3.1. Differential Investment in Translation Machinery
Ribosomes represent parallel protein-production factories and their abundance in
E. coli increases linearly with the growth rate [
117,
118]. Short-generation species may have not only more efficient factories but also more factories than LGB. Each ribosome features a set of 16S, 23S, and 5S rRNAs that are transcribed from the same operon and processed into individual rRNAs [
119].
E. coli has seven rRNA operons (
rrnA to
rrnE,
rrnG,
rrnH) with promotors that are almost identical to the −10 and −35 consensus [
120,
121], suggesting a high demand for rRNA molecules met by both efficient and parallel transcription of multiple rRNA operons. The production of ribosomes in
E. coli is limited by rRNA production [
122], which explains why
E. coli maintains multiple
rrn operons in its genome for parallel transcription. A generalization of this would lead to the prediction that SGB should have more
rrn operons than LGB. This prediction should also apply to tRNA genes because, with more ribosomes, more tRNA molecules are needed [
123].
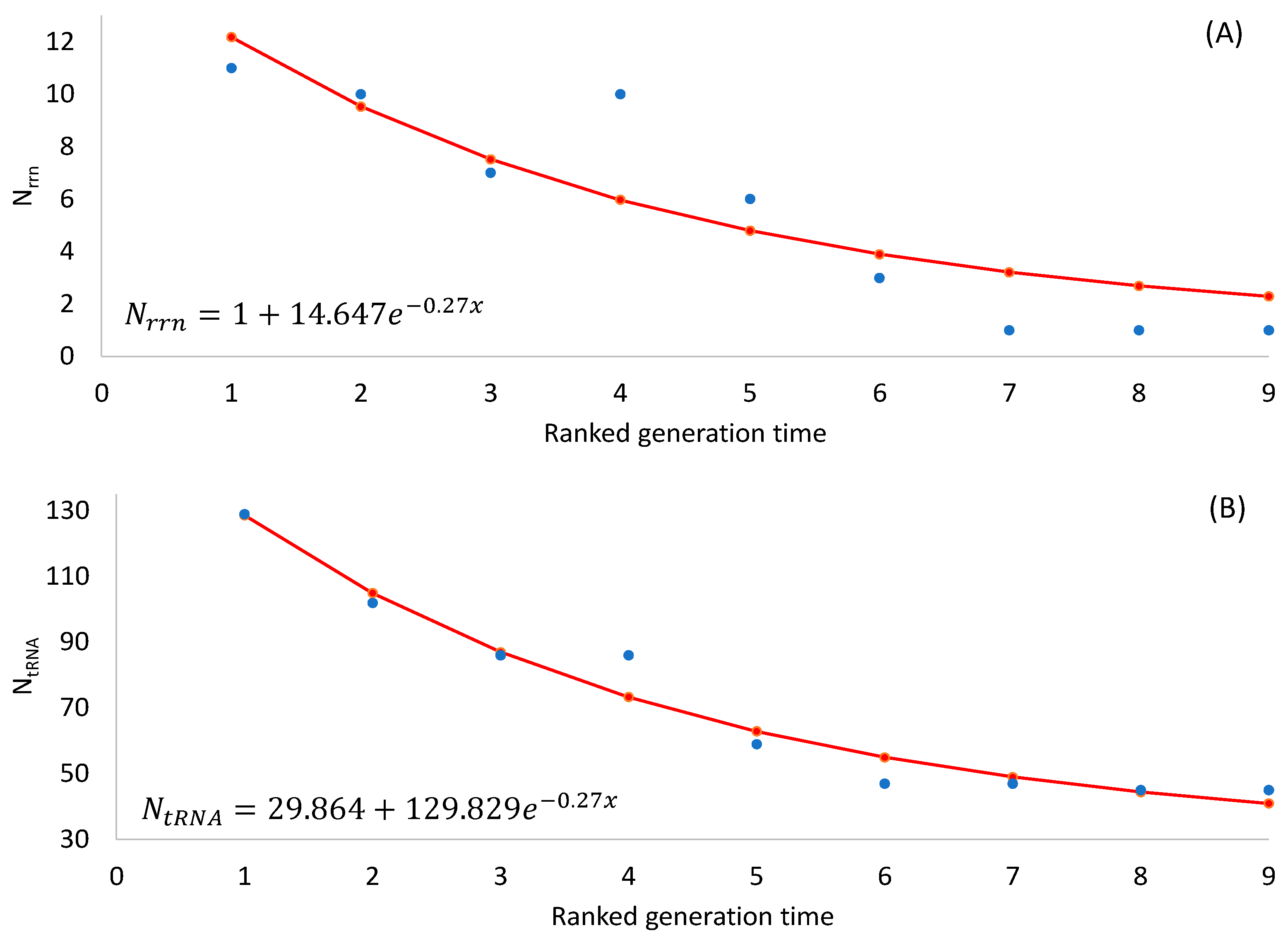
The two predictions are both supported by empirical evidence (
Table 2), with the number of rRNA and tRNA genes decreasing highly significantly with an increasing generation time (
p < 0.0001 for any rank-based nonparametric tests). The observation that SGB maintain more rRNA and tRNA genes in their genomes than LGB is consistent with the interpretation of stronger selection for translation efficiency in SGB than in LGB.
Note that an organism needs at least one
rrn operon for translation. Ribosomal RNAs form the core of ribosomes with all important sites such as A, P, and E sites, with ribosomal proteins padding the surface of a ribosome [
124]. Thus, the number of
rrn operons cannot be less than 1. Also, there should be a minimum set of tRNA genes to decode all 61 sense codons. One might use the following two equations to model the numbers of
rrn operons and tRNA genes, respectively:
where
x is RankGT in
Table 2. Equation (1) ensures a minimum
Nrrn of 1. Equation (2) ensures a minimum
NtRNA of c, which is estimated by the least-squares approach to be 30 (
Figure 1B). The minimum number of tRNA genes required for decoding all sense codons in a natural translation system is observed in a vertebrate mitochondrial genome that encodes 22 tRNA genes.
3.2. Differential Preference for Start Codon AUG
Among the three canonical start codons (AUG, GUG, and UUG), the translation initiation efficiency is consistently in the order of AUG > GUG > UUG [
125,
126]. HEGs in bacteria and bacteriophages tend to prefer AUG as a start codon [
5,
9,
18,
125,
126,
127]. This is true for all nine bacterial species in which the percentage of AUG (AUG%,
Table 3) is consistently higher in HEGs than those in the REST (
Table 3), which includes all genes not in the HEG group and consequently contains both highly expressed and low-expressed genes. This suggests that AUG is the most efficient start codon, as is consistent with previous empirical studies based on within-species comparisons [
5,
8,
9].
Given that AUG is the most efficient start codon, one would predict that SGB should use more AUG codons as start codons than LGB. The AUG% in
Table 3 is indeed strongly associated with the ranked generation time in
Table 1, as predicted. However, the AUG% is also affected by the genomic GC% because GC-rich genomes tend to have more GC-rich codons [
31]. This is best illustrated by focusing on start codons AUG and GUG. AT-rich genomes tend to have AT-biased mutations, which favor AUG over GUG. Among the nine species, the
H. influenzae genome is the most AT-rich and is expected to have a high AUG% because both mutation and selection favor AUG over GUG. In contrast, GC-rich genomes have GC-biased mutations, which will favor GUG over AUG. Because the four LGB all have a higher GC% than the five species with relatively short generations, they may use fewer AUG and more GUG codons as start codons simply because of their genomic GC-richness. For this reason, it is necessary to include genomic GC as a control variable. Also, HEGs and REST genes may differ in the relationship between AUG% and the generation time. A conceptually more comprehensive and explicit model is, therefore, needed.
Here, the dependent variable is AUG% and independent variables include ranked generation time (RankGT), genomic GC content (GC%, which is taken as a proxy for a genomic mutation shared by both HEGs and REST genes), and gene expression (GE with two categories, HEGs and REST genes, encoded as 0 and 1, respectively). The input data (
Table 4) are used to fit the model.
A regression analysis of the input data in
Table 4 showed that all three independent variables were statistically significant, but their interaction terms were not (
Table 5). The regression model was then fitted without interaction terms. This reduced model accounted for 82.0% of the variation in the dependent variable AUG%. The two regression equations derived from the regression coefficients in
Table 5, one for HEGs and the other for the REST, are
Equations (3) and (4) and
Table 5 show that (1) the AUG% decreases highly significantly with the generation time, and (2) the AUG% is significantly higher in HEGs than in REST genes (
Table 5). However, the GC% also has a significant effect on the AUG% (
p = 0.01825,
Table 5), with the AUG% decreasing with an increasing GC%. This is consistent with the interpretation of stronger selection operating on SGB than on LGB. That is, a non-AUG start codon mutating to AUG is more strongly favored by natural selection in SGB than in LGB.
We should emphasize that Equations (3) and (4) are descriptive models. They do not explicitly prevent AUG% from taking values smaller than 0 or larger than 1. A sigmoidal function would have been more appropriate if there were enough data for parameter estimation.
3.3. Differential Preference for Stop Codon UAA
As reviewed previously, the stop codon UAA exhibits the smallest readthrough error rate among the three nonsense codons [
57,
61,
62,
63,
64,
65,
66]. Consequently, HEGs favor stop codon UAA over other stop codons in multiple bacterial species [
9,
67,
74]. This is also true for all nine bacterial species. UAA was preferred by HEGs in
E. coli,
B. subtilis,
M. tuberculosis [
67], and
M. abscessus [
9]. Of the five remaining species, the proportions of UAA in REST genes and HEGs were 0.653 and 0.8816, respectively, in
V. natriegens; 0.6365 and 0.9474 in
V. cholerae; 0.7602 and 0.9429 in
H. influenzae; 0.0585 and 0.2222 in
M. smegmatis; and 0.2364 and 0.2836 in
M. leprae. Thus, UAA is consistently preferred in HEGs relative to REST genes.
Given that UAA is the best stop codon [
128,
129], we predicted that SGB should use more UAA codons as stop codons than LGB. Similar to our analysis of the start codon AUG usage, the dependent variable now was UAA% and the independent variables included the ranked generation time (RankGT), genomic GC content (GC%), and gene expression (GE with two categories, HEGs and REST genes, encoded as 0 and 1, respectively). The genomic GC% is particularly relevant in studying UAA usage because protein-coding genes in a GC-rich genome tend to use more UGA and UAG stop codons than those in an AT-rich genome [
67].
A regression analysis showed that all three independent variables were statistically significant (
Table 6). The model accounted for 94.7% of the total variation in UAA%. The two regression equations derived from the regression coefficients in
Table 6, one for HEGs and the other for the REST, are
Equations (5) and (6) and
Table 6 show that the UAA% decreases highly significantly with an increasing generation time, and that HEGs use UAA significantly more frequently than REST genes (
Table 6, a 17.35326% difference between the two). This is consistent with the interpretation of stronger selection operating on SGB than on LGB. A non-UAA stop codon mutating to UAA is more strongly favored by natural selection in SGB than in LGB.
UAA usage also decreases significantly with an increasing genomic GC% (
Table 6), which is understandable. As the genomic GC% increases, GC-biased mutations will favor UAG and UGA codons over UAA codons [
67]. It is for this reason that the genomic GC content needs to be taken into consideration when assessing codon usage bias.
3.5. Differential Selection Drives tRNA Adaptation
Codons and tRNAs are expected to coevolve and adapt to each other [
2,
6,
30,
31], especially in highly expressed genes [
6,
7,
30,
31,
32,
33,
34,
35,
36,
133]. Given the better codon optimization in SGB than in LGB (
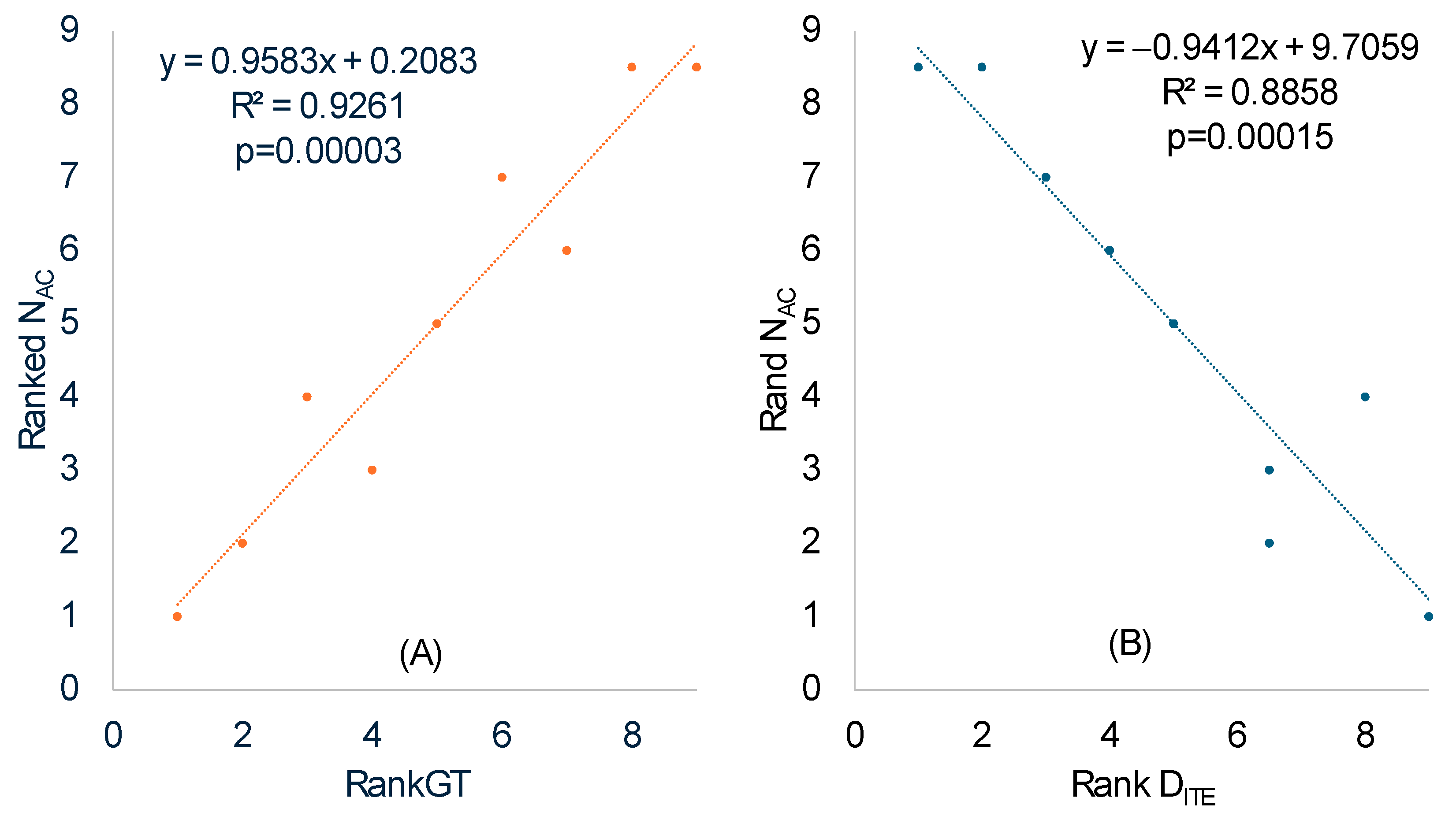
Figure 2 and
Table 7), one would predict more tRNA genes for highly used codons than rarely used codons. If we focus on the anticodons of tRNA genes, then the prediction above implies a smaller effective number of anticodons (N
AC), equivalent to the concept of the effective number of codons [
52,
130], in SGB than in LGB. Specifically, N
AC should increase with RankGT (ranked generation time).
We calculated N
AC in the same way the effective number of codons is calculated [
52], except that codons in coding sequences were replaced by anticodons in tRNA genes. The ranked N
AC increased highly significantly with ranked RankGT (
Table 8 and
Figure 3A), consistent with our prediction that SGB should have a smaller N
AC than LGB. One should note the difference between a codon replacement and an anticodon replacement. A codon replacement may have only a minor effect on the translation of a single gene, but an anticodon replacement will affect the translation of numerous codons. For this reason, anticodons are strongly constrained and much less affected by genomic GC%.
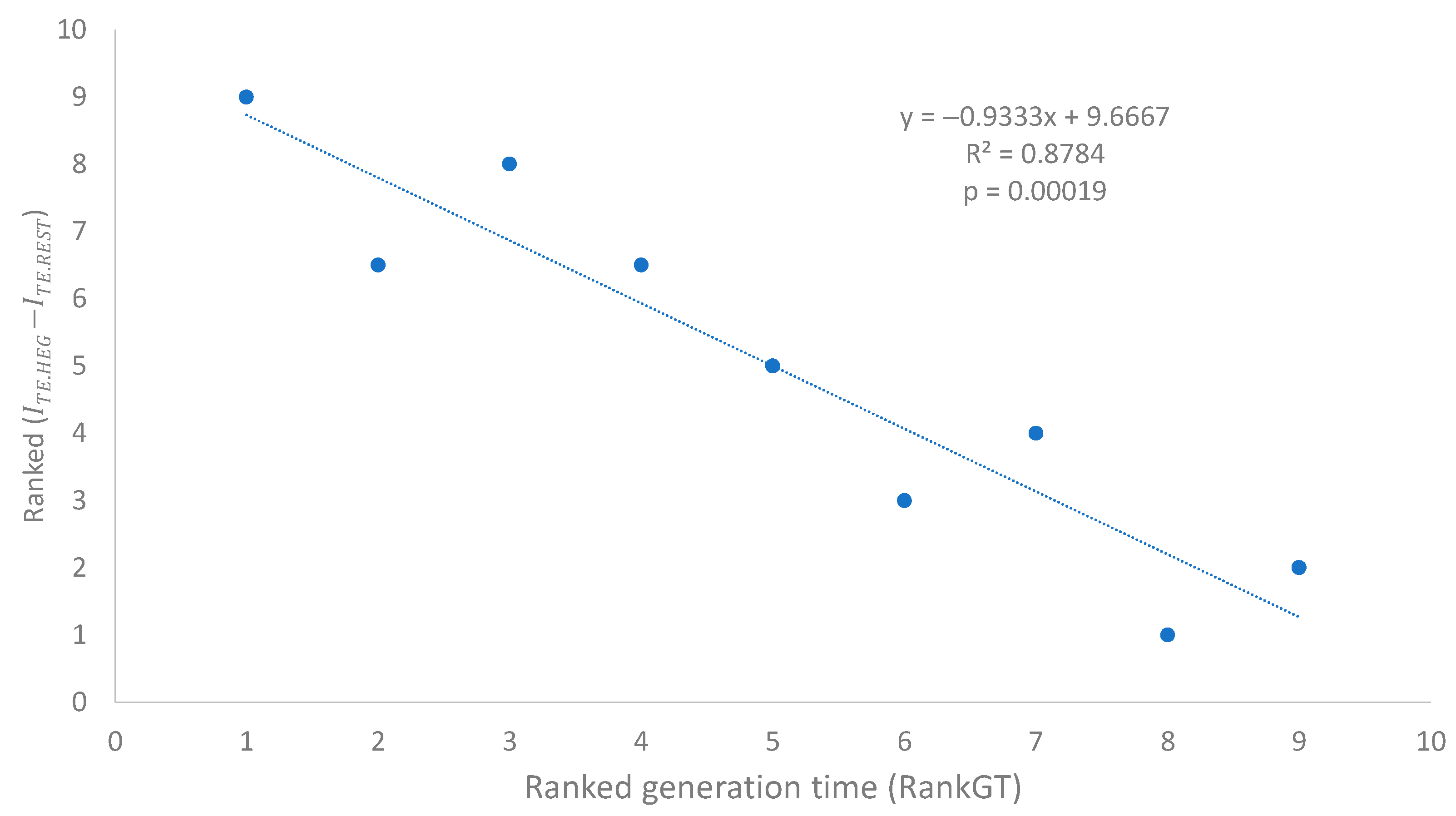
We have previously used Rank
(
Table 7) as a species-level measure of codon adaptation. This
is expected to be negatively associated with the ranked N
AC for the following reason. Better codon adaptation (high Rank
) implies higher usage of major codons, which requires more tRNAs with the corresponding decoding anticodon to translate these overused major codons, leading to a decreased N
AC in species with a high degree of codon adaptation. This expected relationship between N
AC and
was substantiated empirically (
Figure 3B,
p = 0.00015).
The analysis above assumes that tRNA gene copy numbers in bacterial genomes are proportional to the abundances of tRNA molecules in the cell. With the availability of transcriptomic data, it has been found that the assumption is generally true, i.e., the copy number of a tRNA is highly correlated with the transcriptomic representation of the tRNA [
134].
3.6. Secondary Structural Stability near the Start and Stop Codons
Because the 30S ribosomal subunit requires a single-stranded mRNA region for binding [
28,
29], sequences immediately flanking translation signals in bacteria (e.g., Shine–Dalgarno sequence and start and stop codons) are expected to have reduced secondary structures, to avoid embedding translation signals in a stable secondary structure [
18,
27,
116,
126,
135,
136]. This pattern has also been observed in bacteriophage genes [
5,
9]. The weakening of the secondary structure near, or immediately upstream of, the start codon has also been observed in highly expressed eukaryotic mRNAs [
8,
137], especially in mRNAs requiring internal ribosome entry for translation [
138].
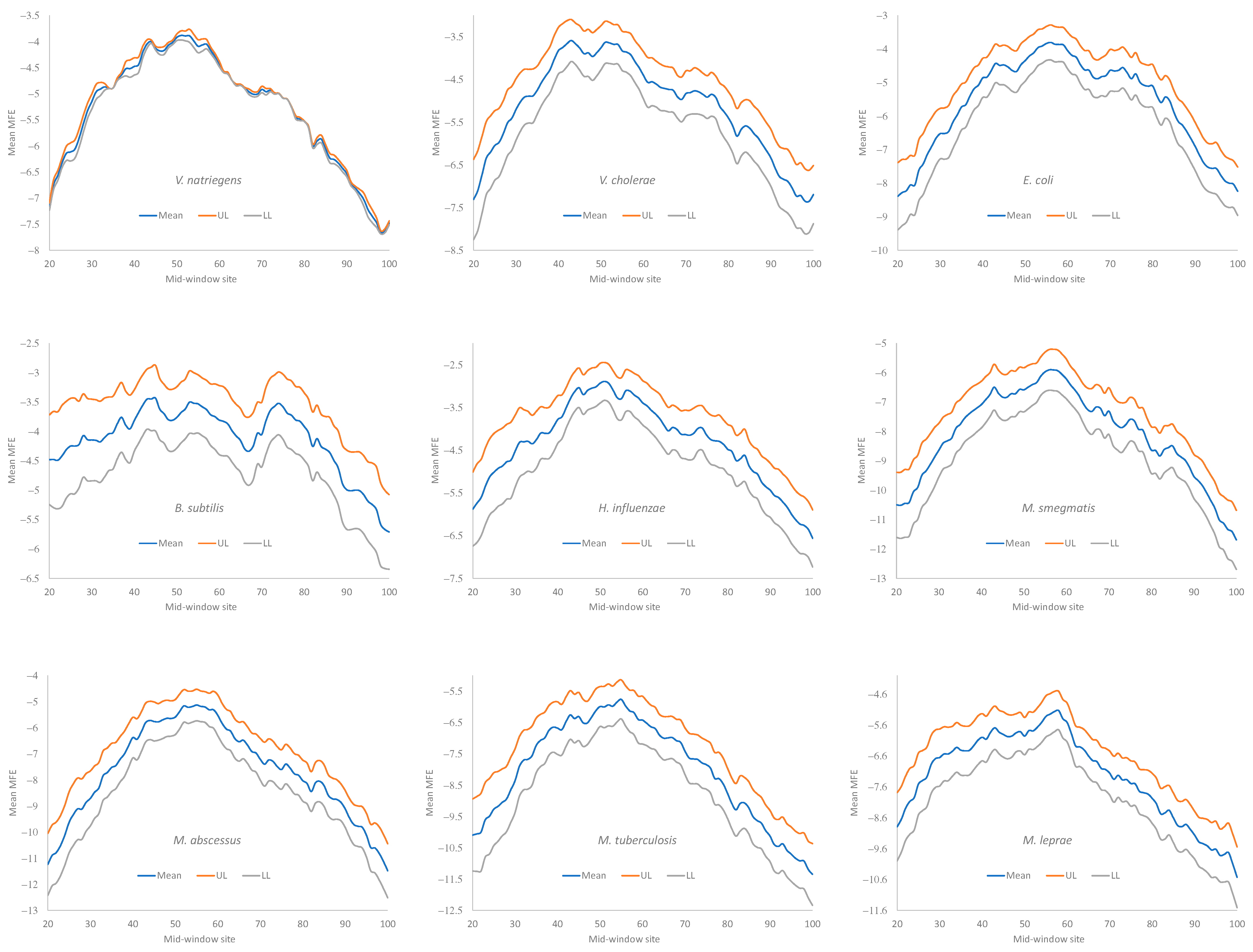
Secondary structural stability in RNA is typically measured by the minimum folding energy (MFE). An MFE equal to 0 means no secondary structure, and a stronger secondary structure corresponds to a more negative MFE value. Experiments involving engineered
E. coli genes have shown that the translation initiation efficiency depends heavily on the MFE of the sequence upstream and including the start codon [
3,
7,
14]. We followed the convention of previous studies [
5,
9] and measured the MFE with a sliding window of 40 nt along mRNA sequences, to quantify the change in secondary structural stability.
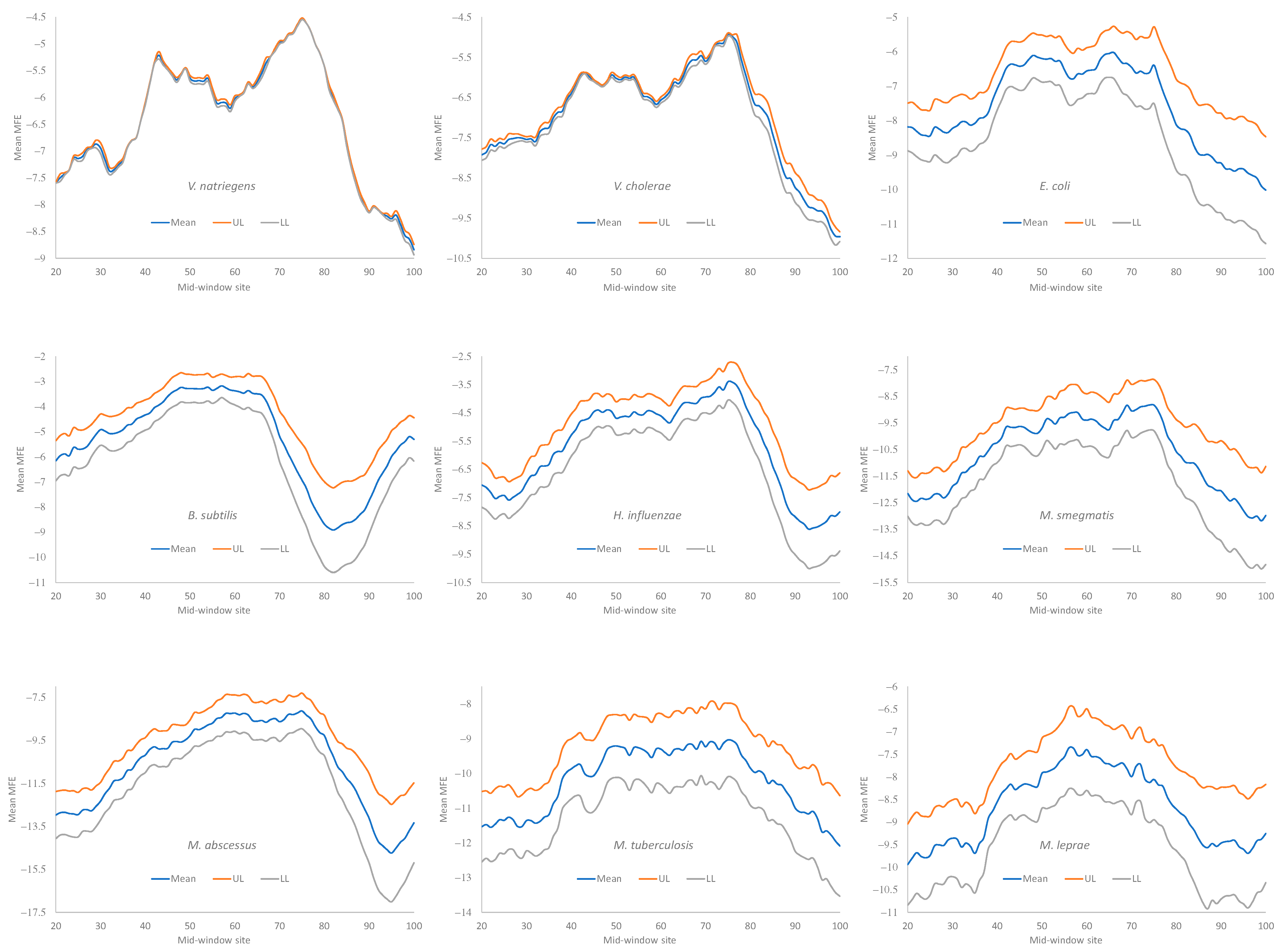
Secondary structural stability, as measured by MFE, decreased near the start codon, but the weakest secondary structure was observed slightly upstream of the start codon (
Figure 4), corresponding to the SD sequence. This pattern has been observed before in bacteriophage genes and their host genes [
5,
9] and is consistent with the interpretation that a strong secondary structure embedding the SD sequence or the start codon is selected against because it prevents the translation initiation signal (SD and start codon) from being decoded by the aSD sequence and the initiation tRNA, respectively.
One might argue against the interpretation that the reduced secondary structure near the translation initiation serves to avoid embedding crucial translation initiation signals such as SD sequences and start codons in a stable secondary structure. The SD sequences are purine-rich and cannot form a secondary structure within them. Thus, the reduced secondary structure near the SD sequence (
Figure 4) could be a direct consequence of the purine-richness in the SD sequences, with nothing to do with the hypothesized avoidance of the secondary structure embedding important translation initiation signals. We thus have two hypotheses. Hypothesis 1 invokes selection against secondary structural stability near the translation initiation signals. Hypothesis 2 states that the weakening of the secondary structure near the translation initiation signal is a simple consequence of the purine-rich SD, with no selection specifically against the secondary structure.
One way to differentiate these two hypotheses is to consider the observation that SD sequences are mainly G-rich. Because G will base-pair with C, Hypothesis 1 (invoking selection against secondary structural stability) will predict a stronger avoidance of nucleotide C relative to nucleotide U. In contrast, Hypothesis 2 gives no reason to expect an avoidance of nucleotide C relative to U near the translation initiation signals.
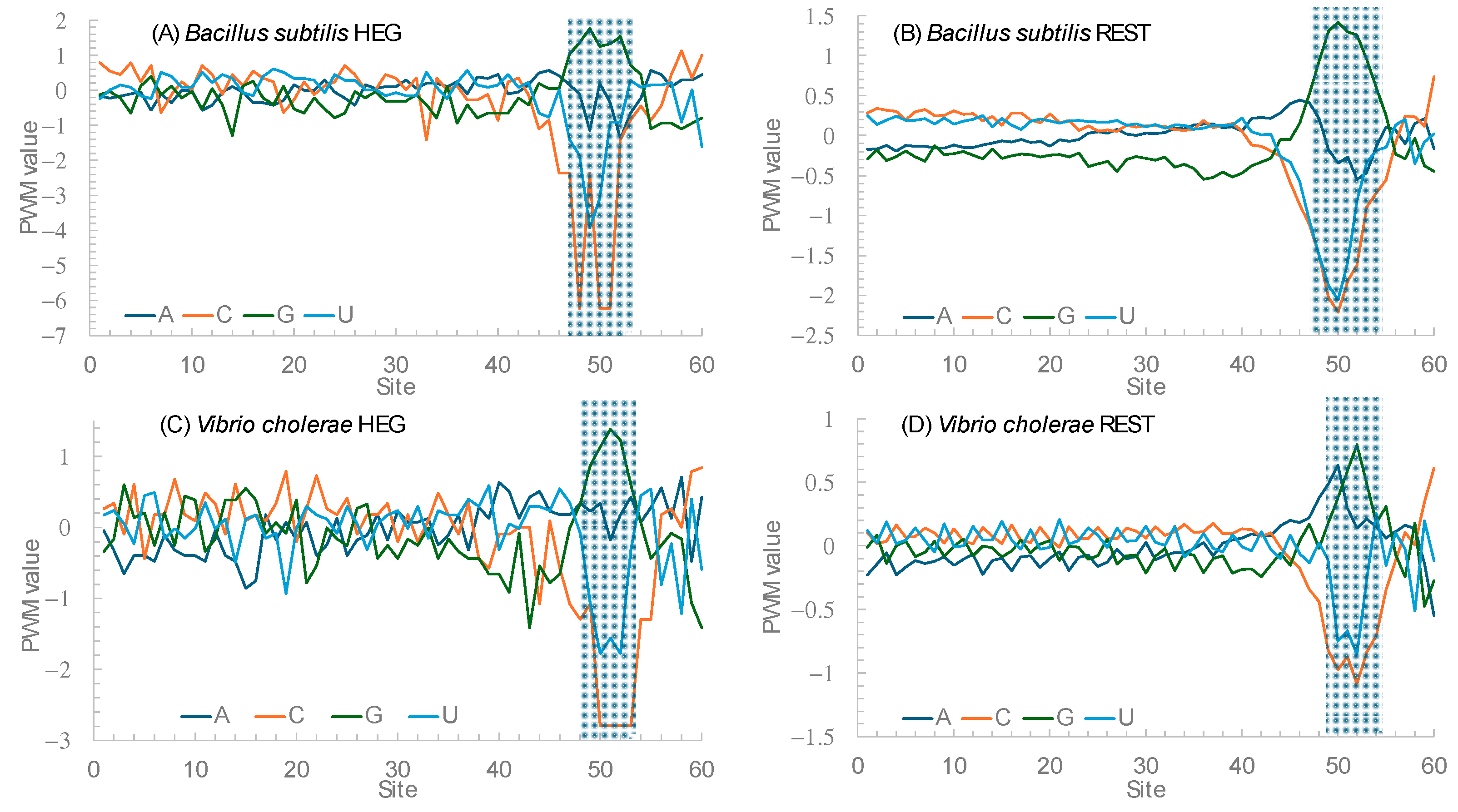
Figure 5 plots the position weight matrix (PWM) scores for the 60 nucleotides immediately upstream of the start codon for two species,
Bacillus subtilis and
Vibrio cholarae. PWM scores measure nucleotide usage bias relative to the background nucleotide frequencies. A value of 0 means unbiased usage, a positive value means overuse, and a negative value means avoidance.
Figure 5A,B contrast the HEGs and the REST genes in
B. subtilis. If Hypothesis 1 is correct, then we expect stronger avoidance of C relative to U in HEGs than in the REST genes. The nucleotide usage patterns in
Figure 5A,B are consistent with the prediction from Hypothesis 1. An approximate statistical test can be performed as follows. Within sites 45 to 55, there 36 nucleotide Cs and 129 nucleotide Us in the HEGs. The corresponding numbers are 2952 C and 7069 U. The percent of C is 21.818% in the former and 29.458% in the latter. The two are significantly different (likelihood ratio chi-square = 4.84, DF = 1,
p = 0.0278). If we narrow the range to sites 48 to 53, then the difference becomes more significant. The nucleotide usage patterns for HEGs and REST genes in
V. cholerae are similar (
Figure 5C,D).
All nine bacterial species exhibited a decreased secondary structure near the translation initiation signals (
Figure 4). However, we did not take into account the effect of the GC%. Increasing the GC% is expected to increase secondary structural stability in mRNA. As we can see from
Table 9,
H. influenzae has the lowest genomic GC% (38.2%), and its mean MFE is the closest to 0 (which means no secondary structure). In contrast,
M. smegmatis and
M. tuberculosis have the highest genomic GC%, and their mRNAs tend to have more negative MFE values (
Figure 4). Accordingly, without controlling for GC%, the observation that the LGB have a stronger secondary structure than the SGB cannot be attributed to a reduced selection in these LGB against a stable secondary structure. Note that the MFE and GC% were calculated for each sequence, and their averages are presented in
Table 9 as the MeanMFE and GC%, respectively.
Given that a weak secondary structure near the SD sequence and the start codon is favorable (
Figure 4), one would predict that SGB should have weaker secondary structures (larger MFE values) than LGB. In order to test this prediction while accommodating the effect of the GC%, we characterized the MFE plots of sliding windows for each species using a single value for each. That is, for each plot in
Figure 4, we calculated the mean value for mid-window sites 46–65 (with the start codon occupying sites 58–60). These sites included both the SD sequence and the start codon. The mean MFE (
Table 9) was now the dependent variable. It was expected to (1) decrease (i.e., more stable secondary structure) with an increasing generation time (RankGT) and increasing GC%, and (2) increase with gene expression (GE, i.e., be greater for HEGs than for REST genes). These three independent variables are also included in
Table 9.
The best model, which accounts for 98.2% of the total variation in MeanMFE in
Table 9, included the three dependent variables and an interaction term (
Table 10). The two-tailed p for RankGT was 0.064 (
Table 10). However, because we had an explicit one-tailed prediction of a negative slope (i.e., MeanMFE should decrease with increasing RankGT), p was half of 0.064, i.e., 0.032. Other regression terms for GC%, GE, and their interaction were highly significant and consistent with the predictions (
Table 10).
As before, we give the two regression equations separately for HEGs and the REST from the regression coefficients in
Table 10:
Equations (8) and (9) show that, for both HEGs and REST genes, secondary structural stability increases with the generation time (MFE becomes more negative with an increasing generation time). This is consistent with our prediction that selection against the secondary structure near the translation start signals (SD sequence and start codon) is stronger in SGB than LGB.
MeanMFE decreases more sharply with the GC% in Equation (9) than in Equation (8), i.e., secondary structural stability increases more rapidly with the GC% for REST genes than for HEGs. This is easy to understand if the selection against secondary structural stability is on average stronger in HEGs than in REST genes. The MeanMFE decreases by only 0.10038 (Equation (8)) for a unit increase in the GC% with the strong selection of HEGs, but decreases by 0.21009 (Equation (9)) for the same unit change in the GC% with the relatively weak selection of REST genes.
The selectionist interpretation above does not consider the effect of mutations, which offers an alternative interpretation. In general, spontaneous mutations in AT-rich genomes tend to be AT-biased, based on (1) comparisons between pseudogenes and their functional counterparts [
139,
140], (2) the mutation patterns of pathogenic bacteria with relaxed selection [
141,
142], and (3) nucleotide bias at the three codon sites across multiple bacterial species [
143].
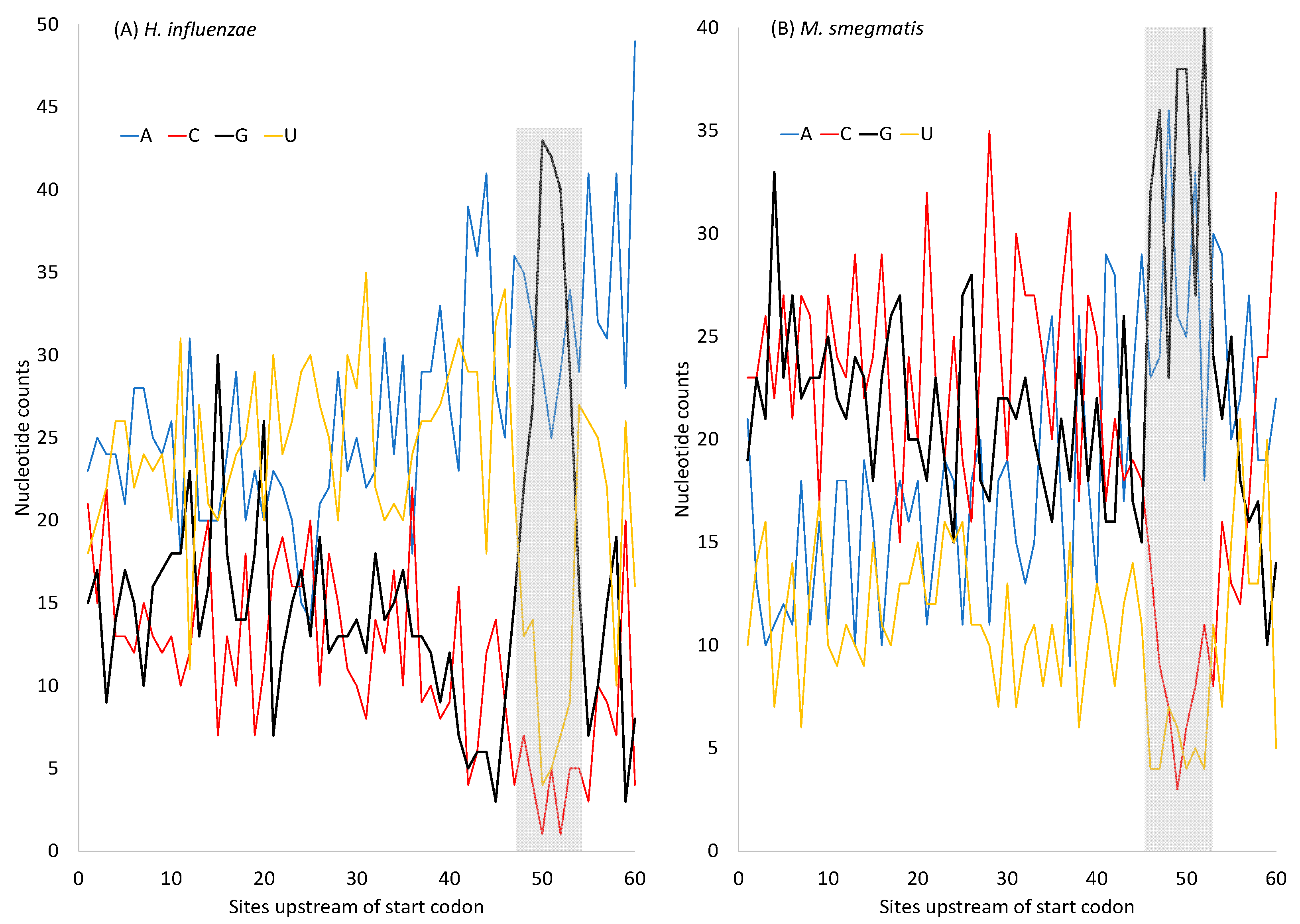
H. influenzae has an AT-rich genome, suggesting AT-biased mutation, in contrast to
M. smegmatis, which has a GC-rich genome. However, protein-coding genes in both species have SD sequences that are purine-rich (especially G-rich) (
Figure 6). The G-rich SD will form base-pairs with nearby C nucleotides, so the MFE will not fall to 0. In pseudogenes where selection for maintaining the G-rich SD sequence is absent, or in low-expressed genes where the selection is weak, the AT-rich
H. influenzae will lose these G nucleotides in the SD sequence, leading to an MFE closer to 0. Indeed, the MeanMFE value for the 19 pseudogenes in
H. influenzae is −2.2887, closer to 0 than all MeanMFE values in
Table 9. Similarly, the MeanMFE is −3.2660 for HEGs and −2.5466 for REST genes (
Table 9). This is consistent with the interpretation that the G-rich SD is more likely to be hit by G→A and G→T mutations and lose G/C base-pairs in REST genes than in HEGs. For example,
H. influenzae has a GC% of 37.9329% for HEGs but only 33.7986% for REST genes, leading to a MeanMFE value closer to 0 in REST genes than in HEGs.
In short, although the purine-rich SD sequences (
Figure 6) can hardly form secondary structures within themselves, the dramatically increased G nucleotides within the SD sequence could base-pair with the neighboring C nucleotides and contribute to secondary structural stability. If there is no selection maintaining the G-richness in the SD sequences, then these G nucleotides may be replaced by A and T, leading to a further decrease in secondary structural stability. Thus, both selection and mutation could contribute to secondary structural stability in sequences near the translation initiation signals (the SD sequence and the start codon). The models in Equations (8) and (9) are, therefore, oversimplified and should be interpreted with caution. The secondary structure in sequences near the stop codon exhibits a similar pattern to those near the start codon (
Figure 6).
The decrease in secondary structural stability may not necessarily be related to the avoidance of embedding SD sequences and start codons. Efficiently translated yeast mRNAs (i.e., mRNAs in polysomes with high ribosome densities) often have a short poly(A) tract before the start codon [
137], with the poly(A) interpreted as long enough to recruit translation initiation factors but short enough to avoid binding by the poly(A)-binding proteins. However, the presence of poly(A) also weakens the secondary structural stability as a secondary consequence.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}