, metals bind with hydrogen to form stable metal hydrides, which can be used for convenient hydrogen storage [27]. Metal hydrides have been known throughout history dating back to the 19th century. Potassium hydride was the first to be synthesised in 1811 by chemically reacting and heating metal potassium with gaseous hydrogen molecules [28]. The equilibrium pressure plateau for the alkali metals (potassium, sodium and lithium) reacting with hydrogen was first measured in 1874 [29]. Metal hydrides possess reversible hydrogen absorption-desorption ability under suitable reaction conditions [30,31]. As a result, they can be applied in H2 storage and in other fields such as in hydrogen-involved catalytic reactions [32]. Metal hydrides play a fundamental role in the environmental and energy catalytic reactions associated with hydrogenolysis, dehydrogenation, reforming, and catalysed hydrogenation [30]. They chemically bind hydrogen and form a dynamic boundary interface. This allows hydrogen to be absorbed, diffuse and interacts with the metal lattice. The abundant bonding sites on metal hydride’s surface and bulk phase distinguishes them from conventional metal catalysts. Furthermore, amongst the advantages of nanostructured metal hydrides in comparison to bulk catalysts are their tuneable heats of absorption-desorption of hydrogen, enhanced hydrogen absorption-desorption reversibility, and newly developed active surface sites proficient in activating stable chemical bonds and accelerate reaction rates [33]. Attributed to the differences and variations in electronegativities between metal elements and hydrogen, metal hydrides have been reported to have different types of hydrogen bonding and are classified into five categories (Table 1). In a recent review article, a summary of the different types of metal-hydrogen bonding was discussed, highlighting the different classification of metal hydrides [31]. It is noteworthy that the different classes are not fixed; some of the metal hydrides can merge between two classes.

Table 1.
Classification of metal hydrides by metal-hydrogen bonding with some selected examples.
Ionic, interstitial and covalent hydrides have been grouped and regarded as binary and/or intermetallic hydrides by other researchers. On the other hand, Wietelmann et al. [34] classified the metal hydrides based on the metal’s position on the periodic table (Figure 3). Each category of metal hydrides has been reported to exhibit varying strengths and weaknesses. Refer to Table 2 for a summary of the benefits and challenges of the various categories of metal hydrides [33,35].
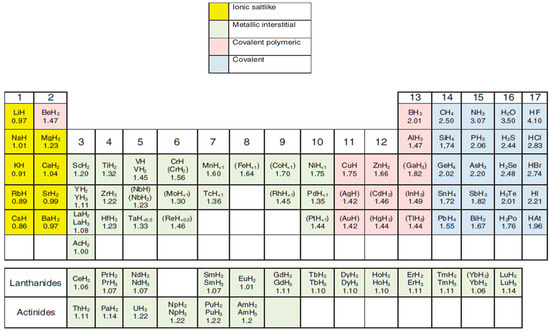
Figure 3.
Periodic table classifying metal hydrides according to bonding types. Note: The hydrides that are unstable at ambient temperatures are in parentheses and are formed at very high H2 pressure. Adapted from Wietelmann et al. [34].
Table 2.
A generalised summary of the drawbacks and benefits of categories of the metal hydrides. Note MHx: M = main group or transition metal; ABxHy: A = hydriding metal, B = non hydriding metal; MEHx: E = boron, nitrogen or aluminium [33,35].
Higher temperatures are required to break and dissociate the metal-hydrogen bond during desorption of hydrogen or hydrogenation. This is attributed to the covalent and ionic bonds between the transition and main group metals with hydrogen [35]. Moreover, transition metals such as platinum group (Ir, Pt, Pd and Rh) metals were noted for their reversible hydrogen absorption-desorption at ambient temperature due to their higher density states at the Fermi level [36]. During the hydriding process, the dissociative adsorption of H2 on the metal oxide (M-O) heterolytically splits H2 into metal hydride (M-H) and O-H (hydroxide) [37,38]. Similarly, Serykh [39] demonstrated that during the dissociative adsorption of H2 on Al2O3-supported In2O3, H2 splits into and forms indium hydride (In-H) and hydroxyl (O-H) sites on the In2O3/Al2O3. A better discussion and illustration of the hydride formation on supported and unsupported metal oxides will be reported later in this review. Although a variety of metal hydrides are regarded as basic and nucleophilic, the hydrides of group 13 metals from Figure 3 are electron deficient and stronger Brønsted acids [34]. Analogous to metal hydrides, metal oxyhydride is a form of a hydride where a metal oxide is bonded to hydrogen through the metal center with a similar nature of chemical bonding to the complex hydride [31]. Amongst the different anion dopants (H−, N3−, F−, e−, and S2−) to modify the electronic, physical and chemical structure of the oxide, H− is a better dopant and/or substitute for the O2− sites. This is attributed to a similar ionic radius between O2− (135 pm) and H− (134 ± 2 pm), and one additional electron which maintains the lattice charge neutrality [40]. Therefore, the most important step during the catalytic hydrogenation reaction on metal hydrides is the transfer of hydride anion from the parent metal hydride complex to the unsaturated substrate, such as CO2, alkyne, and N2 [41].
Hydrogen-involved catalysis over metal hydrides has been reported in recent years, principally on the alkyne hydrogenation, and ammonia synthesis reactions. For example, the Pd phase transition between α- and β-PdHx/ZnO was evidenced from Pd/ZnO during the acetylene hydrogenation to exhibit excellent catalytic activity [42]. The role of hydride at the subsurface of Pd was studied by Teschner et al. [43] for the reduction of 1-Pentyne. As mentioned above, both selective and non-selective hydrogenation reactions were observed due to bulk properties decoupling from the Pd subsurface and the presence of high hydrogen β-hydride, respectively. A 5 wt.% PdHx/Al2O3 was applied on a liquid phase furfural hydrogenation [44]. Although the hydride was quickly consumed and reduced to metallic Pd and Pd carbide, it was revealed that the hydride phase (bulk and surface hydrogen atoms) was easily consumed only in the presence of furfural as a reactant. However, when cyclohexene was used, the bulk hydrogen atoms of PdH0.4 phase were not consumed. The activation and hydrogenation of N2 have also received attention in recent years using alkali, alkaline earth and transition metal hydrides as heterogeneous catalysts in ammonia synthesis [45,46,47,48,49].
The dissociated lattice H− from the MgH2 metal hydride enhanced the hydrogenation of CO2 by interacting with the C of the CO2 to form Mg formate as an intermediate, and subsequently hydrogenating into C2=-C4= lower olefins [50]. Metal hydrides based on Mg were applied in the hydrogenation of dibenzyltoluene and exhibited the best catalytic activity which was attributed to the presence of formed superficial active centers of MgNiH4 on the matrix of MgH2 [51]. The hydrogenation process is a selective reaction on the surface of a heterogeneous catalyst.
The extent and type of the substrate’s reduction depends heavily on the functional groups of the reactant involved such as alkene C=C, alkyne C≡C triple bond, carbonyl C=O, and N≡N triple bond. Based on the above-mentioned reactions using metal hydrides, inspiration can be drawn to synthesise a novel metal hydride that can actively hydrogenate CO2. As a result, it can be concluded that metal hydrides could be applied in CO2 reduction to C1 chemicals, namely, methane, methanol and formic acid. This can result in abundant CO2 resource utilisation and environmental sustainability by continuously mitigating the excessive emissions. Moreover, this can replace the depleting fossil carbon resources and sustain the energy security. The advances in CO2 hydrogenation using metal hydride catalysts will be discussed in the next section.
2.2. CO2 Hydrogenation over Metal Hydrides
2.2.1. Synthesis of CH4 over Metal Hydrides
The formation of metal hydrides involves the conventional synthesis of a metal precursor followed by a hydriding process. Table 3 lists the metal hydrides employed in the hydrogenation of CO2 to a C1 chemical (methane).
Table 3.
Metal hydrides employed in the catalytic hydrogenation of CO2 to methane.
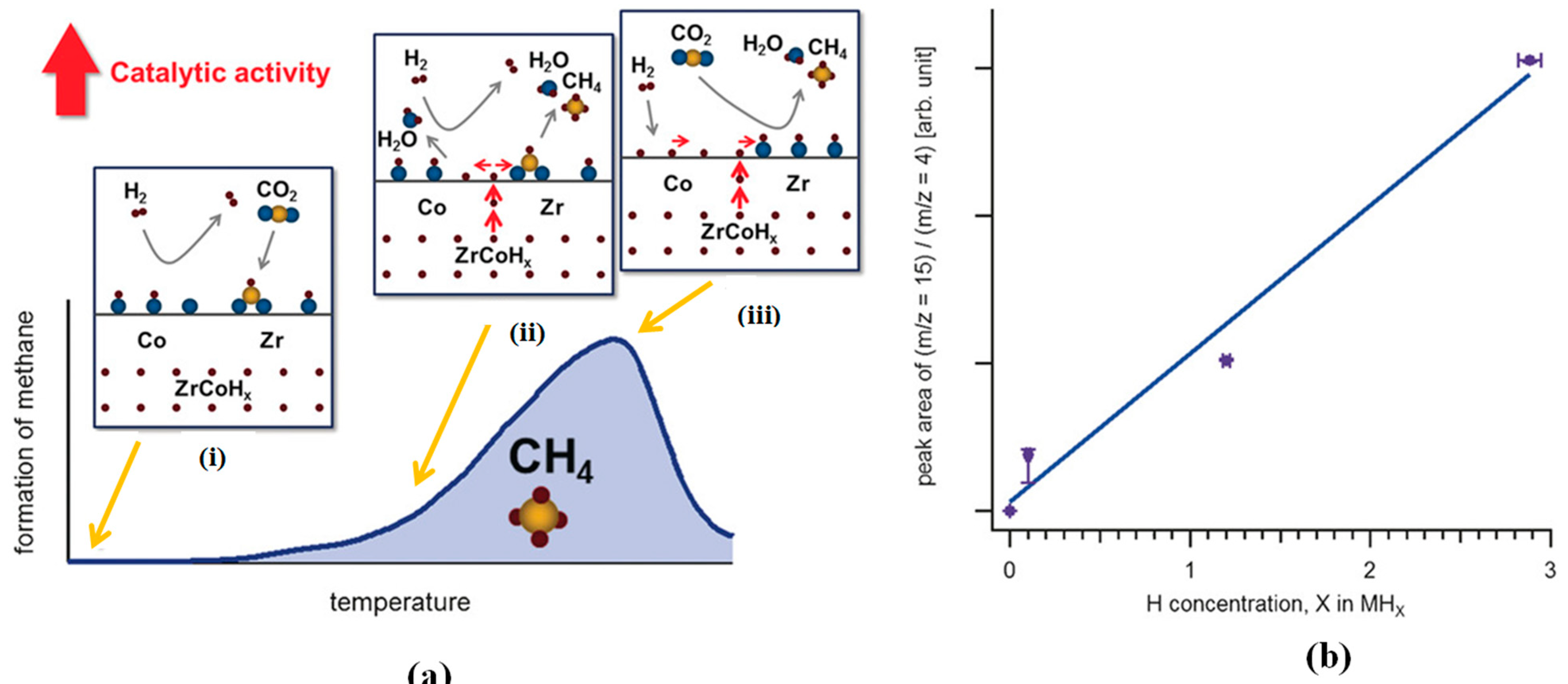
Kato et al. [61] studied the behavior of H2 desorption on bimetallic hydride ZrCoHx and elucidated its catalytic activity in CO2 hydrogenation. The atomic flux of hydrogen desorbed from ZrCoHx facilitated CO2 reduction at the interfacial sites (Figure 4a) with a linear proportionality between the total amounts formed CH4 and hydride concentration in ZrCoHx (Figure 4b). The diffusion of hydrogen atoms from the metal hydride enhanced the CO2 methanation. This behavior was not found on the pristine intermetallic ZrCo.
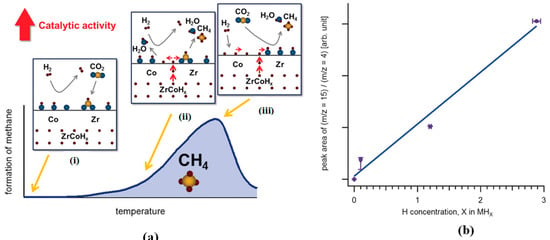
Figure 4.
(a) The schematic depiction of the CO2 reduction over ZrCoHx. (i) The surface coverage of zirconia by CO2 to form formates, (ii) the diffusion of H atoms from the ZrCoHx reacting with formate intermediate producing initially H2O and CH4, and simultaneous reduction of cobalt (Co), (iii) H2 molecules impinged on the interface dissociates on the reduced Co to form H atoms, and together with the H atoms diffusing from the metal hydride spill over the formate to generate H2O and CH4. (b) The peak area of ion current ratio (m/z =15 (CH3+): m/z = 4 (carrier gas He+)) as a function of the initial concentration of hydrogen atom in ZrCoHx (x= 0, 0.1, 1.2, 2.9). Adapted from Kato et al. [61].
In another example, Ni hydride as an active phase, helped to dissociate the highly absorbed CO2 molecules on the surface of Mg2NiH4 hydride, leading to a selective formation of CH4 [62]. In this case, the dissociative adsorption of CO2 on the hydride was the rate determining step of methanation. In a different study that utilised Mg2NiH4 hydride catalysts for the Sabatier type methanation reaction [52], it was found that whilst there were differences the in crystallisation of MgNi2 and MgNiH4 (low-temperature monoclinic and high-temperature pseudo-cubic phases), heating at 330–380 °C under H2 and CO2 gas flow initiated Mg2NiH4 hydride decompositions, disproportionation, and oxidation, empowering and promoting the methanation of CO2. In addition, CO2 methanation at 10 bars started at 50 °C lower than under 1 bar reaching 100% CO2 conversion and 80% CH4 selectivity at 420–430 °C. This demonstrates that a high H2 partial pressure is a stronger factor for the Mg2NiH4 stabilisation over the oxidative potential by CO2. A study comprising both an experimental and DFT calculations was conducted to evaluate the changes in the structure and reduction mechanism of Ni-incorporated MgO/MgH2 for CO2 methanation [54]. A very high CH4 selectivity of 99.5% and space-time yield of 944.6 g kg−1h−1 were obtained at 300 °C and 1 MPa N2/CO2/H2. These results corroborated the theoretical DFT studies. They both suggested that the formation of H+ (from the Ni incorporated in MgO(110)) is necessary to activate the terminal O from the CO2 to form CO* species. Meanwhile, the hydridic hydrogen (H−) from MgH2 hydrogenates the terminal C of the CO2 to CH4. In a different study, carbon nanotubes (CNTs) served as H− protector of MgH2-5wt.%Ni-5wt.%CNT preventing it from oxidation during the CO2 reduction at different reaction temperatures [53]. This enhanced the catalytic activity of the metal hydride catalyst leading to a CH4 yield and selectivity of 79 and 70.9%, respectively, at 350 °C. High catalytic CO2 conversion (100%) and CH4 selectivity (95%) of cubic Mg2NiH4 complex hydride were achieved at 400 °C in a time of stream of 5 h, and its activity was strongly dependent on the ball-milling time before the CO2 reduction [55].
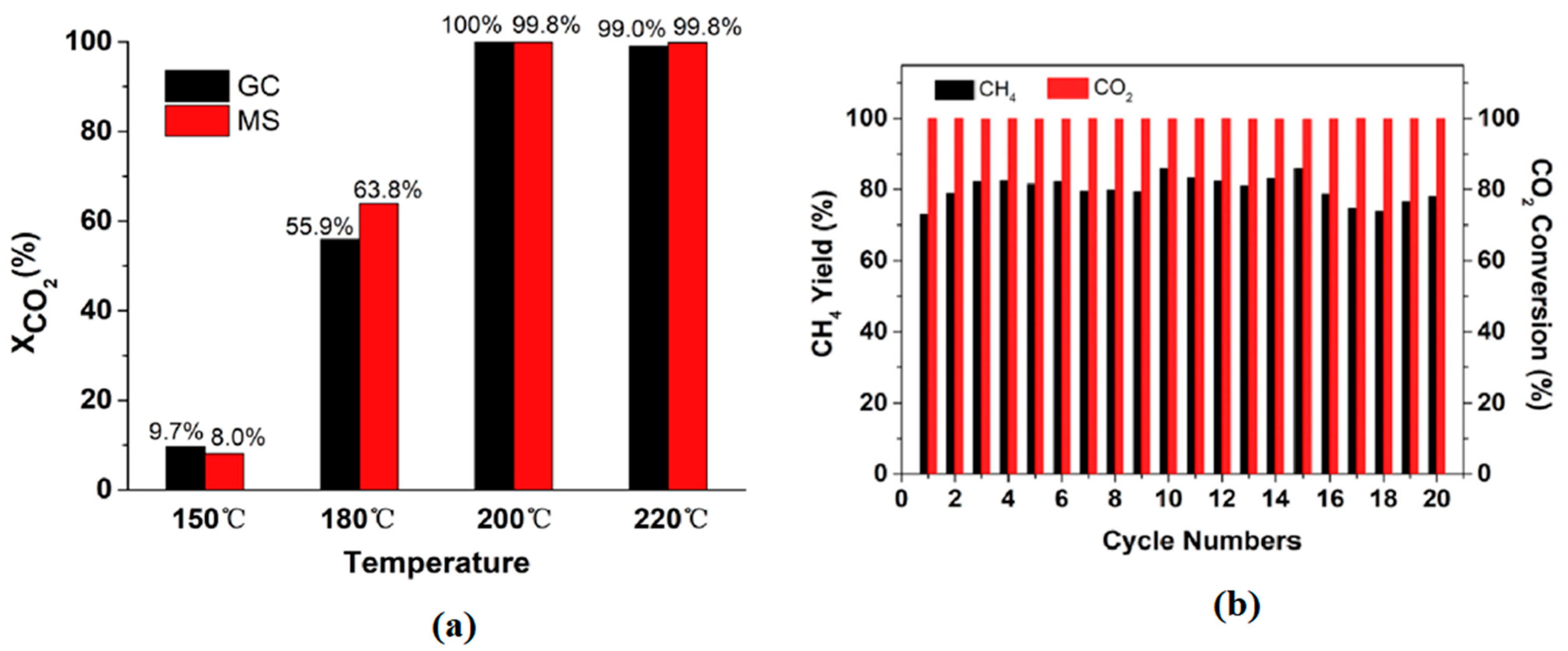
Evenly dispersed Ni nanoparticles and a shortened distance of hydridic hydrogen H− mobility on the LaNi5H5 hydride surface led to a superior catalytic activity of 99% CO2 conversion and 72% CH4 yield at a time of stream of 400 h (Figure 5) [56]. In addition, the presence of La in LaNi5H5 hydride and the lowered reaction temperature inhibited the agglomeration of Ni nanoparticles (NPs). The basic nucleophilic H− of the metal hydride can activate the acidic and electrophilic CO2 during catalytic reactions. The dissociated lattice H− from the Al-doped MgH2 hydride nano-catalyst achieved 27.1% of CO2 conversion and 88.4% of CH4 selectivity under the reactions conditions at 320 °C and 1 MPa of 5:1 H2/CO2 ration [57].
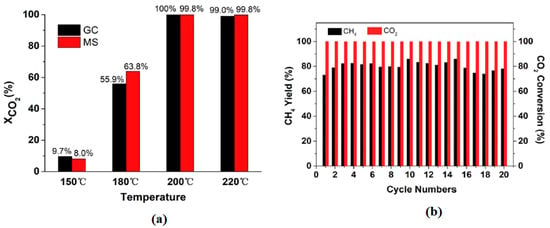
Figure 5.
Catalytic activity of LaNi5H5 (a) CO2 conversion at varying temperatures on a 20 h time of stream measured by GC and MS, and (b) CO2 conversion and methane yield during the 20-cycle stability test. Adapted from Zhong et al. [56].
MgH2/CuxO composite with a defect-rich surface was found to exhibit a 20.7% CO2 conversion at 350 °C when using a high 5:1 ratio of CO2:H2 with high hydrogen utilisation efficiency. Although the composite hydride promoted the CO2 conversion, a very low CH4 selectivity of 45% was observed and attributed to the high selectivity towards C2=–C4= olefins. In contrast, the CH4 selectivity increased to 90% upon decreasing the CO2:H2 ratio to 1:10. The catalytic activity was reported to be a result of defective rich oxygen vacant CuxO and MgH2 surfaces which acted as active sites and facilitated the adsorption and activation of CO2, and subsequent CO2 hydrogenation by lattice H− [50]. Metal active phase can be necessary to enhance the activity of MgH2 hydride as well as lowering the reaction temperature according to Le Chatelier’s principle for CO2 hydrogenation, while maintaining high CH4 yield. Amica et al. [58] achieved a CH4 yield of 78% over MgH2-doped with 10 wt.% Co at 350 °C for 48 h and a 4:1 molar ratio of MgH2:CO2 as optimum reaction conditions. However, a 44.6% CH4 yield was obtained on the MgH2 without cobalt (Co) as a catalyst. A high CH4 yield on the Co-doped catalyst was reported to be due to the presence of the Co, which favors the prevailing methanation mechanism responding to the Sabatier process. MgNiH4 and Mg2FeH6 complex hydrides were also applied as hydrogen sources and promoters for the CO2 hydrogenation to methane [59]. Both complex hydrides initiated and facilitated different reaction pathways (reverse water gas shift followed by CO methanation for Mg2FeH6-CO2 in the presence of steam and the global Sabatier methanation process for MgNiH4-CO2). In addition, they achieved 100% CO2 conversion at 400 °C after 10 and 5 h, respectively, with a high yield of CH4 as was detected by the Fourier transform infrared (FTIR) spectrometer and the formation of MgO by powder X-ray diffraction (PXRD) spectroscopy. CO2 methanation reaction was conducted using activated MgH2 and CaH2 alkaline earth metal hydrides [60]. Although, both metal hydrides exhibited better activity, the highest yield (88%) and productivity (89%) of CH4 was achieved with CaH2 at 450 °C after 48 h. This indicated that the productivity of CH4 can be largely influenced by the choice of alkaline earth metal hydride, reaction time and reaction temperature during CO2 hydrogenation reaction.
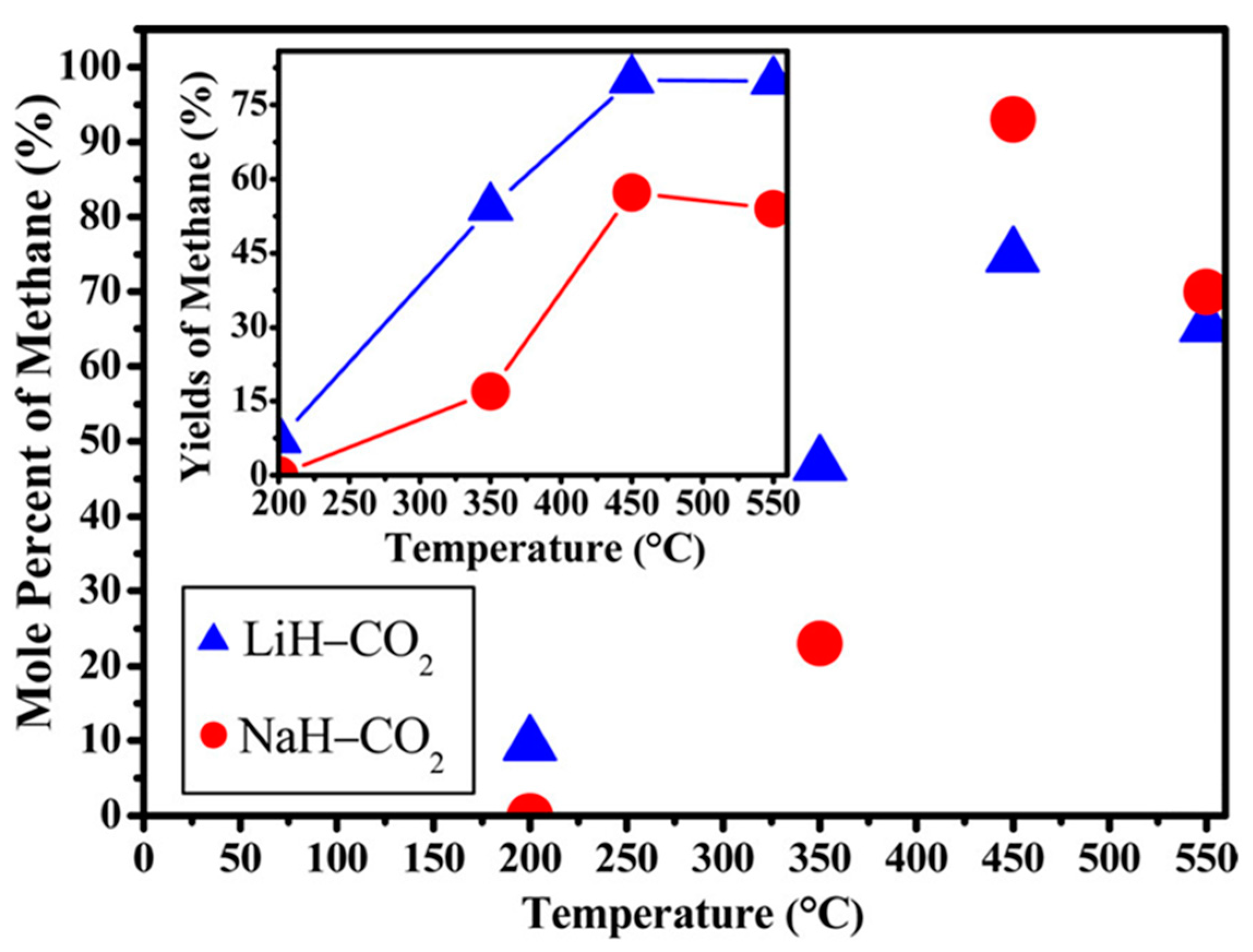
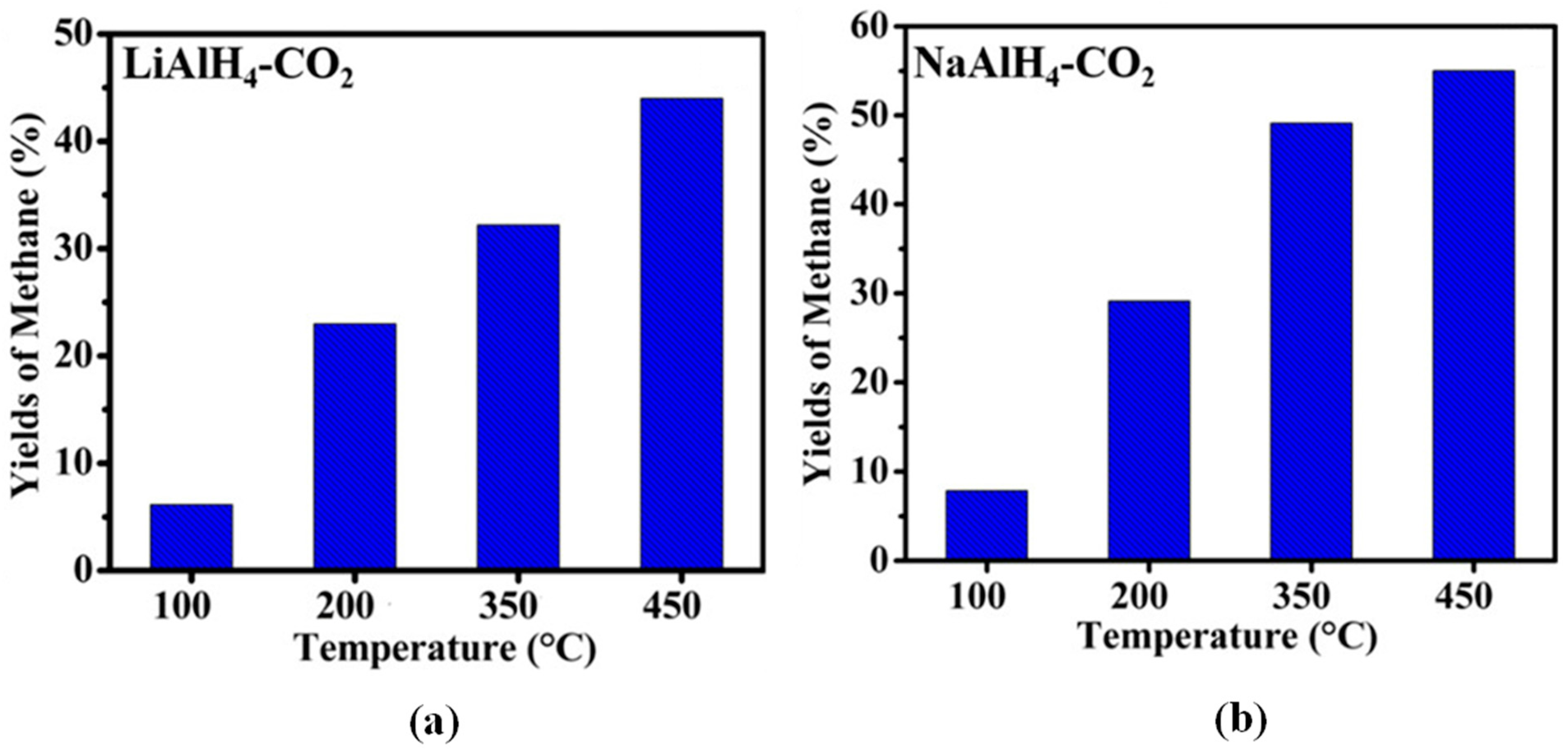
Ball-milled alkali metal hydrides also start to emerge in the CO2 hydrogenation process. On one hand, the highest CH4 mole percentages of 74.7 and 90.3% were reported for LiH and NaH, respectively, in the hydrogenation of CO2 over alkali metal hydrides (Figure 6). However, LiH achieved the maximum CH4 yield of 80.15%, meanwhile 57.3% CH4 yield was observed for NaH [63]. This indicated that the yield and mole % of CH4 in both systems depended on the reaction temperature and time. On the other hand, Zhao et al. [64] studied the CO2 hydrogenation using activated LiAlH4 and NaAlH4 alkali metal aluminium hydrides. Using the same reaction conditions, the maximum yield of 55% CH4 was reached at 450 °C with NaAlH4, in comparison to 45% over LiAlH4. This is elucidated in Figure 7.
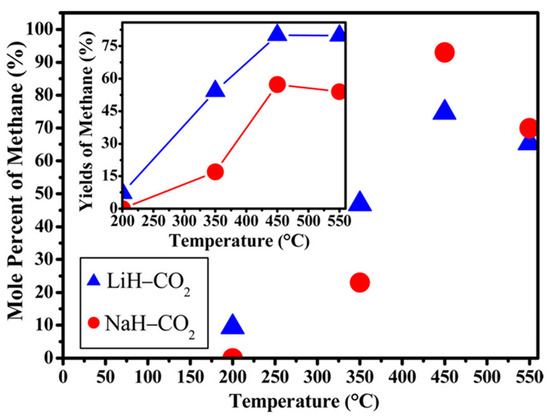
Figure 6.
Reaction yields and mole percent of methane for the ball-milled LiH and NaH with CO2 [MH/CO2 = 4:1 mol/mol and 0.25 MPa, MH (M = Li or Na)] at range of temperatures; 200, 350, 450, and 550 °C for TOS of 48 h. Adapted from Dong et al. [63].
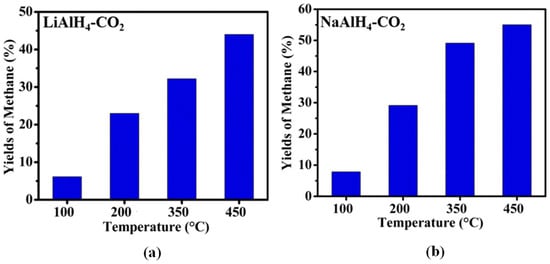
Figure 7.
Yield of methane produced from the reactions between the ball-milled (a) LiAlH4, and (b) NaAlH4 with CO2 [AAlH4/CO2 = 1/1 molar ratio and T = 48 h]. Adapted from Zhao et al. [64].
2.2.2. Synthesis of Formic Acid and Methanol over Metal Hydrides
Nilwanna et al. [65] used density functional theory (DFT) to study the reaction mechanism of aluminium-based metal–organic framework (Al-MOF) supported metal (II) hydrides (metal = Fe, Ni, Mn, Cu and Co) on the hydrogenation of CO2 to formic acid. Although all the 5 Al-MOF supported metal (II) hydrides (MH-Al-MOF) have proven hydrogenation of formate to formic acid to be the rate-determining step, the most active catalyst for hydrogenation of CO2 to formic acid was the Al-MOF supported Ni hydride. Note that biphenyl-4,4′-dicarboxylate (DUT-5) is used as one of Al-MOFs. Refer to Table 4 for rate constants for the rate-determining step and relative Gibbs free energies for the CO2 hydrogenation to formic acid on biphenyl-4,4′-dicarboxylate aluminium-based metal–organic framework supported metal hydrides (MH-DUT-5).
Table 4.
Rate constants for the rate-determining step and relative Gibbs free energies for the CO2 hydrogenation to formic acid on MH-DUT-5 (M = Mn, Fe, Co. Ni, and Cu).
Zhang et al. [66] studied the activation and hydrogenation of CO2 over PtHn− clusters, and the formation of intermediates such as HPtCO2− and H2Pt(HCO2)− was observed, suggesting the formate reaction pathway is energetically favorable. This can favor and promote the formic acid and/or methanol synthesis with subsequent hydrogenation of the formate species. A similar study by DFT calculations indicated that metal hydride MH (M = Cu, Ag, and Au) can initially lead to the formation of formate HCO2− with AgH− as the most effective metal hydride anion followed by CuH− [67]. Since the methanol steam reforming (MSR) is a reverse reaction from the CO2-hydrogenated methanol synthesis, it is paramount to study and understand the catalyst that can actively convert methanol and selectively produce H2 and CO2. In addition, it has to exhibit a better CO2 absorption ability. Kadeer et al. [68] coupled the MSR with the hydrolysis of metal hydride. The employed CaH2-CuO composite achieved a high hydrogen density of 6.5 wt. % (with 99% purity) at 250 °C. This is achieved by shifting the water gas shift reaction equilibrium towards the production of H2 and CO2 (Equation (6)) wherein the produced CO2 from MSR could be absorbed by the hydroxide produced from hydrolysis of the metal hydride (Equation (7)). This results in the production of highly purified H2 and the suppression of CO formation. The high absorption capacity of the hydroxide component is an indication of the coupling effect of the MSR and hydrolysis of the metal hydride. In addition, the H2 generated from the CaH2-coupled MSR exhibited high H2 density in comparison to conventional hydrolysis.
CO + H2O  CO2 + H2
CO2 + H2
CO2 + H2
2 CaH2 + 4 H2O + CH3OH 2 Ca(OH)2 + CaCO3 + 7 H2
Ca(OH)2 + CaCO3 + 7 H2
Kim et al. [69] studied the surface chemistry of CH3OH on Pd(111)-H and Pd(111) to elucidate the effect of metal hydrides and C-O bond cleavage by decomposing CH3OH as a function of temperature (300–750 K) using synchrotron-based ambient-pressure X-ray photoelectron spectroscopy (AP-XPS). They observed the surface and subsurface hydrides from Pd reacting with the decomposed CHx and C/PdCx species to produce a clean surface at 750 K. In contrast, a pristine Pd(111) led to the decomposition of the carbonaceous layer due to the lack of a hydride.
Pd is one of the most studied metal hydrides attributed to its high hydrogen storage capabilities by reversibly absorbing and desorbing H2 under favorable conditions [70]. They can also act as a hydride of AB-type intermetallic alloy when doped with other transitional metals such as Cu, Ir, Ag, and Pt to form Pd-Cu-H, Pd-Ir-H, Pd-Ag-H, and Pd-Pt-H, respectively [71]. The reaction pathways were theoretically calculated for the hydrogenation of CO2. Based on the energy barriers of the rate-limiting steps, it was clear that CH3OH can be selectively produced through the formate pathway on the pristine MgH2(001). On the other hand, the cluster Cu7-MgH2(001) favored the CH4 through the RWGS pathway [72].
2.3. Effect of H2 Dissociation and Formation of Metal Hydrides over Catalysts During CO2 Hydrogenation to C1 Chemicals
During a catalytic reduction reaction, both CO2 and H2 molecule must undergo adsorption, activation and conversion on the surface of a heterogeneous catalyst. In addition to the study of CO2 dissociation, which has been thoroughly reported [26,27], it is significant to understand the dissociation of the H2 and how it affects the catalytic activity and selectivity during a catalytic reaction. Furthermore, it also gives insights in the formation and effect of in situ generated metal hydrides on catalysis. This will aid in selecting the best catalyst for the CO2 reduction reaction. The cleavage of H2 over a metal nanoparticle is broadly homolytic dissociative without an energy barrier. The low activation energy for the homolytic dissociation of H2 on highly catalytic metals such as Ni, Cu, Pt and Pd, leads to low CO2 hydrogenation selectivity due to the over-hydrogenation of CO2 and its reaction intermediates on the metallic sites [73,74]. On the other hand, heterolytic dissociation presents limitations over the metal oxide for subsequent hydrogenation reactions, attributed to insufficient hydridic hydrogen when multiple hydrogen atoms and electrons are transferred, as well as an increase in the possibility of eliminating hydridic hydrogen to hydroxyls as a result of hydridic hydrogen migrating along the lattice oxygen [75]. In addition, it has much higher activation energy when compared to the H2 homolytic dissociation. This leads to slower kinetics, making H2 dissociation the rate-determining step with the necessity to employ high H2 pressure and reaction temperature during reduction reactions [76]. It should be noted that both homolytic and heterolytic dissociation pathways of H2 can occur consecutively on the surface of a metal. However, this depends on the effect of coordination with the support or metal cation. The H2 heterolytic dissociation pathway is favored for the hydrogenation of C=O, N=O, and C=N bonds due to the nature and better capability of polar bonds to accept hydridic hydrogen and protons [77,78,79]. Due to the nature of metal cations to be separated by counter-anions on the surface or sub-surface of bulk metal oxides, they resemble the Lewis acid-base (acid = metal, base = oxide) pair criterion. Metal oxides heterolytically dissociates the H2 molecule into a proton and a hydridic hydrogen, and can exhibit excellent catalytic performance in the hydrogenation of CO2 [79]. The active sites responsible for the H2 heterolytic dissociation are attributed to the defect sites formed on the surface of metal oxide as well as their Lewis acid-base property.
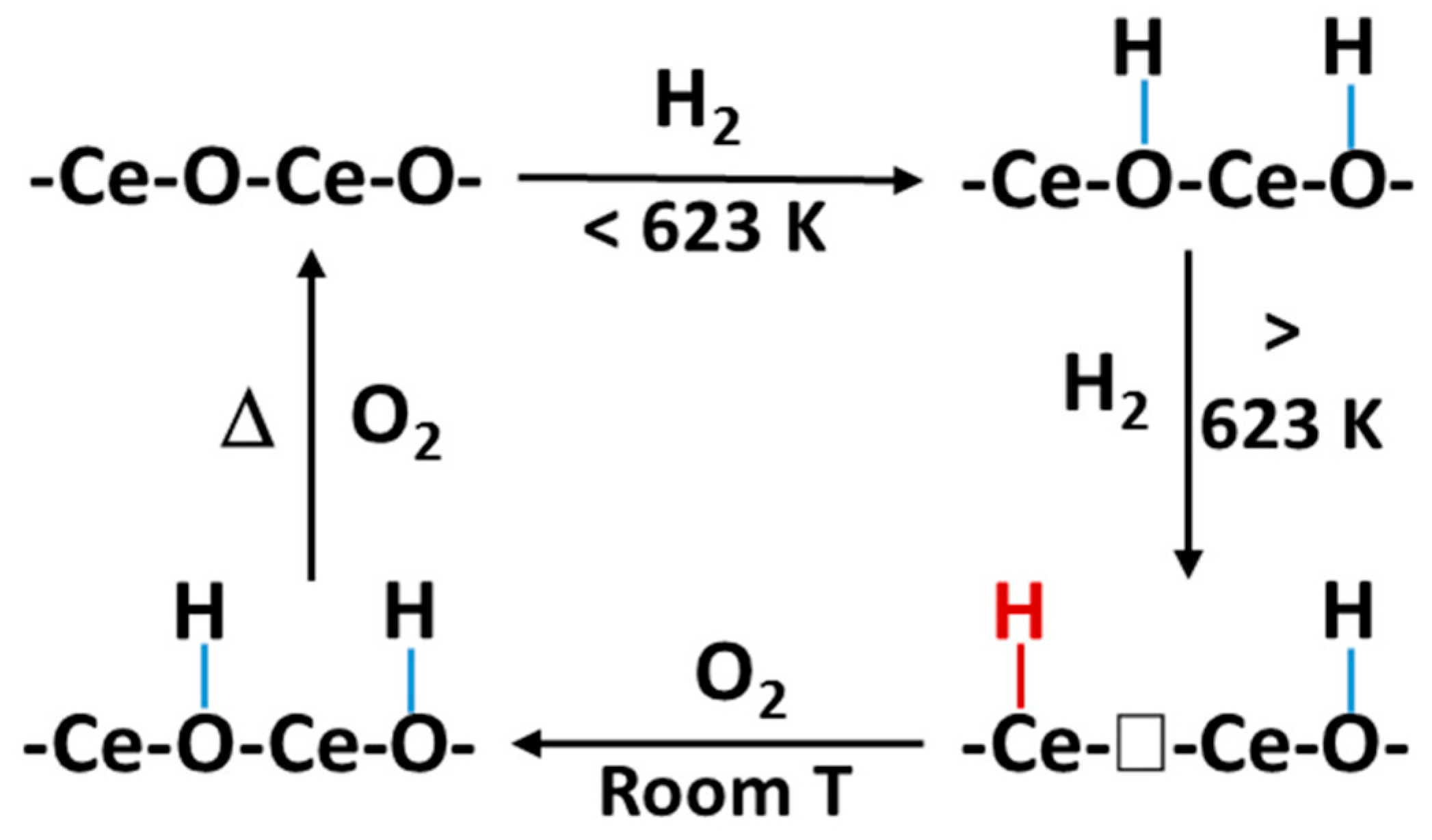
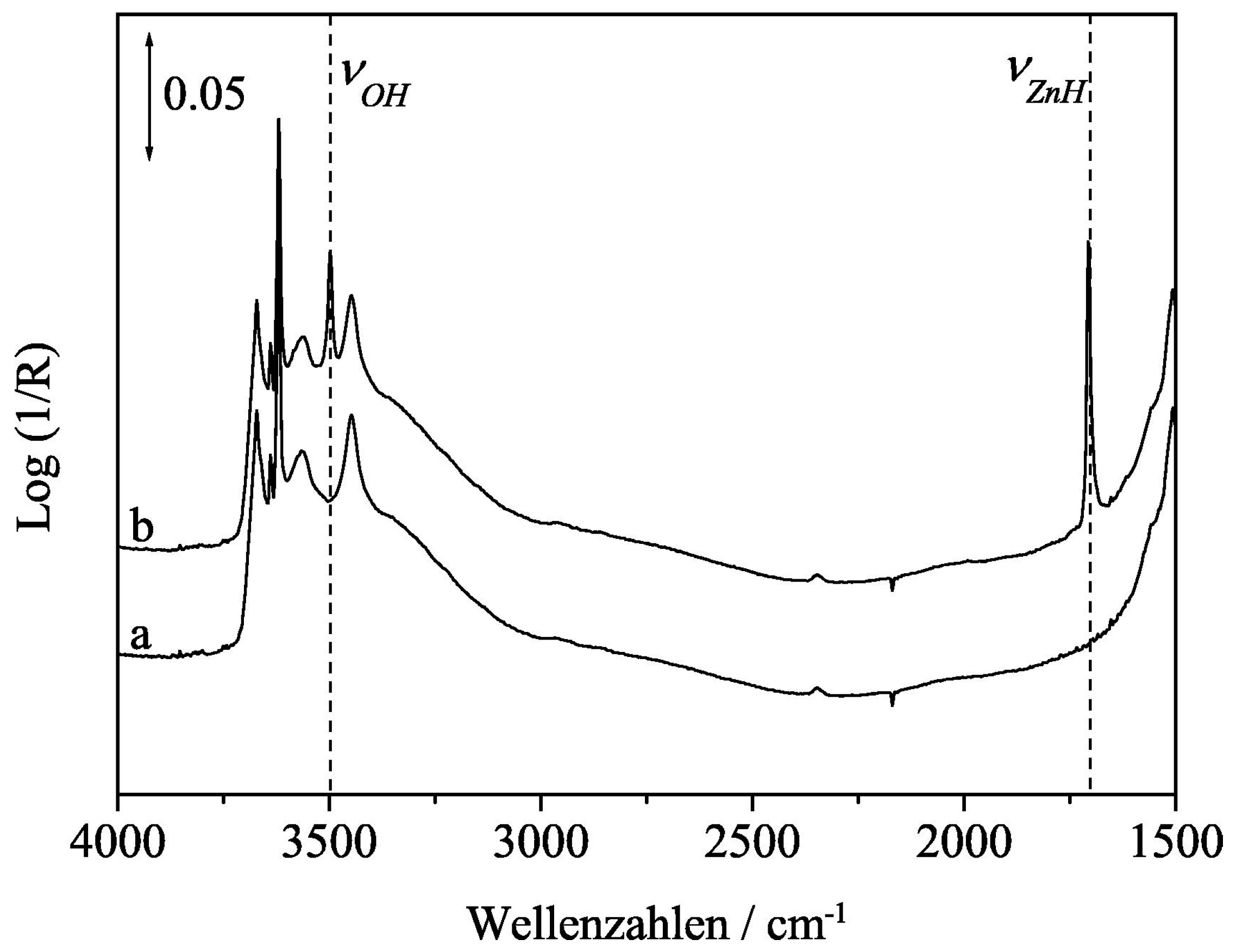
To have a better understanding of the interaction between H2 and a metal oxide during a hydrogenation reaction, Wu et al. [80] employed in situ inelastic neutron scattering spectroscopy (INS) and evidenced the heterolytic dissociation of H2 into Ce-H hydride and O-H hydroxyls over defective ceria, with induced vacant sites of oxygen. Furthermore, they evidenced surface OHs on an oxygen vacant free or perfect ceria through electron and hydride transfer (Figure 8). This highlighted that both hydrides and oxygen vacancies over ceria can be formed via H2 dissociation during reduction. Zinc oxide (ZnO) can serve as an active phase, promoter or support in the synthesis of methanol during CO2 hydrogenation; therefore, a review on the surface chemistry of ZnO was conducted. From the study, the Zn-H hydride bands were observed using infrared spectroscopy when the surface of ZnO was exposed to gaseous hydrogen gas [81]. Refer to Figure 9 for the H2 dissociation into a proton-hydride pair over ZnO.
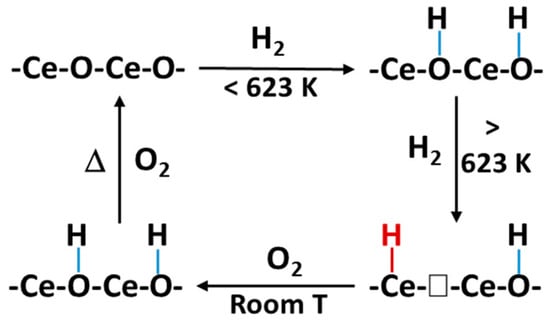
Figure 8.
A schematic diagram of the H2 interacting with CeO2 forming surface Ce−H hydride and OH hydroxyl. Adapted from Wu et al. [80].
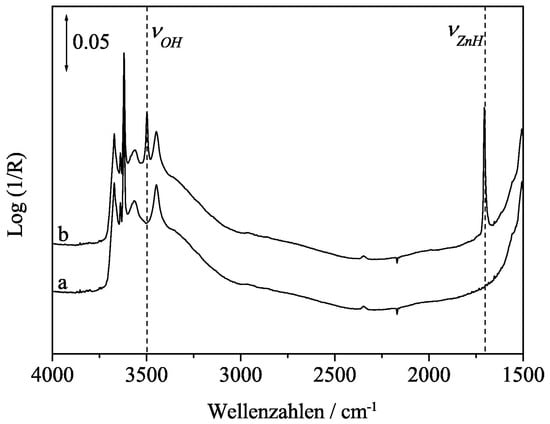
Figure 9.
IR spectra of ZnO (a) exposed to He (b) Zn−H hydride formed upon H2 exposure. Adapted from Strunk et al. [81].
The dissociation of a polarised H2 molecule into hydridic hydrogen (H−) and proton (H+) adsorbates on the surface of a catalyst can lead to better catalytic activity and selectivity for CO2 hydrogenation. During the heterolytic H2 dissociation over the surface of a defective or perfect Indium oxide (In2O3), the H− binds to the In cation and the H+ to the O anion. Similarly to the mechanism of CO2 hydrogenation using ex situ synthesised metal hydrides, the hydridic hydrogen of the in situ generated metal hydride reacts with the C atom of the CO2 adsorbed on the surface of the catalyst to activate and convert it into the desired C1 chemical such as methanol. On the other hand, the O in CO2 reacts with the H+ during protonation [81,82,83].
DFT calculations indicated that the oppositely charged Lewis acid-base pair sites of In and O atoms of the partially and perfectly reduced In2O3 can enhance the heterolytic H2 dissociation. This promoted the hydride formation on In site which can be significant for methanol synthesis via the CO2 hydrogenation [84]. Similarly, it was reported by Jia et al. [85] that oxygen vacant site and defects over the surface of In2O3 facilitate the heterolytic dissociation of H2, which can contribute to the CO2 reduction with enhanced catalytic activity and selectivity. They clearly compare the H2 activation and dissociation on both stoichiometric and non-stoichiometric metal oxides, showing both the heterolytic and homolytic H2 dissociation pathways on the surface of the defect and non-defect metal oxides. It can be noted that metal oxides have the compass to sequentially dissociate H2 into both protons and proton-hydridic hydrogen pairs, absorb and react with CO2. As a result, the defect sites generated by high-temperature activation and the Lewis acid-base pair of the metal oxides serve as active sites for CO2 catalytic hydrogenation reactions. Albeit, dissociative adsorption of H2 was found to be facile via the heterolytic dissociation with moderate energy barrier on both In2O3(111) and In2O3(110) to form In-H and OH via DFT calculations, it was discovered that the diffusion of hydrogen facilitates homolytic adsorption positioning, with In2O3 surfaces hydroxylated by distinct types of OH groups. This resulted in changes in In oxidation states, suggesting that oxidation-reduction properties are also vital for the catalytic hydrogenation of CO2 [86]. On the other hand, despite the protonation and reduction of CO2 via the surface bicarbonate species being more energetically favorable (−0.78 eV adsorption energy) in comparison to hydridic hydrogen transfer (+0.33 eV adsorption energy) on the surface of In2O3(110), it was indicated that the formate route by hydride transfer (InH + CO2) was more favorable over the bicarbonate species route [87]. This will lead to an increment in the selectivity of formate and its subsequent products such as formic acid and methanol. Meanwhile, the subsequent steps in the bicarbonate species route are highly endothermic (+1.07 eV adsorption energy) and could react with surface hydroxyls with ease and exhibit high H2O and CO2 selectivities. Juárez et al. [88] conducted an in situ IR and INS spectroscopic study of H2 species present over Au/CeO2 and evidenced the formation of Au-H hydride and bridging hydroxyl group on the supported catalyst upon exposure to H2 gas. Furthermore, they observed that bridging hydroxyl group bands at 400 to 650 cm−1 and 3690 cm−1, and hydride band at 2126 cm−1 disappeared upon subsequent treatment with CO2. The disappearance of hydrogen species indicates consumption and catalytic activity with CO2. However, it was not possible to observe the Au-H band in INS due to the low percentage of Au (0.48 wt. %) on the supported catalyst, and lower sensitivity of the INS.
In order to promote energy efficiency during CO2 hydrogenation, Mao et al. [89] successfully employed an alkaline earth metal (Ca) to selectively activate and hydrogenate solid CO2 captured by CaO in the form of CaCO3 using ball milling at room temperature. A methane yield and selectivity of 56.1 and 100% were achieved, respectively. Furthermore, the activity was increased upon doping the Ca-CaCO3-H2 with 9 wt. % Ni to 59.05% CH4 yield and 95.35% CaCO3 conversion.
2.4. Oxyhydrides for the CO2 Hydrogenation
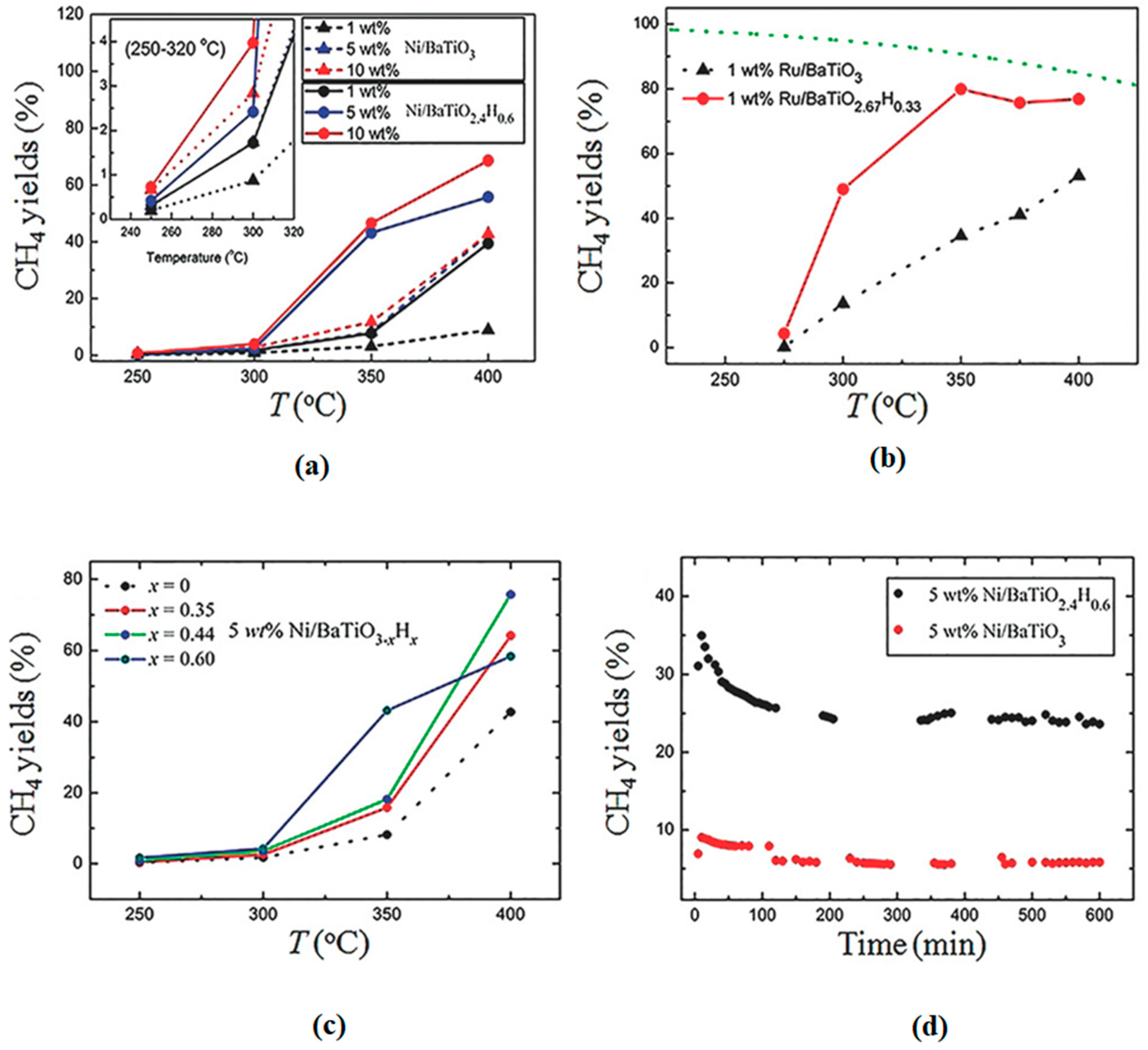
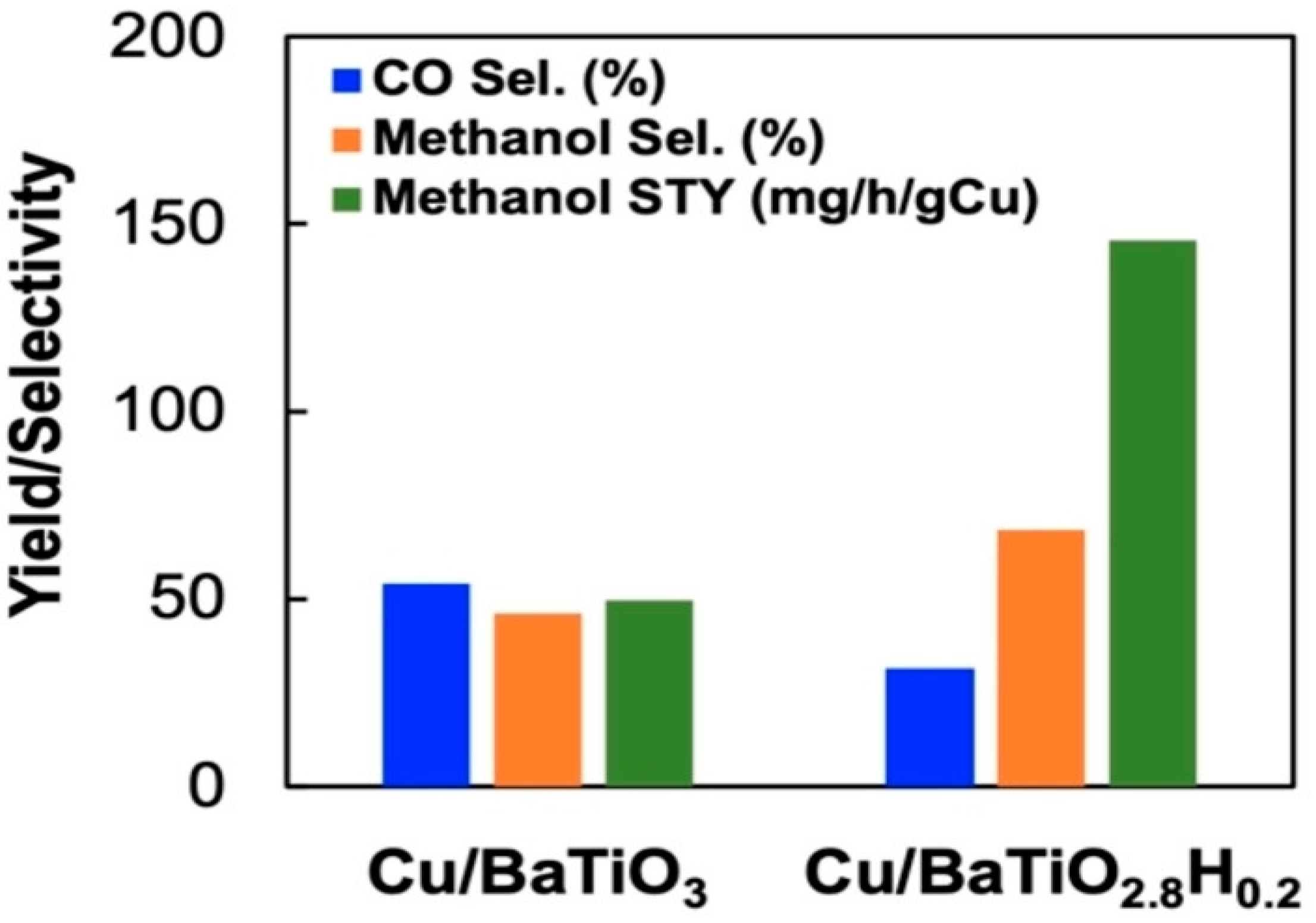
Attributed to their abilities to act as efficient active centers and/or supports, metal oxyhydride can also serve as catalysts for energy- and environmental-related catalytic reactions. Oxyhydrides are also starting to be explored in the CO2 reduction. The catalytic hydrogenation of CO2 into CH4 was enhanced when BaTiO2.4H0.6 and BaTiO2.67H0.33 oxyhydrides were used as a support for Ni and Ru, respectively, in comparison to the Ru/BaTiO3 and Ni/BaTiO3 supported perovskite catalysts [90]. The catalytic activity increased by 2–7 folds over the perovskite-type oxyhydrides supported Ni and Ru (Figure 10) at 350 °C and 1 atm, under the 90 mL.min−1 flow-rate (H2:CO2 = 4:1) optimum reaction conditions. The improved activity was attributed to the reduction in H2 poisoning, possibly influenced by the presence of hydridic hydrogen on oxyhydride (TiO2.4H0.6 and BaTiO2.67H0.33) support. Moreover, Ru/BaTiO3 has been reported to suffer from H2 poisoning. The space-time yield and selectivity of methanol over Cu/BaTiO3 increased significantly from 50 to 146 mg.g−1cat.h−1 and 48 to 68%, respectively (Figure 11), upon partially replacing the O2- anion of the BaTiO3 perovskite with hydride to Cu/BaTiO2.8H0.2 [91]. This was attributed to their ability to increase the local electronic density of the active phase on the metal support interface, which can lead to better promotion of CO2 activation and conversion. Based on recent reported studies, it is clear that metal oxyhydrides are starting to receive attention in the CO2 hydrogenation to C1 value-added products [90,91]. As a result, this can assist in mitigating the ever-rising emissions of CO2 and sustain the energy supply by selectively producing synthetic gas CH4 which can be used as an energy carrier or directly as a renewable fuel. However, not much has been reported on CO2 hydrogenation to formic acid and methanol using metal hydrides. Therefore, a good opportunity is presented to further explore the chemistry of metal hydride heterogeneous catalysts on the synthesis of other C1 chemicals. The next chapter briefly highlights carbon dioxide reduction over homogeneous metal hydride catalysts. This is to elucidate the catalytic importance of hydrides on methanol synthesis via CO2 hydrogenation.
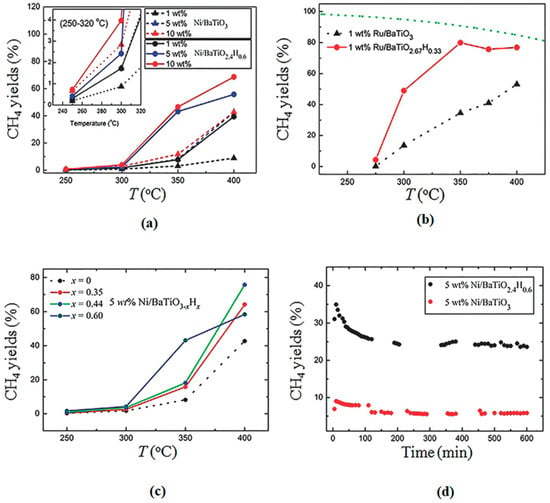
Figure 10.
CH4 yield at different reaction temperatures of (a) Ni and (b) Ru supported pristine perovskite and oxyhydride catalysts. (c) CH4 yield over various contents (x) of Ni/BaTiO3−xHx and (d) CH4 yield over 10 h stability test. Adapted from Tang et al. [90].
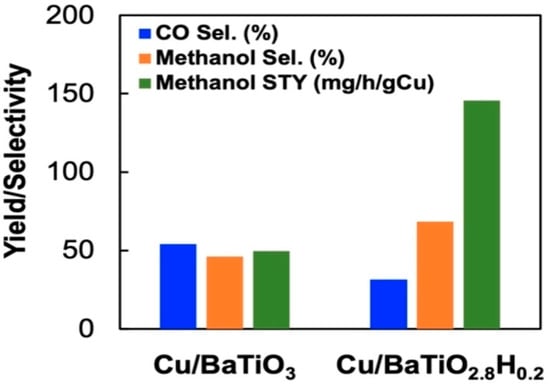
Figure 11.
CO2 hydrogenation over Cu/BaTiO3 and Cu/BaTiO2.8H0.2. Adapted from He at al. [91].
3. Hydrogenation of CO2 over Homogeneous Transition Metal Hydrides Catalysts
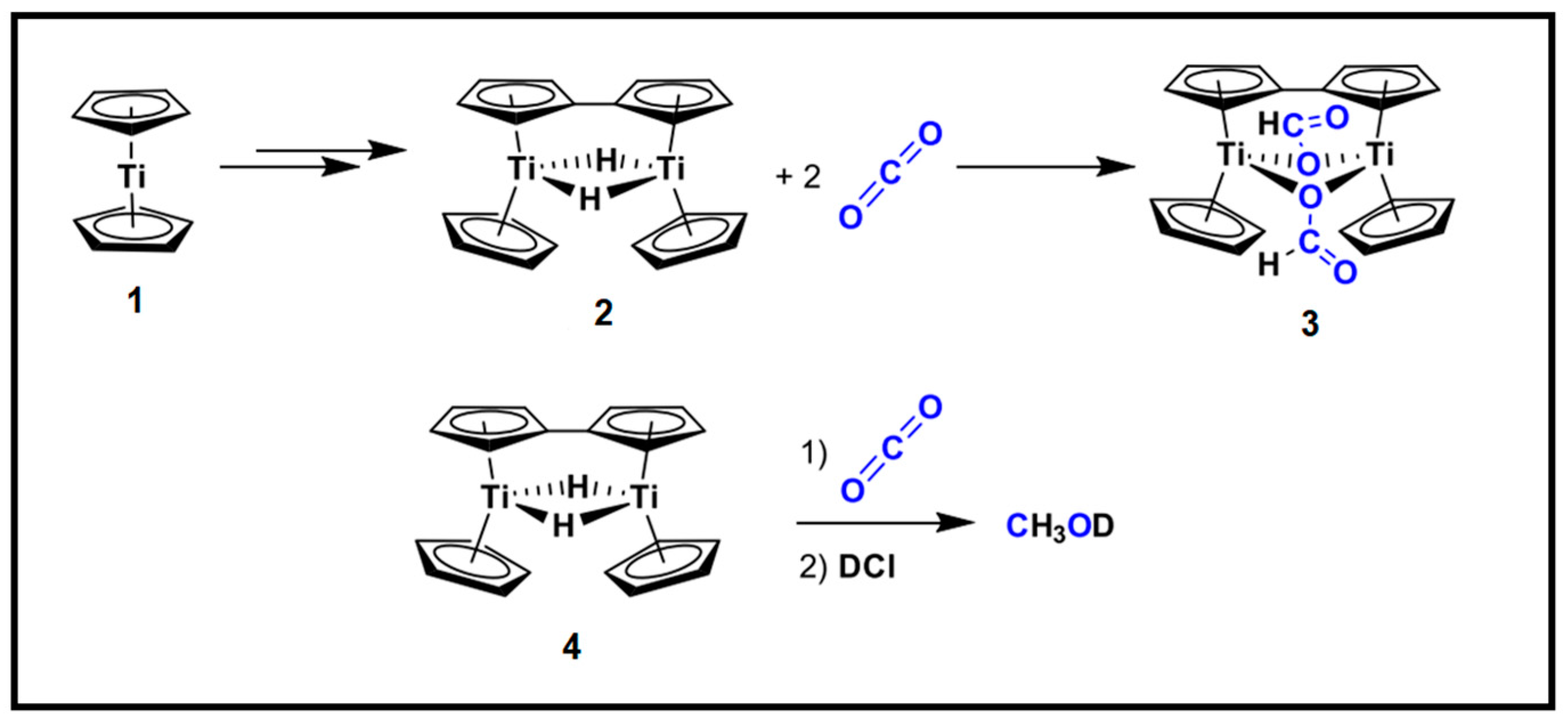
In comparison to CO2 hydrogenation by late (groups 8–10) transition metal complexes which suffer from the production of 2e− reduction products such as formate and CO, a review was conducted for the CO2 reduction with early (groups 3–7) transition metal complexes to form formaldehyde, methoxide species and their subsequent hydrogenated products [92]. Methanol can be observed in the stoichiometric and catalytic conversion attributed to the ability of groups 3–7 metal complexes to pass electrons and the Lewis acidity (of the early metals) to activate CO2 (Figure 12). On the other hand, earth-abundant 3d transition metals have shown significant increases in catalytic activity in comparison to second (4d) and third (5d) row transition metal complexes in CO2 reduction into formic acid and methanol [93]. The conversion of CO2 into methanol via hydrogenation with renewable hydrogen over homogeneous metal hydrides is an emerging field of research. This is due to the high activity of homogeneous metal hydride catalysts. The chemistry of hydride transfers from transition metal complex families and applications on CO2 reduction to fuels and chemicals such as; methanol and formates equivalents have been well documented [93,94,95,96]. Known hydricity values and free energy of the electron reduction (E1/2(Mn+/(n−1)+)) of the parent complex from the sets of data consisting of 51 transition metal hydride complexes were established [41]. From the data sets, a linear trend was observed which promotes hydricity estimation on the parent complex’s reduction potential and guides the design and optimisation of the metal hydride complex for CO2 hydrogenation to lower C-chemicals. Although transition metal complexes and their respective hydrides have a better CO2 catalytic activity towards methanol, transition metal complexes suffer from poor product and catalyst separation, and purification. This leads to their limitation to be applied at a large industrial scale.
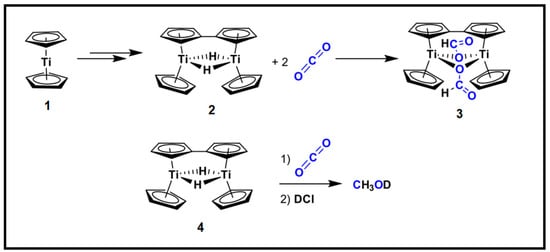
Figure 12.
Schematic representation of the methanol synthesis from CO2 over group 4′s titanium hydride complex. Adapted from Grice. [92].
4. Conclusions
Continuous utilisation of fossil fuels has led to an accelerated rate of global warming beyond the pre-industrial stage. This is due to the excessive emissions of anthropogenic CO2 released into the environment. This review presents how CO2 can be converted into C1 chemicals namely, CH4, HCOOH, and CH3OH using renewable green hydrogen. The CO2 hydrogenation conversion can mitigate CO2 emissions, maintain energy security, and replace depleting fossil resources. Catalysts are applied and play a very important role in CO2 hydrogenation into C1 chemicals. The produced C1 chemicals can be directly used as substitute fuels and carriers or storage for clean energy. Metal hydrides are starting to emerge in the thermo-catalytic hydrogenation of CO2. This is attributed to their enhanced hydrogen adsorption–desorption reversibility, and newly developed active and abundant sites. They can be classified into five different categories based on the type of metal–hydrogen bonding namely, ionic, interstitial, covalent, complex, and high-pressure metal hydrides. Each of these classes have benefits and drawbacks. Various types of complex and ionic metal hydrides applied in the CO2 methanation are reviewed. Wherein, a high CO2 conversion and CH4 selectivity are reported. This is attributed to their high reductibility by hydridic hydrogen (H−) during hydrogenation reactions at different temperatures, pressures, and space velocities. The hydridic hydrogen can activate and convert CO2 through the reverse water gas shift (RWGS) followed by CO methanation and the global Sabatier reactions. The introduction of transition, such as Co and Ni to the metal hydrides, enhances the CH4 selectivity. Furthermore, alkali and alkaline earth metals can act as promoters and hydridic hydrogen sources during the CO2 methanation.
Although, metal hydrides have been thoroughly reported regarding CO2 methanation, few studies have been conducted on the CO2 hydrogenation to other C1 (CH3OH and HCOOH) chemicals. We review the application of metal hydrides on the non-catalytic process for pure H2 and CO2 production from CH3OH. This highlights the importance and role of metal hydrides on the reverse pathway of CO2 hydrogenation to CH3OH. The application of metal hydrides on the methanol steam reforming and decomposition reactions enhanced the production and selectivity of both H2 and CO2, suppressing the RWGS products (CO + H2O).
Hydridic hydrogen formed in situ during catalytic hydrogenation reaction is also reviewed. Both homolytic and heterolytic H2 dissociation can occur simultaneously during hydrogenation. This was observed from spectroscopy experiments. However, the H2 heterolytic dissociation into proton a (H+) and hydridic hydrogen (H−) is favoured for the hydrogenation of C=O, N=O, and C=N polar bonds. Metal oxides heterolytically dissociate H2 into H+ and H− and positively influence the catalytic performance. DFT calculations also suggest that heterolytic dissociation on metal oxide promotes hydridic hydrogen-proton pairs. The hydridic hydride binds to the metal cation, while the proton binds to the oxygen anion (M-H and O-H).
Analogous to metal hydrides, metal oxyhydrides are also starting to find a way into CO2 hydrogenation. Perovskite-type oxyhydride-supported transition metals enhanced the selectivity and space time yield of methanol.
We further elucidated the catalytic importance of metal hydrides on the synthesis of C1 chemicals—mainly CH3OH—by briefly introducing homogeneous transition metal hydrides. Significant increases in catalytic activity and CH3OH selectivity are reported due to the presence of a 3d and group 3–7 metal complexes ability to surpass 2 e- reduction of CO2. Although homogeneous complex metal hydrides have a better catalytic activity, they suffer from poor product and catalyst separation. Therefore, their application is limited at a large scale in industry.
5. Future Outlook and Perspective
The thermo-catalytic hydrogenation of CO2 into fuels and chemicals has been studied for many years and has a substantial technical flexibility. Significant progress has been made in CO2 hydrogenation to C1 chemicals over traditional heterogeneous catalysts. However, metal hydrides are only starting to emerge in the area of CO2 hydrogenation. As of yet, there is very limited research on the formic acid and methanol synthesis over metal hydrides. The emergence of advanced in situ characterisation techniques and artificial intelligence (AI) aided probing or predictions, is anticipated to promote discoveries in metal hydride catalysed reactions. In this case, the nature of chemical bonding during the heterolytic dissociation of H2, hydride transfer and phase transitions, and changes in local electron density of metal hydrides that significantly influence catalytic performance (activity, selectivity and stability) will be well-understood. Moreover, the different reaction pathways over metal hydrides that give a proper understanding of the effect of the hydrides in selectively producing desired products of interest will be unravelled. Therefore, it is anticipated that the rational design and proper synthesis of supported or unsupported metal hydride catalysts will aid in the adsorption, activation and conversion of the thermodynamically stable and kinetically inert CO2 at a much faster reaction rate, as well as producing highly selective methane, formic acid and methanol products.
Author Contributions
Conceptualization, F.C., N.M.M. and X.O.; writing—original draft preparation, M.A.M.; writing—review and editing, M.A.M., X.O., N.M.M. and F.C.; supervision, F.C., N.M.M. and X.O.; funding acquisition, F.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Zhejiang Provincial Natural Science Foundation of China under grant No. LR24B030002.
Data Availability Statement
Data are available on request.
Acknowledgments
The authors gratefully acknowledge the Yongjiang Laboratory and the Nottingham Ningbo China Beacons of Excellence Research and Innovation Institute, and the faculty of science and engineering of the University of Nottingham Ningbo China for the administrative support in writing this review.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| MH | Metal hydrides |
| GHG | Greenhouse gas |
| INS | Inelastic neutron scattering spectroscopy |
| TOS | Time on stream |
| WHSV | Weight hourly space velocity |
| AP-XPS | Ambient-pressure X-ray photoelectron spectroscopy |
| XRD | X-ray spectroscopy |
| IR | Infrared spectroscopy |
| DFT | Density functional theory |
References
- Khan, A.A.; Tahir, M. Recent advancements in engineering approach towards design of photo-reactors for selective photocatalytic CO2 reduction to renewable fuels. J. CO2 Util. 2019, 29, 205–239. [Google Scholar] [CrossRef]
- Zhong, J.; Yang, X.; Wu, Z.; Liang, B.; Huang, Y.; Zhang, T. State of the art and perspectives in heterogeneous catalysis of CO2 hydrogenation to methanol. Chem. Soc. Rev. 2020, 49, 1385–1413. [Google Scholar] [CrossRef]
- Wei, L.; Yu, C.; Zhang, Q.; Liu, H.; Wang, Y. TiO2-based heterojunction photocatalysts for photocatalytic reduction of CO2 into solar fuels. J. Mater. Chem. A 2018, 6, 22411–22436. [Google Scholar] [CrossRef]
- Li, K.; Teng, C.; Wang, S.; Min, Q. Recent Advances in TiO2-Based Heterojunctions for Photocatalytic CO2 Reduction with Water Oxidation: A Review. Front. Chem. 2021, 9, 637501. [Google Scholar] [CrossRef]
- Setia, D.; Roithová, J. Trends in the hydricities of iron, cobalt, and nickel complexes and the metal-hydride reactivities with CO2. Int. J. Mass Spectrom. 2024, 504, 117310. [Google Scholar] [CrossRef]
- Younas, M.; Loong Kong, L.; Bashir, M.J.K.; Nadeem, H.; Shehzad, A.; Sethupathi, S. Recent Advancements, Fundamental Challenges, and Opportunities in Catalytic Methanation of CO2. Energy Fuels 2016, 30, 8815–8831. [Google Scholar] [CrossRef]
- Frei, M.S.; Mondelli, C.; García-Muelas, R.; Kley, K.S.; Puértolas, B.; López, N.; Safonova, O.V.; Stewart, J.A.; Ferré, D.C.; Peréz-Ramírez, J. Atomic-scale engineering of indium oxide promotion by palladium for methanol production via CO2 hydrogenation. Nat. Commun. 2019, 10, 3377. [Google Scholar] [CrossRef]
- Lv, X.; Lu, G.; Wang, Z.Q.; Xu, Z.N.; Guo, G.C. Computational Evidence for Lewis Base-Promoted CO2 Hydrogenation to Formic Acid on Gold Surfaces. ACS Catal. 2017, 7, 4519–4526. [Google Scholar] [CrossRef]
- Reutemann, W.; Kieczka, H. Formic acid. Ullmann’s Encycl. Ind. Chem. 2012, 16, 13–31. [Google Scholar] [CrossRef]
- Pérez-Fortes, M.; Schöneberger, J.C.; Boulamanti, A.; Tzimas, E. Methanol synthesis using captured CO2 as raw material: Techno-economic and environmental assessment. Appl. Energy 2016, 161, 718–732. [Google Scholar] [CrossRef]
- Cordero-Lanzac, T.; Ramirez, A.; Navajas, A.; Gevers, L.; Brunialti, S.; Gandía, L.M.; Aguayo, A.T.; Sarathy, S.M.; Gascon, J. A techno-economic and life cycle assessment for the production of green methanol from CO2: Catalyst and process bottlenecks. J. Energy Chem. 2022, 68, 255–266. [Google Scholar] [CrossRef]
- Iribarren, D.; Calvo-Serrano, R.; Martín-Gamboa, M.; Galán-Martín, A.; Guillén-Gosálbez, G. Social life cycle assessment of green methanol and benchmarking against conventional fossil methanol. Sci. Total Environ. 2022, 824, 153840. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, G.; Song, C.; Guo, X. Catalytic Conversion of Carbon Dioxide to Methanol: Current Status and Future Perspective. Front. Energy Res. 2021, 8, 621119. [Google Scholar] [CrossRef]
- Cheng, D.; Negreiros, F.R.; Aprà, E.; Fortunelli, A. Computational approaches to the chemical conversion of carbon dioxide. ChemSusChem. 2013, 6, 944–965. [Google Scholar] [CrossRef]
- Lykaki, M.; Mandela, E.; Varvoutis, G.; Lampropoulos, A.; Marnellos, G.E.; Konsolakis, M. State-of-the-art thermocatalytic systems for CH4 and CO production via CO2 hydrogenation: Critical comparison, mechanistic considerations and structure-performance insights. Discov. Chem. Eng. 2024, 4, 11. [Google Scholar] [CrossRef]
- Wang, M.; Luo, L.; Wang, C.; Du, J.; Li, H.; Zeng, J. Heterogeneous Catalysts toward CO2 Hydrogenation for Sustainable Carbon Cycle. Accounts Mater. Res. 2022, 3, 565–571. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the Greener Production of Formates/Formic Acid, Methanol, and DME by Heterogeneously Catalyzed CO2 Hydrogenation Processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Jacquemin, M.; Beuls, A.; Ruiz, P. Catalytic production of methane from CO2 and H2 at low temperature: Insight on the reaction mechanism. Catal. Today 2010, 157, 462–466. [Google Scholar] [CrossRef]
- Maru, M.S.; Ram, S.; Shukla, R.S.; Khan, N.H. Ruthenium-hydrotalcite (Ru-HT) as an effective heterogeneous catalyst for the selective hydrogenation of CO2 to formic acid. Mol. Catal. 2018, 446, 23–30. [Google Scholar] [CrossRef]
- Shen, C.; Sun, K.; Zou, R.; Wu, Q.; Mei, D.; Liu, C.J. CO2 Hydrogenation to Methanol on Indium Oxide-Supported Rhenium Catalysts: The Effects of Size. ACS Catal. 2022, 12, 12658–12669. [Google Scholar] [CrossRef]
- Monene, T.J.; Cele, M.N. The activities of selected metals and supports for CO2 hydrogenation to methanol: A review. Appl. Catal. O Open 2024, 197, 207019. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, G.; Zhu, J.; Zhang, X.; Ding, F.; Zhang, A.; Guo, X.; Song, C. CO2 Hydrogenation to Methanol over In2O3-Based Catalysts: From Mechanism to Catalyst Development. ACS Catal. 2021, 11, 1406–1423. [Google Scholar] [CrossRef]
- Grinter, D.C.; Graciani, J.; Palomino, R.M.; Xu, F.; Waluyo, I.; Sanz, J.F.; Senanayake, S.D.; Rodriguez, J.A. Adsorption and activation of CO2 on Pt/CeOx/TiO2(110): Role of the Pt-CeOx interface. Surf. Sci. 2021, 710, 121852. [Google Scholar] [CrossRef]
- Chinchen, G.C.; Spencer, M.S.; Waugh, K.C.; Whan, D.A. Promotion of methanol synthesis and the water-gas shift reactions by adsorbed oxygen on supported copper catalysts. J. Chem. Soc. Faraday Trans. 1 1987, 83, 2193–2212. [Google Scholar] [CrossRef]
- Eren, B.; Weatherup, R.S.; Liakakos, N.; Somorjai, G.A.; Salmeron, M. Dissociative Carbon Dioxide Adsorption and Morphological Changes on Cu(100) and Cu(111) at Ambient Pressures. J. Am. Chem. Soc. 2016, 138, 8207–8211. [Google Scholar] [CrossRef]
- Su, H.; Ye, Y.; Lee, K.J.; Zeng, J.; Mun, B.S.; Crumlin, E.J. Probing the surface chemistry for reverse water gas shift reaction on Pt(111) using ambient pressure X-ray photoelectron spectroscopy. J. Catal. 2020, 391, 123–131. [Google Scholar] [CrossRef]
- Sheffield, J.; Martin, K.B. Alternative Fuels and Advanced Vehicle Technologies for Improved Environmental Performance; Woodhead Publishing Ltd.: Sawston, UK, 2014; pp. 117–135. [Google Scholar]
- Gay-Lussac, J.L.; Thenard, L.J. Recherches physico-chimiques. Paris 1811, 2, 1–57. [Google Scholar]
- Hautefeuille, P.; Troost, L. Alliages de l’hydrogèke avec les métaux alcalins. Ann. Chim. Phys. 1874, 2, 273. [Google Scholar]
- Maeno, Z.; Yasumura, S.; Wu, X.; Huang, M.; Liu, C.; Toyao, T.; Shimizu, K. Isolated Indium Hydrides in CHA Zeolites: Speciation and Catalysis for Nonoxidative Dehydrogenation of Ethane. J. Am. Chem. Soc. 2020, 142, 4820–4832. [Google Scholar] [CrossRef]
- Yu, H.; Li, X.; Zheng, J. Beyond Hydrogen Storage: Metal Hydrides for Catalysis. ACS Catal. 2024, 14, 3139–3157. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, J.; Chen, P. Complex transition metal hydrides for heterogeneous catalysis. Chem Catal. 2023, 3, 100524. [Google Scholar] [CrossRef]
- Schneemann, A.; White, J.L.; Kang, S.Y.; Jeong, S.; Wan, L.F.; Cho, E.S.; Heo, T.W.; Prendergast, D.; Urban, J.J.; Wood, B.C.; et al. Nanostructured Metal Hydrides for Hydrogen Storage. Chem. Rev. 2018, 118, 10775. [Google Scholar] [CrossRef] [PubMed]
- Wietelmann, U.; Felderhoff, M.; Rittmeyer, P. Hydrides. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Berlin, Germany, 2016; pp. 1–36. [Google Scholar]
- Luo, Y.; Wang, Q.; Li, J.; Xu, F.; Sun, L.; Zou, Y.; Chu, H.; Li, B.; Zhang, K. Enhanced hydrogen storage/sensing of metal hydrides by nanomodification. Mater. Today Nano 2020, 9, 100071. [Google Scholar] [CrossRef]
- Dekura, S.; Kobayashi, H.; Kusada, K.; Kitagawa, H. Hydrogen in Palladium and Storage Properties of Related Nanomaterials: Size, Shape, Alloying, and Metal–Organic Framework Coating Effects. Phys. Chem. Chem. Phys. 2019, 20, 1158–1176. [Google Scholar] [CrossRef]
- Copéret, C. C-H bond activation and organometallic intermediates on isolated metal centers on oxide surfaces. Chem. Rev. 2010, 110, 656–680. [Google Scholar] [CrossRef] [PubMed]
- Copéret, C.; Estes, D.P.; Larmier, K.; Searles, K. Isolated Surface Hydrides: Formation, Structure, and Reactivity. Chem. Rev. 2016, 116, 8463–8505. [Google Scholar] [CrossRef]
- Serykh, A.I. Low-Dimensional Indium Oxo-Species on the Surface of In2O3/Al2O3 Catalytic Material: The Sites of Dissociative Adsorption of Hydrogen. J. Phys. Chem. C 2016, 120, 21436–21440. [Google Scholar] [CrossRef]
- Miyazaki, M.; Ogasawara, K.; Nakao, T.; Sasase, M.; Kitano, M.; Hosono, H. Hexagonal BaTiO(3-x)Hx Oxyhydride as a Water-Durable Catalyst Support for Chemoselective Hydrogenation. J. Am. Chem. Soc. 2022, 144, 6453–6464. [Google Scholar] [CrossRef]
- Waldie, K.M.; Ostericher, A.L.; Reineke, M.H.; Sasayama, A.F.; Kubiak, C.P. Hydricity of Transition-Metal Hydrides: Thermodynamic Considerations for CO2 Reduction. ACS Catal. 2018, 8, 1313–1324. [Google Scholar] [CrossRef]
- Niu, Y.; Borsodi, J.; Wootsch, A.; Révay, Z.; Hävecker, M.; Knop-Gericke, A.; Jackson, S.D.; Schlögl, R. Visualizing Formation of Intermetallic PdZn in a Palladium/Zinc Oxide Catalyst: Interfacial Fertilization by PdHx. Angew. Chemie—Int. Ed. 2019, 58, 4232–4237. [Google Scholar] [CrossRef] [PubMed]
- Teschner, D.; Borsodi, J.; Wootsch, A.; Révay, Z.; Hävecker, M.; Knop-Gericke, A.; Jackson, S.D.; Schlögl, R. The Roles of Subsurface Carbon and Hydrogen in Palladium-Catalyzed Alkyne Hydrogenation. Science 2008, 320, 86–89. [Google Scholar] [CrossRef]
- Fovanna, T.; Nachtegaal, M.; Clark, A.H.; Kröcher, O.; Ferri, D. Preparation, Quantification, and Reaction of Pd Hydrides on Pd/Al2O3 in Liquid Environment. ACS Catal. 2023, 13, 3323–3332. [Google Scholar] [CrossRef]
- Chang, F.; Tezsevin, I.; de Rijk, J.W.; Meeldijk, J.D.; Hofmann, J.P.; Er, S.; Ngene, P.; de Jongh, P.E. Potassium hydride-intercalated graphite as an efficient heterogeneous catalyst for ammonia synthesis. Nat. Catal. 2022, 5, 222–230. [Google Scholar] [CrossRef]
- Chang, F.; Guan, Y.; Chang, X.; Guo, J.; Wang, P.; Gao, W.; Wu, G.; Zheng, J.; Li, X.; Chen, P. Alkali and Alkaline Earth Hydrides-Driven N2 Activation and Transformation over Mn Nitride Catalyst. J. Am. Chem. Soc. 2018, 140, 14799–14806. [Google Scholar] [CrossRef]
- Wang, P.; Chang, F.; Gao, W.; Guo, J.; Wu, G.; He, T.; Chen, P. Breaking scaling relations to achieve low-temperature ammonia synthesis through LiH-mediated nitrogen transfer and hydrogenation. Nat. Chem. 2017, 9, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Kobayashi, Y.; Masuda, N.; Uchida, Y.; Okamoto, H.; Kageyama, T.; Hosokawa, S.; Loyer, F.; Mitsuhara, K.; Yamanaka, K.; et al. Metal-Dependent Support Effects of Oxyhydride-Supported Ru, Fe, Co Catalysts for Ammonia Synthesis. Adv. Energy Mater. 2018, 8, 1801772. [Google Scholar] [CrossRef]
- Kitano, M.; Inoue, Y.; Ishikawa, H.; Yamagata, K.; Nakao, T.; Tada, T.; Matsuishi, S.; Yokoyama, T.; Hara, M.; Hosono, H. Essential role of hydride ion in ruthenium-based ammonia synthesis catalysts. Chem. Sci. 2016, 7, 4036–4043. [Google Scholar] [CrossRef]
- Chen, H.; Liu, P.; Li, J.; Wang, Y.; She, C.; Liu, J.; Zhang, L.; Yang, Q.; Zhou, S.; Feng, X. MgH2/CuxO Hydrogen Storage Composite with Defect-Rich Surfaces for Carbon Dioxide Hydrogenation. ACS Appl. Mater. Interfaces 2019, 11, 31009–31017. [Google Scholar] [CrossRef]
- Feng, X.; Jiang, L.; Li, Z.; Wang, S.; Ye, J.; Wu, Y.; Yuan, B. Boosting the hydrogenation activity of dibenzyltoluene catalyzed by Mg-based metal hydrides. Int. J. Hydrogen Energy 2022, 47, 23994–24003. [Google Scholar] [CrossRef]
- Lelis, M.; Varnagiris, S.; Urbonavicius, M.; Zakarauskas, K. Investigation of catalyst development from Mg2NiH4 hydride and its application for the CO2 methanation reaction. Coatings 2020, 10, 1178. [Google Scholar] [CrossRef]
- Amica, G.; Gennari, F.C. Synergistic effect of MgH2 doping with Ni and carbon nanotubes on thermochemical CO2 recycling process for CH4-H2 mixtures production. Int. J. Hydrogen Energy 2022, 47, 428–442. [Google Scholar] [CrossRef]
- Chen, H.; Liu, P.; Liu, J.; Feng, X.; Zhou, S. Mechanochemical in-situ incorporation of Ni on MgO/MgH2 surface for the selective O-/C-terminal catalytic hydrogenation of CO2 to CH4. J. Catal. 2021, 394, 397–405. [Google Scholar] [CrossRef]
- Grasso, M.L.; Puszkiel, J.; Gennari, F.C.; Santoru, A.; Dornheim, M.; Pistidda, C. CO2 reactivity with Mg2NiH4 synthesized by in situ monitoring of mechanical milling. Phys. Chem. Chem. Phys. 2020, 22, 1944–1952. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.; Ouyang, L.; Liu, J.; Wang, H. Metallic Ni nanocatalyst in situ formed from LaNi5H5 toward efficient CO2 methanation. Int. J. Hydrogen Energy 2019, 44, 29068–29074. [Google Scholar] [CrossRef]
- Chen, H.; Ma, N.; Cheng, C.; Zhang, H.; Yuan, W.; Liu, P. Hydrogen activation on aluminium-doped magnesium hydride surface for methanation of carbon dioxide. Appl. Surf. Sci. 2020, 515, 146038. [Google Scholar] [CrossRef]
- Amica, G.; Azcona, R.; Aparicio, S.; Gennari, F.C. Catalysis effect on CO2 methanation using MgH2 as a portable hydrogen medium. Phys. Chem. Chem. Phys. 2020, 22, 14720–14730. [Google Scholar] [CrossRef]
- Grasso, M.L.; Puszkiel, J.; Albanesi, F.; Dornheim, L.M.; Pistidda, C.; Gennari, F.C. CO2 reutilization for methane production via a catalytic process promoted by hydrides. Phys. Chem. Chem. Phys. 2019, 21, 19825–19834. [Google Scholar] [CrossRef]
- Zhao, J.; Wei, Y.F.; Cai, Y.L.; Wang, L.Z.; Xie, J.; Teng, Y.L.; Zhu, W.; Shen, M.; Dong, B.X. Highly Selective and Efficient Reduction of CO2 to Methane by Activated Alkaline Earth Metal Hydrides without a Catalyst. ACS Sustain. Chem. Eng. 2019, 7, 4831–4841. [Google Scholar] [CrossRef]
- Kato, S.; Matam, S.K.; Kerger, P.; Bernard, L.; Battaglia, C.; Vogel, D.; Rohwerder, M.; Zuttel, A. The Origin of the Catalytic Activity of a Metal Hydride in CO2 Reduction. Angew. Chemie—Int. Ed. 2016, 55, 6028–6032. [Google Scholar] [CrossRef]
- Kato, S.; Borgschulte, A.; Ferri, D.; Bielmann, M.; Crivello, J.C.; Wiedenmann, D.; Parlinska-Wojtan, M.; Rossbach, P.; Lu, Y.; Remhof, A.; et al. CO2 hydrogenation on a metal hydride surface. Phys. Chem. Chem. Phys. 2012, 14, 5518–5526. [Google Scholar] [CrossRef]
- Dong, B.X.; Wang, L.Z.; Song, L.; Zhao, J.; Teng, Y.L. Thermochemical Reduction of Carbon Dioxide with Alkali Metal Hydrides, Producing Methane and Hydrogen Fuels at Moderate Temperatures. Energy Fuels 2016, 30, 6620–6625. [Google Scholar] [CrossRef]
- Zhao, J.; Teng, Y.L.; Dong, B.X. Thermal Reduction of CO2 with Activated Alkali Metal Aluminum Hydrides for Selective Methanation. Energy Fuels 2020, 34, 11210–11218. [Google Scholar] [CrossRef]
- Nilwanna, K.; Sittiwong, J.; Boekfa, B.; Treesukol, P.; Boonya-udtayan, S.; Probst, M.; Maihom, T.; Limtrakul, J. Aluminum-based metal-organic framework support metal(II)-hydride as catalyst for the hydrogenation of carbon dioxide to formic acid: A computational study. Mol. Catal. 2023, 541, 113116. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, G.; Meiwes-Broer, K.H.; Gantefor, G.; Bowen, K. CO2 Activation and Hydrogenation by PtHn- Cluster Anions. Angew. Chemie—Int. Ed. 2016, 55, 9644–9647. [Google Scholar] [CrossRef] [PubMed]
- Habib, M.; Sarkar, R.; Biswas, S.; Pramanik, A.; Sarkar, P.; Pal, S. Unambiguous hydrogenation of CO2 by coinage-metal hydride anions: An intuitive idea based on in silico experiments. Phys. Chem. Chem. Phys. 2019, 21, 7483–7490. [Google Scholar] [CrossRef] [PubMed]
- Kadeer, K.; Li, X.; Zheng, J. Hydrogen generation by coupling methanol steam reforming with metal hydride hydrolysis. Chem. Commun. 2023, 59, 5443–5446. [Google Scholar] [CrossRef]
- Kim, J.; Lim, H.; Tian, Y.; Piliai, L.; Hunt, A.; Waluyo, I.; Senanayake, S.D.; Rodriguez, J.A. The Surface Chemistry of Methanol on Pd(111) and H-Pd(111) Surfaces: C-O Bond Cleavage and the Effects of Metal Hydride Formation. J. Phys. Chem. Lett. 2024, 15, 6209–6215. [Google Scholar] [CrossRef]
- Rusman, N.A.A.; Dahari, M. A review on the current progress of metal hydrides material for solid-state hydrogen storage applications. Int. J. Hydrogen Energy 2016, 41, 12108–12126. [Google Scholar] [CrossRef]
- Akamaru, S.; Hara, M.; Matsuyama, M. Alloying effects on the hydrogen-storage capability of Pd-TM-H (TM = Cu, Au, Pt, Ir) systems. J. Alloys Compd. 2014, 614, 238–243. [Google Scholar] [CrossRef]
- Yao, J.; Wang, B.; Chen, H.; Han, Z.; Wu, Y.; Cai, Z.; Manggada, G.W.; Elsayed, M.A.; Zhou, S. Effect of copper cluster on reaction pathways of carbon dioxide hydrogenation on magnesium hydride surface. Int. J. Hydrogen Energy. 2024, 78, 1089–1098. [Google Scholar] [CrossRef]
- Jin, P.; Luo, N.; Wang, F. Analogy in the Mechanism of Heterolytic H2 Dissociation. ACS Catal. 2024, 14, 18639–18650. [Google Scholar] [CrossRef]
- Kyriakou, G.; Boucher, M.B.; Jewell, A.D.; Lewis, E.A.; Lawton, T.J.; Baber, A.E.; Tierney, H.L.; Flytzani-Stephanopoulos, M.; Sykes, E.C.H. Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations. Science 2012, 335, 1209–1212. [Google Scholar] [CrossRef]
- Yang, C.; Ma, S.; Liu, Y.; Wang, L.; Yuan, D.; Shao, W.; Zhang, L.; Yang, F.; Lin, T.; Ding, H.; et al. Homolytic H2 dissociation for enhanced hydrogenation catalysis on oxides. Nat. Commun. 2024, 15, 540. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Dong, L.; Wang, Z.; Han, X.; Daemen, L.L.; Li, J.; Cheng, Y.; Guo, Y.; Liu, X.; Hu, Y.; et al. A unique Co@CoO catalyst for hydrogenolysis of biomass-derived 5-hydroxymethylfurfural to 2,5-dimethylfuran. Nat. Commun. 2022, 13, 3657. [Google Scholar] [CrossRef]
- Vilé, G.; Albani, D.; Nachtegaal, M.; Chen, Z.; Dontsova, D.; Antonietti, M.; López, N.; Pérez-Ramírez, J. A Stable Single-Site Palladium Catalyst for Hydrogenations. Angew. Chemie—Int. Ed. 2015, 54, 11265–11269. [Google Scholar] [CrossRef]
- Eisenstein, O.; Crabtree, R.H. Outer sphere hydrogenation catalysis. New J. Chem. 2013, 37, 21–27. [Google Scholar] [CrossRef]
- Aireddy, D.R.; Ding, K. Heterolytic Dissociation of H2 in Heterogeneous Catalysis. ACS Catal. 2022, 12, 4707–4723. [Google Scholar] [CrossRef]
- Wu, Z.; Cheng, Y.; Tao, F.; Daemen, L.; Foo, G.S.; Nguyen, L.; Zhang, X.; Beste, A.; Ramirez-Cuesta, A.J. Direct Neutron Spectroscopy Observation of Cerium Hydride Species on a Cerium Oxide Catalyst. J. Am. Chem. Soc. 2017, 139, 9721–9727. [Google Scholar] [CrossRef]
- Strunk, J.; Kähler, K.; Xia, X.; Muhler, M. The surface chemistry of ZnO nanoparticles applied as heterogeneous catalysts in methanol synthesis. Surf. Sci. 2009, 603, 1776–1783. [Google Scholar] [CrossRef]
- Ye, J.; Liu, C.; Mei, D.; Ge, Q. Active Oxygen Vacancy Site for Methanol Synthesis from CO2 Hydrogenation on In2O3(110): A DFT Study. Am. Chem. Soc. 2013, 3, 1296–1306. [Google Scholar] [CrossRef]
- Frei, M.S.; Capdevila-Cortada, M.; García-Muelas, R.; Mondello, C.; López, N.; Stewart, J.A.; Ferré, D.C.; Pérez-Ramírez, J. Mechanism and microkinetics of methanol synthesis via CO2 hydrogenation on indium oxide. J. Catal. 2018, 361, 313–321. [Google Scholar] [CrossRef]
- Qin, B.; Li, S. First principles investigation of dissociative adsorption of H2 during CO2 hydrogenation over cubic and hexagonal In2O3 catalysts. Phys. Chem. Chem. Phys. 2020, 22, 3390–3399. [Google Scholar] [CrossRef]
- Jia, J.; Qian, C.; Dong, Y.; Li, Y.F.; Wang, H.; Ghoussoub, M.; Butler, K.T.; Walsh, A.; Ozin, G.A. Heterogeneous catalytic hydrogenation of CO2 by metal oxides: Defect engineering-perfecting imperfection. Chem. Soc. Rev. 2017, 46, 4631–4644. [Google Scholar] [CrossRef]
- Posada-Borbón, A.; Grönbeck, H. Hydrogen adsorption on In2O3(111) and In2O3(110). Phys. Chem. Chem. Phys. 2020, 22, 16193–16202. [Google Scholar] [CrossRef]
- Ye, J.; Ge, Q.; Liu, C. DFT study of CO2 adsorption and hydrogenation on the In2O3 surfaces. J. Phys. Chem. 2012, 116, 7817–7825. [Google Scholar] [CrossRef]
- Juárez, R.; Parker, S.F.; Concepción, P.; Corma, A.; García, H. Heterolytic and heterotopic dissociation of hydrogen on ceria-supported gold nanoparticles. Combined inelastic neutron scattering and FT-IR spectroscopic study on the nature and reactivity of surface hydrogen species. Chem. Sci. 2010, 1, 731–738. [Google Scholar] [CrossRef]
- Mao, G.C.; Kan, X.T.; Xiao, M.X.; Liu, W.L.; Dong, B.X.; Teng, Y.L. Alkaline Earth Metal-Induced Hydrogenation of the CaO-Captured CO2 to Methane at Room Temperature. Ind. Eng. Chem. Res. 2022, 61, 10124–10132. [Google Scholar] [CrossRef]
- Tang, Y.; Kobayashi, Y.; Tassel, C.; Yamamoto, T.; Kageyama, H. Hydride-Enhanced CO2 Methanation: Water-Stable BaTiO2.4H0.6 as a New Support. Adv. Energy Mater. 2018, 8, 1800800. [Google Scholar] [CrossRef]
- He, Y.; Li, Y.; Lei, M.; Polo-Garzon, F.; Perez-Aguilar, J.; Bare, S.R.; Formo, E.; Kim, H.; Daemen, L.; Cheng, Y.; et al. Significant Roles of Surface Hydrides in Enhancing the Performance of Cu/BaTiO2.8H0.2 Catalyst for CO2 Hydrogenation to Methanol. Angew. Chemie—Int. Ed. 2024, 63, e202313389. [Google Scholar] [CrossRef]
- Grice, K.A. Carbon dioxide reduction with homogenous early transition metal complexes: Opportunities and challenges for developing CO2 catalysis. Coord. Chem. Rev. 2017, 336, 78–95. [Google Scholar] [CrossRef]
- Das, C.; Grover, J.; Tannu; Das, A.; Maiti, D.; Dutta, A.; Lahiri, G.K. Recent developments in first-row transition metal complex-catalyzed CO2 hydrogenation. Dalt. Trans. 2022, 51, 8160–8168. [Google Scholar] [CrossRef] [PubMed]
- Fong, H.; Peters, J.C. Hydricity of an Fe-H species and catalytic CO2 hydrogenation. Inorg. Chem. 2015, 54, 5124–5135. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Waldie, K.M.; Warnke, I.; De Crisci, A.G.; Batista, V.S.; Waymouth, R.M.; Chidsey, C.E.D. Experimental and Theoretical Study of CO2 Insertion into Ruthenium Hydride Complexes. Inorg. Chem. 2016, 55, 1623–1632. [Google Scholar] [CrossRef]
- Hadlington, T.J.; Kefalidis, C.E.; Maron, L.; Jones, C. Efficient Reduction of Carbon Dioxide to Methanol Equivalents Catalyzed by Two-Coordinate Amido-Germanium(II) and -Tin(II) Hydride Complexes. ACS Catal. 2017, 7, 1853–1859. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).