

Familial Alzheimer’s Disease Neurons Bearing Mutations in PSEN1 Display Increased Calcium Responses to AMPA as an Early Calcium Dysregulation Phenotype


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Neuronal Differentiation: Induced Pluripotent Stem Cell Culture
2.2. Neuronal Differentiation
2.3. Lentivirus Production
2.4. Neuronal Characterization by Immunocytochemistry
2.5. Amyloid-β Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. Western Blotting for Tau: Protein Extraction
2.7. Detergent Compatible (DC) Protein Assay
2.8. Western Blotting
2.9. Flexstation to Measure Calcium Responses to Glutamate, NMDA, AMPA and Kainate
2.10. RT-qPCR for AMPAR Subunits: RNA Extraction and Purification
2.11. cDNA Synthesis
2.12. qPCR
2.13. Data Analysis
3. Results
3.1. Generation of iPSC-Derived Neurons from FAD and Control Lines
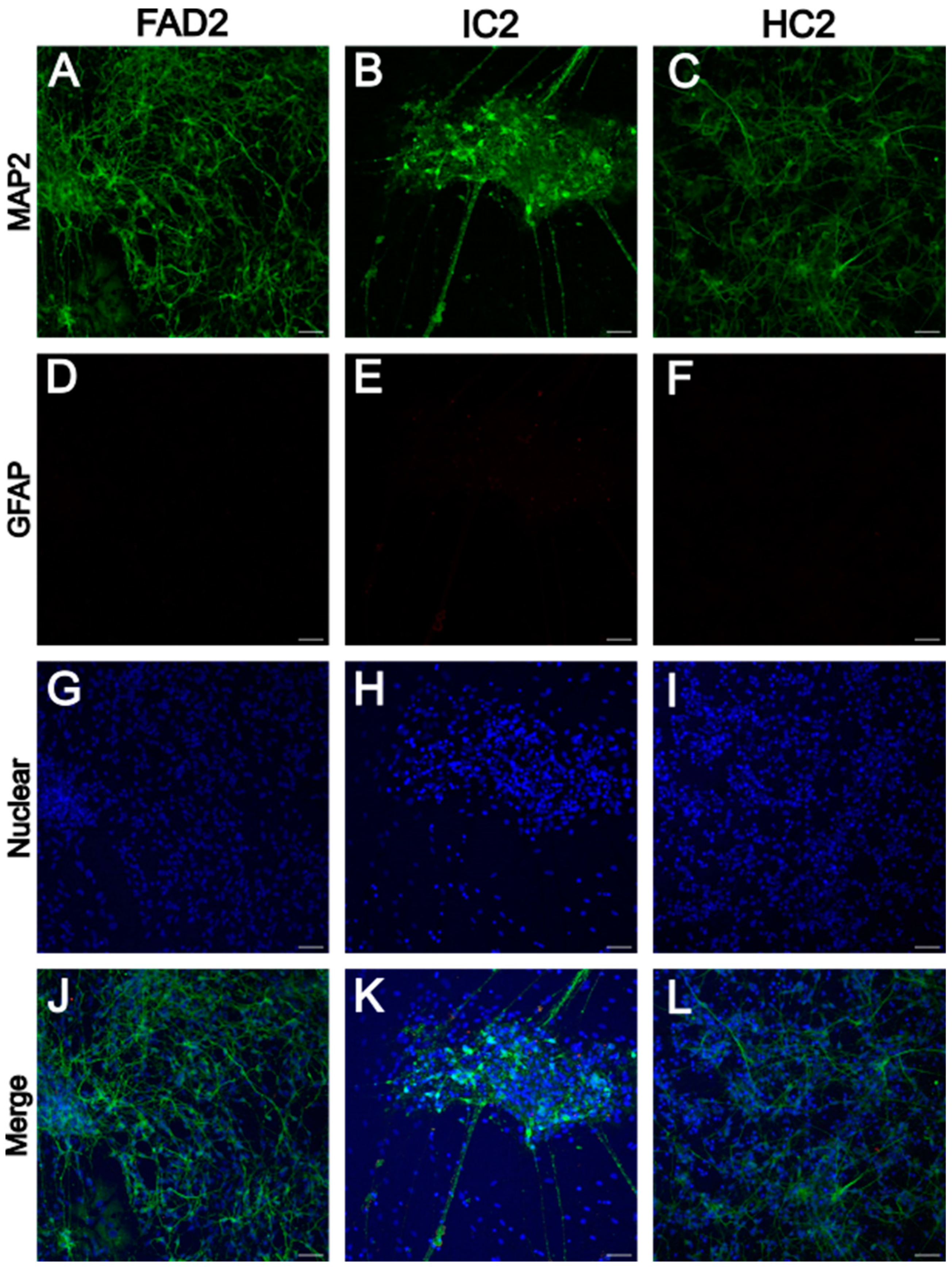
iPSC-Derived Neurons Express Neuronal Marker MAP2
3.2. FAD NGN2-Derived iPSC Neurons Did Not Show Evidence of Aβ Pathology at Day 35 of Maturation
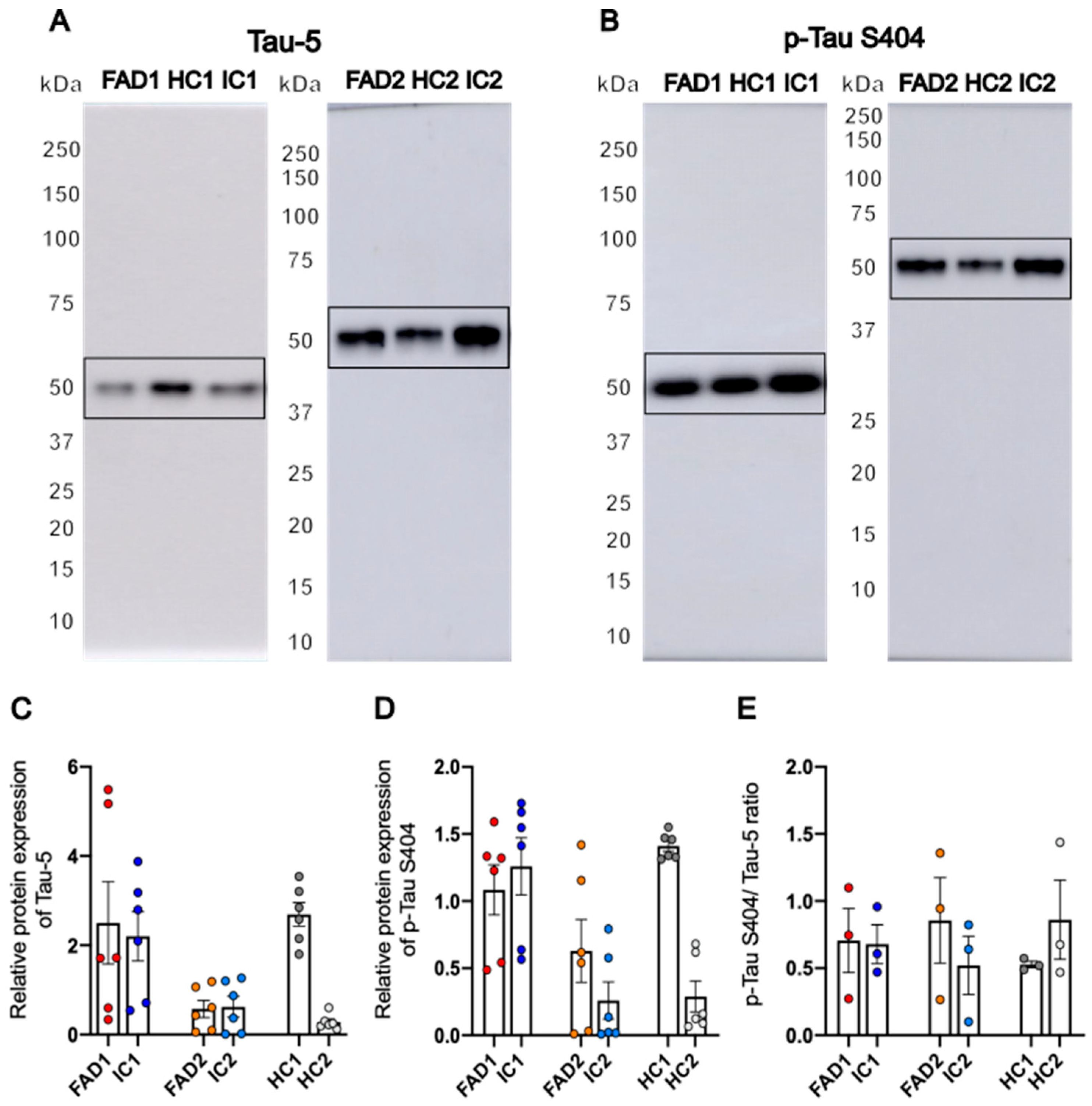
3.3. FAD NGN2 Derived iPSC Neurons Did Not Show Tau Pathology at Day 35 of Maturation
3.4. FAD Neurons Demonstrated Increased AMPAR Ca2+ Signalling Compared to Isogenic Controls
3.5. The iPSC-Derived Neurons Displayed Ca2+ Responses to High K+
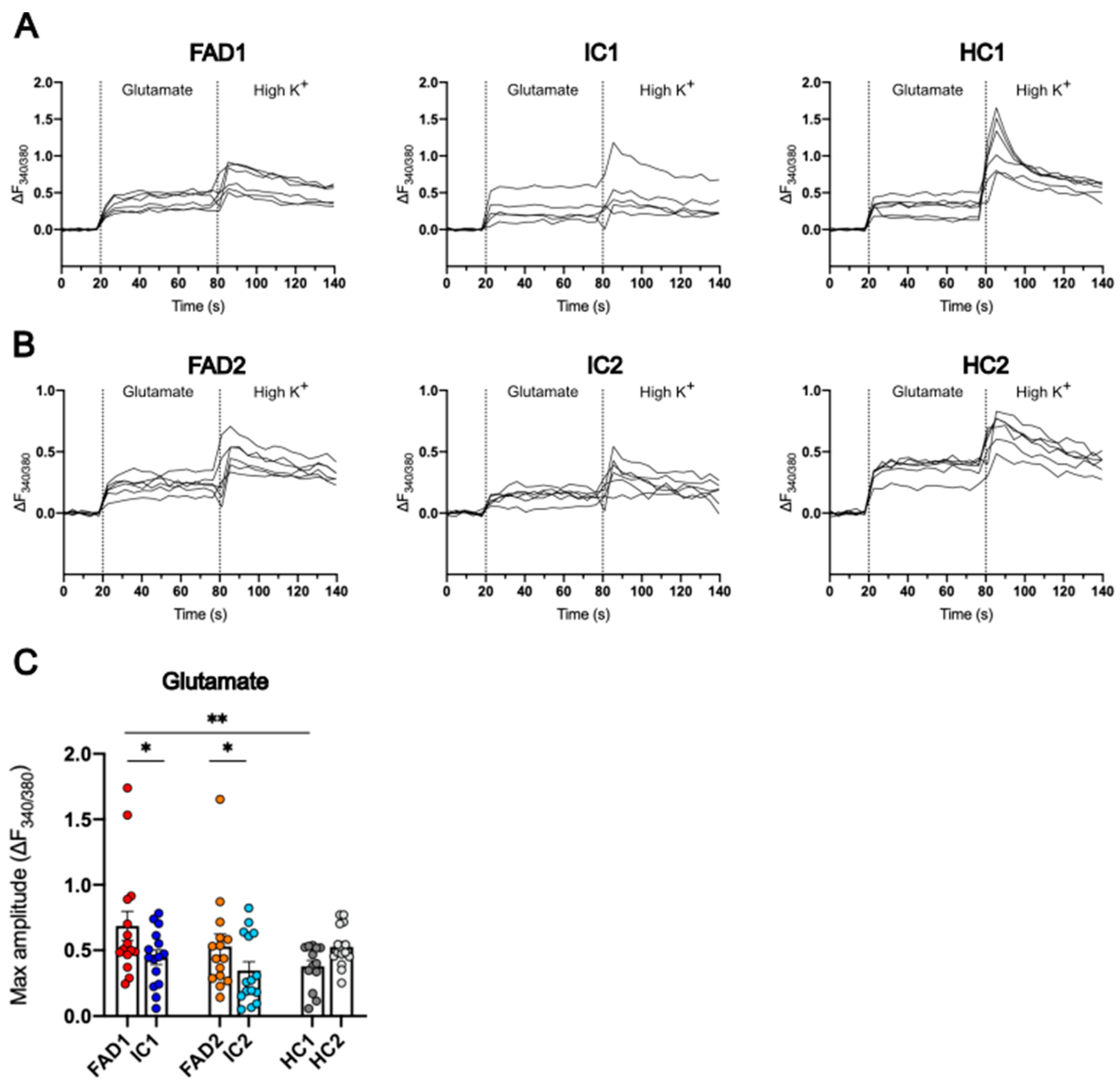
3.6. FAD Neurons Displayed Increased Ca2+ Responses to Glutamate
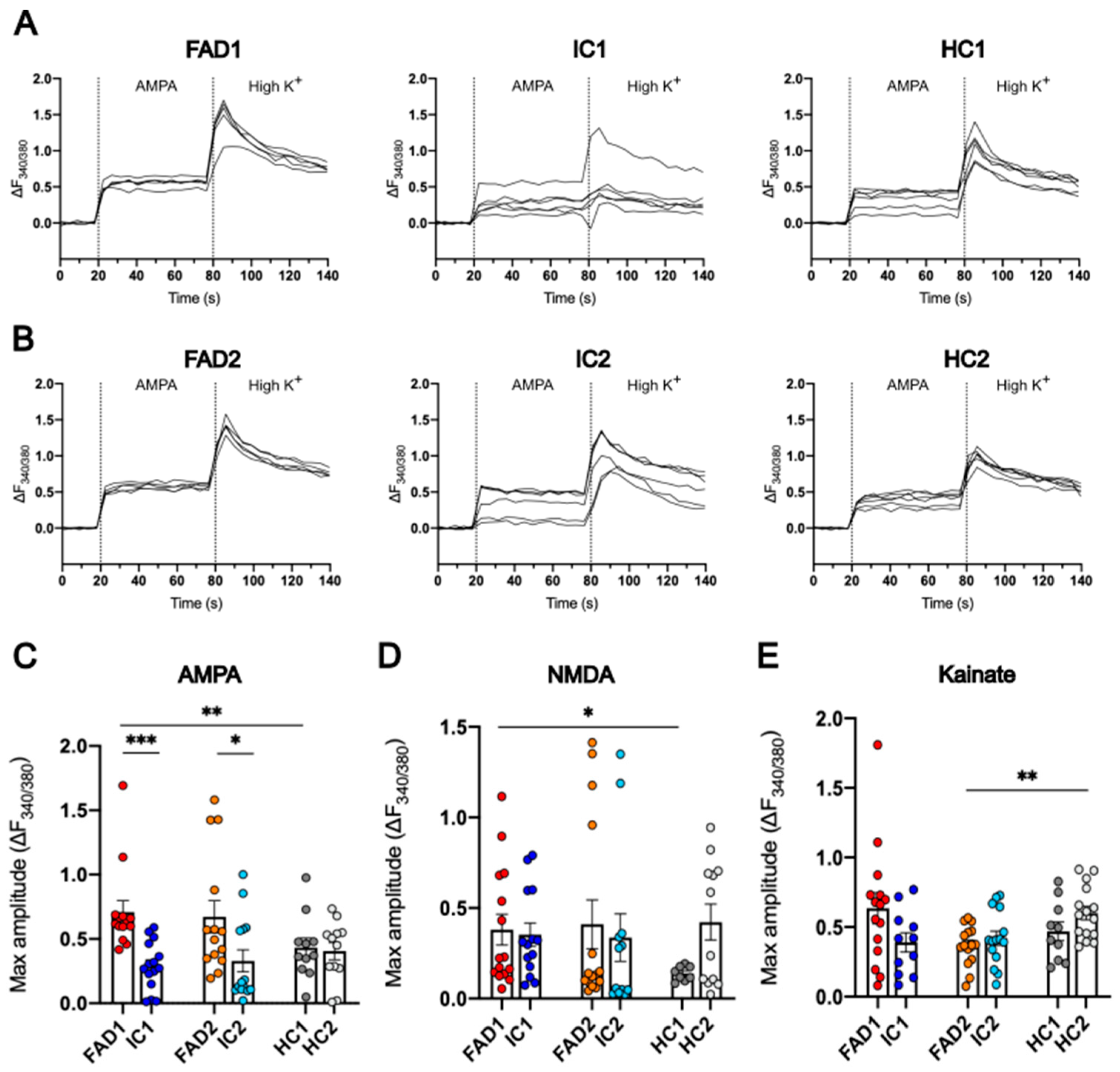
3.7. FAD Neurons Displayed Increased Ca2+ Responses to AMPA but Not NMDA or Kainate Compared to Their Isogenic Control Lines
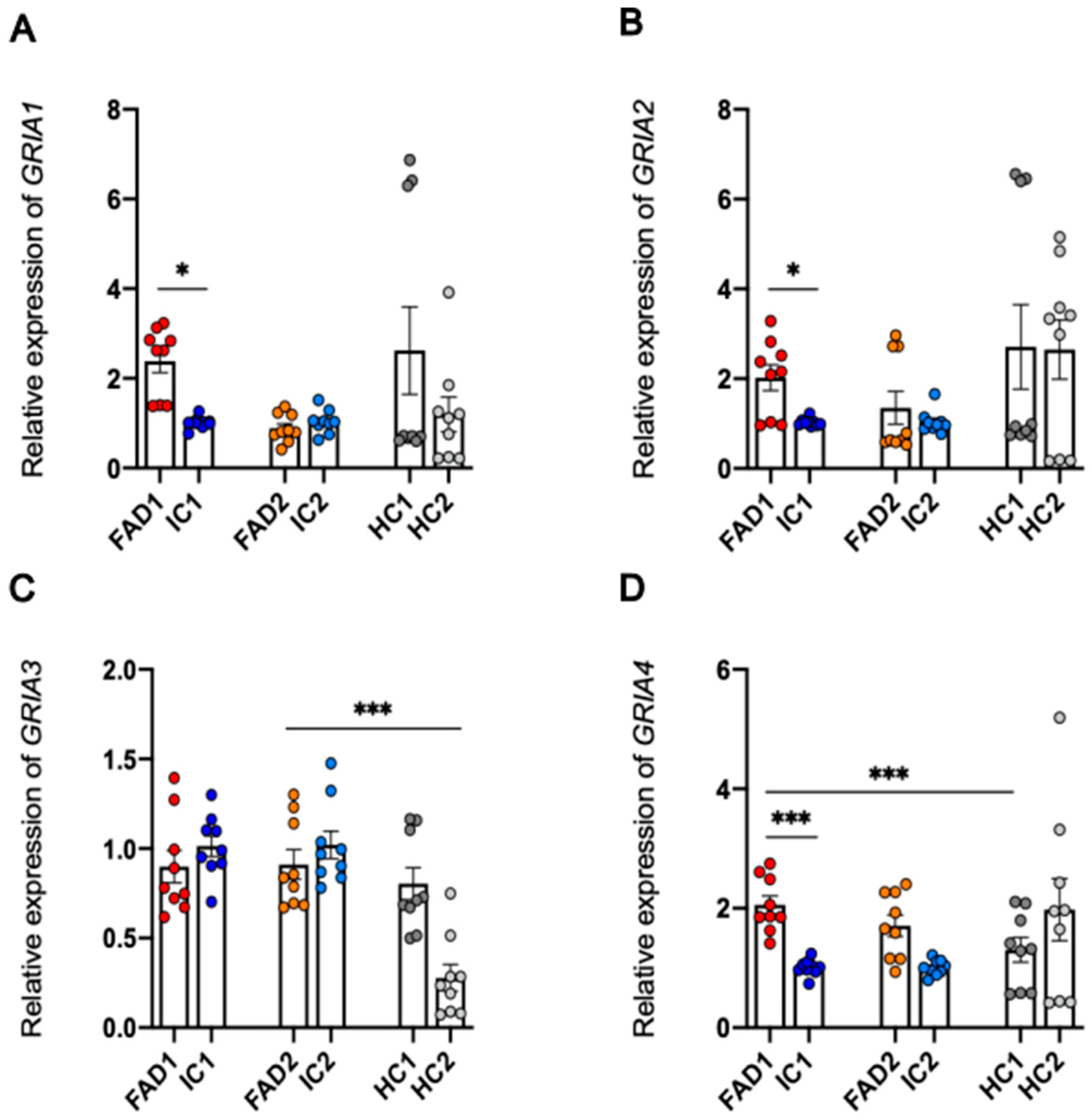
3.8. Regulation at the Level of mRNA or Protein of the AMPA Receptor Subunits Does Not Explain the Increased Calcium Responses to AMPA in FAD Neurons Compared to Isogenic Control Neurons
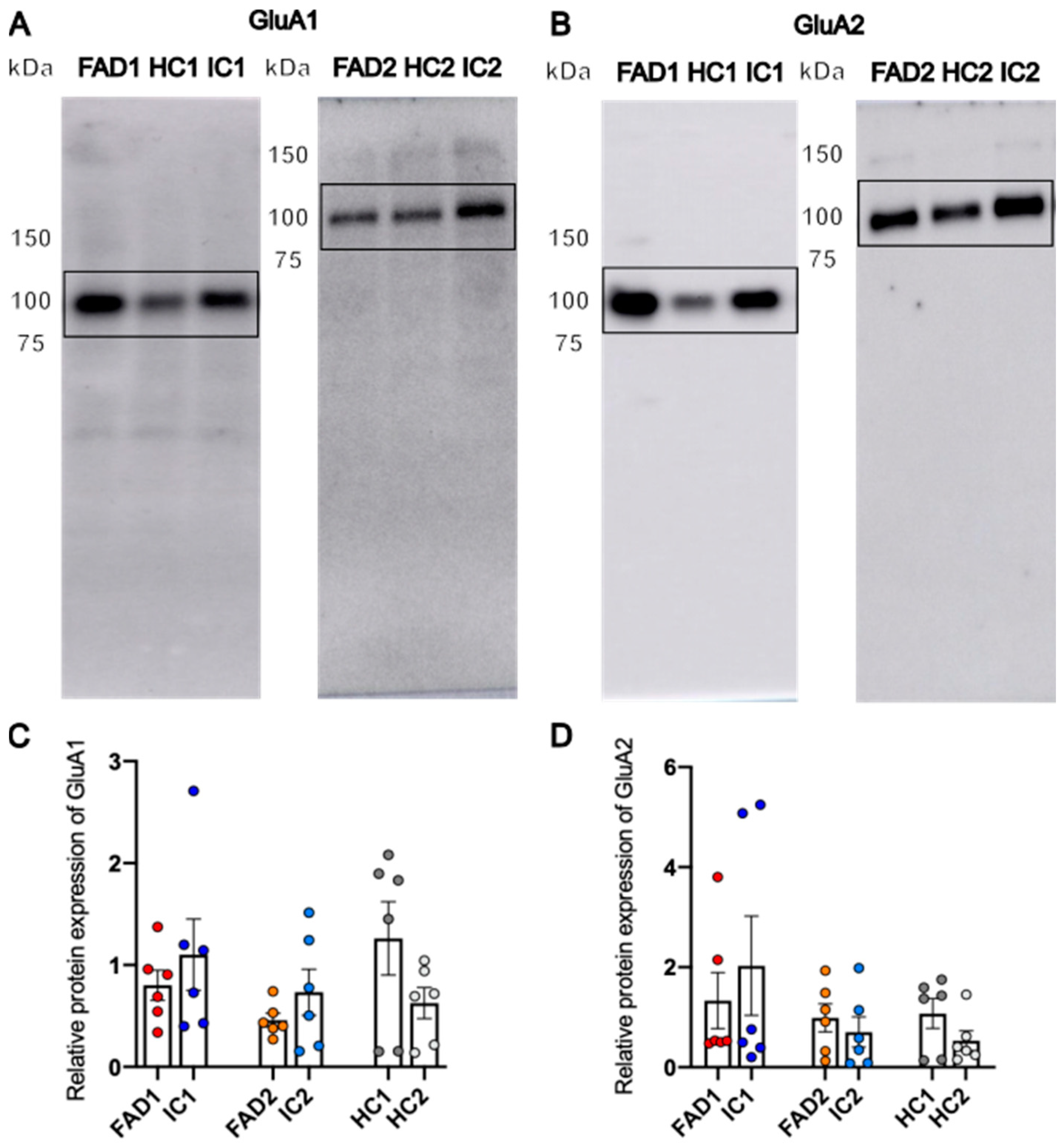
Protein Expression of GluA1 and GluA2 Is Not Significantly Different between FAD and Control Neurons
4. Discussion
4.1. FAD Neurons Lacking Aβ and Tau Pathology Show Elevated Ca2+ Responses to Glutamate and AMPA Compared to Isogenic Controls
4.2. Aberrant Ca2+ Signalling of FAD Neurons Occurs Independently of Changes in GluA1 and GluA2 Protein Expression
5. Future Directions
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bird, T.D. Alzheimer Disease Overview. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 1998. [Google Scholar]
- Blacker, D.; Tanzi, R.E. The Genetics of Alzheimer Disease: Current Status and Future Prospects. Arch. Neurol. 1998, 55, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Brion, J.P.; Couck, A.M.; Passareiro, E.; Flament-Durand, J. Neurofibrillary Tangles of Alzheimer’s Disease: An Immunohistochemical Study. J. Submicrosc. Cytol. 1985, 17, 89–96. [Google Scholar] [PubMed]
- Ge, M.; Zhang, J.; Chen, S.; Huang, Y.; Chen, W.; He, L.; Zhang, Y. Role of Calcium Homeostasis in Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2022, 18, 487–498. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Mattson, M.P. Neuronal Calcium Mishandling and the Pathogenesis of Alzheimer’s Disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, S.; Herreman, A.; De Strooper, B.; Bezprozvanny, I. Role of Presenilins in Neuronal Calcium Homeostasis. J. Neurosci. 2010, 30, 8566–8580. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins form ER Ca2+ Leak Channels, a Function Disrupted by Familial Alzheimer’s Disease-Linked Mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef]
- Green, K.N.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA Pump Activity Is Physiologically Regulated by Presenilin and Regulates Amyloid β Production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef]
- Cheung, K.-H.; Shineman, D.; Müller, M.; Cárdenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.-Y.; Foskett, J.K. Supplemental Data—Mechanism of Ca2+ Disruption in Alzheimer’s Disease by Presenilin Regulation of InsP3 Receptor Channel Gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.Y.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca2+ Homeostasis by Regulating VATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430. [Google Scholar] [CrossRef]
- Wisden, W.; Seeburg, P.H. Mammalian Ionotropic Glutamate Receptors. Curr. Opin. Neurobiol. 1993, 3, 291–298. [Google Scholar] [CrossRef]
- Schwenk, J.; Baehrens, D.; Haupt, A.; Bildl, W.; Boudkkazi, S.; Roeper, J.; Fakler, B.; Schulte, U. Regional Diversity and Developmental Dynamics of the AMPA-Receptor Proteome in the Mammalian Brain. Neuron 2014, 84, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR Removal Underlies Aβ-Induced Synaptic Depression and Dendritic Spine Loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef]
- Whitcomb, D.J.; Hogg, E.L.; Regan, P.; Piers, T.; Narayan, P.; Whitehead, G.; Winters, B.L.; Kim, D.H.; Kim, E.; St George-Hyslop, P.; et al. Intracellular Oligomeric Amyloid-Beta Rapidly Regulates GluA1 Subunit of AMPA Receptor in the Hippocampus. Sci. Rep. 2015, 5, 10934. [Google Scholar] [CrossRef]
- Hoover, B.R.; Reed, M.N.; Su, J.; Penrod, R.D.; Kotilinek, L.A.; Grant, M.K.; Pitstick, R.; Carlson, G.A.; Lanier, L.M.; Yuan, L.L.; et al. Tau Mislocalization to Dendritic Spines Mediates Synaptic Dysfunction Independently of Neurodegeneration. Neuron 2010, 68, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P. Molecular Basis of NMDA Receptor Functional Diversity. Eur. J. Neurosci. 2011, 33, 1351–1365. [Google Scholar] [CrossRef]
- Sun, X.-Y.; Tuo, Q.-Z.; Liuyang, Z.-Y.; Xie, A.-J.; Feng, X.-L.; Yan, X.; Qiu, M.; Li, S.; Wang, X.-L.; Cao, F.-Y.; et al. Extrasynaptic NMDA Receptor-Induced Tau Overexpression Mediates Neuronal Death through Suppressing Survival Signaling ERK Phosphorylation. Cell Death Dis. 2016, 7, e2449. [Google Scholar] [CrossRef] [PubMed]
- Lesné, S.; Ali, C.; Gabriel, C.; Croci, N.; MacKenzie, E.T.; Glabe, C.G.; Plotkine, M.; Marchand-Verrecchia, C.; Vivien, D.; Buisson, A. NMDA Receptor Activation Inhibits α-Secretase and Promotes Neuronal Amyloid-β Production. J. Neurosci. 2005, 25, 9367–9377. [Google Scholar] [CrossRef]
- Bordji, K.; Becerril-Ortega, J.; Nicole, O.; Buisson, A. Activation of Extrasynaptic, But Not Synaptic, NMDA Receptors Modifies Amyloid Precursor Protein Expression Pattern and Increases Amyloid-β Production. J. Neurosci. 2010, 30, 15927–15942. [Google Scholar] [CrossRef]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble Oligomers of Amyloid β Protein Facilitate Hippocampal Long-Term Depression by Disrupting Neuronal Glutamate Uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef]
- Arias, C.; Arrieta, I.; Tapia, R. Β-Amyloid Peptide Fragment 25–35 Potentiates the Calcium-dependent Release of Excitatory Amino Acids from Depolarized Hippocampal Slices. J. Neurosci. Res. 1995, 41, 561–566. [Google Scholar] [CrossRef]
- Parpura-Gill, A.; Beitz, D.; Uemura, E. The Inhibitory Effects of β-Amyloid on Glutamate and Glucose Uptakes by Cultured Astrocytes. Brain Res. 1997, 754, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA Receptor Trafficking by Amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Winslow, B.T.; Onysko, M.K.; Stob, C.M.; Hazlewood, K.A. Treatment of Alzheimer Disease. Am. Fam. Physician 2011, 83, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Falcón-Moya, R.; Sihra, T.S.; Rodríguez-Moreno, A. Kainate Receptors: Role in Epilepsy. Front. Mol. Neurosci. 2018, 11, 217. [Google Scholar] [CrossRef]
- Barthet, G.; Moreira-De-Sá, A.; Zhang, P.; Deforges, S.; Castanheira, J.; Gorlewicz, A.; Mulle, C. Presenilin and APP Regulate Synaptic Kainate Receptors. J. Neurosci. 2022, 42, 9253–9262. [Google Scholar] [CrossRef]
- Malenka, R.C.; Bear, M.F. LTP and LTD: An Embarrassment of Riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef]
- Askenazi, M.; Kavanagh, T.; Pires, G.; Ueberheide, B.; Wisniewski, T.; Drummond, E. Compilation of All Known Protein Changes in the Human Alzheimer’s Disease Brain. bioRxiv 2023. [Google Scholar] [CrossRef]
- Choi, D.W. Glutamate Neurotoxicity and Diseases of the Nervous System. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.F.; Hascup, E.R.; Bartke, A.; Hascup, K.N. Friend or Foe? Defining the Role of Glutamate in Aging and Alzheimer’s Disease. Front. Aging 2022, 3, 65. [Google Scholar] [CrossRef]
- Targa Dias Anastacio, H.; Matosin, N.; Ooi, L. Neuronal Hyperexcitability in Alzheimer’s Disease: What Are the Drivers behind This Aberrant Phenotype? Transl. Psychiatry 2022, 12, 257. [Google Scholar] [CrossRef] [PubMed]
- Celone, K.A.; Calhoun, V.D.; Dickerson, B.C.; Atri, A.; Chua, E.F.; Miller, S.L.; DePeau, K.; Rentz, D.M.; Selkoe, D.J.; Blacker, D.; et al. Alterations in Memory Networks in Mild Cognitive Impairment and Alzheimer’s Disease: An Independent Component Analysis. J. Neurosci. 2006, 26, 10222–10231. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, B.C.; Salat, D.H.; Greve, D.N.; Chua, E.F.; Rand-Giovannetti, E.; Rentz, D.M.; Bertram, L.; Mullin, K.; Tanzi, R.E.; Blacker, D.; et al. Increased Hippocampal Activation in Mild Cognitive Impairment Compared to Normal Aging and AD. Neurology 2005, 65, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Balez, R.; Ooi, L. Getting to NO Alzheimer’s Disease: Neuroprotection versus Neurotoxicity Mediated by Nitric Oxide. Oxid. Med. Cell. Longev. 2016, 2016, 3806157. [Google Scholar] [CrossRef]
- Ghatak, S.; Dolatabadi, N.; Trudler, D.; Zhang, X.; Wu, Y.; Mohata, M.; Ambasudhan, R.; Talantova, M.; Lipton, S.A. Mechanisms of Hyperexcitability in Alzheimer’s Disease HiPSC-Derived Neurons and Cerebral Organoids vs. Isogenic Control. eLife 2019, 8, e50333. [Google Scholar] [CrossRef] [PubMed]
- Šišková, Z.; Justus, D.; Kaneko, H.; Friedrichs, D.; Henneberg, N.; Beutel, T.; Pitsch, J.; Schoch, S.; Becker, A.; VonderKammer, H.; et al. Dendritic Structural Degeneration Is Functionally Linked to Cellular Hyperexcitability in a Mouse Model of Alzheimer’s Disease. Neuron 2014, 84, 1023–1033. [Google Scholar] [CrossRef]
- Lerdkrai, C.; Asavapanumas, N.; Brawek, B.; Kovalchuk, Y.; Mojtahedi, N.; Del Moral, M.O.; Garaschuk, O. Intracellular Ca2+ Stores Control in Vivo Neuronal Hyperactivity in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1279–E1288. [Google Scholar] [CrossRef] [PubMed]
- Maksour, S.; Finol-Urdaneta, R.K.; Hulme, A.J.; Castro Cabral-da-Silva, M.; Targa Dias Anastacio, H.; Balez, R.; Berg, T.; Turner, C.; Sanz, S.; Engel, M.; et al. Alzheimer’s Disease Induced Neurons Bearing PSEN1 Mutations Exhibit Reduced Excitability. bioRxiv 2024. [Google Scholar] [CrossRef]
- Engel, M.; Balez, R.; Muñoz, S.S.; Cabral-da-Silva, M.C.; Stevens, C.H.; Bax, M.; Do-Ha, D.; Sidhu, K.; Sachdev, P.; Ooi, L. Viral-Free Generation and Characterization of a Human Induced Pluripotent Stem Cell Line from Dermal Fibroblasts. Stem Cell Res. 2018, 32, 135–138. [Google Scholar] [CrossRef]
- Muñoz, S.S.; Balez, R.; Castro Cabral-da-Silva, M.e.; Berg, T.; Engel, M.; Bax, M.; Do-Ha, D.; Stevens, C.H.; Greenough, M.; Bush, A.; et al. Generation and Characterization of Human Induced Pluripotent Stem Cell Lines from a Familial Alzheimer’s Disease PSEN1 A246E Patient and a Non-Demented Family Member Bearing Wild-Type PSEN1. Stem Cell Res. 2018, 31, 227–230. [Google Scholar] [CrossRef]
- Nehme, R.; Zuccaro, E.; Ghosh, S.D.; Li, C.; Sherwood, J.L.; Pietilainen, O.; Barrett, L.E.; Limone, F.; Worringer, K.A.; Kommineni, S.; et al. Combining NGN2 Programming with Developmental Patterning Generates Human Excitatory Neurons with NMDAR-Mediated Synaptic Transmission. Cell Rep. 2018, 23, 2509–2523. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Lehmann, S.; Otto, M.; Zetterberg, H.; Lewczuk, P. Advantages and Disadvantages of the Use of the CSF Amyloid β (Aβ) 42/40 Ratio in the Diagnosis of Alzheimer’s Disease. Alzheimer’s Res. Ther. 2019, 11, 34. [Google Scholar] [CrossRef]
- Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Li, Y.; Gordon, B.A.; Holtzman, D.M.; Morris, J.C.; Benzinger, T.L.S.; Xiong, C.; et al. High-Precision Plasma β-Amyloid 42/40 Predicts Current and Future Brain Amyloidosis. Neurology 2019, 93, e1647. [Google Scholar] [CrossRef] [PubMed]
- Amft, M.; Ortner, M.; Eichenlaub, U.; Goldhardt, O.; Diehl-Schmid, J.; Hedderich, D.M.; Yakushev, I.; Grimmer, T. The Cerebrospinal Fluid Biomarker Ratio Aβ42/40 Identifies Amyloid Positron Emission Tomography Positivity Better than Aβ42 Alone in a Heterogeneous Memory Clinic Cohort. Alzheimer’s Res. Ther. 2022, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Zetterberg, H.; Buchhave, P.; Andreasson, U.; Londos, E.; Minthon, L.; Blennow, K. Prediction of Alzheimer’s Disease Using the CSF Aβ42/Aβ40 Ratio in Patients with Mild Cognitive Impairment. Dement. Geriatr. Cogn. Disord. 2007, 23, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Balez, R.; Steiner, N.; Engel, M.; Muñoz, S.S.; Lum, J.S.; Wu, Y.; Wang, D.; Vallotton, P.; Sachdev, P.; O’Connor, M.; et al. Neuroprotective Effects of Apigenin against Inflammation, Neuronal Excitability and Apoptosis in an Induced Pluripotent Stem Cell Model of Alzheimer’s Disease. Sci. Rep. 2016, 6, 31450. [Google Scholar] [CrossRef] [PubMed]
- Balez, R.; Stevens, C.H.; Lenk, K.; Sidhu, K.; Sutherland, G.; Ooi, L. Increased Neuronal Nitric Oxide Synthase in Alzheimer’s Disease Mediates Spontaneous Calcium Signalling and Divergent Glutamatergic Calcium Responses. Antioxid. Redox Signal. 2024, 1–23. [Google Scholar] [CrossRef]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human IPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, H.; Ma, Y.; Shi, G.; Song, J.; Tang, Y.; Li, S.; Li, T.; Liu, N.; Tang, F.; et al. Early Pathogenic Event of Alzheimer’s Disease Documented in IPSCs from Patients with PSEN1 Mutations. Oncotarget 2017, 8, 7900–7913. [Google Scholar] [CrossRef]
- Mondragón-Rodríguez, S.; Perry, G.; Luna-Muñoz, J.; Acevedo-Aquino, M.C.; Williams, S. Phosphorylation of Tau Protein at Sites Ser396-404 Is One of the Earliest Events in Alzheimer’s Disease and Down Syndrome. Neuropathol. Appl. Neurobiol. 2014, 40, 121–135. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s Disease-Linked Presenilin I Variants Elevate Aβ1- 42/1-40 Ratio in Vitro and in Vivo. Neuron 1996, 17, 1005–1013. [Google Scholar] [CrossRef]
- Steiner, H.; Romig, H.; Grim, M.G.; Philipp, U.; Pesold, B.; Citron, M.; Baumeister, R.; Haass, C. The Biological and Pathological Function of the Presenilin-1 Δexon 9 Mutation Is Independent of Its Defect to Undergo Proteolytic Processing. J. Biol. Chem. 1999, 274, 7615–7618. [Google Scholar] [CrossRef] [PubMed]
- Murayama, O.; Tomita, T.; Nihonmatsu, N.; Murayama, M.; Sun, X.; Honda, T.; Iwatsubo, T.; Takashima, A. Enhancement of Amyloid β 42 Secretion by 28 Different Presenilin 1 Mutations of Familial Alzheimer’s Disease. Neurosci. Lett. 1999, 265, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Berezovska, O.; Lleo, A.; Herl, L.D.; Frosch, M.P.; Stern, E.A.; Bacskai, B.J.; Hyman, B.T. Familial Alzheimer’s Disease Presenilin 1 Mutations Cause Alterations in the Conformation of Presenilin and Interactions with Amyloid Precursor Protein. J. Neurosci. 2005, 25, 3009–3017. [Google Scholar] [CrossRef]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging–Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease. Alzheimers. Dement. 2012, 8, 1. [Google Scholar] [CrossRef]
- Ries, M.; Sastre, M. Mechanisms of Aβ clearance and degradation by glial cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef]
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent Developments in Clinical Biomarkers and Therapies Targeting Tau Phosphorylation in Alzheimer’s Disease and Other Tauopathies. Mol. Neurodegener. 2021, 16, 37. [Google Scholar] [CrossRef]
- Wesseling, H.; Mair, W.; Kumar, M.; Schlaffner, C.N.; Tang, S.; Beerepoot, P.; Fatou, B.; Guise, A.J.; Cheng, L.; Takeda, S.; et al. Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell 2020, 183, 1699–1713.e13. [Google Scholar] [CrossRef]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-Independent Regulators of Tau Pathology in Alzheimer Disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.S.; Washicosky, K.J.; Brand, E.; von Maydell, D.; Aronson, J.; Kim, S.; Capen, D.E.; Cetinbas, M.; Sadreyev, R.; Ning, S.; et al. Amyloid-Β42/40 Ratio Drives Tau Pathology in 3D Human Neural Cell Culture Models of Alzheimer’s Disease. Nat. Commun. 2020, 11, 1377. [Google Scholar] [CrossRef]
- Spitzer, N.C.; Kingston, P.A.; Manning, T.J.; Conklin, M.W. Outside and in: Development of Neuronal Excitability. Curr. Opin. Neurobiol. 2002, 12, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Bartlett, P.F.; Adams, D.J. Kir and Kv Channels Regulate Electrical Properties and Proliferation of Adult Neural Precursor Cells. Mol. Cell. Neurosci. 2008, 37, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Das, B.; Yao, A.Y.; Yan, R. Metabotropic Glutamate Receptors in Alzheimer’s Disease Synaptic Dysfunction: Therapeutic Opportunities and Hope for the Future. J. Alzheimer’s Dis. 2020, 78, 1345–1361. [Google Scholar] [CrossRef] [PubMed]
- Jurado, S. AMPA Receptor Trafficking in Natural and Pathological Aging. Front. Mol. Neurosci. 2018, 10, 446. [Google Scholar] [CrossRef]
- Guntupalli, S.; Widagdo, J.; Anggono, V. Amyloid- β -Induced Dysregulation of AMPA Receptor Trafficking. Neural Plast. 2016, 2016, 3204519. [Google Scholar] [CrossRef] [PubMed]
- Greenough, M.A.; Lane, D.J.R.; Balez, R.; Anastacio, H.T.D.; Zeng, Z.; Ganio, K.; McDevitt, C.A.; Acevedo, K.; Belaidi, A.A.; Koistinaho, J.; et al. Selective Ferroptosis Vulnerability Due to Familial Alzheimer’s Disease Presenilin Mutations. Cell Death Differ. 2022, 29, 2123–2136. [Google Scholar] [CrossRef] [PubMed]
- Lasky, J.L.; Wu, H. Notch Signaling, Brain Development, and Human Disease. Pediatr. Res. 2005, 57, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.; Gourmaud, S.; Li, X.; Stewart, D.; Irwin, D.; Talos, D.; Jensen, F. Seizures Exacerbate Excitatory: Inhibitory Imbalance in Alzheimer’s Disease with Attenuation after Rapamycin Treatment in 5XFAD Mice. bioRxiv 2023. [Google Scholar] [CrossRef]
- Guo, C.; Ma, Y.Y. Calcium Permeable-AMPA Receptors and Excitotoxicity in Neurological Disorders. Front. Neural Circuits 2021, 15, 711564. [Google Scholar] [CrossRef]
- Kawahara, Y.; Ito, K.; Sun, H.; Kanazawa, I.; Kwak, S. Low Editing Efficiency of GluR2 MRNA Is Associated with a Low Relative Abundance of ADAR2 MRNA in White Matter of Normal Human Brain. Eur. J. Neurosci. 2003, 18, 23–33. [Google Scholar] [CrossRef]
- Wright, A.; Vissel, B. The Essential Role of AMPA Receptor GluA2 Subunit RNA Editing in the Normal and Diseased Brain. Front. Mol. Neurosci. 2012, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Gaisler-Salomon, I.; Kravitz, E.; Feiler, Y.; Safran, M.; Biegon, A.; Amariglio, N.; Rechavi, G. Hippocampus-Specific Deficiency in RNA Editing of GluA2 in Alzheimer’s Disease. Neurobiol. Aging 2014, 35, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Khermesh, K.; D’Erchia, A.M.; Barak, M.; Annese, A.; Wachtel, C.; Levanon, E.Y.; Picardi, E.; Eisenberg, E. Reduced Levels of Protein Recoding by A-to-I RNA Editing in Alzheimer’s Disease. RNA 2016, 22, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, S.; Smith, M.A.; Jones, E.G. Editing for an AMPA Receptor Subunit RNA in Prefrontal Cortex and Striatum in Alzheimer’s Disease, Huntington’s Disease and Schizophrenia. Brain Res. 1995, 699, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Konen, L.M.; Wright, A.L.; Royle, G.A.; Morris, G.P.; Lau, B.K.; Seow, P.W.; Zinn, R.; Milham, L.T.; Vaughan, C.W.; Vissel, B. A New Mouse Line with Reduced GluA2 Q/R Site RNA Editing Exhibits Loss of Dendritic Spines, Hippocampal CA1-Neuron Loss, Learning and Memory Impairments and NMDA Receptor-Independent Seizure Vulnerability. Mol. Brain 2020, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Pachernegg, S.; Münster, Y.; Muth-Köhne, E.; Fuhrmann, G.; Hollmann, M. GluA2 Is Rapidly Edited at the Q/R Site during Neural Differentiation in Vitro. Front. Cell. Neurosci. 2015, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Shi, Y.; Jackson, A.C.; Bjorgan, K.; During, M.J.; Sprengel, R.; Seeburg, P.H.; Nicoll, R.A. Subunit Composition of Synaptic AMPA Receptors Revealed by a Single-Cell Genetic Approach. Neuron 2009, 62, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Wenthold, R.J.; Petralia, R.S.; Blahos, J.; Niedzielski, A.S. Evidence for Multiple AMPA Receptor Complexes in Hippocampal CA1/CA2 Neurons. J. Neurosci. 1996, 16, 1982–1989. [Google Scholar] [CrossRef]
- Zhang, Q.; Ma, C.; Gearing, M.; Wang, P.G.; Chin, L.S.; Li, L. Integrated Proteomics and Network Analysis Identifies Protein Hubs and Network Alterations in Alzheimer’s Disease. Acta Neuropathol. Commun. 2018, 6, 19. [Google Scholar] [CrossRef]
- Mendonça, C.F.; Kuras, M.; Nogueira, F.C.S.; Plá, I.; Hortobágyi, T.; Csiba, L.; Palkovits, M.; Renner, É.; Döme, P.; Marko-Varga, G.; et al. Proteomic Signatures of Brain Regions Affected by Tau Pathology in Early and Late Stages of Alzheimer’s Disease. Neurobiol. Dis. 2019, 130, 104509. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Yin, L.; Thambisetty, M.; Troncoso, J.C.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Deep Proteomic Network Analysis of Alzheimer’s Disease Brain Reveals Alterations in RNA Binding Proteins and RNA Splicing Associated with Disease. Mol. Neurodegener. 2018, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Patassini, S.; Rustogi, N.; Riba-Garcia, I.; Hale, B.D.; Phillips, A.M.; Waldvogel, H.; Haines, R.; Bradbury, P.; Stevens, A.; et al. Regional Protein Expression in Human Alzheimer’s Brain Correlates with Disease Severity. Commun. Biol. 2019, 2, 43. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.H.Y.; Walby, J.L.; Palpagama, T.H.; Turner, C.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Glutamatergic Receptor Expression Changes in the Alzheimer’s Disease Hippocampus and Entorhinal Cortex. Brain Pathol. 2021, 31, e13005. [Google Scholar] [CrossRef] [PubMed]
- Stepler, K.E.; Mahoney, E.R.; Kofler, J.; Hohman, T.J.; Lopez, O.L.; Robinson, R.A.S. Inclusion of African American/Black Adults in a Pilot Brain Proteomics Study of Alzheimer’s Disease. Neurobiol. Dis. 2020, 146, 105129. [Google Scholar] [CrossRef]
- Sweet, R.A.; MacDonald, M.L.; Kirkwood, C.M.; Ding, Y.; Schempf, T.; Jones-Laughner, J.; Kofler, J.; Ikonomovic, M.D.; Lopez, O.L.; Garver, M.E.; et al. Apolipoprotein E∗4 (APOE∗4) Genotype Is Associated with Altered Levels of Glutamate Signaling Proteins and Synaptic Coexpression Networks in the Prefrontal Cortex in Mild to Moderate Alzheimer Disease. Mol. Cell. Proteom. 2016, 15, 2252–2262. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.B.; Carter, E.K.; Dammer, E.B.; Duong, D.M.; Gerasimov, E.S.; Liu, Y.; Liu, J.; Betarbet, R.; Ping, L.; Yin, L.; et al. Large-Scale Deep Multi-Layer Analysis of Alzheimer’s Disease Brain Reveals Strong Proteomic Disease-Related Changes Not Observed at the RNA Level. Nat. Neurosci. 2022, 25, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Roche, K.W. Posttranslational Regulation of AMPA Receptor Trafficking and Function. Curr. Opin. Neurobiol. 2012, 22, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Roche, K.W.; O’Brien, R.J.; Mammen, A.L.; Bernhardt, J.; Huganir, R.L. Characterization of Multiple Phosphorylation Sites on the AMPA Receptor GluR1 Subunit. Neuron 1996, 16, 1179–1188. [Google Scholar] [CrossRef]
- Derkach, V.; Barria, A.; Soderling, T.R. Ca2+/Calmodulin-Kinase II Enhances Channel Conductance of α-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionate Type Glutamate Receptors. Proc. Natl. Acad. Sci. USA 1999, 96, 3269–3274. [Google Scholar] [CrossRef]
- Banke, T.G.; Bowie, D.; Lee, H.K.; Huganir, R.L.; Schousboe, A.; Traynelis, S.F. Control of GluR1 AMPA Receptor Function by CAMP-Dependent Protein Kinase. J. Neurosci. 2000, 20, 89–102. [Google Scholar] [CrossRef]
- Fox, C.J.; Russell, K.; Titterness, A.K.; Yu, T.W.; Christie, B.R. Tyrosine Phosphorylation of the GluR2 Subunit Is Required for Long-Term Depression of Synaptic Efficacy in Young Animals in Vivo. Hippocampus 2007, 17, 600–605. [Google Scholar] [CrossRef]
- Ahmadian, G.; Ju, W.; Liu, L.; Wyszynski, M.; Lee, S.H.; Dunah, A.W.; Taghibiglou, C.; Wang, Y.; Lu, J.; Wong, T.P.; et al. Tyrosine Phosphorylation of GluR2 Is Required for Insulin-Stimulated AMPA Receptor Endocytosis and LTD. EMBO J. 2004, 23, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Widagdo, J.; Chai, Y.J.; Ridder, M.C.; Chau, Y.Q.; Johnson, R.C.; Sah, P.; Huganir, R.L.; Anggono, V. Activity-Dependent Ubiquitination of GluA1 and GluA2 Regulates AMPA Receptor Intracellular Sorting and Degradation. Cell Rep. 2015, 10, 783–795. [Google Scholar] [CrossRef]
- Schwarz, L.A.; Hall, B.J.; Patrick, G.N. Activity-Dependent Ubiquitination of GluA1 Mediates a Distinct AMPA Receptor Endocytosis and Sorting Pathway. J. Neurosci. 2010, 30, 16718–16729. [Google Scholar] [CrossRef] [PubMed]
- Widagdo, J.; Guntupalli, S.; Jang, S.E.; Anggono, V. Regulation of AMPA Receptor Trafficking by Protein Ubiquitination. Front. Mol. Neurosci. 2017, 10, 347. [Google Scholar] [CrossRef]
- Lin, A.; Hou, Q.; Jarzylo, L.; Amato, S.; Gilbert, J.; Shang, F.; Man, H.Y. Nedd4-Mediated AMPA Receptor Ubiquitination Regulates Receptor Turnover and Trafficking. J. Neurochem. 2011, 119, 27–39. [Google Scholar] [CrossRef]
- Huo, Y.; Khatri, N.; Hou, Q.; Gilbert, J.; Wang, G.; Man, H.Y. The Deubiquitinating Enzyme USP46 Regulates AMPA Receptor Ubiquitination and Trafficking. J. Neurochem. 2015, 134, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Lussier, M.P.; Herring, B.E.; Nasu-Nishimura, Y.; Neutzner, A.; Karbowski, M.; Youle, R.J.; Nicoll, R.A.; Roche, K.W. Ubiquitin Ligase RNF167 Regulates AMPA Receptor-Mediated Synaptic Transmission. Proc. Natl. Acad. Sci. USA 2012, 109, 19426–19431. [Google Scholar] [CrossRef]
- Hayashi, T.; Rumbaugh, G.; Huganir, R.L. Differential Regulation of AMPA Receptor Subunit Trafficking by Palmitoylation of Two Distinct Sites. Neuron 2005, 47, 709–723. [Google Scholar] [CrossRef]
- Lin, D.T.; Makino, Y.; Sharma, K.; Hayashi, T.; Neve, R.; Takamiya, K.; Huganir, R.L. Regulation of AMPA Receptor Extrasynaptic Insertion by 4.1N, Phosphorylation and Palmitoylation. Nat. Neurosci. 2009, 12, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Yamashita, M.; Kaneko, M.; Okuno, H.; Abe, M.; Yamazaki, M.; Natsume, R.; Yamada, D.; Kaizuka, T.; Suwa, R.; et al. Deficiency of AMPAR–Palmitoylation Aggravates Seizure Susceptibility. J. Neurosci. 2018, 38, 10220–10235. [Google Scholar] [CrossRef] [PubMed]
- Baudry, M.; Massicotte, G.; Hauge, S. Phosphatidylserine Increases the Affinity of the AMPA/Quisqualate Receptor in Rat Brain Membranes. Behav. Neural Biol. 1991, 55, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Rufini, S.; Tancredi, V.; Forcina, R.; Grossi, D.; D’Arcangelo, G. Cholesterol Depletion Inhibits Synaptic Transmission and Synaptic Plasticity in Rat Hippocampus. Exp. Neurol. 2008, 212, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Korinek, M.; Gonzalez-Gonzalez, I.M.; Smejkalova, T.; Hajdukovic, D.; Skrenkova, K.; Krusek, J.; Horak, M.; Vyklicky, L. Cholesterol Modulates Presynaptic and Postsynaptic Properties of Excitatory Synaptic Transmission. Sci. Rep. 2020, 10, 12651. [Google Scholar] [CrossRef]
- Korinek, M.; Vyklicky, V.; Borovska, J.; Lichnerova, K.; Kaniakova, M.; Krausova, B.; Krusek, J.; Balik, A.; Smejkalova, T.; Horak, M.; et al. Cholesterol Modulates Open Probability and Desensitization of NMDA Receptors. J. Physiol. 2015, 593, 2279–2293. [Google Scholar] [CrossRef]
- Antonini, A.; Caioli, S.; Saba, L.; Vindigni, G.; Biocca, S.; Canu, N.; Zona, C. Membrane Cholesterol Depletion in Cortical Neurons Highlights Altered NMDA Receptor Functionality in a Mouse Model of Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta—Mol. Basis Dis. 2018, 1864, 509–519. [Google Scholar] [CrossRef]
- Sanchez-Mejia, R.O.; Newman, J.W.; Toh, S.; Yu, G.-Q.; Zhou, Y.; Halabisky, B.; Cissé, M.; Scearce-Levie, K.; Cheng, I.H.; Gan, L.; et al. Phospholipase A2 Reduction Ameliorates Cognitive Deficits in a Mouse Model of Alzheimer’s Disease. Nat. Neurosci. 2008, 11, 1311–1318. [Google Scholar] [CrossRef]
- Bernard, J.; Chabot, C.; Gagné, J.; Baudry, M.; Massicotte, G. Melittin Increases AMPA Receptor Affinity in Rat Brain Synaptoneurosomes. Brain Res. 1995, 671, 195–200. [Google Scholar] [CrossRef]
- Ménard, C.; Patenaude, C.; Massicotte, G. Phosphorylation of AMPA Receptor Subunits Is Differentially Regulated by Phospholipase A2 Inhibitors. Neurosci. Lett. 2005, 389, 51–56. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| iPSC Name | Disease Status | WT/Mutation | APOE Genotype | Age at Collection | Sex | iPSC Line Characterisation |
|---|---|---|---|---|---|---|
| FAD1 | Familial AD | PSEN1S290C | ε3/3 | 47 | M | [39] |
| IC1 | Isogenic control | PSEN1WT | ε3/3 | 47 | M | [39] |
| HC1 | Healthy control | PSEN1WT | ε2/4 | 57 | F | [40] |
| FAD2 | Familial AD | PSEN1A246E | ε3/4 | 56 | F | [41] |
| IC2 | Isogenic control | PSEN1WT | ε3/4 | 56 | F | Anastacio et al., 2024 in revision |
| HC2 | Healthy control | PSEN1WT | ε2/3 | 75 | F | [41] |
| Target | Sequence | Tm |
|---|---|---|
| B2M | F: AAGGACTGGTCTTTCTATCTC R: GATCCCACTTAACTATCTTGG | 55 °C |
| GAPDH | F: GAGCACAAGAGGAAGAGAGAGACCC R: GTTGAGCACAGGGTACTTTATTGATGGTACATG | 58 °C |
| HPRT1 | F: TGACACTGGCAAAACAATGCA R: GGTCCTTTTCACCAGCAAGCT | 58 °C |
| GRIA1 | F: CTAGAAGATCCTTATGTGATGC R: CTCCGTATTTTCCATCACTG | 58 °C |
| GRIA2 | F: GGAATCTCCGTATGTTATGATG R: TTGTACTTGAACCCACAATG | 55 °C |
| GRIA3 | F: TATTGTATCTGGGGCGTTAC R: TTGAGAACTCAAGAAGGGAG | 55 °C |
| GRIA4 | F: GGTACGATAAAGGTGAATGTG R: AAAAGGTCAGCTTCATTCTC | 58 °C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Targa Dias Anastacio, H.; Matosin, N.; Ooi, L. Familial Alzheimer’s Disease Neurons Bearing Mutations in PSEN1 Display Increased Calcium Responses to AMPA as an Early Calcium Dysregulation Phenotype. Life 2024, 14, 625. https://doi.org/10.3390/life14050625
Targa Dias Anastacio H, Matosin N, Ooi L. Familial Alzheimer’s Disease Neurons Bearing Mutations in PSEN1 Display Increased Calcium Responses to AMPA as an Early Calcium Dysregulation Phenotype. Life. 2024; 14(5):625. https://doi.org/10.3390/life14050625
Chicago/Turabian StyleTarga Dias Anastacio, Helena, Natalie Matosin, and Lezanne Ooi. 2024. "Familial Alzheimer’s Disease Neurons Bearing Mutations in PSEN1 Display Increased Calcium Responses to AMPA as an Early Calcium Dysregulation Phenotype" Life 14, no. 5: 625. https://doi.org/10.3390/life14050625
APA StyleTarga Dias Anastacio, H., Matosin, N., & Ooi, L. (2024). Familial Alzheimer’s Disease Neurons Bearing Mutations in PSEN1 Display Increased Calcium Responses to AMPA as an Early Calcium Dysregulation Phenotype. Life, 14(5), 625. https://doi.org/10.3390/life14050625