
Bacterial and Archaeal Communities in Erhai Lake Sediments: Abundance and Metabolic Insight into a Plateau Lake at the Edge of Eutrophication



Abstract
1. Introduction
2. Materials and Methods
2.1. Site Description and Sample Collection
2.2. Environmental Factors Analysis
2.3. DNA Extraction and PCR Amplification
2.4. Sequence Analysis
2.5. Definition of Abundant and Rare Taxa
2.6. Co-Occurrence Network Analyses and Keystone Species Definition
2.7. Other Statistical Analyses
3. Results
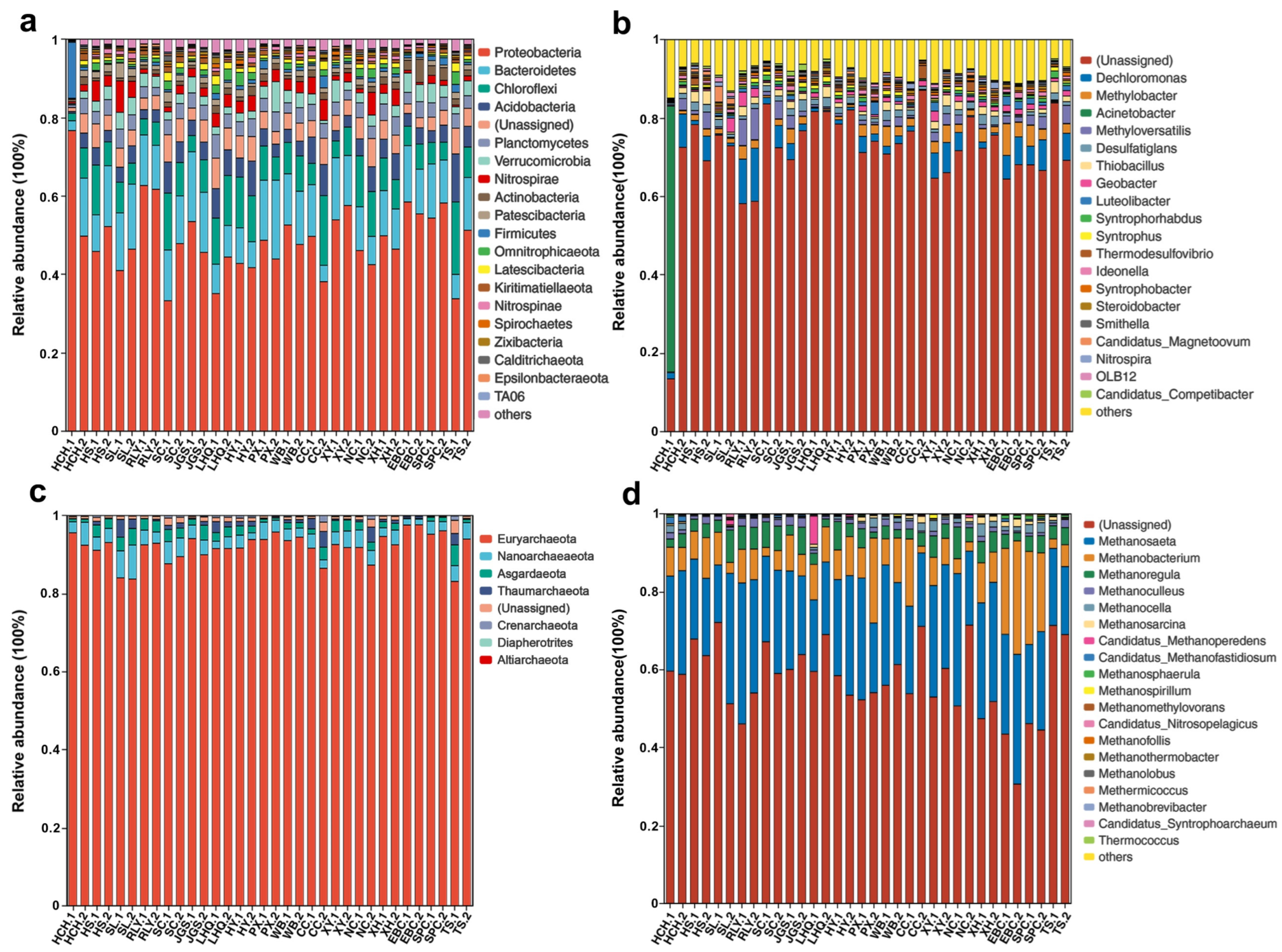
3.1. Overall Microbial Community Compositions and Diversity
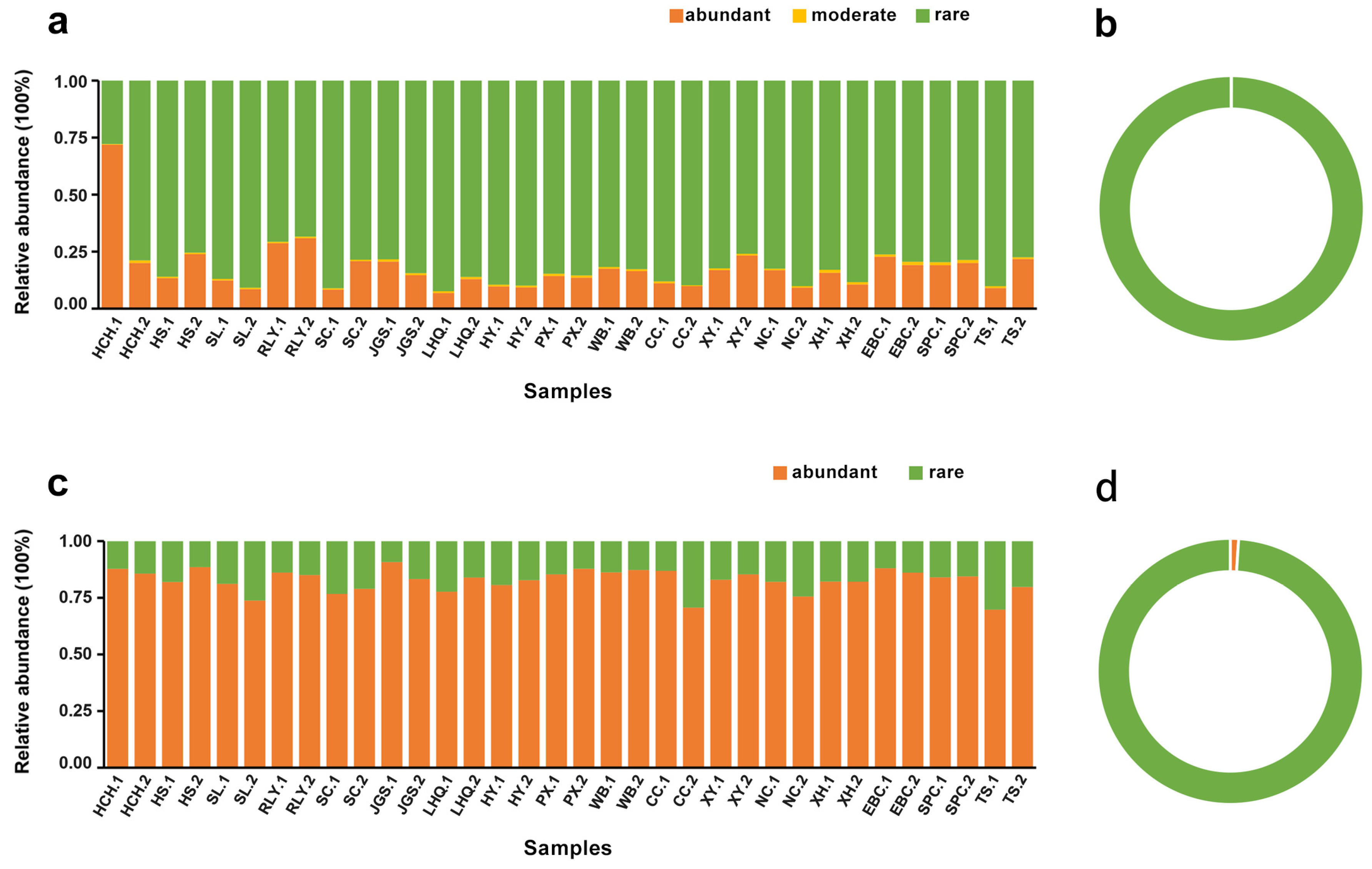
3.2. Identification of Abundant and Rare Sub-Communities
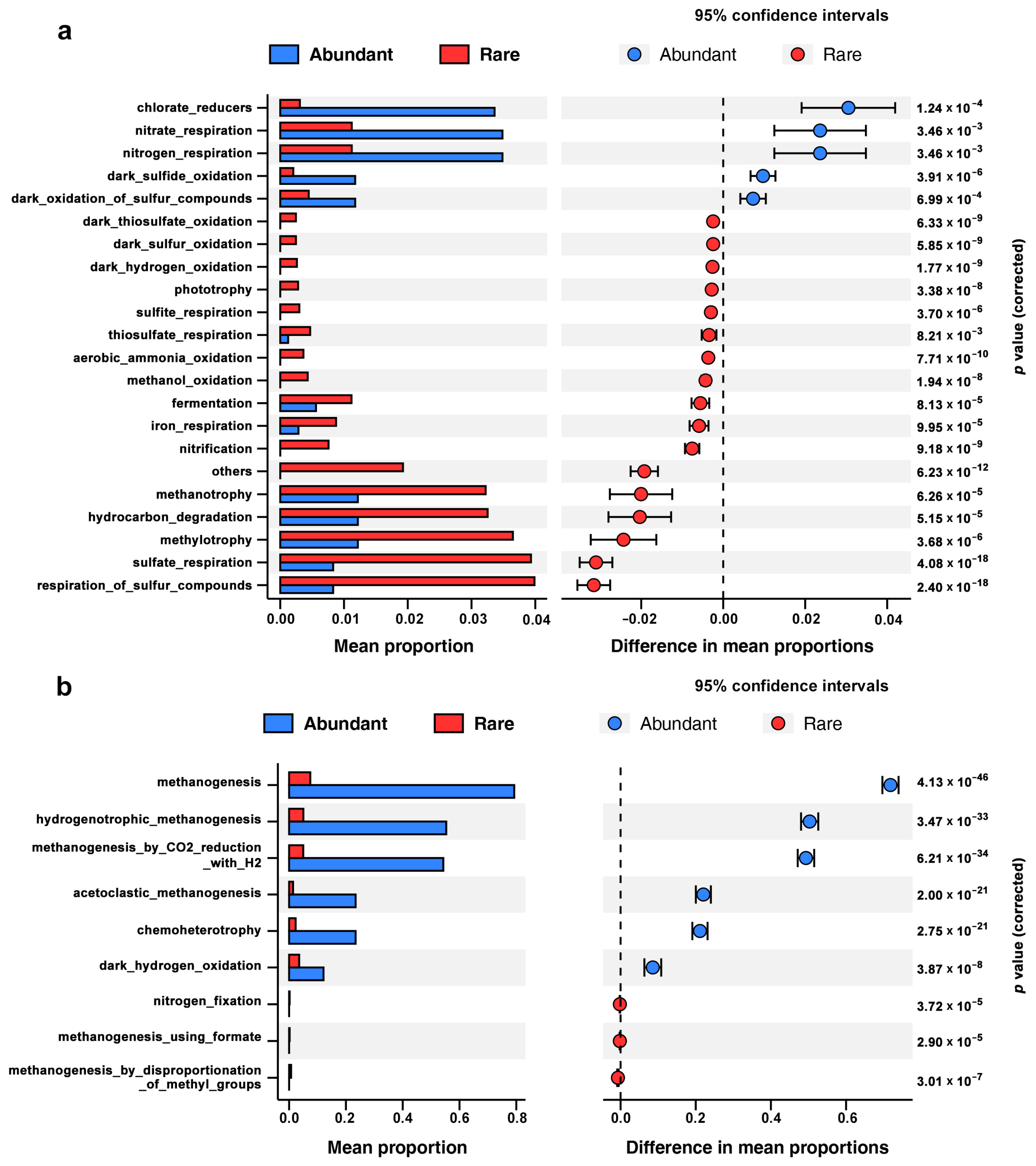
3.3. Metabolic Potential of Abundant and Rare Sub-Communities
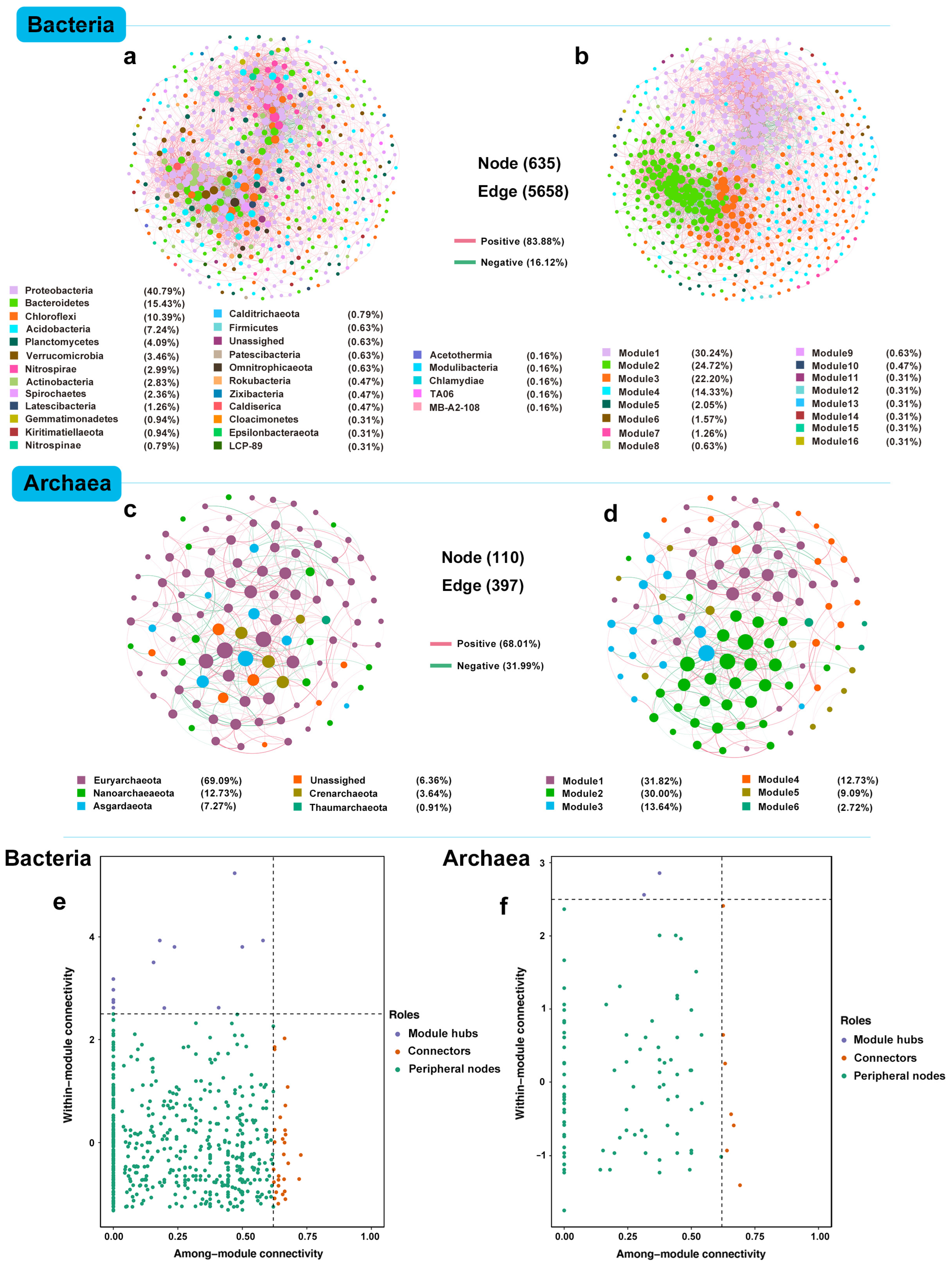
3.4. Overall Microbial Network Co-Existence Patterns
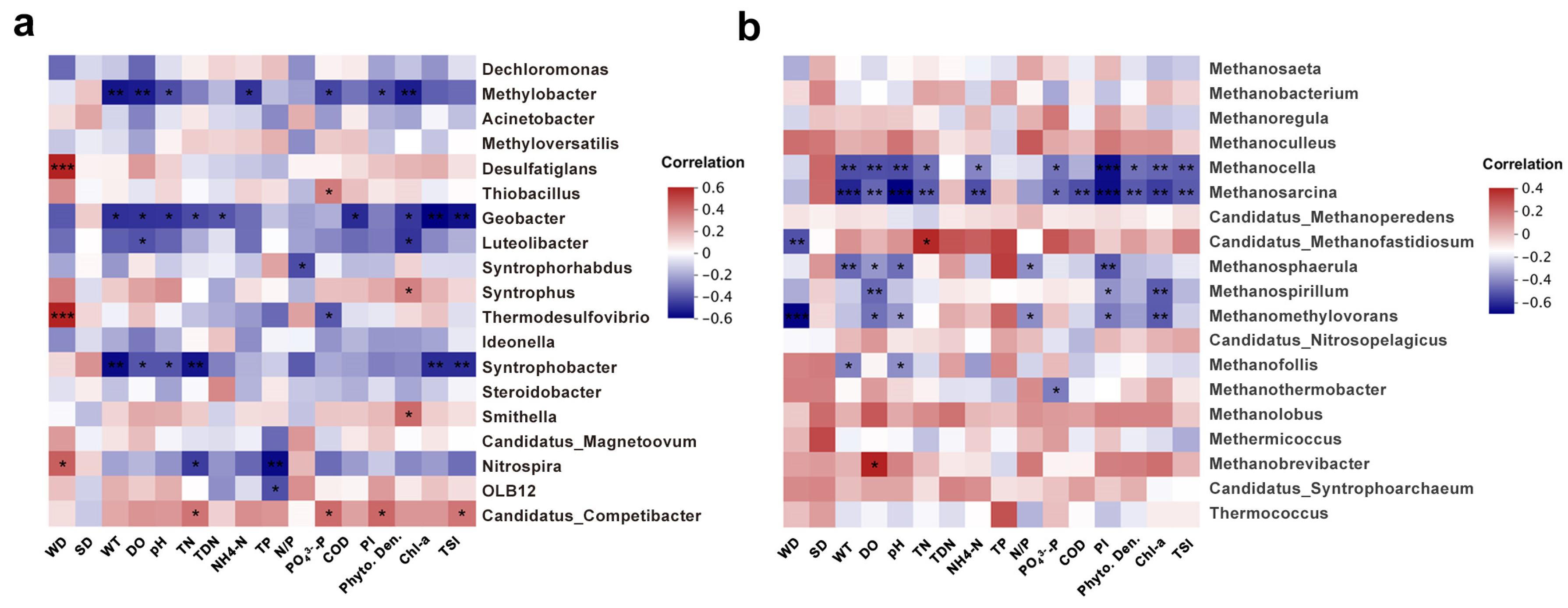
3.5. Correlation between Microbial Community and Environmental Factors
4. Discussion
4.1. Characteristics of the Overall Microbial Community
4.2. Rare and Abundant Taxa Dominated Co-Occurrence Networks
4.3. Rare and Abundant Taxa Play Different Potential Functions
4.4. Effects of Environmental Factors and Potential Microbial Indicators of Lake Eutrophication
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alexander, T.J.; Vonlanthen, P.; Seehausen, O. Does eutrophication-driven evolution change aquatic ecosystems? Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160041. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.Q.; Bachand, S.C.; McIntyre, P.B.; Kraemer, B.M.; Vadeboncoeur, Y.; Kimirei, I.A.; Tamatamah, R.; McMahon, K.D.; Anantharaman, K. Depth-discrete metagenomics reveals the roles of microbes in biogeochemical cycling in the tropical freshwater Lake Tanganyika. ISME J. 2021, 15, 1971–1986. [Google Scholar] [CrossRef] [PubMed]
- Nowlin, W.H.; Evarts, J.L.; Vanni, M.J. Release rates and potential fates of nitrogen and phosphorus from sediments in a eutrophic reservoir. Freshw. Biol. 2005, 50, 301–322. [Google Scholar] [CrossRef]
- Huang, W.; Dong, X.; Tu, C.; Yang, H.; Chang, Y.; Yang, X.; Chen, H.; Che, F. Response mechanism of sediment endogenous phosphorus release to functional microorganisms and its cyanobacterial growth and disappearance effects. Sci. Total Environ. 2024, 906, 167676. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, L.D.; de Faria, D.M.; Crossetti, L.O.; Marques, D.D. Phytoplankton, periphyton, and zooplankton patterns in the pelagic and littoral regions of a large subtropical shallow lake. Hydrobiologia 2019, 831, 119–132. [Google Scholar] [CrossRef]
- Wang, H.J.; Lu, J.W.; Wang, W.D.; Yang, L.Y.; Yin, C.Q. Methane fluxes from the littoral zone of hypereutrophic Taihu Lake, China. J. Geophys. Res.-Atmos. 2006, 111, D17109. [Google Scholar] [CrossRef]
- Sala, M.M.; Güde, H. Seasonal dynamics of pelagic and benthic (littoral and profundal) bacterial abundances and activities in a deep prealpine lake (L. Constance). Arch. Fur Hydrobiol. 2006, 167, 351–369. [Google Scholar] [CrossRef]
- Falkowski, P.G.; Fenchel, T.; Delong, E.F. The microbial engines that drive Earth’s biogeochemical cycles. Science 2008, 320, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Paerl, H.W.; Pinckney, J.L. A mini-review of microbial consortia: Their roles in aquatic production and biogeochemical cycling. Microb. Ecol. 1996, 31, 225–247. [Google Scholar] [CrossRef]
- Fuhrman, J.A. Microbial community structure and its functional implications. Nature 2009, 459, 193–199. [Google Scholar] [CrossRef]
- Chen, J.; Wu, J.L.; Liu, M.; Li, L.Q.; Zhang, W.J.; Wang, D.S.; Ma, T. Bacterial community structure in the surface sediments of different habitats of Baiyangdian Lake, Northern China: Effects of nutrient conditions. J. Soils Sediments 2021, 21, 1866–1874. [Google Scholar] [CrossRef]
- Fagervold, S.K.; Bourgeois, S.; Pruski, A.M.; Charles, F.; Kerhervé, P.; Vétion, G.; Galand, P.E. River organic matter shapes microbial communities in the sediment of the Rhone prodelta. ISME J. 2014, 8, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Liang, J.C.; Ma, Y.Q.; Zhu, L.; Liu, Y. Bacterial community and eutrophic index analysis of the East Lake. Environ. Pollut. 2019, 252, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Pearman, J.K.; Wood, S.A.; Vandergoes, M.J.; Atalah, J.; Waters, S.; Adamson, J.; Thomson-Laing, G.; Thompson, L.; Howarth, J.D.; Hamilton, D.P.; et al. A bacterial index to estimate lake trophic level: National scale validation. Sci. Total Environ. 2022, 812, 152385. [Google Scholar] [CrossRef] [PubMed]
- Galand, P.E.; Casamayor, E.O.; Kirchman, D.L.; Lovejoy, C. Ecology of the rare microbial biosphere of the Arctic Ocean. Proc. Natl. Acad. Sci. USA 2009, 106, 22427–22432. [Google Scholar] [CrossRef] [PubMed]
- Jousset, A.; Bienhold, C.; Chatzinotas, A.; Gallien, L.; Gobet, A.; Kurm, V.; Küsel, K.; Rillig, M.C.; Rivett, D.W.; Salles, J.F.; et al. Where less may be more: How the rare biosphere pulls ecosystems strings. ISME J. 2017, 11, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Pedrós-Alió, C. The rare bacterial biosphere. Annu. Rev. Mar. Sci. 2012, 4, 449–466. [Google Scholar] [CrossRef] [PubMed]
- He, Z.B.; Liu, D.; Shi, Y.; Wu, X.J.; Dai, Y.X.; Shang, Y.W.; Peng, J.J.; Cui, Z.L. Broader environmental adaptation of rare rather than abundant bacteria in reforestation succession soil. Sci. Total Environ. 2022, 828, 154364. [Google Scholar] [CrossRef] [PubMed]
- Sauret, C.; Séverin, T.; Vétion, G.; Guigue, C.; Goutx, M.; Pujo-Pay, M.; Conan, P.; Fagervold, S.K.; Ghiglione, J.-F. ‘Rare biosphere’ bacteria as key phenanthrene degraders in coastal seawaters. Environ. Pollut. 2014, 194, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.Q.; Cao, X.F.; Wang, J.; Zhao, L.; Sun, J.H.; Jiang, D.L.; Huang, Y. Similar community assembly mechanisms underlie similar biogeography of rare and abundant bacteria in lakes on Yungui Plateau, China. Limnol. Oceanogr. 2017, 62, 723–735. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Wu, G.; Jiang, H.C.; Yang, J.; She, W.Y.; Khan, I.; Li, W.J. Abundant and rare microbial miospheres respond differently to environmental and spatial factors in Tibetan hot springs. Front. Microbiol. 2018, 9, 345048. [Google Scholar] [CrossRef]
- Gong, X.Z.; Chen, Z.Y.; Deng, Y.; Zhao, D.; Gao, P.; Zhang, L.; Tu, Q.C.; Qu, L.Y.; Zheng, L.W.; Zhang, Y.; et al. Contrasting archaeal and bacterial community assembly processes and the importance of rare taxa along a depth gradient in shallow coastal sediments. Sci. Total Environ. 2022, 852, 158411. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Wang, J.M.; Wei, G.H.; Chen, W.M.; Lu, Y.H. Dominant role of abundant rather than rare bacterial taxa in maintaining agro-soil microbiomes under environmental disturbances. Chemosphere 2019, 235, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Brearley, F.Q.; Huang, L.; Tang, J.; Xu, Q.; Li, X.; Huang, Y.; Zou, S.; Chen, X.; Hou, W. Abundant and rare taxa of planktonic fungal community exhibit distinct assembly patterns along coastal eutrophication gradient. Microb. Ecol. 2023, 85, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Geng, M.; Zhang, W.; Hu, T.; Wang, R.; Cheng, X.; Wang, J. Eutrophication causes microbial community homogenization via modulating generalist species. Water Res. 2022, 210, 118003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.Q.; Duan, S.Q. Lakes as sentinels of climate change on the Tibetan Plateau. All Earth 2021, 33, 161–165. [Google Scholar] [CrossRef]
- Ni, Z.K.; Wang, S.R. Historical accumulation and environmental risk of nitrogen and phosphorus in sediments of Erhai Lake, Southwest China. Ecol. Eng. 2015, 79, 42–53. [Google Scholar] [CrossRef]
- Pan, X.; Lin, L.; Huang, Z.; Liu, M.; Dong, L.; Chen, J.; Crittenden, J. Distribution characteristics and pollution risk evaluation of the nitrogen and phosphorus species in the sediments of Lake Erhai, Southwest China. Environ. Sci. Pollut. Res. 2019, 26, 22295–22304. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Yang, Y.Y.; Wu, Z.; Feng, Q.Y.; Xie, S.G.; Liu, Y. Spatiotemporal variation of planktonic and sediment bacterial assemblages in two plateau freshwater lakes at different trophic status. Appl. Microbiol. Biotechnol. 2016, 100, 4161–4175. [Google Scholar] [CrossRef]
- Xue, Y.Y.; Chen, H.H.; Yang, J.R.; Liu, M.; Huang, B.Q.; Yang, J. Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. ISME J. 2018, 12, 2263–2277. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. In Proceedings of the International AAAI Conference on Web and Social Media, San Jose, CA, USA, 17–20 May 2009; pp. 361–362. [Google Scholar]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.D.J.; Neufeld, J.D. Ecology and exploration of the rare biosphere. Nat. Rev. Microbiol. 2015, 13, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yao, P.; Sun, C.; Li, S.; Shi, X.; Zhang, X.H.; Liu, J. Vertical diversity and association pattern of total, abundant and rare microbial communities in deep-sea sediments. Mol. Ecol. 2021, 30, 2800–2816. [Google Scholar] [CrossRef] [PubMed]
- Kuang, B.; Xiao, R.; Wang, C.; Zhang, L.; Wei, Z.; Bai, J.; Zhang, K.; Campos, M.; Jorquera, M.A. Bacterial community assembly in surface sediments of a eutrophic shallow lake in northern China. Ecohydrol. Hydrobiol. 2022. [Google Scholar] [CrossRef]
- Jung, J.; Park, W. species as model microorganisms in environmental microbiology: Current state and perspectives. Appl. Microbiol. Biotechnol. 2015, 99, 2533–2548. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Yang, Y.Y.; Zhao, L.; Li, Y.Z.; Xie, S.G.; Liu, Y. Distribution of sediment bacterial and archaeal communities in plateau freshwater lakes. Appl. Microbiol. Biotechnol. 2015, 99, 3291–3302. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, S.; Méndez, V.; Aguila, P.; Seeger, M. Bioremediation of petroleum hydrocarbons: Catabolic genes, microbial communities, and applications. Appl. Microbiol. Biotechnol. 2014, 98, 4781–4794. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.C.; Adorno, M.A.T.; Okada, D.Y.; Delforno, T.P.; Gomes, P.C.F.L.; Sakamoto, I.K.; Varesche, M.B.A. Analysis of a microbial community associated with polychlorinated biphenyl degradation in anaerobic batch reactors. Biodegradation 2014, 25, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Cupples, A.M. RDX degrading microbial communities and the prediction of microorganisms responsible for RDX bioremediation. Int. Biodeterior. Biodegrad. 2013, 85, 260–270. [Google Scholar] [CrossRef]
- Li, J.F.; Zhang, J.Y.; Liu, L.Y.; Fan, Y.C.; Li, L.S.; Yang, Y.F.; Lu, Z.H.; Zhang, X.G. Annual periodicity in planktonic bacterial and archaeal community composition of eutrophic Lake Taihu. Sci. Rep. 2015, 5, 15488. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Chen, J.F.; Chen, X.L.; Jiang, Q.S.; Liu, Y.; Xie, S.G. Cyanobacterial bloom induces structural and functional succession of microbial communities in eutrophic lake sediments. Environ. Pollut. 2021, 284, 117157. [Google Scholar] [CrossRef] [PubMed]
- Haller, L.; Tonolla, M.; Zopfi, J.; Peduzzi, R.; Wildi, W.; Poté, J. Composition of bacterial and archaeal communities in freshwater sediments with different contamination levels (Lake Geneva, Switzerland). Water Res. 2011, 45, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Alegría-Gómez, J.; Castañón-González, J.H.; Hernández-García, J.A.; González-Terreros, E.; Velázquez-Ríos, I.O.; Ruíz-Valdiviezo, V.M. Changes in the abundance and diversity of bacterial and archaeal communities at different depths in a eutrophic freshwater lake in southwestern Mexico. Environ. Sci. Pollut. Res. 2023, 30, 98362–98376. [Google Scholar] [CrossRef] [PubMed]
- Xing, P.; Tao, Y.; Luo, J.; Wang, L.; Li, B.; Li, H.; Wu, Q.L. Stratification of microbiomes during the holomictic period of Lake Fuxian, an alpine monomictic lake. Limnol. Oceanogr. 2019, 65, S134–S148. [Google Scholar] [CrossRef]
- Offre, P.; Spang, A.; Schleper, C. Archaea in biogeochemical cycles. Annu. Rev. Microbiol. 2013, 67, 437–457. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, D.Y.; Makarova, K.S.; Abbas, B.; Ferrer, M.; Golyshin, P.N.; Galinski, E.A.; Ciordia, S.; Mena, M.C.; Merkel, A.Y.; Wolf, Y.I.; et al. Discovery of extremely halophilic, methyl-reducing euryarchaea provides insights into the evolutionary origin of methanogenesis. Nat. Microbiol. 2017, 2, 17081. [Google Scholar] [CrossRef] [PubMed]
- Conrad, R.; Klose, M.; Enrich-Prast, A. Acetate turnover and methanogenic pathways in Amazonian lake sediments. Biogeosciences 2020, 17, 1063–1069. [Google Scholar] [CrossRef]
- Coyte, K.Z.; Schluter, J.; Foster, K.R. The ecology of the microbiome: Networks, competition, and stability. Science 2015, 350, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Dini-Andreote, F.; Stegen, J.C.; van Elsas, J.D.; Salles, J.F. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc. Natl. Acad. Sci. USA 2015, 112, E1326–E1332. [Google Scholar] [CrossRef]
- Wang, J.L.; Wang, C.J.; Hu, M.; Bian, L.H.; Qu, L.N.; Sun, H.M.; Wu, X.F.; Ren, G.L. Bacterial co-occurrence patterns are more complex but less stable than archaea in enhanced oil recovery applied oil reservoirs. Process Biochem. 2023, 130, 40–49. [Google Scholar] [CrossRef]
- Hou, D.W.; Zhou, R.J.; Wei, D.D.; Zeng, S.Z.; Weng, S.P.; Yan, Q.Y.; He, J.G.; Huang, Z.J. Abundant and rare microbial communities respectively contribute to an aquaculture pond ecosystem. Front. Mar. Sci. 2022, 9, 856126. [Google Scholar] [CrossRef]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-González, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.H.; Dong, P.C.; Zhao, T.; Deng, Y.T.; Li, J.; Song, L.R.; Wang, J.N.; Zhou, L.; Shi, J.Q.; Wu, Z.X. Strategies for regulating the intensity of different cyanobacterial blooms: Insights from the dynamics and stability of bacterioplankton communities. Sci. Total Environ. 2024, 918, 170707. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Li, M.Y.; Yang, Y.C.; Yu, H.; Xiao, F.S.; Mao, C.Z.; Huang, J.; Yu, Y.H.; Wang, Y.F.; Wu, B.; et al. Nitrite and nitrate reduction drive sediment microbial nitrogen cycling in a eutrophic lake. Water Res. 2022, 220, 118637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, T.T.; Wang, Q.; Li, L.; Shen, T.T.; Gao, G. Bacterial community composition in aquatic and sediment samples with spatiotemporal dynamics in large, shallow, eutrophic Lake Chaohu, China. J. Freshw. Ecol. 2019, 34, 575–589. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, Y.; Gao, G.; Jiang, J.H. Spatial-temporal variation of bacterial communities in sediments in Lake Chaohu, a large, shallow eutrophic lake in China. Int. J. Environ. Res. Public Health 2019, 16, 3966. [Google Scholar] [CrossRef]
- Te, S.H.; Tan, B.F.; Thompson, J.R.; Gin, K.Y.H. Relationship of microbiota and cyanobacterial secondary metabolites in dominated bloom. Environ. Sci. Technol. 2017, 51, 4199–4209. [Google Scholar] [CrossRef]
- He, S.M.; Stevens, S.L.R.; Chan, L.K.; Bertilsson, S.; del Rio, T.G.; Tringe, S.G.; Malmstrom, R.R.; McMahon, K.D. Ecophysiology of Freshwater Verrucomicrobia Inferred from Metagenome-Assembled Genomes. Msphere 2017, 2, e00277-17. [Google Scholar] [CrossRef]
- Ayala-Muñoz, D.; Macalady, J.L.; Sánchez-España, J.; Falagán, C.; Couradeau, E.; Burgos, W.D. Microbial carbon, sulfur, iron, and nitrogen cycling linked to the potential remediation of a meromictic acidic pit lake. ISME J. 2022, 16, 2666–2679. [Google Scholar] [CrossRef]
- Gao, P.F.; Wang, P.; Ding, M.J.; Zhang, H.; Huang, G.X.; Nie, M.H.; Wang, G.W. A meta-analysis reveals that geographical factors drive the bacterial community variation in Chinese lakes. Environ. Res. 2023, 224, 115561. [Google Scholar] [CrossRef]
- Shang, Y.; Wu, X.; Wang, X.; Wei, Q.; Ma, S.; Sun, G.; Zhang, H.; Wang, L.; Dou, H.; Zhang, H. Factors affecting seasonal variation of microbial community structure in Hulun Lake, China. Sci. Total Environ. 2022, 805, 150294. [Google Scholar] [CrossRef]
- McIlroy, S.J.; Albertsen, M.; Andresen, E.K.; Saunders, A.M.; Kristiansen, R.; Stokholm-Bjerregaard, M.; Nielsen, K.L.; Nielsen, P.H. ‘Candidatus Competibacter’-lineage genomes retrieved from metagenomes reveal functional metabolic diversity. ISME J. 2014, 8, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Westerholm, M.; Calusinska, M.; Dolfing, J. Syntrophic propionate-oxidizing bacteria in methanogenic systems. FEMS Microbiol. Rev. 2022, 46, fuab057. [Google Scholar] [CrossRef] [PubMed]
- Lovley, D.R.; Ueki, T.; Zhang, T.; Malvankar, N.S.; Shrestha, P.M.; Flanagan, K.A.; Aklujkar, M.; Butler, J.E.; Giloteaux, L.; Rotaru, A.E.; et al. Geobacter: The microbe electric’s physiology, ecology, and practical applications. Adv. Microb. Physiol. 2011, 59, 1–100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chang, J.L.; Lin, C.; Pan, Y.R.; Cui, K.P.; Zhang, X.Y.; Liang, P.; Huang, X. Enhancement of methanogenesis via direct interspecies electron transfer between and conducted by granular activated carbon. Bioresour. Technol. 2017, 245, 132–137. [Google Scholar] [CrossRef]
- Li, T.; Zhou, Q.X. The key role of in regulating emissions and biogeochemical cycling of soil-derived greenhouse gases. Environ. Pollut. 2020, 266, 115135. [Google Scholar] [CrossRef]
- Yan, X.C.; Xu, X.G.; Ji, M.; Zhang, Z.Q.; Wang, M.Y.; Wu, S.J.; Wang, G.X.; Zhang, C.; Liu, H.C. Cyanobacteria blooms: A neglected facilitator of CH4 production in eutrophic lakes. Sci. Total Environ. 2019, 651, 466–474. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria | Archaea | |||||
|---|---|---|---|---|---|---|
| ASVs Number | Avg. Relative Abundance | ASVs Number | Avg. Relative Abundance | |||
| All ASVs | 108,543 | 100.00% | 6357 | 100.00% | ||
| Abundant taxa | Always abundant taxa (AAT) | 0 | 0.00% | 2 | 42.85% | |
| Conditionally abundant taxa (CAT) | 10 | 9.93% | 23 | 29.17% | ||
| Conditionally rare and abundant taxa (CRAT) | 30 | 7.71% | 39 | 10.68% | ||
| Total | 40 | 17.63% | 64 | 82.70% | ||
| Rare taxa | Always rare taxa (ART) | 88,143 | 10.82% | 3565 | 0.58% | |
| Conditionally rare taxa (CRT) | 20,354 | 70.70% | 2728 | 16.72% | ||
| Total | 108,497 | 81.52% | 6293 | 17.30% | ||
| Moderate taxa (MT) | 6 | 0.85% | 0 | 0.00% | ||
| Predicted KEGG-Annotated Genes | |||
|---|---|---|---|
| Abundant Sub-Community | Rare Sub-Community | Overall Community | |
| Bacteria | 3611 | 7481 | 7481 |
| Archaea | 3701 | 4064 | 4064 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Z.; Li, W.; Yang, K.; Wang, X.; Xiong, S.; Zhang, X. Bacterial and Archaeal Communities in Erhai Lake Sediments: Abundance and Metabolic Insight into a Plateau Lake at the Edge of Eutrophication. Microorganisms 2024, 12, 1617. https://doi.org/10.3390/microorganisms12081617
Xie Z, Li W, Yang K, Wang X, Xiong S, Zhang X. Bacterial and Archaeal Communities in Erhai Lake Sediments: Abundance and Metabolic Insight into a Plateau Lake at the Edge of Eutrophication. Microorganisms. 2024; 12(8):1617. https://doi.org/10.3390/microorganisms12081617
Chicago/Turabian StyleXie, Zhen, Wei Li, Kaiwen Yang, Xinze Wang, Shunzi Xiong, and Xiaojun Zhang. 2024. "Bacterial and Archaeal Communities in Erhai Lake Sediments: Abundance and Metabolic Insight into a Plateau Lake at the Edge of Eutrophication" Microorganisms 12, no. 8: 1617. https://doi.org/10.3390/microorganisms12081617
APA StyleXie, Z., Li, W., Yang, K., Wang, X., Xiong, S., & Zhang, X. (2024). Bacterial and Archaeal Communities in Erhai Lake Sediments: Abundance and Metabolic Insight into a Plateau Lake at the Edge of Eutrophication. Microorganisms, 12(8), 1617. https://doi.org/10.3390/microorganisms12081617