
Pulmonary Hypertension and Hyperglycemia—Not a Sweet Combination

Abstract
:1. Introduction
2. Pathophysiology of Pulmonary Hypertension
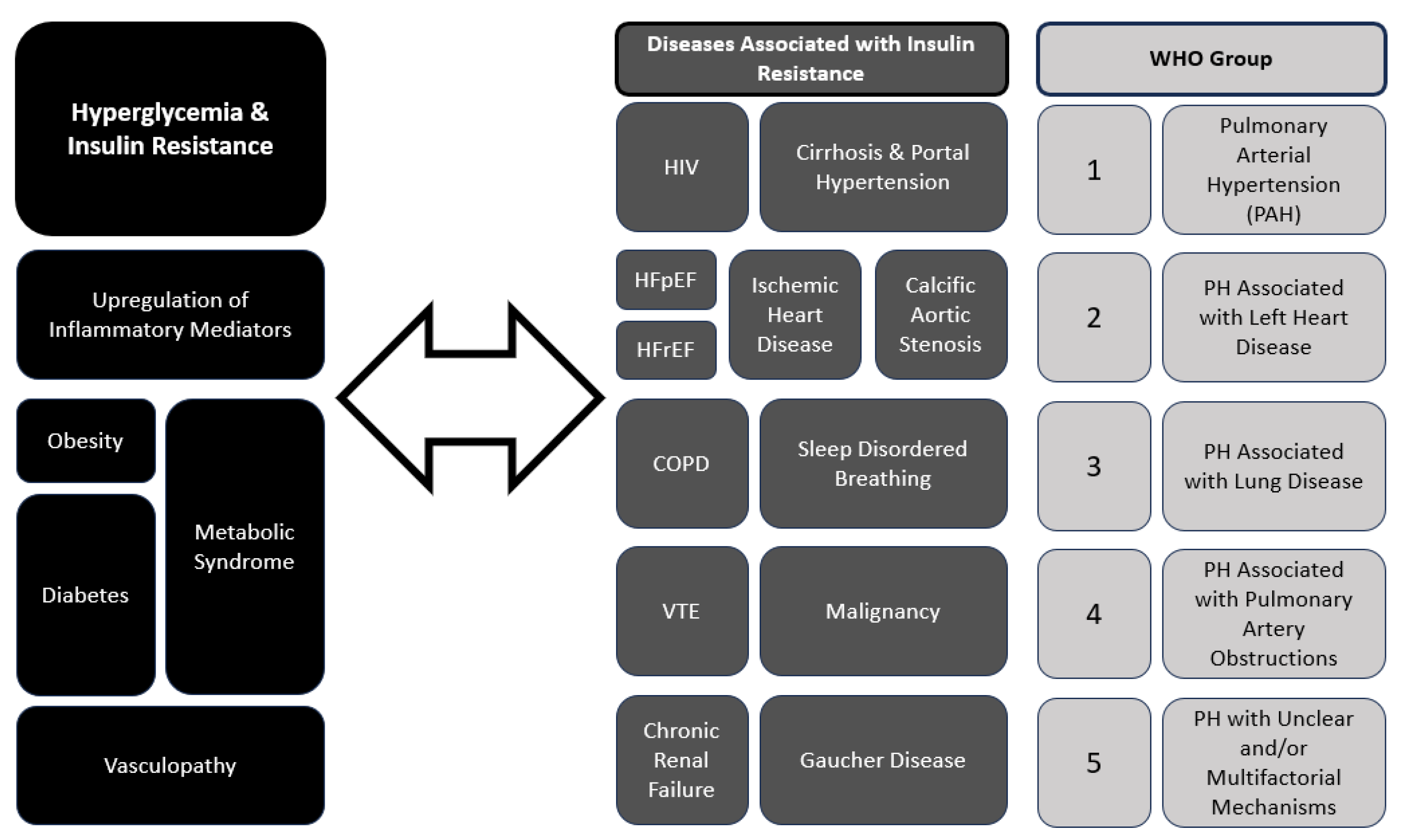
3. The Interplay between Pulmonary Hypertension and Hyperglycemia
4. PH and Diabetes Mellitus Have Shared Pathophysiological Mechanisms
4.1. Tumor Necrosis Factor-alpha (TNF-α)
4.1.1. Role in Insulin Resistance
4.1.2. Role in PH
4.2. Interleukin-6 (IL-6)
4.2.1. Role in Insulin Resistance
4.2.2. Role in PH
4.3. C-Reactive Protein (CRP)
4.3.1. Role in Insulin Resistance
4.3.2. Role in PH
4.4. Nuclear Factor-kappa B (NF-κB)
4.4.1. Role in Insulin Resistance
4.4.2. Role in PH
4.5. Monocyte Chemoattractant Protein-1 (MCP-1)
4.5.1. Role in Insulin Resistance
4.5.2. Role in PH
5. Metabolic Dysregulation in Pulmonary Hypertension
6. Oxidative Stress
7. Hormonal Factors
8. Genetic Susceptibility
9. Shared Risk Factors and Clinical Implications
9.1. Hypoxia
9.2. Obesity and Obstructive Sleep Apnea
Obesity and Inflammation
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.; Brida, M.; Carlsen, J.; Coats, A.J.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2023, 61, 2200879. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Janocha, A.J.; Erzurum, S.C. Metabolism in Pulmonary Hypertension. Annu. Rev. Physiol. 2021, 83, 551–576. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Erzurum, S.C. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Compr. Physiol. 2011, 1, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.F.; Su, Y.C. Vascular Metabolic Mechanisms of Pulmonary Hypertension. Curr. Med. Sci. 2020, 40, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Kaneko, F.T.; Zheng, S.; Comhair, S.A.A.; Janocha, A.J.; Goggans, T.; Thunnissen, F.B.J.M.; Farver, C.; Hazen, S.L.; Jennings, C.; et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004, 18, 1746–1748. [Google Scholar] [CrossRef] [PubMed]
- Willson, C.; Watanabe, M.; Tsuji-Hosokawa, A.; Makino, A. Pulmonary vascular dysfunction in metabolic syndrome. J. Physiol. 2019, 597, 1121–1141. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Keramidas, G.; Gourgoulianis, K.I.; Kotsiou, O.S. Venous Thromboembolic Disease in Chronic Inflammatory Lung Diseases: Knowns and Unknowns. J. Clin. Med. 2021, 10, 2061. [Google Scholar] [CrossRef] [PubMed]
- Condliffe, R.; Durrington, C.; Hameed, A.; Lewis, R.A.; Venkateswaran, R.; Gopalan, D.; Dorfmüller, P. Clinical-radiological-pathological correlation in pulmonary arterial hypertension. Eur. Respir. Rev. 2023, 32, 230138. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, L.; Jia, Y.; Balistrieri, A.; Fraidenburg, D.R.; Wang, J.; Tang, H.; Yuan, J.X. Pathogenic Mechanisms of Pulmonary Arterial Hypertension: Homeostasis Imbalance of Endothelium-Derived Relaxing and Contracting Factors. JACC Asia 2022, 2, 787–802. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Xu, B.T.; Wan, S.R.; Ma, X.M.; Long, Y.; Xu, Y.; Jiang, Z.Z. The role of oxidative stress in diabetes mellitus-induced vascular endothelial dysfunction. Cardiovasc. Diabetol. 2023, 22, 237. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S.; Pausch, C.; Coghlan, J.G.; Huscher, D.; Pittrow, D.; Grünig, E.; Staehler, G.; Vizza, C.D.; Gall, H.; Distler, O.; et al. Risk stratification and response to therapy in patients with pulmonary arterial hypertension and comorbidities: A COMPERA analysis. J. Heart Lung Transplant. 2023, 42, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, I.T.; Stangou, Μ.; Sarafidis, P.; Gouliovaki, A.; Giamalis, P.; Tsouchnikas, I.; Didangelos, T.; Papagianni, A. TNF-α pathway and T-cell immunity are activated early during the development of diabetic nephropathy in Type II Diabetes Mellitus. Clin. Immunol. 2020, 215, 108423. [Google Scholar] [CrossRef] [PubMed]
- Pavkov, M.E.; Weil, E.J.; Fufaa, G.D.; Nelson, R.G.; Lemley, K.V.; Knowler, W.C.; Niewczas, M.A.; Krolewski, A.S. Tumor necrosis factor receptors 1 and 2 are associated with early glomerular lesions in type 2 diabetes. Kidney Int. 2016, 89, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.-R.; Yang, S.-H. The role of tumor necrosis factor alpha (tnf-α) in autoimmune disease and current tnf-α inhibitors in therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhao, Y.; Li, S.; Wu, H.; Ma, D.; Wan, C. TNF-α and IL-8 levels are positively correlated with hypobaric hypoxic pulmonary hypertension and pulmonary vascular remodeling in rats. Open Life Sci. 2023, 18, 20220650. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Chaurasia, B.; Goldau, J.; Vogt, M.C.; Ruud, J.; Nguyen, K.D.; Theurich, S.; Hausen, A.C.; Schmitz, J.; Brönneke, H.S.; et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat. Immunol. 2014, 15, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Song, J.; Gao, J.; Cheng, J.; Xie, H.; Zhang, L.; Wang, Y.-H.; Gao, Z.; Wang, Y.; Wang, X.; et al. Adipocyte-derived kynurenine promotes obesity and insulin resistance by activating the AhR/STAT3/IL-6 signaling. Nat. Commun. 2022, 13, 3489. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, P.; Wang, X.; Liu, Z.; Zhang, Z.; Zhang, Y.; Wang, Z.; Feng, Y.; Wang, Q.; Guo, X.; et al. Inflammation and Insulin Resistance in Diabetic Chronic Coronary Syndrome Patients. Nutrients 2023, 15, 2808. [Google Scholar] [CrossRef] [PubMed]
- Yaku, A.; Inagaki, T.; Asano, R.; Okazawa, M.; Mori, H.; Sato, A.; Hia, F.; Masaki, T.; Manabe, Y.; Ishibashi, T.; et al. Regnase-1 Prevents Pulmonary Arterial Hypertension Through mRNA Degradation of Interleukin-6 and Platelet-Derived Growth Factor in Alveolar Macrophages. Circulation 2022, 146, 1006–1022, Erratum in Circulation 2022, 146, e280. [Google Scholar] [CrossRef] [PubMed]
- Toshner, M.; Rothman, A. IL-6 in pulmonary hypertension: Why novel is not always best. Eur. Respir. J. 2020, 55, 2000314. [Google Scholar] [CrossRef]
- Toshner, M.; Church, C.; Harbaum, L.; Rhodes, C.; Moreschi, S.S.V.; Liley, J.; Jones, R.; Arora, A.; Batai, K.; Desai, A.A.; et al. Mendelian randomisation and experimental medicine approaches to interleukin-6 as a drug target in pulmonary arterial hypertension. Eur. Respir. J. 2022, 59, 2002463, Erratum in Eur. Respir. J. 2022, 60, 2052463. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Bijli, K.M.; Ramirez, A.; Murphy, T.C.; Kleinhenz, J.; Hart, C.M. Hypoxia downregulates PPARγ via an ERK1/2-NF-κB-Nox4-dependent mechanism in human pulmonary artery smooth muscle cells. Free. Radic. Biol. Med. 2013, 63, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Stanimirovic, J.; Radovanovic, J.; Banjac, K.; Obradovic, M.; Essack, M.; Zafirovic, S.; Gluvic, Z.; Gojobori, T.; Isenovic, E.R. Role of C-Reactive Protein in Diabetic Inflammation. Mediat. Inflamm. 2022, 2022, 3706508. [Google Scholar] [CrossRef] [PubMed]
- Ben, J.; Jiang, B.; Wang, D.; Liu, Q.; Zhang, Y.; Qi, Y.; Tong, X.; Chen, L.; Liu, X.; Zhang, Y.; et al. Major vault protein suppresses obesity and atherosclerosis through inhibiting IKK-NF-κB signaling mediated inflammation. Nat. Commun. 2019, 10, 1801. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Huang, Y.; Song, Z.; Zhou, H.J.; Zhang, H.; Perry, R.J.; Shulman, G.I.; Min, W. Mitophagy-mediated adipose inflammation contributes to type 2 diabetes with hepatic insulin resistance. J. Exp. Med. 2021, 218, e20201416. [Google Scholar] [CrossRef] [PubMed]
- Maimaitiaili, N.; Zeng, Y.; Ju, P.; Zhakeer, G.; Guangxi, E.; Yao, H.; Shi, Y.; Zhai, M.; Zhuang, J.; Peng, W.; et al. NLRC3 deficiency promotes hypoxia-induced pulmonary hypertension development via IKK/NF-κB p65/HIF-1α pathway. Exp. Cell Res. 2023, 431, 113755. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wu, X.; Wang, J.; He, M.; Han, H.; Hu, S.; Xu, J.; Yang, M.; Tan, Q.; Wang, Y.; et al. Paeoniflorin attenuates monocrotaline-induced pulmonary arterial hypertension in rats by suppressing TAK1-MAPK/NF-κB pathways. Int. J. Med. Sci. 2022, 19, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Chiu, C.J.; Kuan, C.H.; Chen, F.H.; Shen, Y.C.; Wu, C.H.; Hsu, Y.H. IL-29 promoted obesity-induced inflammation and insulin resistance. Cell Mol. Immunol. 2020, 17, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, D.; Das, S.; Guria, S.; Basu, S.; Mukherjee, S. Fetuin-A regulates adipose tissue macrophage content and activation in insulin resistant mice through MCP-1 and iNOS: Involvement of IFNγ-JAK2-STAT1 pathway. Biochem. J. 2021, 478, 4027–4043. [Google Scholar] [CrossRef] [PubMed]
- Magoń, W.; Stępniewski, J.; Waligóra, M.; Jonas, K.; Przybylski, R.; Podolec, P.; Kopeć, G. Changes in Inflammatory Markers in Patients with Chronic Thromboembolic Pulmonary Hypertension Treated with Balloon Pulmonary Angioplasty. Cells 2022, 11, 1491. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Carmeliet, P. The Link between Angiogenesis and Endothelial Metabolism. Annu. Rev. Physiol. 2017, 79, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Luo, F.; Wu, P.; Huang, Y.; Das, A.; Chen, S.; Chen, J.; Hu, X.; Li, F.; Fang, Z.; et al. Metabolomics reveals metabolite changes of patients with pulmonary arterial hypertension in China. J. Cell Mol. Med. 2020, 24, 2484–2496. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Brittain, E.L.; Trammell, A.W.; Fessel, J.P.; Austin, E.D.; Penner, N.; Maynard, K.B.; Gleaves, L.; Talati, M.; Absi, T.; et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 189, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, M.D.; Marciano, D.P.; Fu, P.; Franco, M.C.; Unwalla, H.; Tieu, K.; Fineman, J.R.; Wang, T.; Black, S.M. Metabolic reprogramming, oxidative stress, and pulmonary hypertension. Redox Biol. 2023, 64, 102797. [Google Scholar] [CrossRef]
- Paulin, R.; Michelakis, E.D. The metabolic theory of pulmonary arterial hypertension. Circ. Res. 2014, 115, 148–164. [Google Scholar] [CrossRef]
- Heresi, G.A.; Mey, J.T.; Bartholomew, J.R.; Haddadin, I.S.; Tonelli, A.R.; Dweik, R.A.; Kirwan, J.P.; Kalhan, S.C. Plasma metabolomic profile in chronic thromboembolic pulmonary hypertension. Pulm. Circ. 2020, 10, 2045894019890553. [Google Scholar] [CrossRef] [PubMed]
- Papachristoforou, E.; Lambadiari, V.; Maratou, E.; Makrilakis, K. Association of Glycemic Indices (Hyperglycemia, Glucose Variability, and Hypoglycemia) with Oxidative Stress and Diabetic Complications. J. Diabetes Res. 2020, 2020, 7489795. [Google Scholar] [CrossRef] [PubMed]
- Poyatos, P.; Gratacós, M.; Samuel, K.; Orriols, R.; Tura-Ceide, O. Oxidative Stress and Antioxidant Therapy in Pulmonary Hypertension. Antioxidants 2023, 12, 1006. [Google Scholar] [CrossRef]
- Fulton, D.J.R.; Li, X.; Bordan, Z.; Haigh, S.; Bentley, A.; Chen, F.; Barman, S.A. Reactive oxygen and nitrogen species in the development of pulmonary hypertension. Antioxidants 2017, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, F.; Nigro, E.; Mollica, M.; Costigliola, A.; D’agnano, V.; Daniele, A.; Bianco, A.; Guerra, G. Pulmonary Hypertension and Obesity: Focus on Adiponectin. Int. J. Mol. Sci. 2019, 20, 912. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, S.; Jankowich, M.; Wu, W.C.; Abbasi, S.; Morrison, A.R.; Choudhary, G. Association of plasma adiponectin with pulmonary hypertension, mortality and heart failure in African Americans: Jackson Heart Study. Pulm. Circ. 2020, 10, 2045894020961242. [Google Scholar] [CrossRef] [PubMed]
- Kraja, A.T.; Liu, C.; Fetterman, J.L.; Graff, M.; Have, C.T.; Gu, C.; Yanek, L.R.; Feitosa, M.F.; Arking, D.E.; Chasman, D.I.; et al. Associations of Mitochondrial and Nuclear Mitochondrial Variants and Genes with Seven Metabolic Traits. Am. J. Hum. Genet. 2019, 104, 112–138. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial DNA variation in human radiation and disease. Cell 2015, 163, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Farha, S.; Hu, B.; Comhair, S.; Zein, J.; Dweik, R.; Erzurum, S.C.; Aldred, M.A. Mitochondrial Haplogroups and Risk of Pulmonary Arterial Hypertension. PLoS ONE 2016, 11, e0156042. [Google Scholar] [CrossRef] [PubMed]
- Aldred, M.A.; Comhair, S.A.; Varella-Garcia, M.; Asosingh, K.; Xu, W.; Noon, G.P.; Thistlethwaite, P.A.; Tuder, R.M.; Erzurum, S.C.; Geraci, M.W.; et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- The International PPH Consortium; Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., 3rd; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef]
- Diebold, I.; Hennigs, J.K.; Miyagawa, K.; Li, C.G.; Nickel, N.P.; Kaschwich, M.; Cao, A.; Wang, L.; Reddy, S.; Chen, P.-I.; et al. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 2015, 21, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Naderi, N.; Boobejame, P.; Bakhshandeh, H.; Amin, A.; Taghavi, S.; Maleki, M. Insulin resistance in pulmonary arterial hypertension, is it a novel disease modifier? Res. Cardiovasc. Med. 2014, 3, e19710. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.A.; Bradley, D. Pulmonary Arterial Hypertension and Insulin Resistance. J. Mol. Genet. Med. 2014, 2 (Suppl. S1), 15. [Google Scholar] [CrossRef] [PubMed]
- Farha, S.; Comhair, S.; Hou, Y.; Park, M.M.; Sharp, J.; Peterson, L.; Willard, B.; Zhang, R.; DiFilippo, F.P.; Neumann, D.R.; et al. Metabolic endophenotype associated with right ventricular glucose uptake in pulmonary hypertension. Pulm. Circ. 2021, 11, 20458940211054325. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Ota, H.; Kimura, Y.; Takasawa, S. Effects of Intermittent Hypoxia on Pulmonary Vascular and Systemic Diseases. Int. J. Environ. Res. Public Health 2019, 16, 3101. [Google Scholar] [CrossRef] [PubMed]
- Mair, K.M.; Gaw, R.; MacLean, M.R. Obesity, estrogens and adipose tissue dysfunction—Implications for pulmonary arterial hypertension. Pulm. Circ. 2020, 10, 2045894020952019. [Google Scholar] [CrossRef] [PubMed]
- McLean, L.L.; Pellino, K.; Brewis, M.; Peacock, A.; Johnson, M.; Church, A.C. The obesity paradox in pulmonary arterial hypertension: The Scottish perspective. ERJ Open Res. 2019, 5, 00241–2019. [Google Scholar] [CrossRef] [PubMed]
- Poms, A.D.; Turner, M.; Farber, H.W.; Meltzer, L.A.; McGoon, M.D. Comorbid conditions and outcomes in patients with pulmonary arterial hypertension: A REVEAL registry analysis. Chest 2013, 144, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Weatherald, J.; Huertas, A.; Boucly, A.; Guignabert, C.; Taniguchi, Y.; Adir, Y.; Jevnikar, M.; Savale, L.; Jaïs, X.; Peng, M.; et al. Association between BMI and obesity with survival in pulmonary arterial hypertension. Chest 2018, 154, 872–881. [Google Scholar] [CrossRef]
- Kawut, S.M.; Archer-Chicko, C.L.; DeMichele, A.; Fritz, J.S.; Klinger, J.R.; Ky, B.; Palevsky, H.I.; Palmisciano, A.J.; Patel, M.; Pinder, D.; et al. Anastrozole in pulmonary arterial hypertension. A randomized, double-blind, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 2017, 195, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.F.; Ewart, M.A.; Mair, K.; Nilsen, M.; Dempsie, Y.; Loughlin, L.; Maclean, M.R. Oestrogen receptor alpha in pulmonary hypertension. Cardiovasc. Res 2015, 106, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Kawut, S.M.; Pinder, D.; Al-Naamani, N.; McCormick, A.; Palevsky, H.I.; Fritz, J.; Smith, K.A.; Mazurek, J.A.; Doyle, M.F.; MacLean, M.R.; et al. Fulvestrant for the treatment of pulmonary arterial hypertension. Ann. Am. Thorac. Soc. 2019, 16, 1456–1459. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Austin, E.D.; Talati, M.; Fessel, J.P.; Farber-Eger, E.H.; Brittain, E.L.; Hemnes, A.R.; Loyd, J.E.; West, J. Oestrogen inhibition reverses pulmonary arterial hypertension and associated metabolic defects. Eur. Respir. J. 2017, 50, 1602337. [Google Scholar] [CrossRef]

{kind=link}
| Inflammatory Mediator | Relation to Hyperglycemia | Role in PH |
|---|---|---|
| TNF-α |
|
|
| IL-6 |
|
|
| CRP |
|
|
| NF-kB |
|
|
| MCP-1 |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruck, O.; Pandit, L.M. Pulmonary Hypertension and Hyperglycemia—Not a Sweet Combination. Diagnostics 2024, 14, 1119. https://doi.org/10.3390/diagnostics14111119
Bruck O, Pandit LM. Pulmonary Hypertension and Hyperglycemia—Not a Sweet Combination. Diagnostics. 2024; 14(11):1119. https://doi.org/10.3390/diagnostics14111119
Chicago/Turabian StyleBruck, Or, and L. M. Pandit. 2024. "Pulmonary Hypertension and Hyperglycemia—Not a Sweet Combination" Diagnostics 14, no. 11: 1119. https://doi.org/10.3390/diagnostics14111119
APA StyleBruck, O., & Pandit, L. M. (2024). Pulmonary Hypertension and Hyperglycemia—Not a Sweet Combination. Diagnostics, 14(11), 1119. https://doi.org/10.3390/diagnostics14111119