

Metagenomic Analysis for Diagnosis of Hemorrhagic Fever in Minas Gerais, Brazil

 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
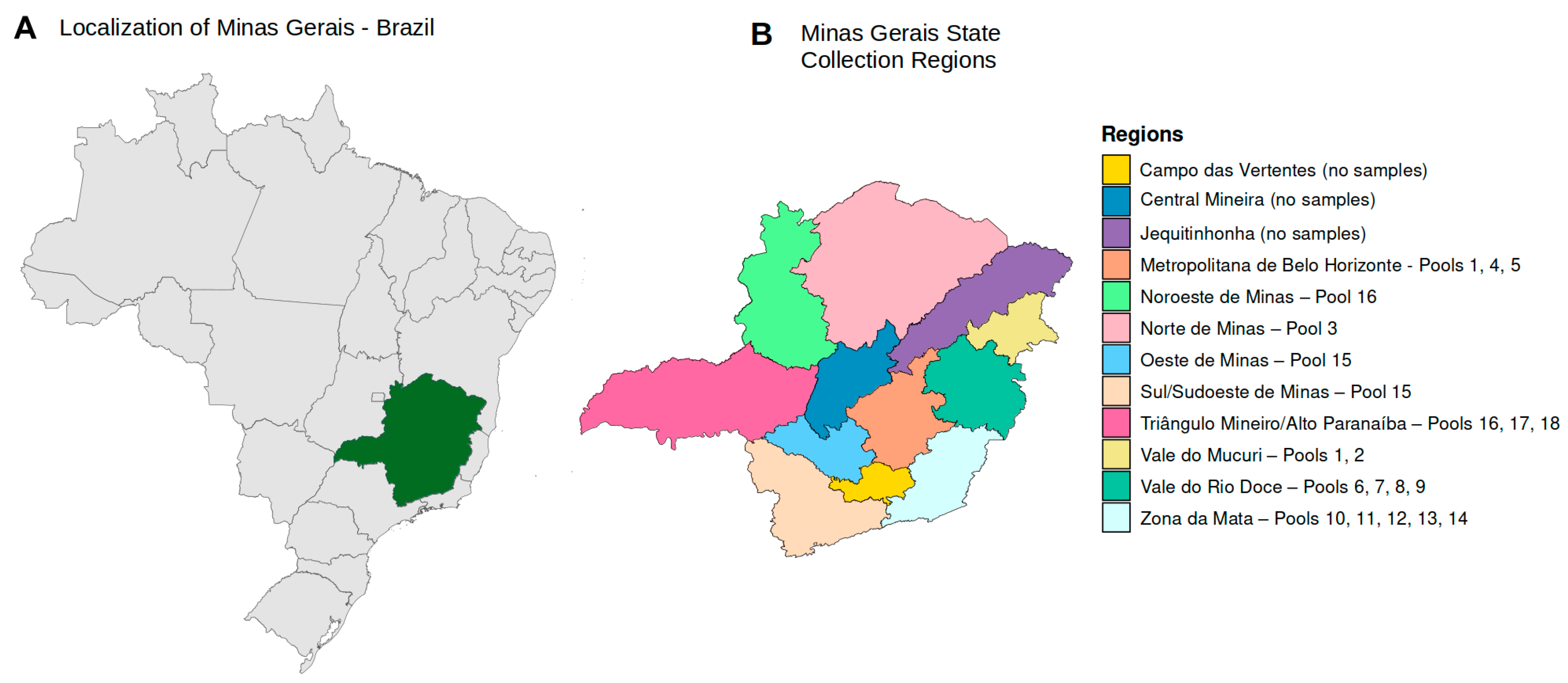
2.1. Clinical Samples
2.2. Nucleic Acids Extraction, Amplification, and Next-Generation Sequencing
2.3. Bioinformatic Pipeline and Analysis
2.4. Viral Confirmation by Molecular Methods
2.5. Genomic Reconstruction
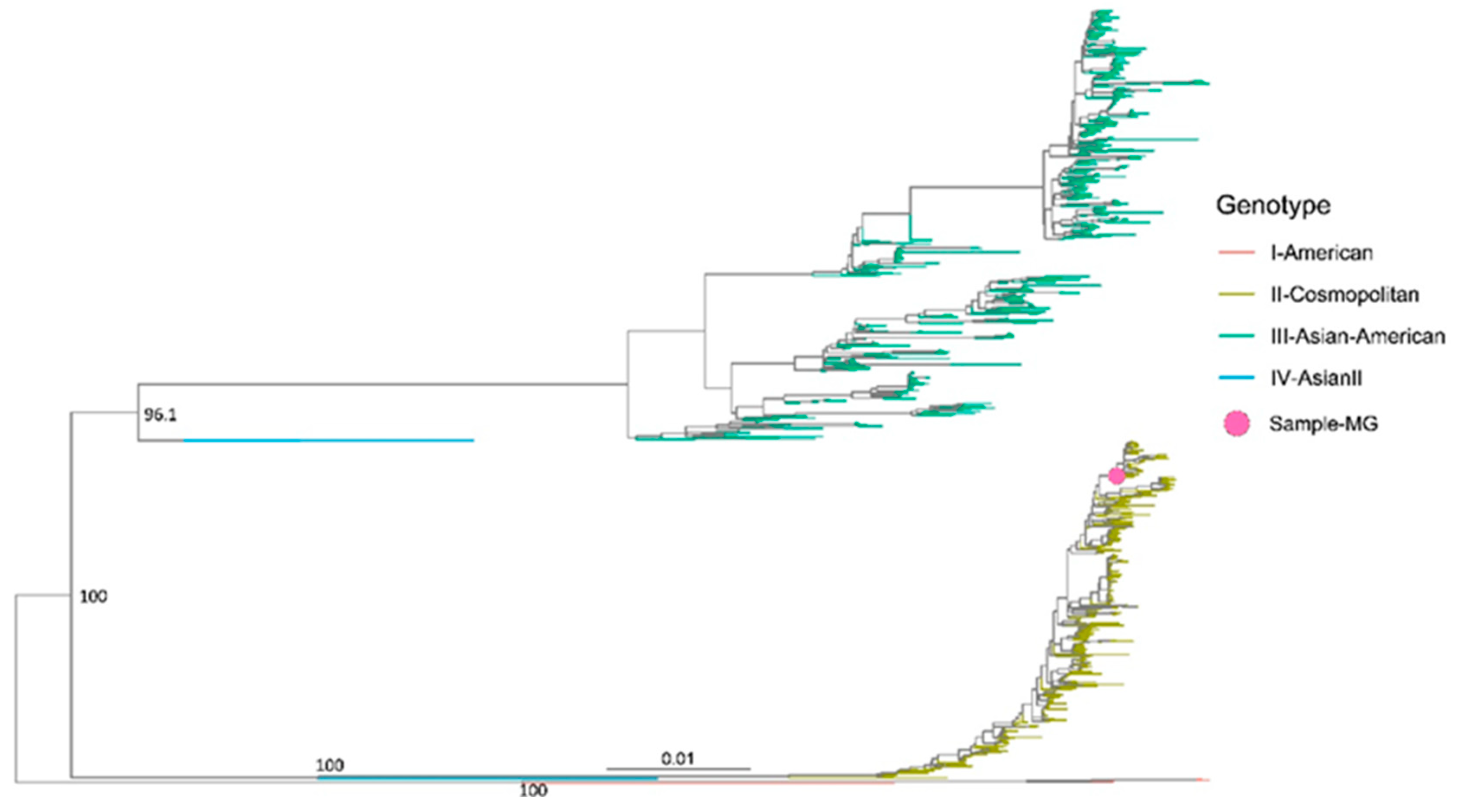
2.6. Phylogenetic Analysis
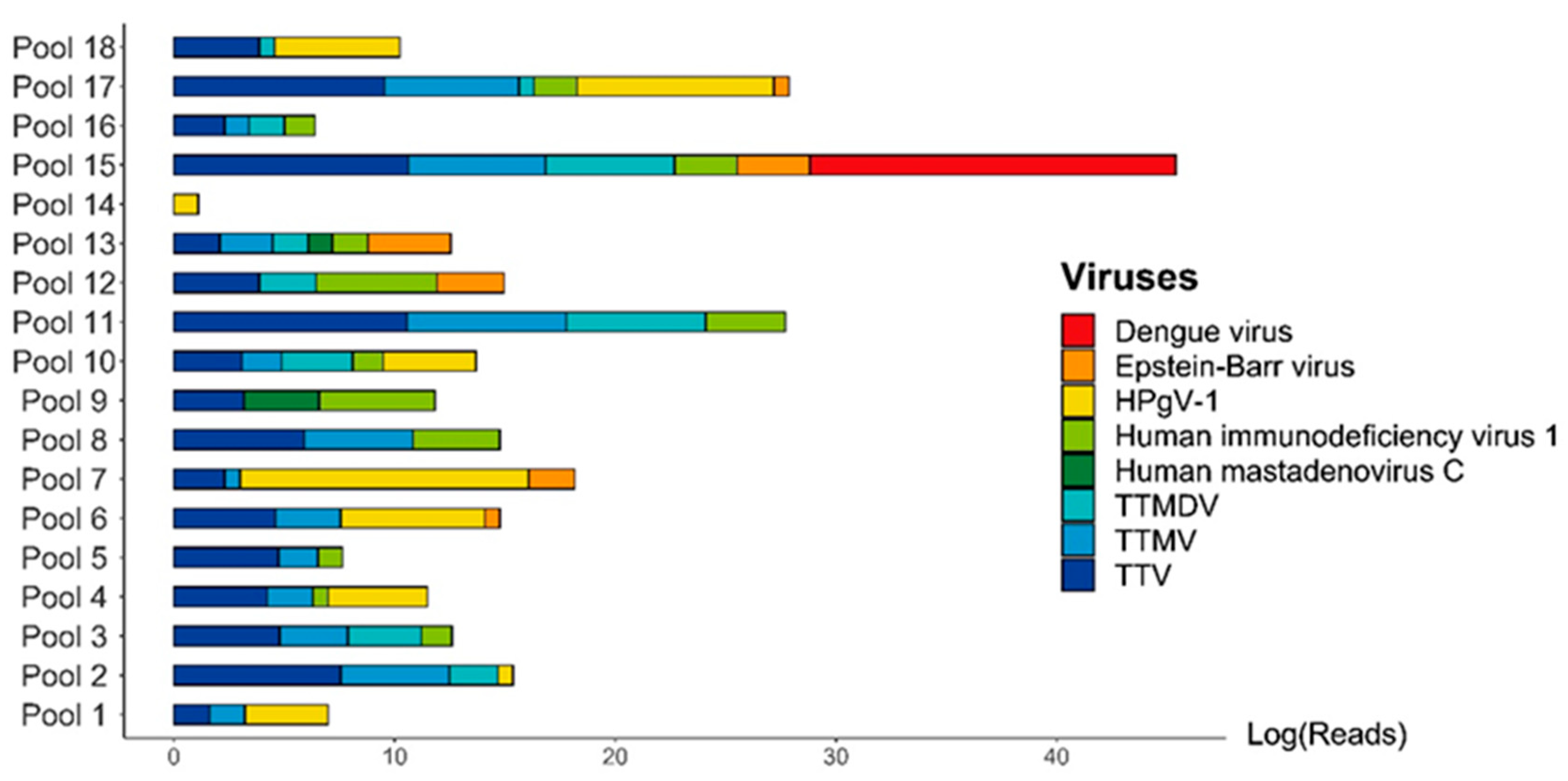
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Figueiredo, L.T. Febres hemorrágicas por vírus no Brasil [Viral hemorrhagic fevers in Brazil]. Rev. Soc. Bras. Med. Trop. 2006, 39, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Flórez-Álvarez, L.; de Souza, E.E.; Botosso, V.F.; de Oliveira, D.B.L.; Ho, P.L.; Taborda, C.P.; Palmisano, G.; Capurro, M.L.; Pinho, J.R.R.; Ferreira, H.L.; et al. Hemorrhagic fever iroses: Pathogenesis, therapeutics, and emerging and re-emerging potential. Front. Microbiol. 2022, 13, 1040093. [Google Scholar] [CrossRef] [PubMed]
- Araújo, L.J.T.; Gonzalez, L.L.; Buss, L.F.; Guerra, J.M.; Gomez, D.S.; Ferreira, C.S.D.S.; Cirqueira, C.S.; Ghillardi, F.; Witkin, S.S.; Sabino, E.C. Surveillance of hemorrhagic fever and/or neuroinvasive disease: Challenges of diagnosis. Rev. Saude Publica 2021, 55, 41. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fan, L.C.; Chai, Y.H.; Xu, J.F. Advantages and challenges of metagenomic sequencing for the diagnosis of pulmonary infectious diseases. Clin. Respir. J. 2022, 16, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Chiu, C. The Role of Metagenomics and Next-Generation Sequencing in Infectious Disease Diagnosis. Clin. Chem. 2021, 68, 115–124. [Google Scholar] [CrossRef]
- Domingo, C.; Patel, P.; Yillah, J.; Weidmann, M.; Méndez, J.A.; Nakouné, E.R.; Niedrig, M. Advanced yellow fever virus ge-nome detection in point-of-care facilities and reference laboratories. J. Clin. Microbiol. 2012, 50, 4054–4060. [Google Scholar] [CrossRef] [PubMed]
- Labruna, M.B.; Whitworth, T.; Horta, M.C.; Bouyer, D.H.; McBride, J.W.; Pinter, A.; Popov, V.; Gennari, S.M.; Walker, D.H. Rickettsia species infecting Amblyomma cooperi ticks from an area in the state of São Paulo, Brazil, where Brazilian spotted fever is endemic. J. Clin. Microbiol. 2004, 42, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, R.A.; Gee, J.E.; Wilkins, P.P.; McCaustland, K.; Hoffmaster, A.R. Detection of pathogenic Leptospira spp. through TaqMan polymerase chain reaction targeting the LipL32 gene. Diagn. Microbiol. Infect. Dis. 2009, 64, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Bourhy, P.; Bremont, S.; Zinini, F.; Giry, C.; Picardeau, M. Comparison of real-time PCR assays for detection of pathogenic Leptospira spp. in blood and identification of variations in target sequences. J. Clin. Microbiol. 2011, 49, 2154–2160. [Google Scholar] [CrossRef]
- FastQC a Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 5 February 2024).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- R: The R Project for Statistical Computing. Available online: https://www.R-project.org (accessed on 15 February 2024).
- Rstudio Team. Rstudio: Integrated Development for R; Rstudio, PBC: Boston, MA, USA, 2024; Available online: http://www.rstudio.com (accessed on 15 February 2024).
- Wickham, H.; Hester, J.; Bryan, J. Readr: Read Rectangular Text Data. 2024. Available online: https://readr.tidyverse.org (accessed on 16 February 2024).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the tidyverse. J. Open-Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org/ (accessed on 16 February 2024).
- Xiao, N. ggsci: Scientific Journal and Sci-Fi Themed Color Palettes for ‘ggplot2′. 2024. Available online: https://github.com/nanxstats/ggsci (accessed on 16 February 2024).
- Huhtamo, E.; Hasu, E.; Uzcátegui, N.Y.; Erra, E.; Nikkari, S.; Kantele, A.; Vapalahti, O.; Piiparinen, H. Early diagnosis of dengue in travelers: Comparison of a novel real-time RT-PCR, NS1 antigen detection and serology. J. Clin. Virol. 2010, 47, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.W.; Russell, B.J.; Lanciotti, R.S. Serotype-specific detection of dengue viruses in a fourplex real-time reverse transcriptase PCR assay. J. Clin. Microbiol. 2005, 43, 4977–4983. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Rincon, N.; Wood, D.E.; Breitwieser, F.P.; Pockrandt, C.; Langmead, B.; Salzberg, S.L.; Steinegger, M. Metagenome analysis using the Kraken software suite. Nat. Protoc. 2022, 17, 2815–2839. [Google Scholar] [CrossRef]
- Moshiri, N. ViralMSA: Massively scalable reference-guided multiple sequence alignment of viral genomes. Bioinformatics 2021, 37, 714–716. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup, The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.; Wong, T.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Singhi, S.; Kissoon, N.; Bansal, A. Dengue and dengue hemorrhagic fever: Management issues in an intensive care unit. J. Pediatr. (Rio J.) 2007, 83, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.H.; Zhang, S.L.; Tan, H.C.; Kwek, S.S.; Sessions, O.M.; Chan, C.Y.; Liu, I.D.; Lee, C.K.; Tambyah, P.A.; Ooi, E.E.; et al. Persistent Dengue Infection in an Immunosuppressed Patient Reveals the Roles of Humoral and Cellular Immune Responses in Virus Clearance. Cell Host Microbe 2019, 26, 601–605.e3. [Google Scholar] [CrossRef] [PubMed]
- Rowe, E.K.; Leo, Y.S.; Wong, J.G.; Thein, T.L.; Gan, V.C.; Lee, L.K.; Lye, D.C. Challenges in dengue fever in the elderly: Atypical presentation and risk of severe dengue and hospital-acquired infection. PLoS Neglected Trop. Dis. 2014, 8, 2777, Correction in: Challenges in Dengue Fever in the Elderly: Atypical Presentation and Risk of Severe Dengue and Hospita-Acquired Infection. PLoS Neglected Trop. Dis. 2014, 8, e2886. [Google Scholar] [CrossRef]
- Suppiah, J.; Ching, S.M.; Amin-Nordin, S.; Mat-Nor, L.A.; Ahmad-Najimudin, N.A.; Low, G.K.; Abdul-Wahid, M.Z.; Thayan, R.; Chee, H.Y. Clinical manifestations of dengue in relation to dengue serotype and genotype in Malaysia: A retrospective observational study. PLoS Neglected Trop. Dis. 2018, 12, e0006817. [Google Scholar] [CrossRef]
- Giovanetti, M.; Pereira, L.A.; Santiago, G.A.; Fonseca, V.; Mendoza, M.P.G.; de Oliveira, C.; de Moraes, L.; Xavier, J.; Tosta, S.; Fristch, H.; et al. Emergence of Dengue Virus Serotype 2 Cosmopolitan Genotype, Brazil. Emerg. Infect. Dis. 2022, 28, 1725–1727. [Google Scholar] [CrossRef]
- Pallen, M.J. Diagnostic metagenomics: Potential applications to bacterial, viral and parasitic infections. Parasitology 2014, 141, 1856–1862. [Google Scholar] [CrossRef] [PubMed]
- Carr, V.R.; Chaguza, C. Metagenomics for surveillance of respiratory pathogens. Nat. Rev. Microbiol. 2021, 19, 285. [Google Scholar] [CrossRef] [PubMed]
- Biggs, H.M.; Behravesh, C.B.; Bradley, K.K.; Dahlgren, F.S.; Drexler, N.A.; Dumler, J.S.; Folk, S.M.; Kato, C.Y.; Lash, R.R.; Levin, M.L.; et al. Diagnosis and Management of Tickborne Rickettsial Diseases: Rocky Mountain Spotted Fever and Other Spotted Fever Group Rickettsioses, Ehrlichioses, and Anaplasmosis. MMWR Recomm. Rep. 2016, 65, 1–44. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Pool Number | Total Number of Reads (Millions) | Number of Reads after Filtering and Trimming (Millions) | Unmapped Reads (Millions) | Viral Reads |
|---|---|---|---|---|
| 1 | 103.010 | 84.575 | 0.183 | 56 (0.03%) |
| 2 | 121.620 | 104.490 | 1.440 | 8275 (0.57%) |
| 3 | 130.640 | 117.370 | 15.600 | 602 (0.003%) |
| 4 | 129.870 | 107.480 | 1.070 | 15,353 (1.44%) |
| 5 | 116.220 | 94.794 | 1.270 | 25,457 (2.00%) |
| 6 | 167.540 | 143.230 | 1.160 | 892 (0.08%) |
| 7 | 176.370 | 154.430 | 1.720 | 485,921 (28.18%) |
| 8 | 103.140 | 90.939 | 21.100 | 20,548 (0.10%) |
| 9 | 129.920 | 113.770 | 9.105 | 758 (0.01%) |
| 10 | 119.890 | 101.820 | 3.400 | 354 (0.01%) |
| 11 | 113.000 | 95.453 | 1.960 | 46,446 (2.37%) |
| 12 | 124.890 | 104.510 | 1.240 | 5930 (0.48%) |
| 13 | 129.450 | 109.390 | 1.060 | 1130 (0.11%) |
| 14 | 146.160 | 120.320 | 0.813 | 128 (0.02%) |
| 15 | 114.320 | 97.523 | 16.600 | 15,844,322 (95.72%) |
| 16 | 51.271 | 44.317 | 0.731 | 96 (0.01%) |
| 17 | 171.710 | 143.690 | 2.340 | 22,799 (0.97%) |
| 18 | 126.580 | 100.410 | 0.925 | 7782 (0.84%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iani, F.C.d.M.; de Campos, G.M.; Adelino, T.E.R.; da Silva, A.S.; Kashima, S.; Alcantara, L.C.J.; Sampaio, S.C.; Giovanetti, M.; Elias, M.C.; Slavov, S.N. Metagenomic Analysis for Diagnosis of Hemorrhagic Fever in Minas Gerais, Brazil. Microorganisms 2024, 12, 769. https://doi.org/10.3390/microorganisms12040769
Iani FCdM, de Campos GM, Adelino TER, da Silva AS, Kashima S, Alcantara LCJ, Sampaio SC, Giovanetti M, Elias MC, Slavov SN. Metagenomic Analysis for Diagnosis of Hemorrhagic Fever in Minas Gerais, Brazil. Microorganisms. 2024; 12(4):769. https://doi.org/10.3390/microorganisms12040769
Chicago/Turabian StyleIani, Felipe Campos de Melo, Gabriel Montenegro de Campos, Talita Emile Ribeiro Adelino, Anielly Sarana da Silva, Simone Kashima, Luiz Carlos Junior Alcantara, Sandra Coccuzzo Sampaio, Marta Giovanetti, Maria Carolina Elias, and Svetoslav Nanev Slavov. 2024. "Metagenomic Analysis for Diagnosis of Hemorrhagic Fever in Minas Gerais, Brazil" Microorganisms 12, no. 4: 769. https://doi.org/10.3390/microorganisms12040769
APA StyleIani, F. C. d. M., de Campos, G. M., Adelino, T. E. R., da Silva, A. S., Kashima, S., Alcantara, L. C. J., Sampaio, S. C., Giovanetti, M., Elias, M. C., & Slavov, S. N. (2024). Metagenomic Analysis for Diagnosis of Hemorrhagic Fever in Minas Gerais, Brazil. Microorganisms, 12(4), 769. https://doi.org/10.3390/microorganisms12040769