

Designing Sustainable Polymer Blends: Tailoring Mechanical Properties and Degradation Behaviour in PHB/PLA/PCL Blends in a Seawater Environment

 , , ,
, , ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Material Extrusion
2.3. Material Moulding
2.4. Seawater Degradation
2.5. Weight Variance
2.6. Mechanical Properties
2.7. Infrared Analysis
2.8. Thermal Analysis
2.9. Morphology Characterisation
3. Results and Discussion
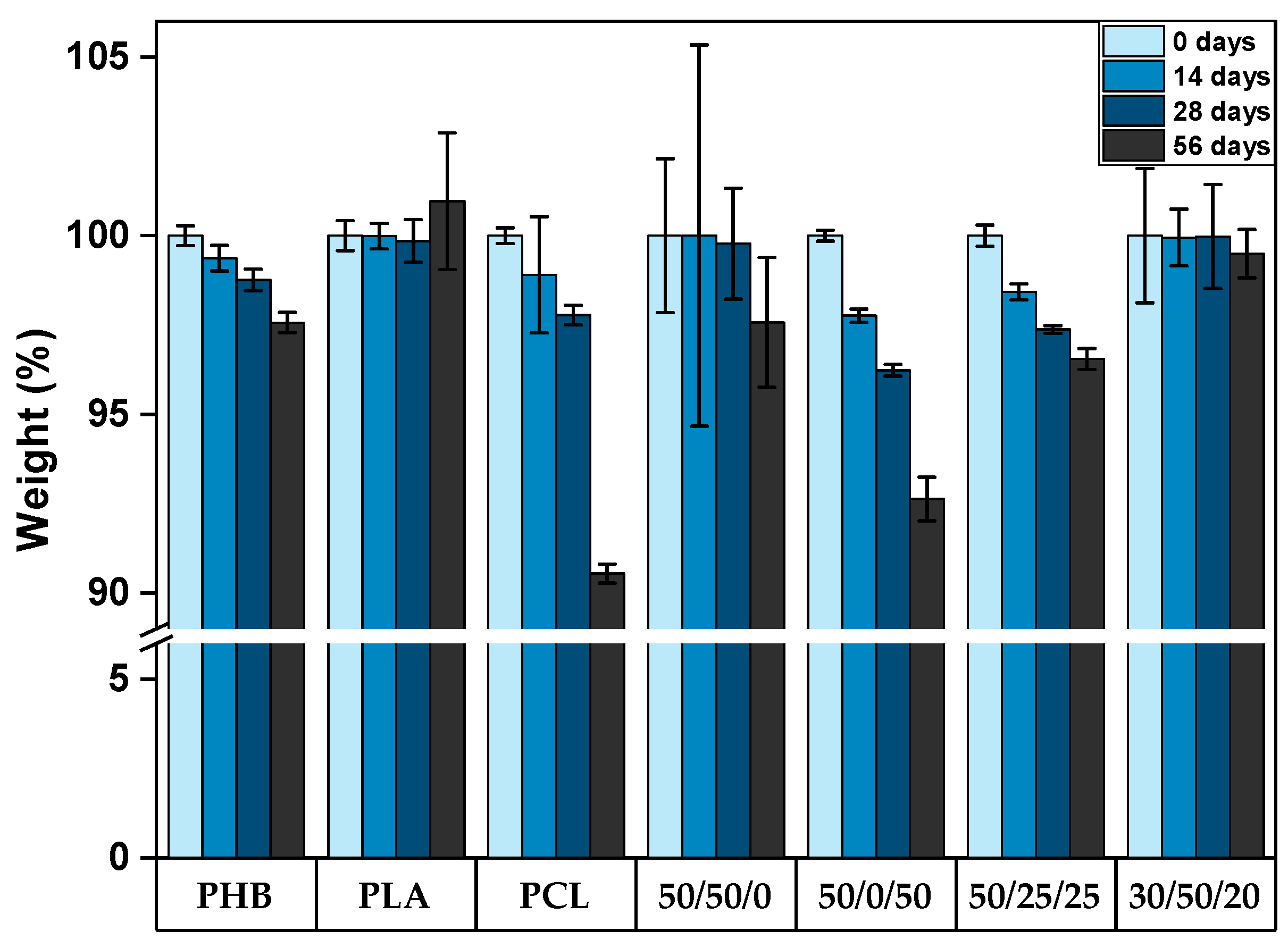
3.1. Seawater Degradation
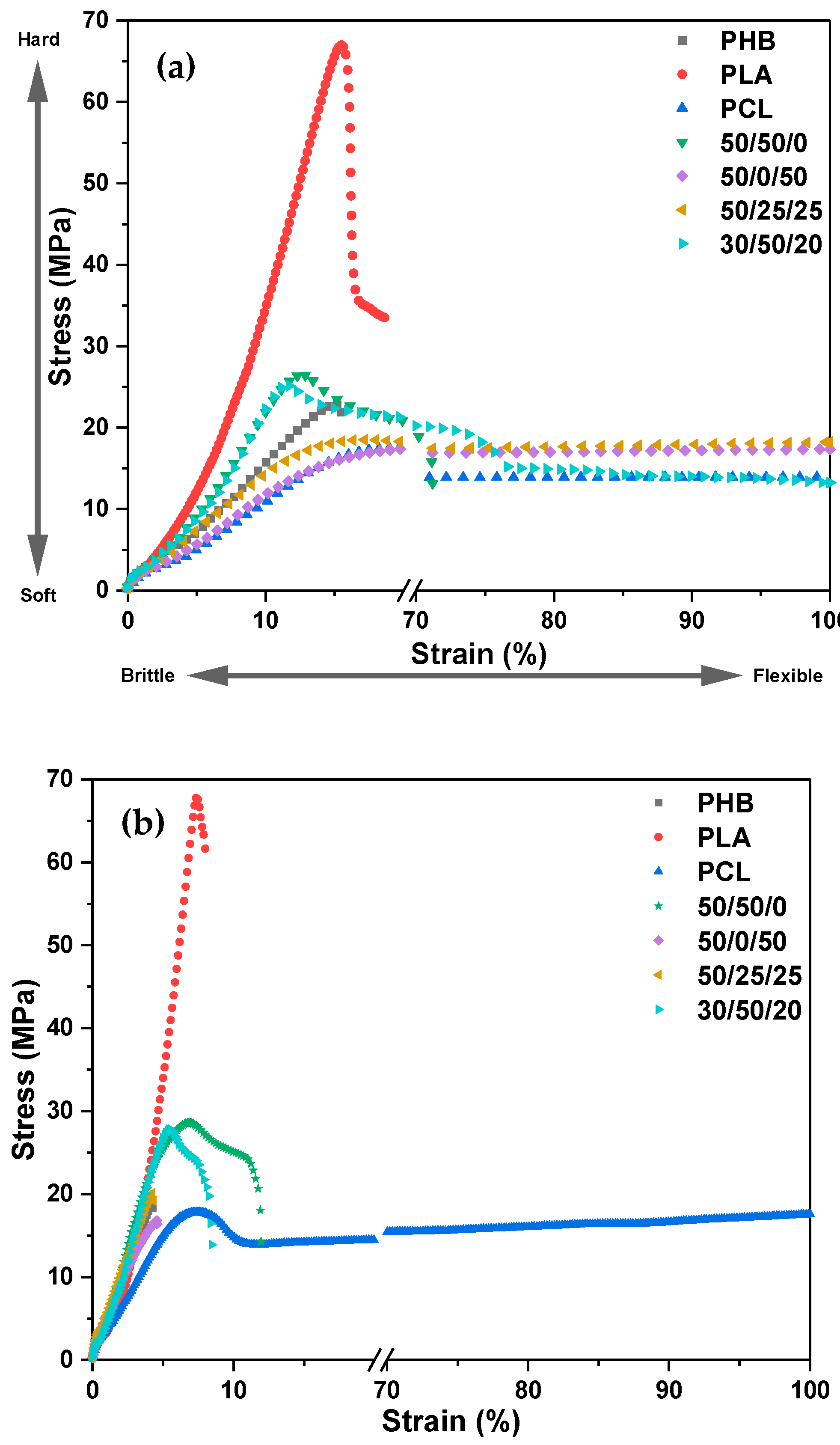
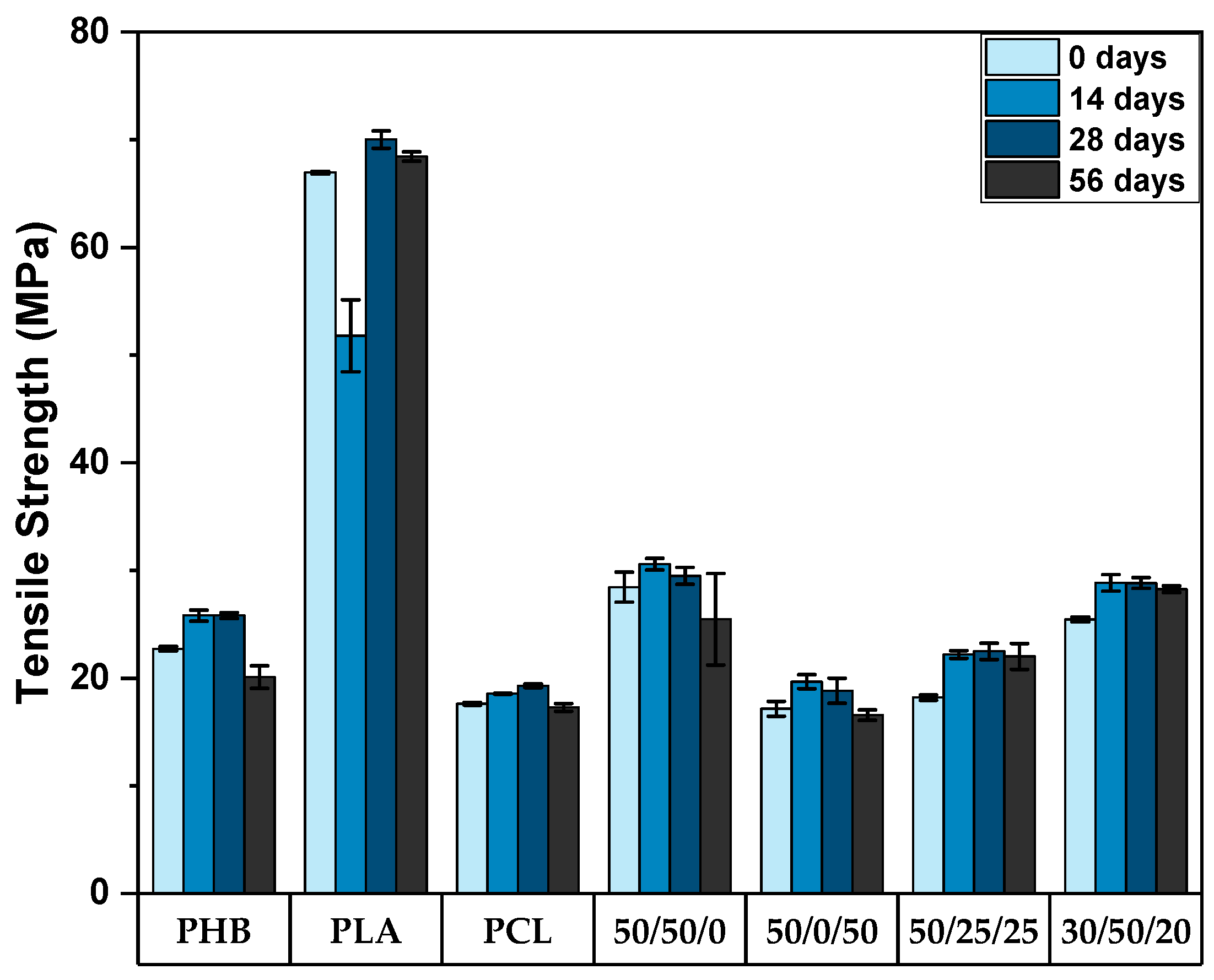
3.2. Mechanical Properties
3.3. Infrared Analysis
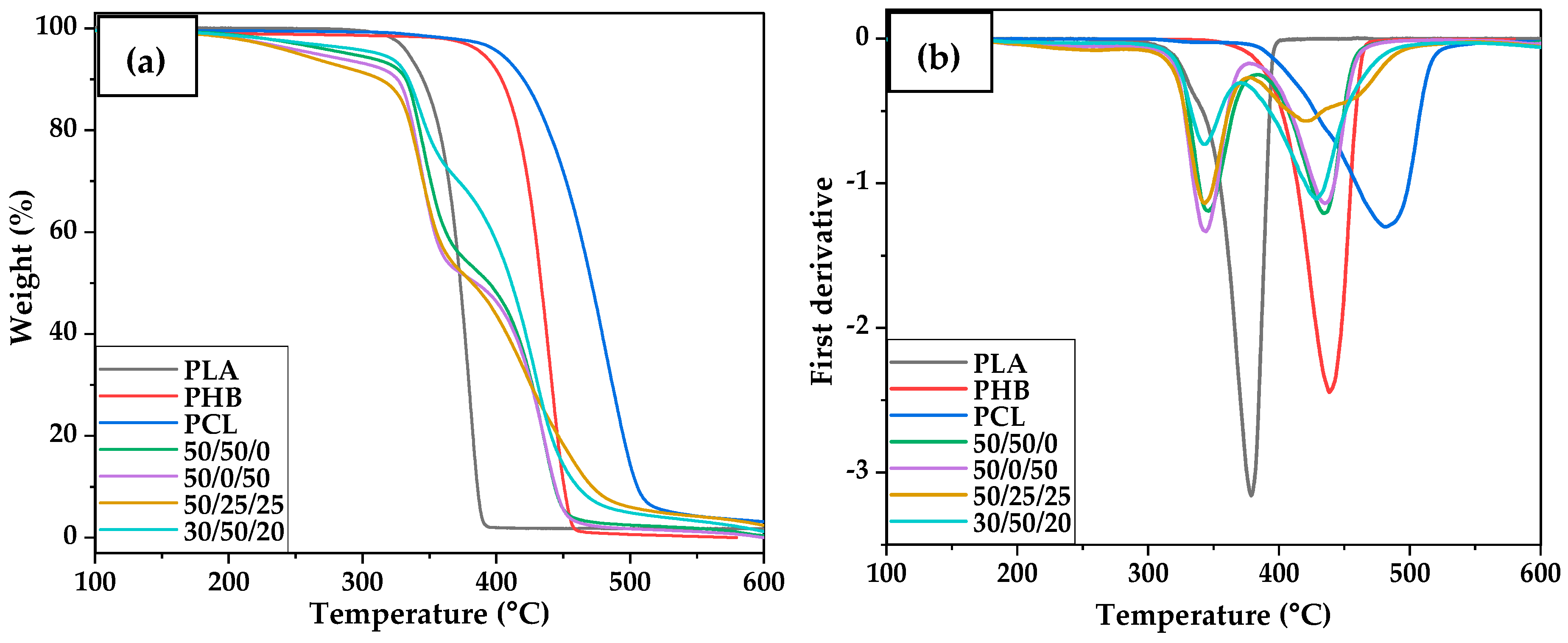
3.4. Thermal Transitions and Degradation of PHB/PLA/PCL Blends
3.5. Morphology of the Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Kliem, S.; Kreutzbruck, M.; Bonten, C. Review on the biological degradation of polymers in various environments. Materials 2020, 13, 4586. [Google Scholar] [CrossRef] [PubMed]
- Leslie, H.A.; van Velzen, M.J.M.; Brandsma, S.H.; Vethaak, A.D.; Garcia-Vallejo, J.J.; Lamoree, M.H. Discovery and quantification of plastic particle pollution in human blood. Environ. Int. 2022, 163, 107199. [Google Scholar] [CrossRef]
- Chahine, M.T. The hydrological cycle and its influence on climate. Nature 1992, 359, 373–380. [Google Scholar] [CrossRef]
- Volova, T.G.; Boyandin, A.N.; Vasiliev, A.D.; Karpov, V.A.; Prudnikova, S.V.; Mishukova, O.V.; Boyarskikh, U.A.; Filipenko, M.L.; Rudnev, V.P.; Bá Xuân, B.; et al. Biodegradation of polyhydroxyalkanoates (PHAs) in tropical coastal waters and identification of PHA-degrading bacteria. Polym. Degrad. Stab. 2010, 95, 2350–2359. [Google Scholar] [CrossRef]
- Heimowska, A.; Morawska, M.; Bocho-Janiszewska, A. Biodegradation of poly(ϵ-caprolactone) in natural water environments. Polish J. Chem. Technol. 2017, 19, 120–126. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, H.; Suzuyoshi, K. Environmental degradation of biodegradable polyesters 2. Poly(ε-caprolactone), poly[(R)-3-hydroxybutyrate], and poly(L-lactide) films in natural dynamic seawater. Polym. Degrad. Stab. 2002, 75, 357–365. [Google Scholar] [CrossRef]
- Bagheri, A.R.; Laforsch, C.; Greiner, A.; Agarwal, S. Fate of So-Called Biodegradable Polymers in Seawater and Freshwater. Glob. Challenges 2017, 1, 1700048. [Google Scholar] [CrossRef]
- Munn, C.B. Marine Microbiology: Ecology & Applications, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2019; pp. 1–27. [Google Scholar]
- Mackenzie, F.T.; Duxbury, A.C.; Byrne, R.H. Seawater. Available online: https://www.britannica.com/science/seawater (accessed on 12 April 2023).
- Bher, A.; Mayekar, P.C.; Auras, R.A.; Schvezov, C.E. Biodegradation of Biodegradable Polymers in Mesophilic Aerobic Environments. Int. J. Mol. Sci. 2022, 23, 12165. [Google Scholar] [CrossRef]
- García-Campo, M.J.; Boronat, T.; Quiles-Carrillo, L.; Balart, R.; Montanes, N. Manufacturing and characterization of toughened poly(lactic acid) (PLA) formulations by ternary blends with biopolyesters. Polymers 2018, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Engler, L.G.; Crespo, J.S.; Gately, N.M.; Major, I.; Devine, D.M. Process Optimization for the 3D Printing of PLA and HNT Composites with Arburg Plastic Freeforming. J. Compos. Sci. 2022, 6, 309. [Google Scholar] [CrossRef]
- Da Silva, D.; Kaduri, M.; Poley, M.; Adir, O.; Krinsky, N.; Shainsky-Roitman, J.; Schroeder, A. Biocompatibility, biodegradation and excretion of polylactic acid (PLA) in medical implants and theranostic systems. Chem. Eng. J. 2018, 340, 9–14. [Google Scholar] [CrossRef]
- Lyu, S.P.; Untereker, D. Degradability of polymers for implantable biomedical devices. Int. J. Mol. Sci. 2009, 10, 4033–4065. [Google Scholar] [CrossRef] [Green Version]
- Narancic, T.; Verstichel, S.; Reddy, C.S.; Morales-Gamez, L.; Kenny, S.T.; De Wilde, B.; Babu, P.R.; O’Connor, K.E. Biodegradable Plastic Blends Create New Possibilities for End-of-Life Management of Plastics but They Are Not a Panacea for Plastic Pollution. Environ. Sci. Technol. 2018, 52, 10441–10452. [Google Scholar] [CrossRef] [PubMed]
- Dalton, B.; Bhagabati, P.; De Micco, J.; Padamati, R.B.; O’Connor, K. A Review on Biological Synthesis of the Biodegradable Polymers Polyhydroxyalkanoates and the Development of Multiple Applications. Catalysts 2022, 12, 319. [Google Scholar] [CrossRef]
- Bhasney, S.M.; Bhagabati, P.; Kumar, A.; Katiyar, V. Morphology and crystalline characteristics of polylactic acid [PLA]/linear low density polyethylene [LLDPE]/microcrystalline cellulose [MCC] fiber composite. Compos. Sci. Technol. 2019, 171, 54–61. [Google Scholar] [CrossRef]
- Siracusa, V.; Karpova, S.; Olkhov, A.; Zhulkina, A.; Kosenko, R.; Iordanskii, A. Gas transport phenomena and polymer dynamics in PHB/PLA blend films as potential packaging materials. Polymers 2020, 12, 647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simão, J.A.; Bellani, C.F.; Branciforti, M.C. Thermal properties and crystallinity of PCL/PBSA/cellulose nanocrystals grafted with PCL chains. J. Appl. Polym. Sci. 2017, 134, 44493. [Google Scholar] [CrossRef]
- Brannigan, R.P.; Dove, A.P. Synthesis, properties and biomedical applications of hydrolytically degradable materials based on aliphatic polyesters and polycarbonates. Biomater. Sci. 2017, 5, 9–21. [Google Scholar] [CrossRef]
- Chen, Y.; Hung, S.T.; Chou, E.; Wu, H.S.; Devine, D.M. Bioresorbable Polymers and their Biomedical Applications; Smithers Rapra: Shrewsbury, UK, 2017; Volume 15, pp. 1–26. [Google Scholar]
- Meereboer, K.W.; Misra, M.; Mohanty, A.K. Review of recent advances in the biodegradability of polyhydroxyalkanoate (PHA) bioplastics and their composites. Green Chem. 2020, 22, 5519–5558. [Google Scholar] [CrossRef]
- Agarwal, M.; Koelling, K.W.; Chalmers, J.J. Characterization of the degradation of polylactic acid polymer in a solid substrate environment. Biotechnol. Prog. 1998, 14, 517–526. [Google Scholar] [CrossRef]
- Tabasi, R.Y.; Ajji, A. Selective degradation of biodegradable blends in simulated laboratory composting. Polym. Degrad. Stab. 2015, 120, 435–442. [Google Scholar] [CrossRef]
- Limsukon, W.; Auras, R.; Selke, S. Hydrolytic degradation and lifetime prediction of poly(lactic acid) modified with a multifunctional epoxy-based chain extender. Polym. Test. 2019, 80, 106108. [Google Scholar] [CrossRef]
- Hoüglund, A.; Odelius, K.; Albertsson, A.C. Crucial differences in the hydrolytic degradation between industrial polylactide and laboratory-scale poly(L-lactide). ACS Appl. Mater. Interfaces 2012, 4, 2788–2793. [Google Scholar] [CrossRef]
- Kolstad, J.J.; Vink, E.T.H.; De Wilde, B.; Debeer, L. Assessment of anaerobic degradation of IngeoTM polylactides under accelerated landfill conditions. Polym. Degrad. Stab. 2012, 97, 1131–1141. [Google Scholar] [CrossRef]
- Elhami, V.; Hempenius, M.A.; Schuur, B. Crotonic Acid Production by Pyrolysis and Vapor Fractionation of Mixed Microbial Culture-Based Poly(3-hydroxybutyrate-co-3-hydroxyvalerate). Ind. Eng. Chem. Res. 2023, 62, 916–923. [Google Scholar] [CrossRef]
- Kasuya, K.I.; Takagi, K.I.; Ishiwatari, S.I.; Yoshida, Y.; Doi, Y. Biodegradabilities of various aliphatic polyesters in natural waters. Polym. Degrad. Stab. 1998, 59, 327–332. [Google Scholar] [CrossRef]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Woodard, L.N.; Grunlan, M.A. Hydrolytic Degradation and Erosion of Polyester Biomaterials. ACS Macro Lett. 2018, 7, 976–982. [Google Scholar] [CrossRef] [Green Version]
- Artsis, M.I.; Bonartsev, A.P.; Iordanskii, A.L.; Bonartseva, G.A.; Zaikov, G.E. Biodegradation and medical application of microbial poly(3-Hydroxybutyrate). Mol. Cryst. Liq. Cryst. 2012, 555, 232–262. [Google Scholar] [CrossRef]
- Pfau, M.R.; McKinzey, K.G.; Roth, A.A.; Grunlan, M.A. PCL-Based Shape Memory Polymer Semi-IPNs: The Role of Miscibility in Tuning the Degradation Rate. Biomacromolecules 2020, 21, 2493–2501. [Google Scholar] [CrossRef]
- Wang, S.; Kempen, D.H.R.; Ruiter, G.C.W.; Cai, L.; Spinner, R.J.; Windebank, A.J.; Yaszemski, M.J.; Lu, L. Molecularly engineered biodegradable polymer networks with a wide range of stiffness for bone and peripheral nerve regeneration. Adv. Funct. Mater. 2015, 25, 2715–2724. [Google Scholar] [CrossRef]
- Bartczak, Z.; Galeski, A.; Kowalczuk, M.; Sobota, M.; Malinowski, R. Tough blends of poly(lactide) and amorphous poly([R,S]-3-hydroxy butyrate)—Morphology and properties. Eur. Polym. J. 2013, 49, 3630–3641. [Google Scholar] [CrossRef]
- Liu, H.; Chen, F.; Liu, B.; Estep, G.; Zhang, J. Super toughened poly(lactic acid) ternary blends by simultaneous dynamic vulcanization and interfacial compatibilization. Macromolecules 2010, 43, 6058–6066. [Google Scholar] [CrossRef]
- Zenkiewicz, M.; Richert, J.; Rytlewski, P.; Moraczewski, K.; Stepczyńska, M.; Karasiewicz, T. Characterisation of multi-extruded poly(lactic acid). Polym. Test. 2009, 28, 412–418. [Google Scholar] [CrossRef]
- Taubner, V.; Shishoo, R. Influence of processing parameters on the degradation of poly(L-lactide) during extrusion. J. Appl. Polym. Sci. 2001, 79, 2128–2135. [Google Scholar] [CrossRef]
- Chen, C.C.; Chueh, J.Y.; Tseng, H.; Huang, H.M.; Lee, S.Y. Preparation and characterization of biodegradable PLA polymeric blends. Biomaterials 2003, 24, 1167–1173. [Google Scholar] [CrossRef]
- Elsawy, M.A.; Kim, K.H.; Park, J.W.; Deep, A. Hydrolytic degradation of polylactic acid (PLA) and its composites. Renew. Sustain. Energy Rev. 2017, 79, 1346–1352. [Google Scholar] [CrossRef]
- Urquijo, J.; Guerrica-Echevarría, G.; Eguiazábal, J.I. Melt processed PLA/PCL blends: Effect of processing method on phase structure, morphology, and mechanical properties. J. Appl. Polym. Sci. 2015, 132, 42641. [Google Scholar] [CrossRef]
- Ferri, J.M.; Fenollar, O.; Jorda-Vilaplana, A.; García-Sanoguera, D.; Balart, R. Effect of miscibility on mechanical and thermal properties of poly(lactic acid)/polycaprolactone blends. Polym. Int. 2016, 65, 453–463. [Google Scholar] [CrossRef]
- Jamshidian, M.; Tehrany, E.A.; Imran, M.; Jacquot, M.; Desobry, S. Poly-Lactic Acid: Production, applications, nanocomposites, and release studies. Compr. Rev. Food Sci. Food Saf. 2010, 9, 552–571. [Google Scholar] [CrossRef]
- Bao, Q.; Wong, W.; Liu, S.; Tao, X. Accelerated Degradation of Poly(lactide acid)/Poly(hydroxybutyrate) (PLA/PHB) Yarns/Fabrics by UV and O2 Exposure in South China Seawater. Polymers 2022, 14, 1216. [Google Scholar] [CrossRef]
- Vogel, C.; Siesler, H.W. Thermal degradation of poly(ε-caprolactone), poly(L-lactic acid) and their blends with poly(3-hydroxy-butyrate) studied by TGA/FT-IR spectroscopy. Macromol. Symp. 2008, 265, 183–194. [Google Scholar] [CrossRef]
- Herrera-Kao, W.A.; Loría-Bastarrachea, M.I.; Pérez-Padilla, Y.; Cauich-Rodríguez, J.V.; Vázquez-Torres, H.; Cervantes-Uc, J.M. Thermal degradation of poly(caprolactone), poly(lactic acid), and poly(hydroxybutyrate) studied by TGA/FTIR and other analytical techniques. Polym. Bull. 2018, 75, 4191–4205. [Google Scholar] [CrossRef]
- Li, S.; He, J.; Yu, P.H.; Cheung, M.K. Thermal Degradation of Poly(3-hydroxybutyrate) and Poly(3-hydroxybutyrate- co -3-hydroxyvalerate) as Studied by TG, TG–FTIR, and Py–GC/MS. J. Appl. Pol. Sci. 2002, 89, 1530–1536. [Google Scholar] [CrossRef]
- Braun, B.; Dorgan, J.R.; Dec, S.F. Infrared spectroscopic determination of lactide concentration in polylactide: An improved methodology. Macromolecules 2006, 39, 9302–9310. [Google Scholar] [CrossRef]
- Mofokeng, J.P.; Luyt, A.S. Morphology and thermal degradation studies of melt-mixed poly(lactic acid) (PLA)/poly(ε-caprolactone) (PCL) biodegradable polymer blend nanocomposites with TiO2 as filler. Polym. Test. 2015, 45, 93–100. [Google Scholar] [CrossRef]
- Patrício, T.; Bártolo, P. Thermal stability of PCL/PLA blends produced by physical blending process. Procedia Eng. 2013, 59, 292–297. [Google Scholar] [CrossRef]
- Arrieta, M.P.; López, J.; López, D.; Kenny, J.M.; Peponi, L. Development of flexible materials based on plasticized electrospun PLA-PHB blends: Structural, thermal, mechanical and disintegration properties. Eur. Polym. J. 2015, 73, 433–446. [Google Scholar] [CrossRef]
- Yeo, J.C.C.; Muiruri, J.K.; Thitsartarn, W.; Li, Z.; He, C. Recent advances in the development of biodegradable PHB-based toughening materials: Approaches, advantages and applications. Mater. Sci. Eng. C 2018, 92, 1092–1116. [Google Scholar] [CrossRef]
- Farias, N.C.; Major, I.; Devine, D.; Fournet, M.B.; Pezzoli, R.; Taghinezhad, S.F.; Hesabi, M. Multiple recycling of a PLA/PHB biopolymer blend for sustainable packaging applications: Rheology-morphology, thermal, and mechanical performance analysis. Polym. Eng. Sci. 2022, 62, 1764–1774. [Google Scholar] [CrossRef]
- Ramezani Dana, H.; Ebrahimi, F. Synthesis, properties, and applications of polylactic acid-based polymers. Polym. Eng. Sci. 2022, 63, 22–43. [Google Scholar] [CrossRef]
- Navarro-Baena, I.; Sessini, V.; Dominici, F.; Torre, L.; Kenny, J.M.; Peponi, L. Design of biodegradable blends based on PLA and PCL: From morphological, thermal and mechanical studies to shape memory behavior. Polym. Degrad. Stab. 2016, 132, 97–108. [Google Scholar] [CrossRef]
- D’Amico, D.A.; Iglesias Montes, M.L.; Manfredi, L.B.; Cyras, V.P. Fully bio-based and biodegradable polylactic acid/poly(3-hydroxybutirate) blends: Use of a common plasticizer as performance improvement strategy. Polym. Test. 2016, 49, 22–28. [Google Scholar] [CrossRef]
- Ma, P.; Spoelstra, A.B.; Schmit, P.; Lemstra, P.J. Toughening of poly (lactic acid) by poly (β-hydroxybutyrate-co-β- hydroxyvalerate) with high β-hydroxyvalerate content. Eur. Polym. J. 2013, 49, 1523–1531. [Google Scholar] [CrossRef]
- Zhang, K.; Mohanty, A.K.; Misra, M. Fully biodegradable and biorenewable ternary blends from polylactide, poly(3-hydroxybutyrate-co-hydroxyvalerate) and poly(butylene succinate) with balanced properties. ACS Appl. Mater. Interfaces 2012, 4, 3091–3101. [Google Scholar] [CrossRef] [PubMed]
- Gerard, T.; Budtova, T.; Podshivalov, A.; Bronnikov, S. Polylactide/poly(hydroxybutyrate-co-hydroxyvalerate) blends: Morphology and mechanical properties. Express Polym. Lett. 2014, 8, 609–617. [Google Scholar] [CrossRef]
- Wei, L.; Stark, N.M.; McDonald, A.G. Interfacial improvements in biocomposites based on poly(3-hydroxybutyrate) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) bioplastics reinforced and grafted with α-cellulose fibers. Green Chem. 2015, 17, 4800–4814. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Temperature (°C) | Shot Size (mm) | Cooling | Pressure (Bar) | ||||
|---|---|---|---|---|---|---|---|---|
| Zone 1 | Zone 2 | Zone 3 | T (°C) | t (s) | 1st | 2nd | ||
| PHB | 185 | 175 | 170 | 35 | 50 | 25 | 40 | 30 |
| PLA | 210 | 200 | 180 | 40 | 35 | 35 | 80 | 70 |
| PCL | 170 | 160 | 150 | 40 | 35 | 35 | 100 | 90 |
| 50/50/0 | 160 | 150 | 140 | 37 | 35 | 35 | 90 | 85 |
| 50/0/50 | 150 | 140 | 130 | 36 | 35 | 35 | 70 | 65 |
| 50/25/25 | 150 | 140 | 130 | 36 | 35 | 35 | 70 | 65 |
| 30/50/20 | 160 | 150 | 140 | 36 | 35 | 35 | 90 | 85 |
| Days | |||||
|---|---|---|---|---|---|
| Ratio | 0 | 14 | 28 | 56 | |
| PHB | (A2/A1) | 5.17 | 3.91 | 14.13 | 3.78 |
| (A3/A1) | 5.28 | 3.49 | 15.27 | 3.66 | |
| PLA | (A2/A1) | 11.68 | 8.37 | 10.52 | 9.09 |
| (A3/A1) | 31.00 | 20.48 | 27.24 | 23.19 | |
| PCL | (A2/A1) | 1.46 | 1.41 | 1.44 | 1.33 |
| (A3/A1) | 1.65 | 2.24 | 1.94 | 2.02 | |
| 50/50/0 | (A2/A1) | 5.35 | 5.19 | 3.18 | 5.63 |
| (A3/A1) | 10.05 | 8.56 | 4.76 | 9.35 | |
| 50/0/50 | (A2/A1) | 2.35 | 2.08 | 2.22 | 4.05 |
| (A3/A1) | 2.43 | 2.15 | 1.88 | 4.72 | |
| 50/25/25 | (A2/A1) | 3.58 | 3.03 | 3.37 | 3.23 |
| (A3/A1) | 4.44 | 3.96 | 3.96 | 3.96 | |
| 30/50/25 | (A2/A1) | 4.03 | 4.46 | 4.13 | 4.49 |
| (A3/A1) | 5.58 | 6.48 | 5.84 | 7.10 | |
| Material | T5 (°C) | T10 (°C) | Tmax (°C) | Residual Weight (%) | ||
|---|---|---|---|---|---|---|
| PLA | PHB | PCL | ||||
| PHB | 389.9 | 403.7 | - | 435.2 ± 0.1 | - | 0.26 ± 0.1 |
| PLA | 247.1 | 292.2 | 339.2 ± 0.0 | - | - | 1.82 ± 0.1 |
| PCL | 403.7 | 420.2 | - | - | 472.0 ± 0.1 | 4.26 ± 0.2 |
| 50/50/0 | 290.6 | 332.4 | 348.0 ± 0.1 | 429.3 ± 0.1 | - | 1.86 ± 0.2 |
| 50/0/50 | 265.3 | 326.5 | - | 344.2 ± 0.1 | 430.4 ± 0.1 | 1.20 ± 0.1 |
| 50/25/25 | 252.2 | 313.3 | 343.6 ± 0.1 | 421.5 ± 0.5 | 464.5 ± 0.5 | 4.21 ± 0.2 |
| 30/50/20 | 312.3 | 334.4 | 343.5 ± 0.1 | 380.6 ± 0.4 | 426.1 ± 0.1 | 3.27 ± 0.3 |
| Sample Name | d | Tg (°C) | Tm (°C) | Tc (°C) | ΔHc (J/g) | ΔHm (J/g) | Crystallinity Index (%) |
|---|---|---|---|---|---|---|---|
| PHB | 0 | 55.3 | 167.0 | 110.6 | −76.8 | 67.2 | 46.0 |
| 56 | 62.9 | 166.8 | 111.2 | −72.2 | 65.2 | 44.7 | |
| PLA | 0 | 66.7 | 149.9 | 59.2 | −5.5 | 6.8 | 7.3 |
| 56 | 67.2 | 150.1 | 59.4 | −4.9 | 7.0 | 7.5 | |
| PCL | 0 | −43.1 | 61.3 | 28.1 | −65.3 | 61.3 | 45.4 |
| 56 | −41.7 | 65.0 | 28.7 | −71.0 | 82.2 | 60.9 | |
| 50/50/0 | 0 | 53.8 | PLA (141.8), PHB (169.8) | PLA (97.2), PHB (106.4) | PLA (−1.3), PHB (−32.2) | PLA (10.8), PHB (27.4) | PLA (11.5), PHB (18.8) |
| 56 | 55.7 | PLA (145.2), PHB (169.8) | PLA (97.9), PHB (107.5) | PLA (−1.8), PHB (−20.0) | PLA (9.9), PHB (29.7) | PLA (10.6), PHB (20.3) | |
| 50/0/50 | 0 | 29.4 | PCL (58.3), PHB (169.0) | PCL (29.4), PHB (105.6) | PCL (−35.6), PHB (−36.7) | PCL (34.4), PHB (34.9) | PCL (25.5), PHB (23.9) |
| 56 | 29.8 | PCL (63.7), PHB (171.4) | PCL (29.8), PHB (107.1) | PCL (−34.2), PHB (−34.9) | PCL (39.7), PHB (34.3) | PCL (29.4), PHB (23.5) | |
| 50/25/25 | 0 | 58.8 | PCL (58.8), PLA (143.5), PHB (168.8) | PCL (29.4), PHB (106.2) | PCL (−18.6), PHB (−29.0) | PCL (15.8), PLA (4.0), PHB (27.1) | PCL (11.7), PLA (4.3), PHB (18.6) |
| 56 | 60.1 | PCL (60.1), PLA (143.8), PHB (168.7) | PCL (29.8), PHB (106.6) | PCL (−21.1), PHB (−31.7) | PCL (26.6), PLA (4.5), PHB (30.4) | PCL (19.7), PLA (4.8), PHB (20.8) | |
| 30/50/20 | 0 | 59.8 | PCL (59.5), PLA (144.5), PHB (172.8) | PCL (30.2), PHB (105.0) | PCL (−11.4), PHB (−16.8) | PCL (9.3), PLA (9.2), PHB (13.7) | PCL (6.9), PLA (9.8), PHB (9.4) |
| 56 | 61.4 | PCL (61.4), PLA (146.4), PHB (171.4) | PCL (30.8), PHB (105.6) | PCL (−15.1), PHB (−21.1) | PCL (21.7), PLA (11.8), PHB (17.8) | PCL (16.1), PLA (12.6), PHB (12.2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
G. Engler, L.; Farias, N.C.; S. Crespo, J.; Gately, N.M.; Major, I.; Pezzoli, R.; Devine, D.M. Designing Sustainable Polymer Blends: Tailoring Mechanical Properties and Degradation Behaviour in PHB/PLA/PCL Blends in a Seawater Environment. Polymers 2023, 15, 2874. https://doi.org/10.3390/polym15132874
G. Engler L, Farias NC, S. Crespo J, Gately NM, Major I, Pezzoli R, Devine DM. Designing Sustainable Polymer Blends: Tailoring Mechanical Properties and Degradation Behaviour in PHB/PLA/PCL Blends in a Seawater Environment. Polymers. 2023; 15(13):2874. https://doi.org/10.3390/polym15132874
Chicago/Turabian StyleG. Engler, Leonardo, Naiara C. Farias, Janaina S. Crespo, Noel M. Gately, Ian Major, Romina Pezzoli, and Declan M. Devine. 2023. "Designing Sustainable Polymer Blends: Tailoring Mechanical Properties and Degradation Behaviour in PHB/PLA/PCL Blends in a Seawater Environment" Polymers 15, no. 13: 2874. https://doi.org/10.3390/polym15132874
APA StyleG. Engler, L., Farias, N. C., S. Crespo, J., Gately, N. M., Major, I., Pezzoli, R., & Devine, D. M. (2023). Designing Sustainable Polymer Blends: Tailoring Mechanical Properties and Degradation Behaviour in PHB/PLA/PCL Blends in a Seawater Environment. Polymers, 15(13), 2874. https://doi.org/10.3390/polym15132874