
Structural and Functional Differences in Small Intestinal and Fecal Microbiota: 16S rRNA Gene Investigation in Rats
 ,
,
Abstract
1. Introduction
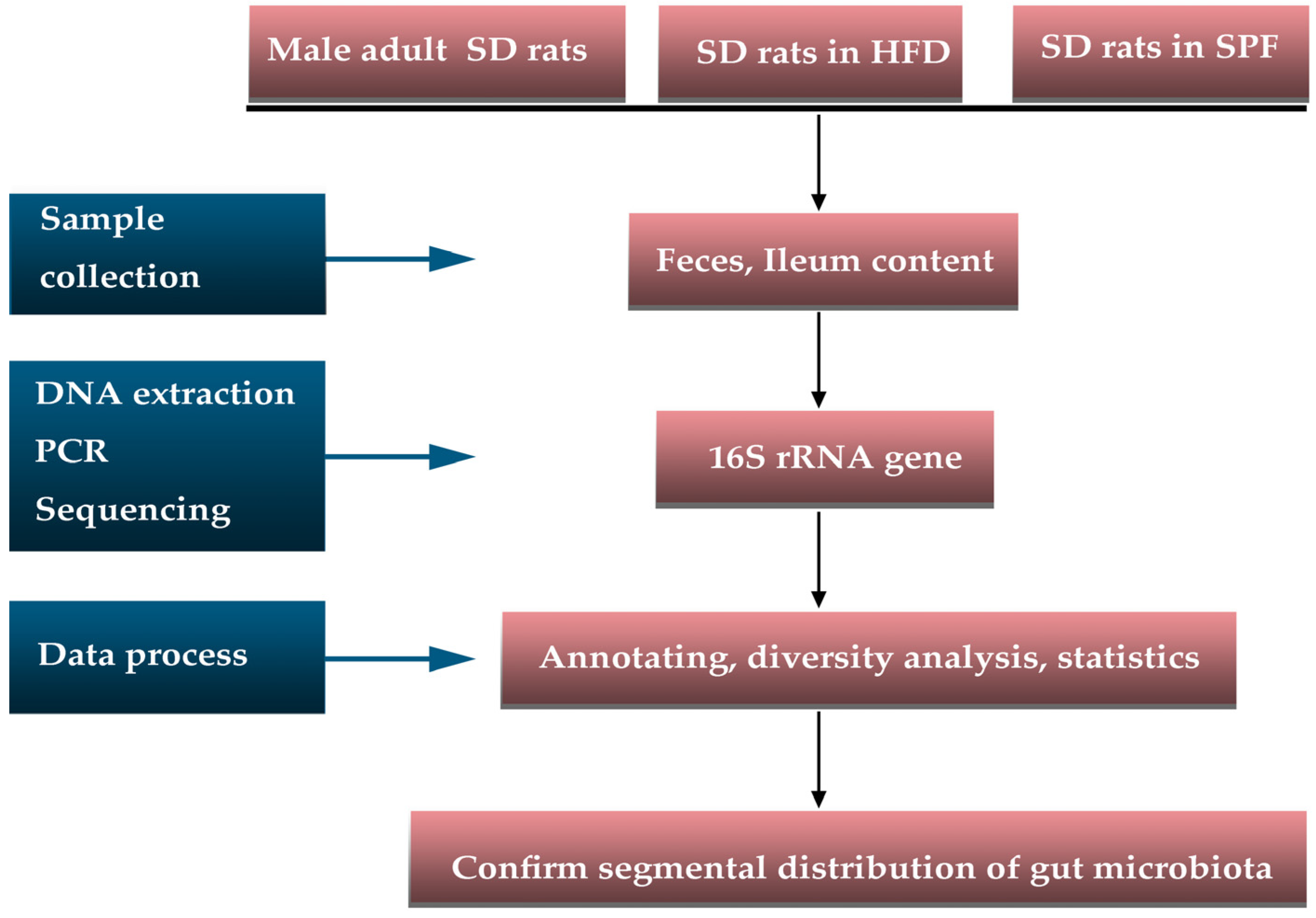
2. Materials and Methods
2.1. Animals
2.2. Sample Collection
2.3. DNA Extraction, PCR, and Sequencing
2.4. Data Preprocessing
2.5. Statistics
3. Results
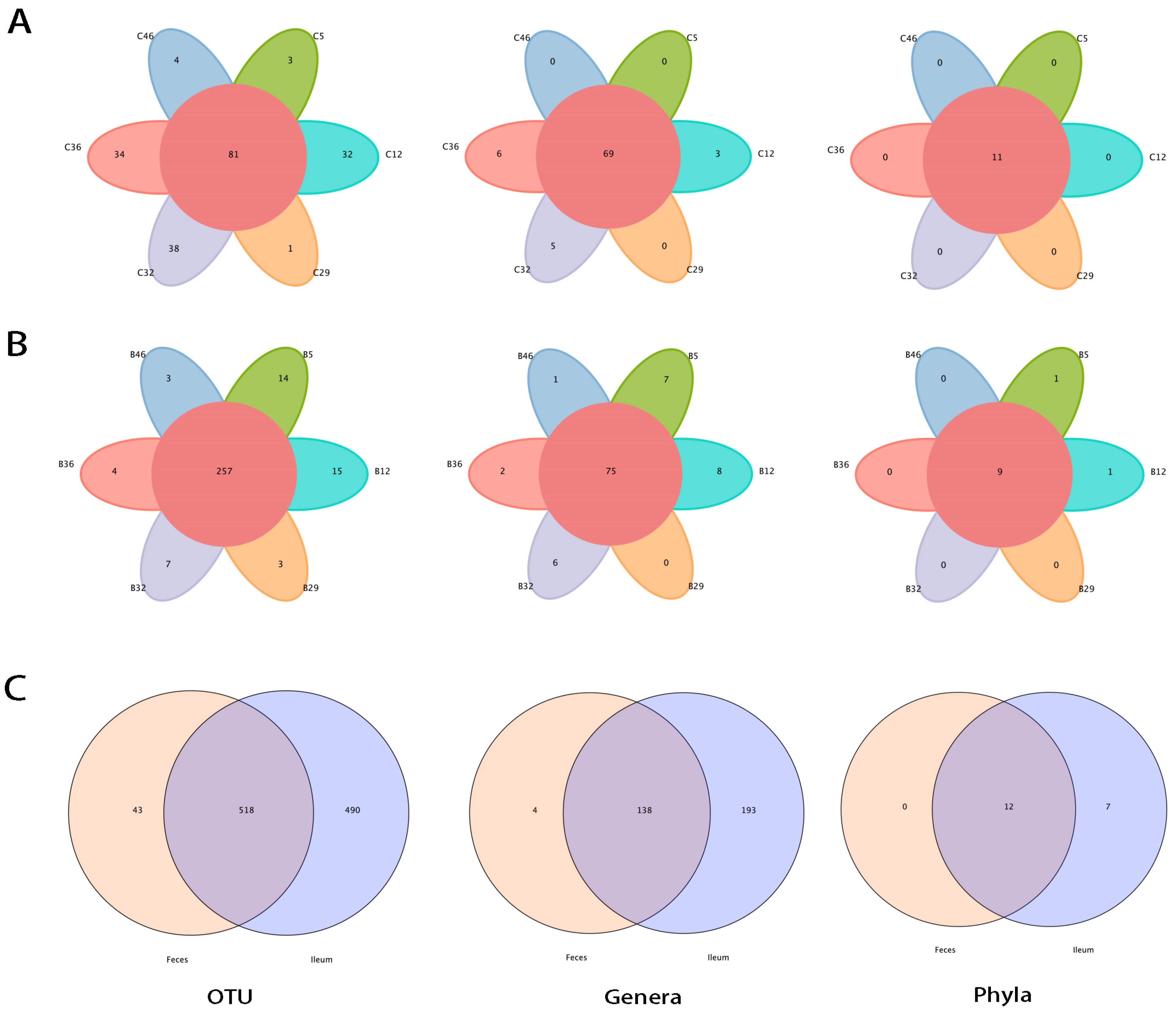
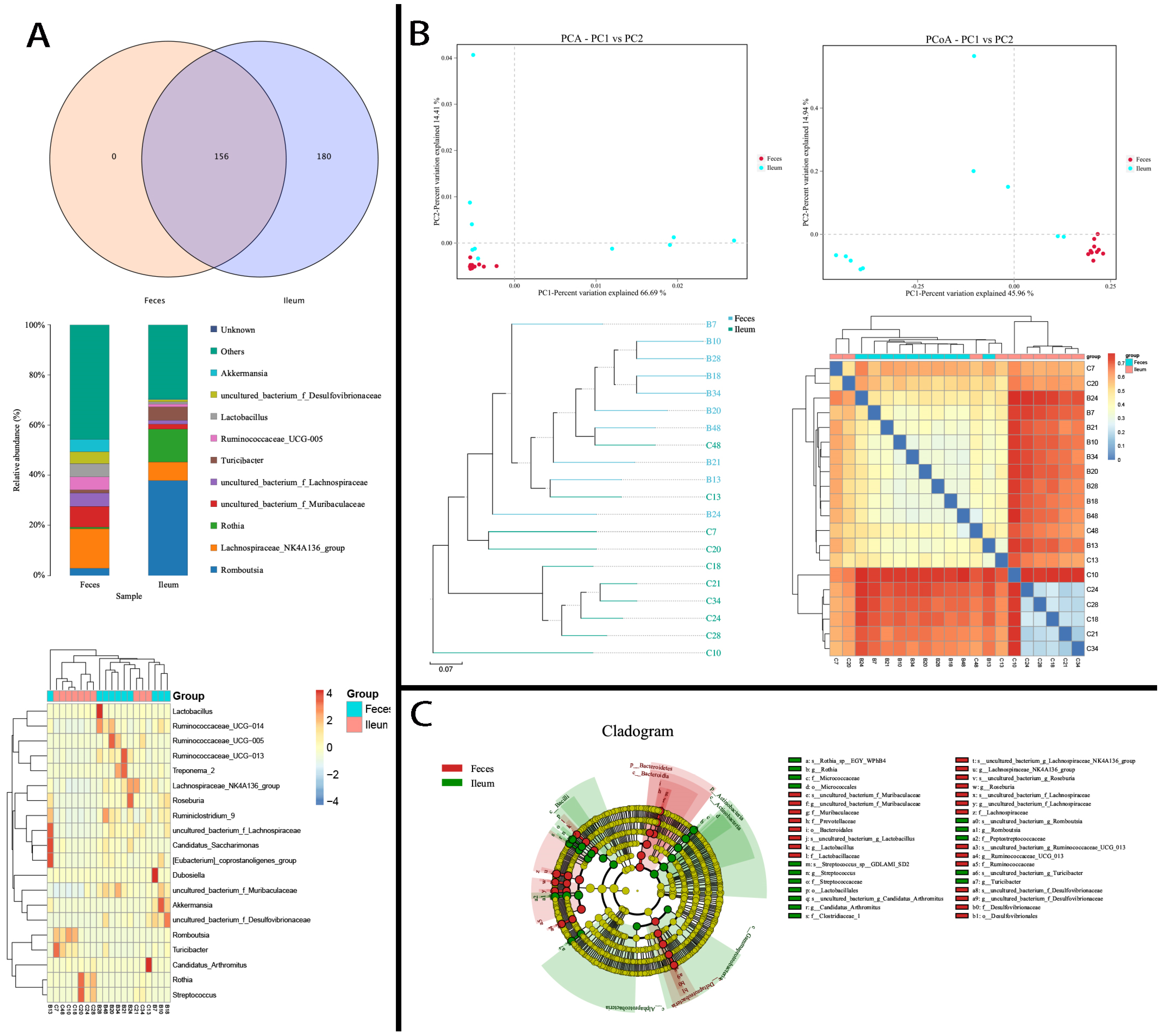
3.1. Large Proportions of Ileal Flora Were Exclusive to the Feces
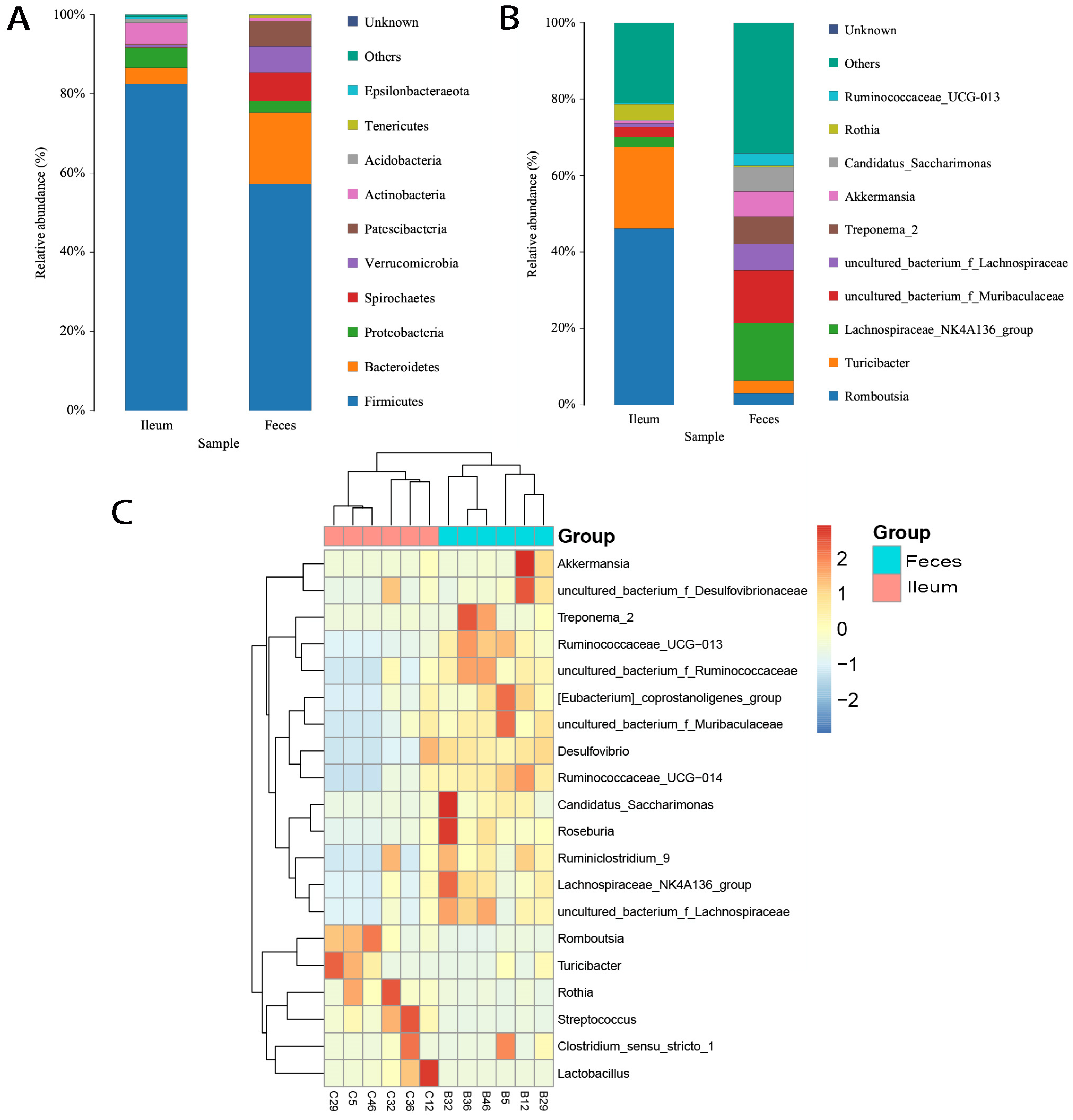
3.2. Shared Bacteria Were Composed of Distinct Floral Structures in the Ileum and Feces
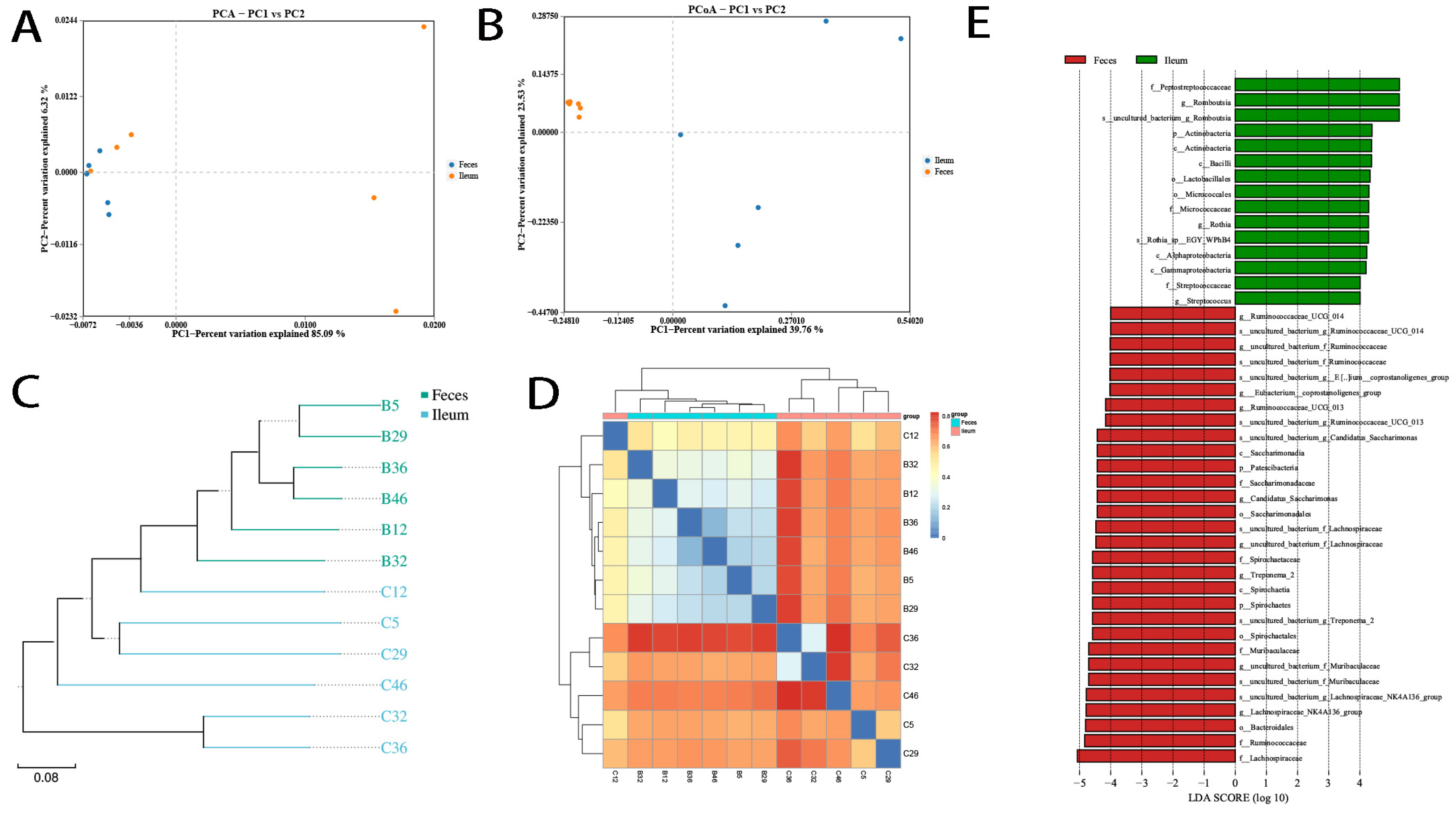
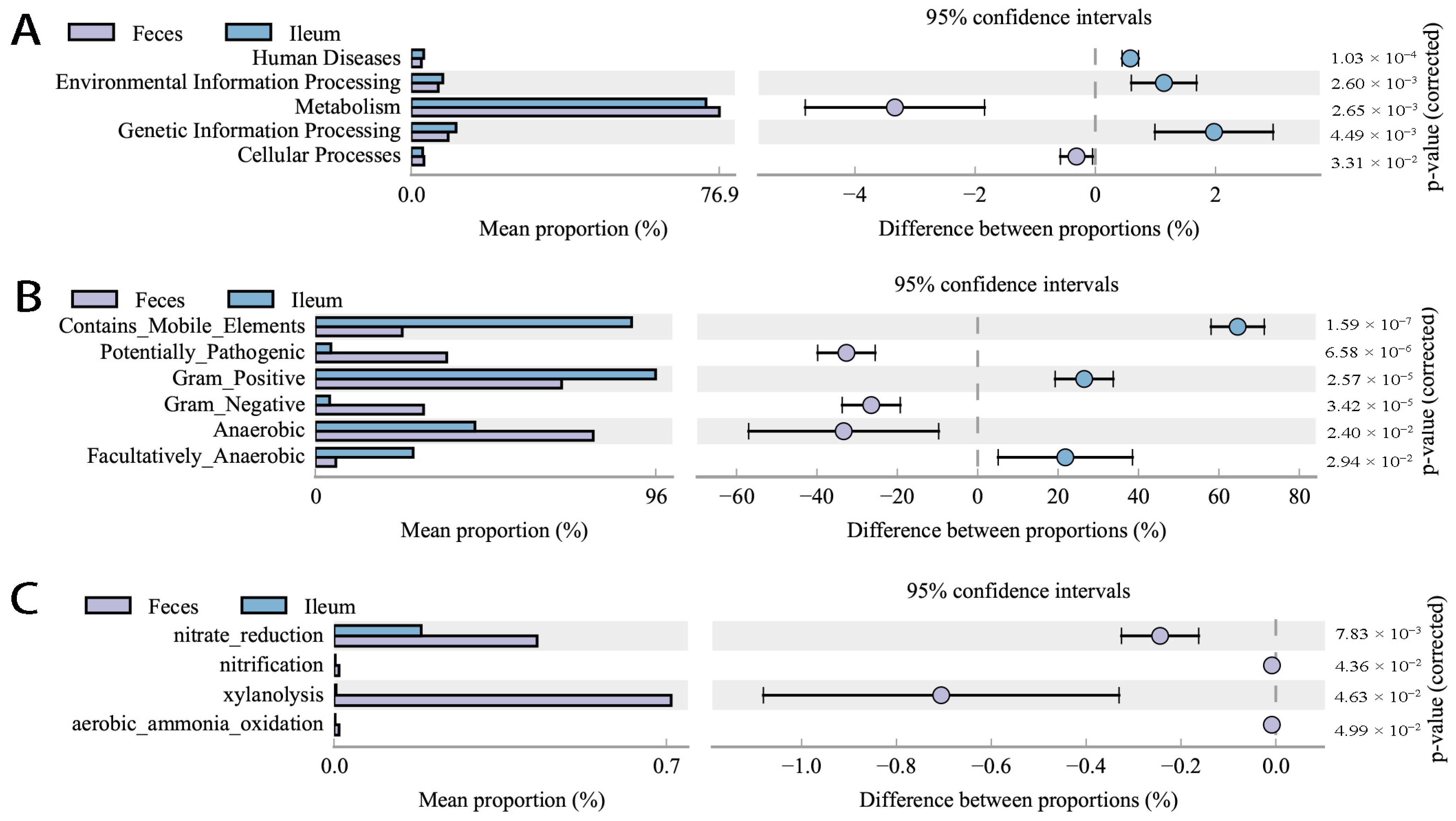
3.3. Differing Diversity, Biomarkers, and Functions in Ileal and Fecal Bacteria
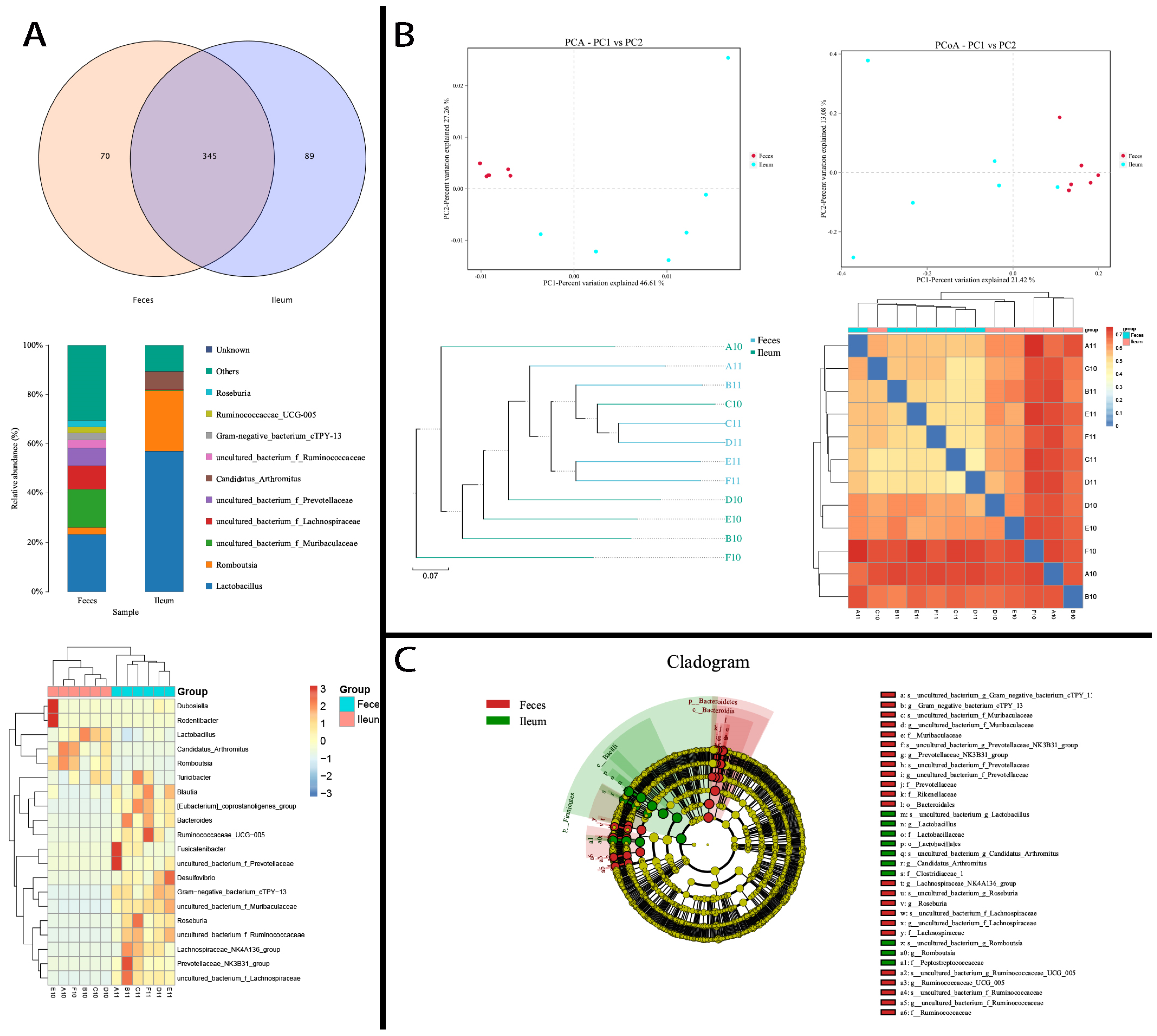
3.4. Ileal and Fecal Microbiota Under High-Fat Diet Intervention
3.5. Ileal Microbiota Under Specific-Pathogen–Free Conditions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kennedy, M.S.; Chang, E.B. The microbiome: Composition and locations. Prog. Mol. Biol. Transl. Sci. 2020, 176, 1–42. [Google Scholar] [PubMed]
- Morgan, X.C.; Huttenhower, C. Meta’omic analytic techniques for studying the intestinal microbiome. Gastroenterology 2014, 146, 1437–1448.e1. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Guryn, K.; Hubert, N.; Frazier, K.; Urlass, S.; Musch, M.W.; Ojeda, P.; Pierre, J.F.; Miyoshi, J.; Sontag, T.J.; Cham, C.M.; et al. Small intestine microbiota regulate host digestive and absorptive adaptive responses to dietary lipids. Cell Host Microbe 2018, 23, 458–469.e5. [Google Scholar] [CrossRef] [PubMed]
- Escobedo, G.; López-Ortiz, E.; Torres-Castro, I. Gut microbiota as a key player in triggering obesity, systemic inflammation and insulin resistance. Rev. Investig. Clin. 2014, 66, 450–459. [Google Scholar]
- Cani, P.D.; Van Hul, M. Gut microbiota and obesity: Causally linked? Expert. Rev. Gastroenterol. Hepatol. 2020, 14, 401–403. [Google Scholar] [CrossRef]
- Kazemian, N.; Mahmoudi, M.; Halperin, F.; Wu, J.C.; Pakpour, S. Gut microbiota and cardiovascular disease: Opportunities and challenges. Microbiome 2020, 8, 36. [Google Scholar] [CrossRef]
- Heiss, C.N.; Olofsson, L.E. Gut microbiota-dependent modulation of energy metabolism. J. Innate Immun. 2018, 10, 163–171. [Google Scholar] [CrossRef]
- Cuevas-Sierra, A.; Ramos-Lopez, O.; Riezu-Boj, J.I.; Milagro, F.I.; Martinez, J.A. Diet, gut microbiota, and obesity: Links with host genetics and epigenetics and potential applications. Adv. Nutr. 2019, 10, S17–S30. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Moran, C.; Sheehan, D.; Shanahan, F. The small bowel microbiota. Curr. Opin. Gastroenterol. 2015, 31, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Hollister, E.B.; Gao, C.; Versalovic, J. Compositional and functional features of the gastrointestinal microbiome and their effects on human health. Gastroenterology 2014, 146, 1449–1458. [Google Scholar] [CrossRef]
- Tomas, J.; Mulet, C.; Saffarian, A.; Cavin, J.B.; Ducroc, R.; Regnault, B.; Kun Tan, C.; Duszka, K.; Burcelin, R.; Wahli, W.; et al. High-fat diet modifies the PPAR-γ pathway leading to disruption of microbial and physiological ecosystem in murine small intestine. Proc. Natl. Acad. Sci. USA 2016, 113, E5934–E5943. [Google Scholar] [CrossRef]
- Vuik, F.; Dicksved, J.; Lam, S.Y.; Fuhler, G.M.; van der Laan, L.; van de Winkel, A.; Konstantinov, S.R.; Spaander, M.; Peppelenbosch, M.P.; Engstrand, L.; et al. Composition of the mucosa-associated microbiota along the entire gastrointestinal tract of human individuals. United Eur. Gastroenterol. J. 2019, 7, 897–907. [Google Scholar] [CrossRef]
- Schwensen, H.F.; Kan, C.; Treasure, J.; Høiby, N.; Sjögren, M. A systematic review of studies on the faecal microbiota in anorexia nervosa: Future research may need to include microbiota from the small intestine. Eat Weight. Disord. 2018, 23, 399–418. [Google Scholar] [CrossRef] [PubMed]
- Leite, G.G.S.; Weitsman, S.; Parodi, G.; Celly, S.; Sedighi, R.; Sanchez, M.; Morales, W.; Villanueva-Millan, M.J.; Barlow, G.M.; Mathur, R.; et al. Mapping the segmental microbiomes in the human small bowel in comparison with stool: A REIMAGINE study. Dig. Dis. Sci. 2020, 65, 2595–2604. [Google Scholar] [CrossRef]
- Su, S.; Zhao, Y.; Liu, Z.; Liu, G.; Du, M.; Wu, J.; Bai, D.; Li, B.; Bou, G.; Zhang, X.; et al. Characterization and comparison of the bacterial microbiota in different gastrointestinal tract compartments of Mongolian horses. Microbiologyopen 2020, 9, 1085–1101. [Google Scholar] [CrossRef]
- Zhang, J.G.; Sun, X.W.; Gao, P.; Xie, L.; Liu, W.Q.; Xiao, H.T.; Ai, H. Food restriction alters villi morphology in obese rats: Gut mechanism for weight regain? Exp. Biol. Med. 2012, 237, 993–999. [Google Scholar] [CrossRef]
- Chen, K.; Luan, X.; Liu, Q.; Wang, J.; Chang, X.; Snijders, A.M.; Mao, J.H.; Secombe, J.; Dan, Z.; Chen, J.H.; et al. Drosophila histone demethylase KDM5 regulates social behavior through immune control and gut microbiota maintenance. Cell Host Microbe 2019, 25, 537–552.e8. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Yan, D.; Xia, P.; Song, X.; Lin, T.; Cao, H. Community structure and functional diversity of epiphytic bacteria and planktonic bacteria on submerged macrophytes in Caohai Lake, southwest of China. Ann. Microbiol. 2019, 69, 933–944. [Google Scholar] [CrossRef]
- Heintz-Buschart, A.; Wilmes, P. Human gut microbiome: Function matters. Trends Microbiol. 2018, 26, 563–574. [Google Scholar] [CrossRef]
- Coker, O.O.; Dai, Z.; Nie, Y.; Zhao, G.; Cao, L.; Nakatsu, G.; Wu, W.K.; Wong, S.H.; Chen, Z.; Sung, J.J.Y.; et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 2018, 67, 1024–1032. [Google Scholar] [CrossRef]
- Vinasco, K.; Mitchell, H.M.; Kaakoush, N.O.; Castaño-Rodríguez, N. Microbial carcinogenesis: Lactic acid bacteria in gastric cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 188309. [Google Scholar] [CrossRef]
- Martinez-Guryn, K.; Leone, V.; Chang, E.B. Regional diversity of the gastrointestinal microbiome. Cell Host Microbe 2019, 26, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Mentula, S.; Harmoinen, J.; Heikkilä, M.; Westermarck, E.; Rautio, M.; Huovinen, P.; Könönen, E. Comparison between cultured small-intestinal and fecal microbiotas in beagle dogs. Appl Environ. Microbiol. 2005, 71, 4169–4175. [Google Scholar] [CrossRef]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]
- Ahn, J.-S.; Lkhagva, E.; Jung, S.; Kim, H.-J.; Chung, H.-J.; Hong, S.-T. Fecal microbiome does not represent whole gut microbiome. Cellular Microbiology 2023, 2023, 1–14. [Google Scholar] [CrossRef]
- Shalon, D.; Culver, R.N.; Grembi, J.A.; Folz, J.; Treit, P.V.; Shi, H.; Rosenberger, F.A.; Dethlefsen, L.; Meng, X.; Yaffe, E.; et al. Profiling the human intestinal environment under physiological conditions. Nature 2023, 617, 581–591. [Google Scholar] [CrossRef]
- Menard, J.; Bagheri, S.; Menon, S.; Yu, Y.T.; Goodman, L.B. Noninvasive sampling of the small intestinal chyme for microbiome, metabolome and antimicrobial resistance genes in dogs, a proof of concept. Anim. Microbiome 2023, 5, 64. [Google Scholar] [CrossRef]
- Boicean, A.; Birlutiu, V.; Ichim, C.; Brusnic, O.; Onișor, D.M. Fecal microbiota transplantation in liver cirrhosis. Biomedicines 2023, 11, 2930. [Google Scholar] [CrossRef]
- Boicean, A.; Birlutiu, V.; Ichim, C.; Anderco, P.; Birsan, S. Fecal microbiota transplantation in inflammatory bowel disease. Biomedicines 2023, 11, 1016. [Google Scholar] [CrossRef] [PubMed]
- Peterson, W.L. Helicobacter pylori and peptic ulcer disease. N. Engl. J. Med. 1991, 324, 1043–1048. [Google Scholar]
- Takeuchi, T.; Kameyama, K.; Miyauchi, E.; Nakanishi, Y.; Kanaya, T.; Fujii, T.; Kato, T.; Sasaki, T.; Tachibana, N.; Negishi, H.; et al. Fatty acid overproduction by gut commensal microbiota exacerbates obesity. Cell Metab. 2023, 35, 361–375.e9. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Sex | Sampling | Analysis Methods | Sampling Locations | Relationship | Year of Reported |
|---|---|---|---|---|---|---|
| Beagle dogs | M | Permanent jejunal fistula | Culture | Jejunum fluid, feces | Fecal samples were unable to represent the microbiota present in the upper gut. | 2005 [32] |
| Human | M/F | Esophagogastro- duodenoscopy | 16S rRNA gene sequencing | Stomach, duodenum, jejunum, feces | The small bowel microbiome was markedly different from that in the stool and varied between segments. | 2020 [17] |
| Mongolian horses | M/F | Postmortem | 16S rRNA gene sequencing | Stomach, foregut (jejunum, ileum), hindgut (cecum, colon) | The microbial community structures were significantly different among the stomach, foregut, and hindgut. | 2020 [18] |
| Pig | M | Postmortem | 16S rRNA gene sequencing | Whole gut | The fecal microbiome was insufficient to represent the whole gut microbiome. | 2023 [34] |
| Human | M/F | Sampling capsule | 16S rRNA gene sequencing | Small intestinal fluid, feces | Analysis of stool provided neither a complete nor an accurate representation of the longitudinal and temporal variability of the microbiota composition, virus activity, host proteome or bile acid contents within the intestines | 2023 [35] |
| Beagle dogs | M/F | SIMBA™ capsule | 16S rRNA gene sequencing | Small intestinal fluid, feces | A statistically significant difference in the microbial composition of capsules and feces was found. | 2023 [36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.-W.; Li, H.-R.; Jin, X.-L.; Tang, X.; Wang, D.-W.; Zhang, X.; Zhang, J.-G. Structural and Functional Differences in Small Intestinal and Fecal Microbiota: 16S rRNA Gene Investigation in Rats. Microorganisms 2024, 12, 1764. https://doi.org/10.3390/microorganisms12091764
Sun X-W, Li H-R, Jin X-L, Tang X, Wang D-W, Zhang X, Zhang J-G. Structural and Functional Differences in Small Intestinal and Fecal Microbiota: 16S rRNA Gene Investigation in Rats. Microorganisms. 2024; 12(9):1764. https://doi.org/10.3390/microorganisms12091764
Chicago/Turabian StyleSun, Xiao-Wei, Hong-Rui Li, Xiao-Lei Jin, Xiao Tang, Da-Wen Wang, Xiao Zhang, and Jian-Gang Zhang. 2024. "Structural and Functional Differences in Small Intestinal and Fecal Microbiota: 16S rRNA Gene Investigation in Rats" Microorganisms 12, no. 9: 1764. https://doi.org/10.3390/microorganisms12091764
APA StyleSun, X.-W., Li, H.-R., Jin, X.-L., Tang, X., Wang, D.-W., Zhang, X., & Zhang, J.-G. (2024). Structural and Functional Differences in Small Intestinal and Fecal Microbiota: 16S rRNA Gene Investigation in Rats. Microorganisms, 12(9), 1764. https://doi.org/10.3390/microorganisms12091764