The Presence of Cholesteryl Ester Transfer Protein (CETP) in Endothelial Cells Generates Vascular Oxidative Stress and Endothelial Dysfunction

 ,
,  ,
,  , , , , and
, , , , and 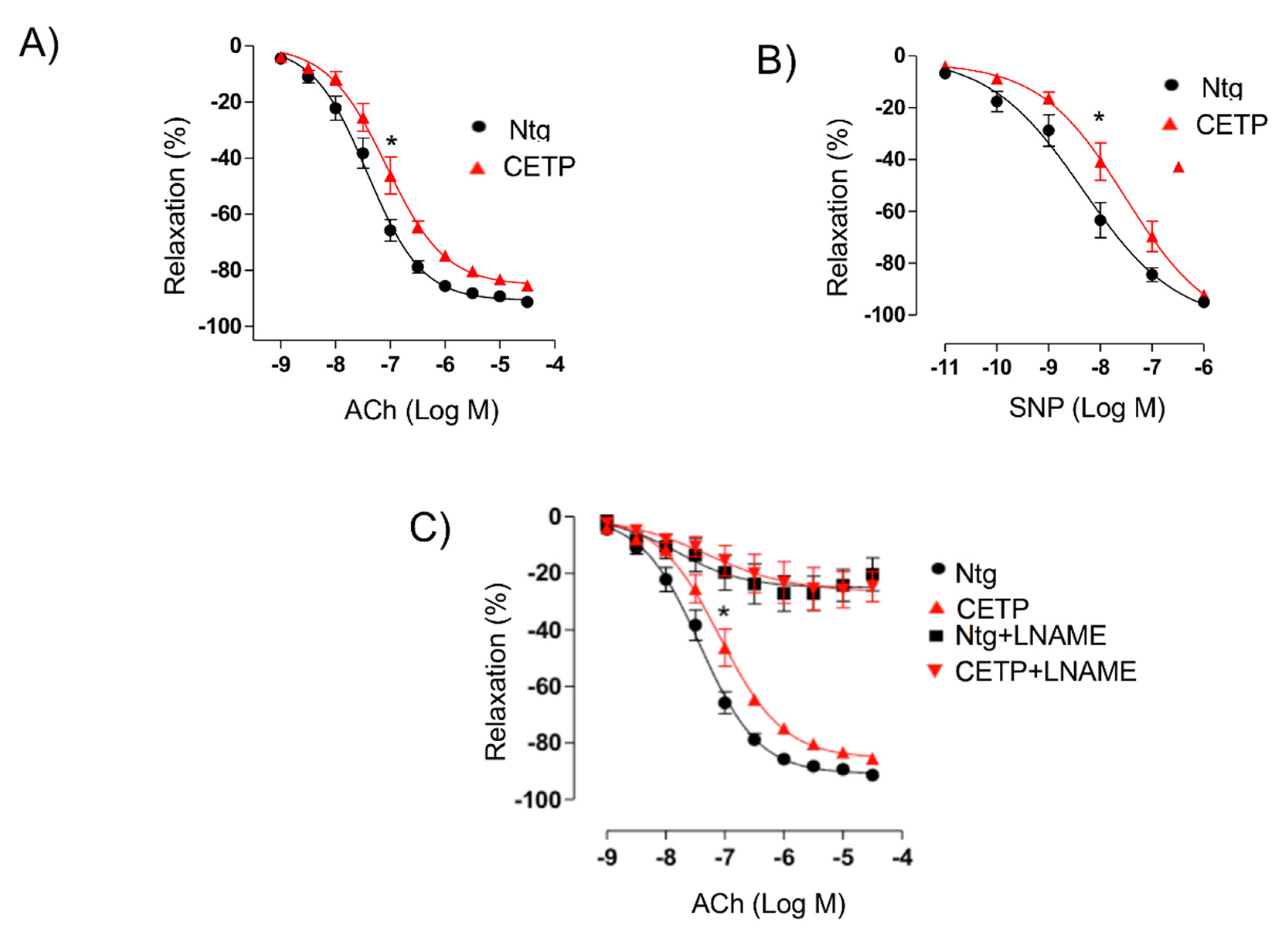{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Aortic Endothelial Cell Culture (HAECs)
2.2. Human Monocytic Cells
2.3. Preparation of Fluorescent THP-1 Cells
2.4. Adhesion Experiment of THP-1 Cells/Monocyte-EC adhesion
2.5. Animals
2.6. Vascular Reactivity
2.7. Gene-Expression Analysis
2.8. Subcellular Fractionation and Western Blot Analysis
2.9. Measurements of Superoxide in HAECs
2.10. Measurements of Superoxide in Aortas
2.11. Production of H2O2 by HAECs
2.12. Production of H2O2 by Aortas
2.13. Reduced (GSH) and Oxidized (GSSG) Glutathione Levels
2.14. Immunofluorescence Staining
2.15. Measurement of Mitochondrial Superoxide Production in HAECs
2.16. Flow Cytometer Analysis
2.17. Statistical Analysis
3. Results
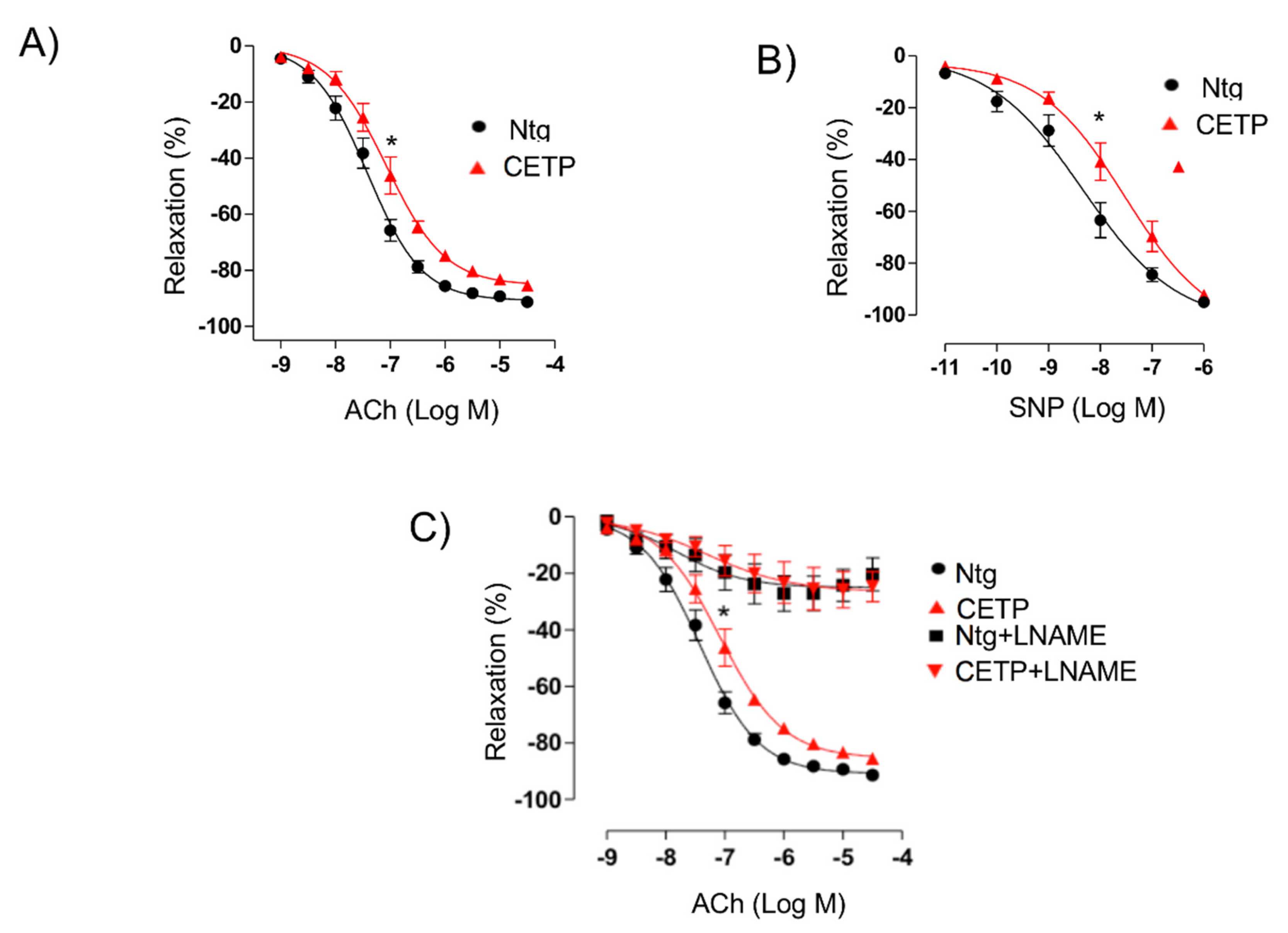
3.1. CETP Impairs the Endothelium-Dependent Relaxation
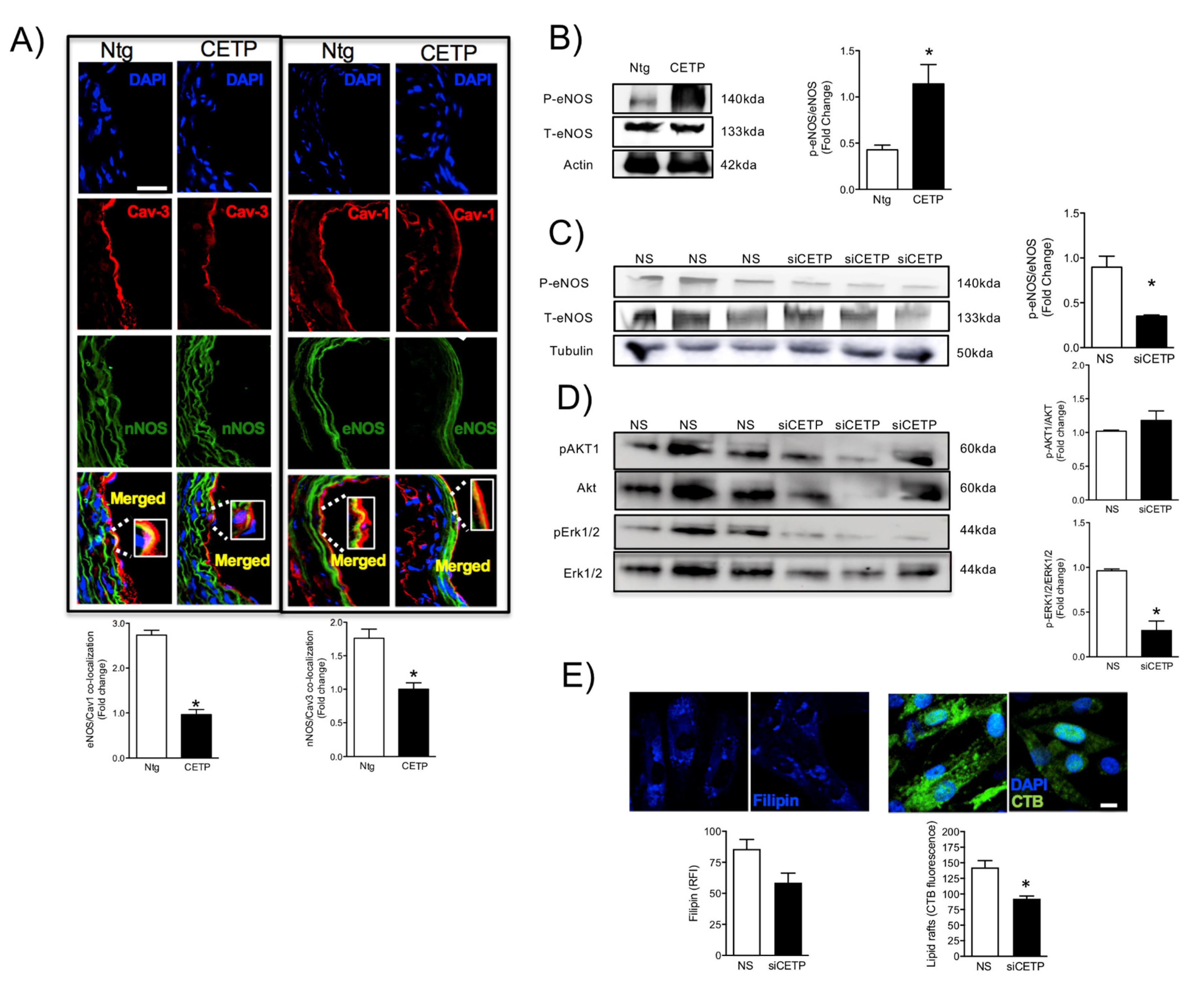
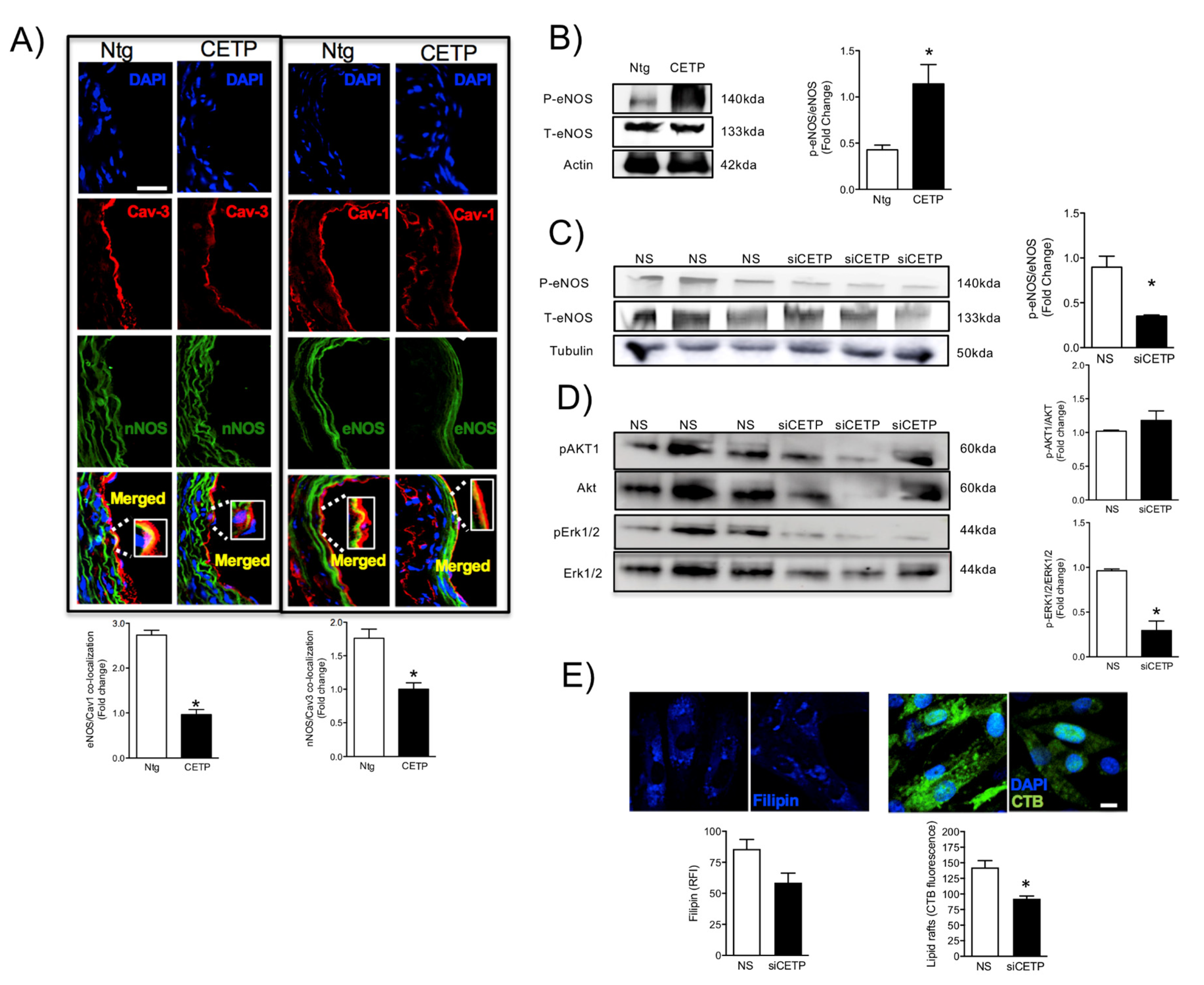
3.2. Effects of CETP on Caveolin/NOS Association, eNOS Activation
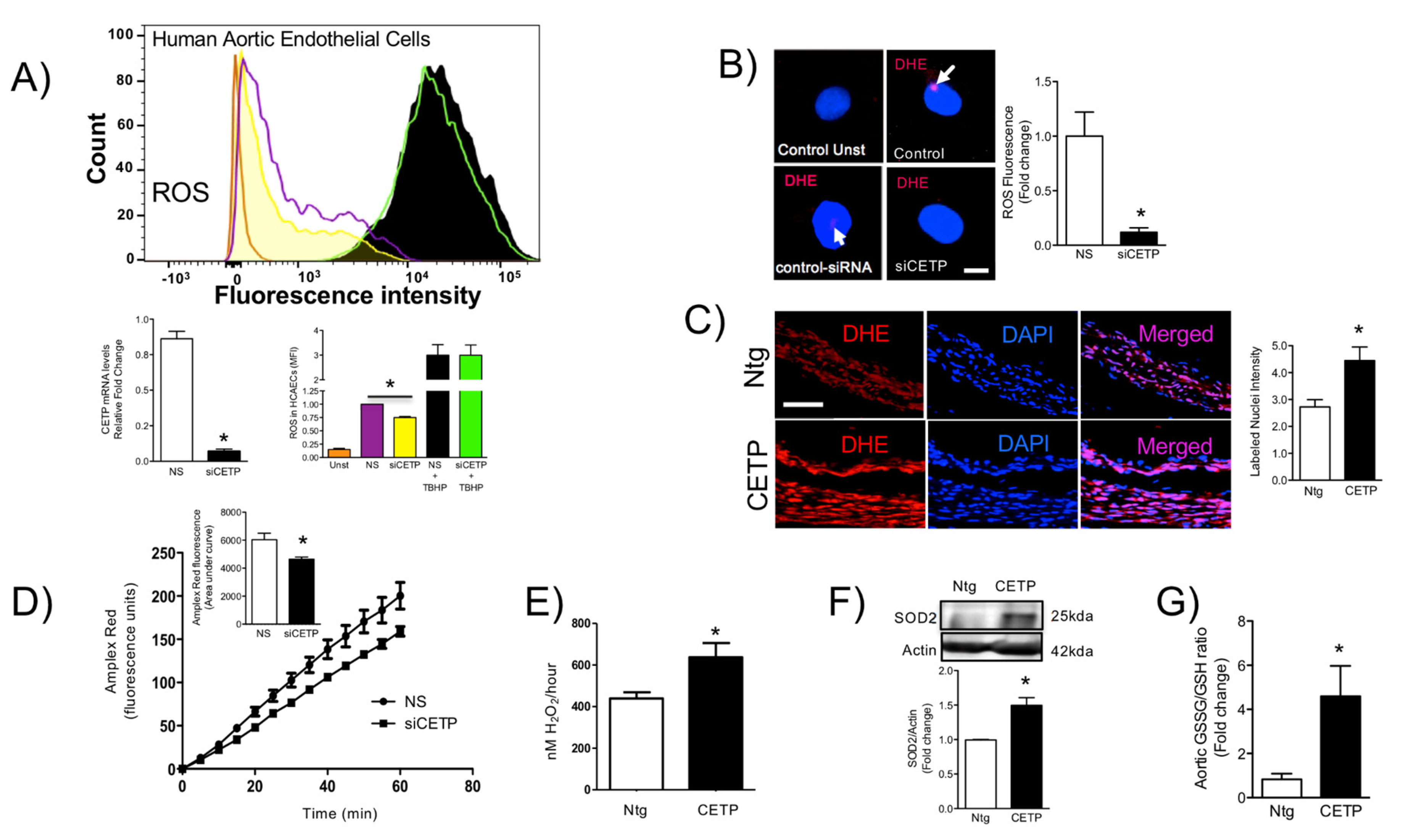
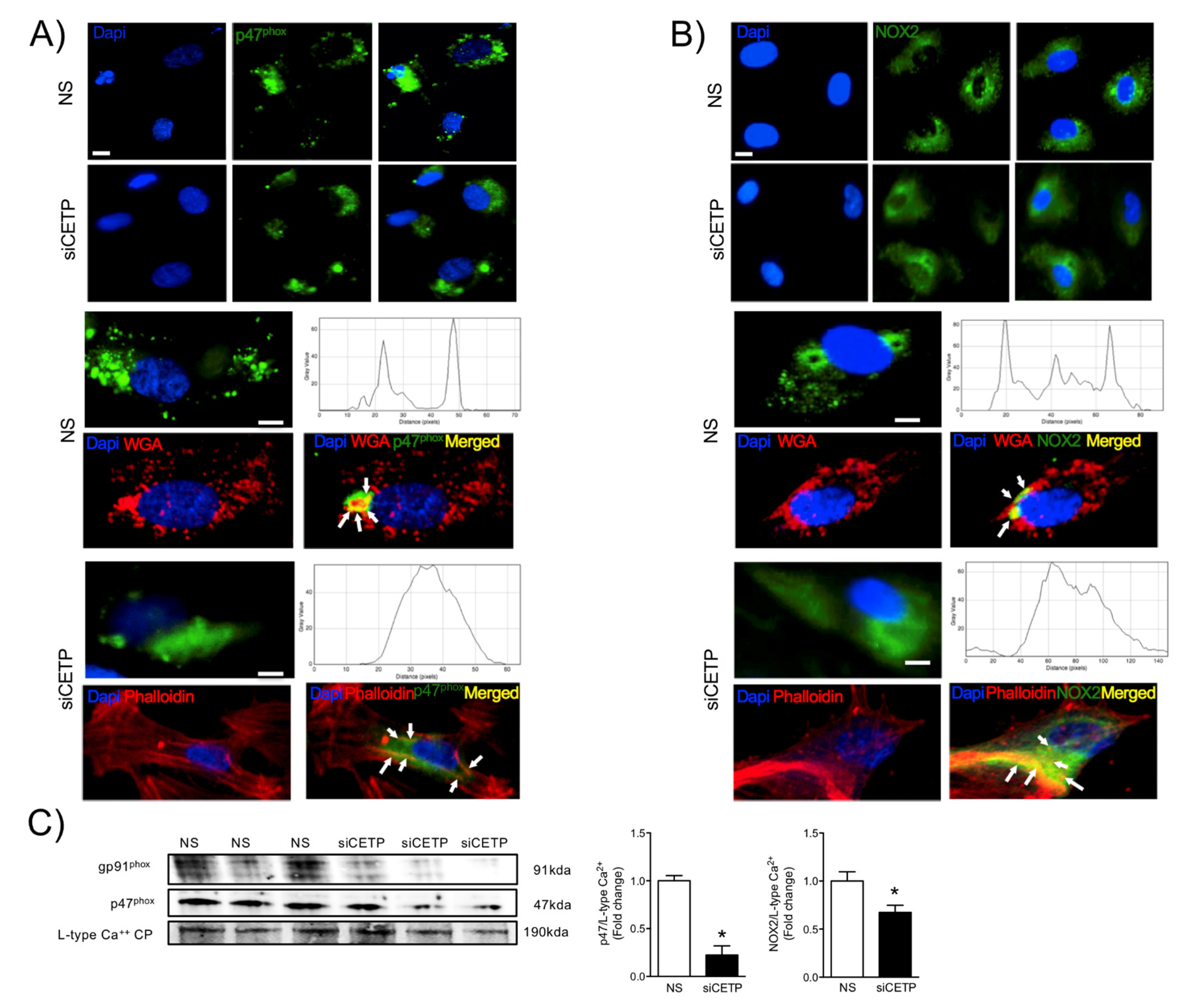
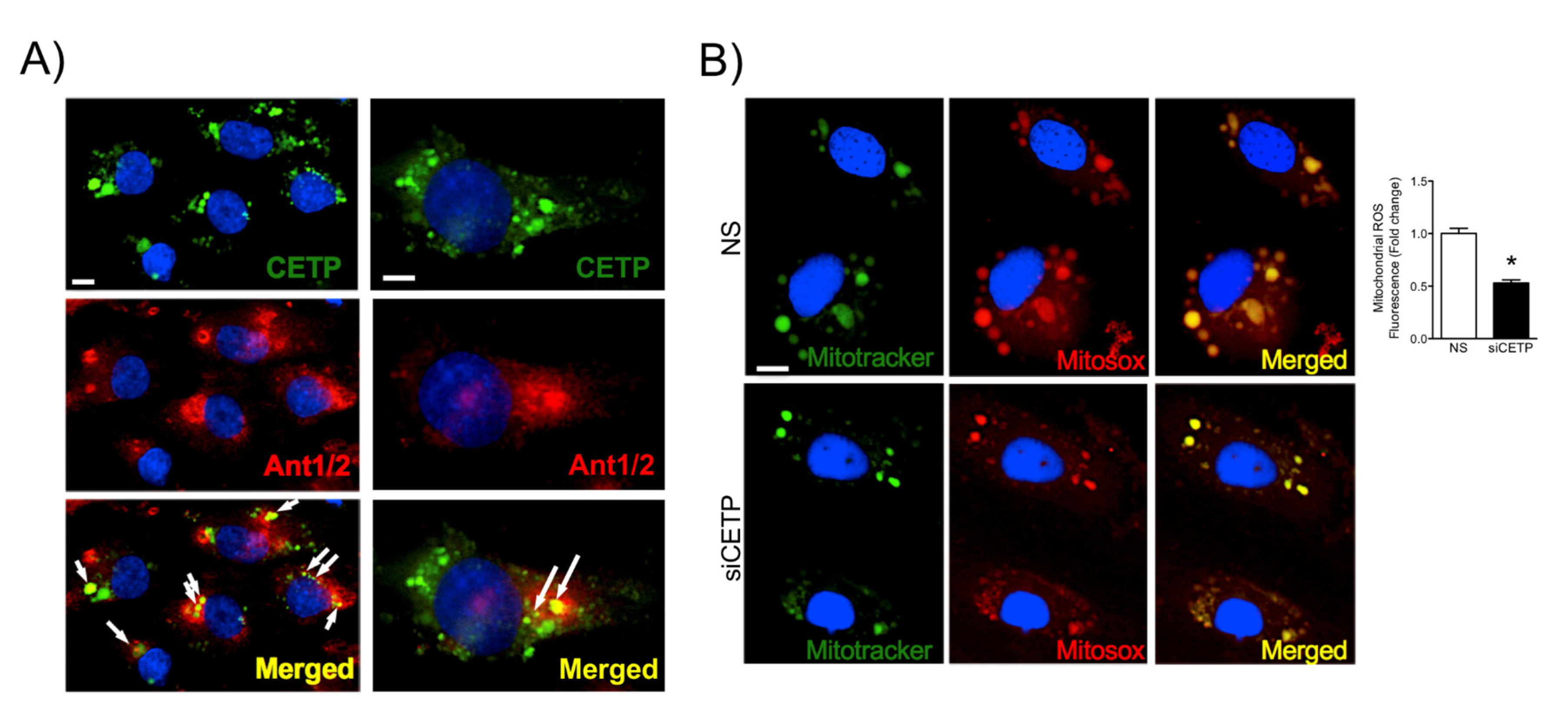
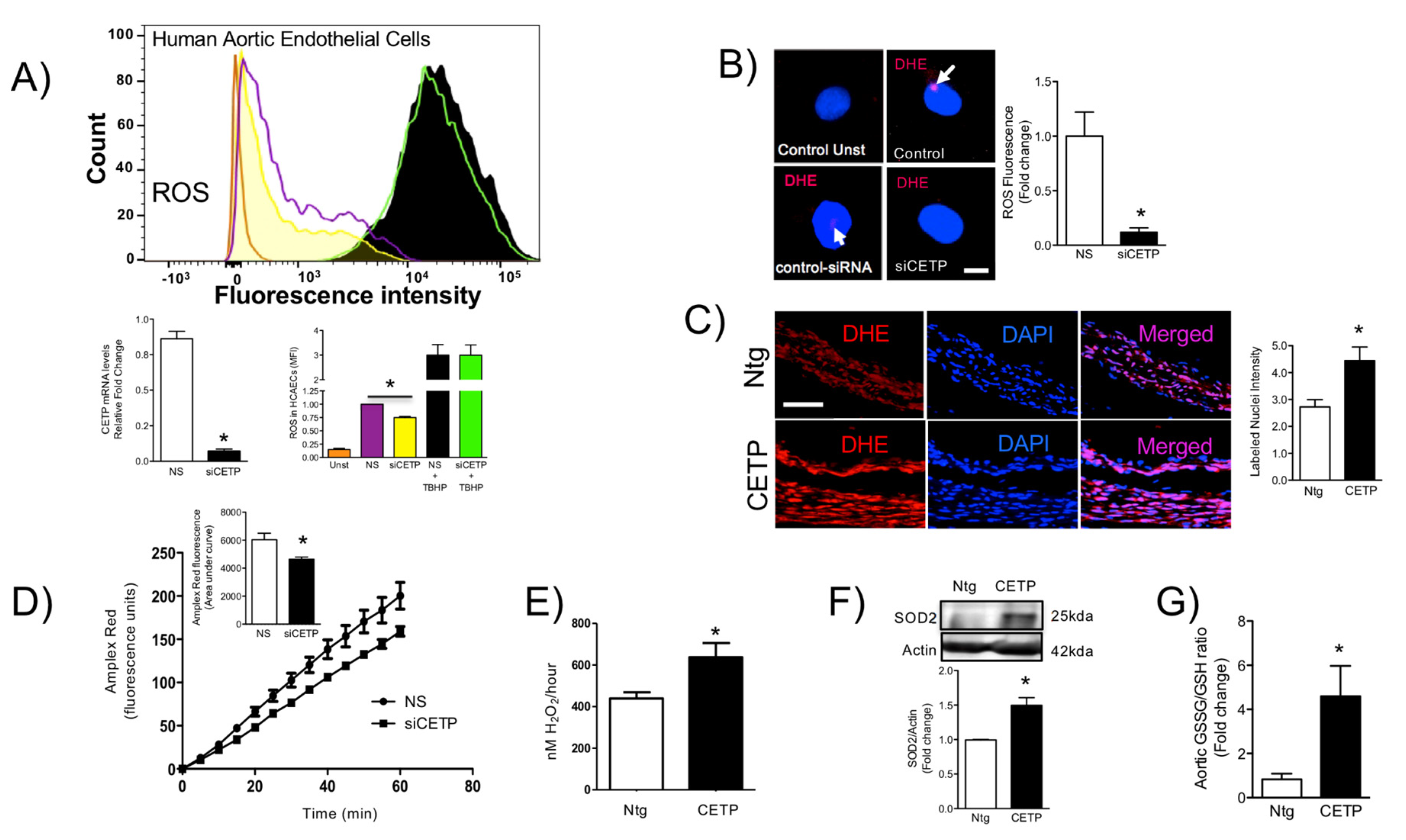
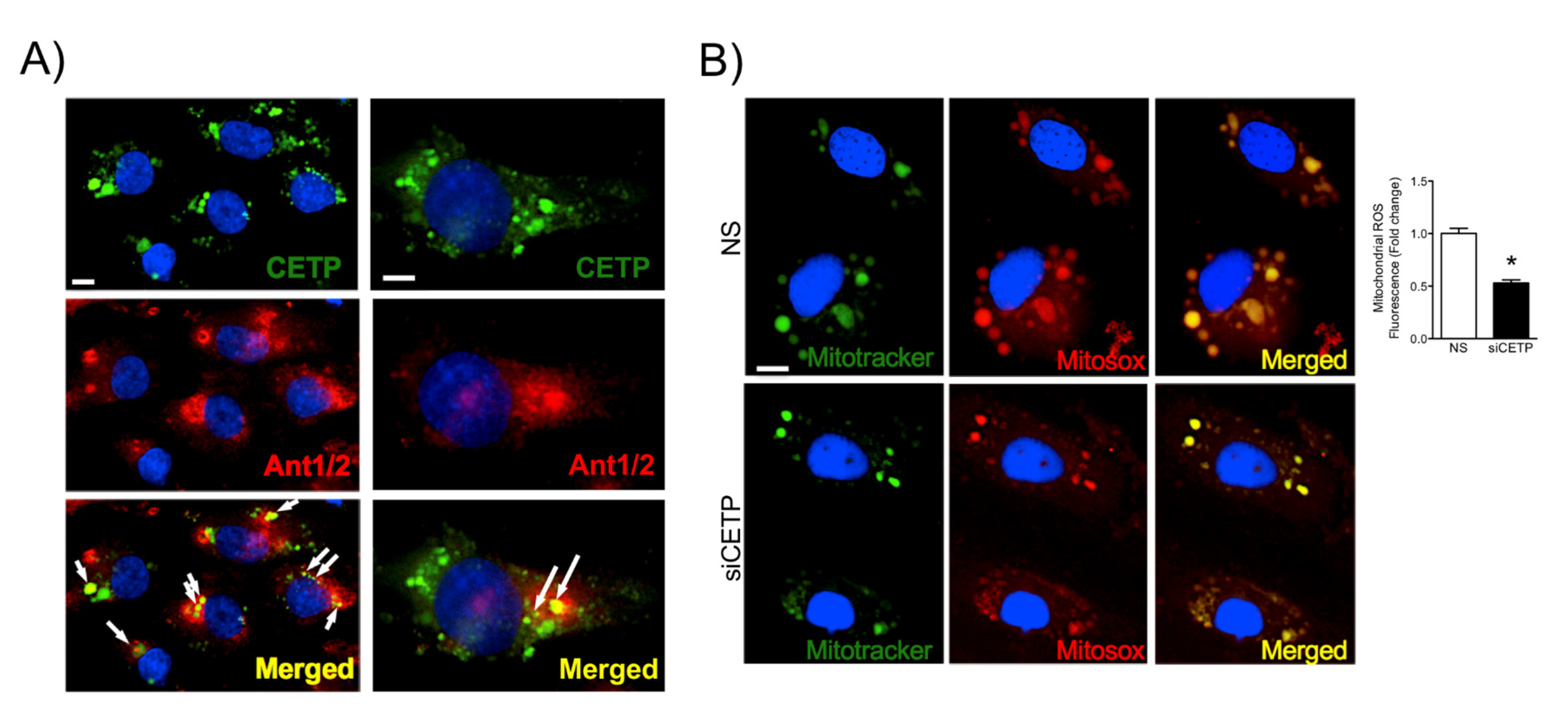
3.3. CETP is Involved with Increased Vascular ROS
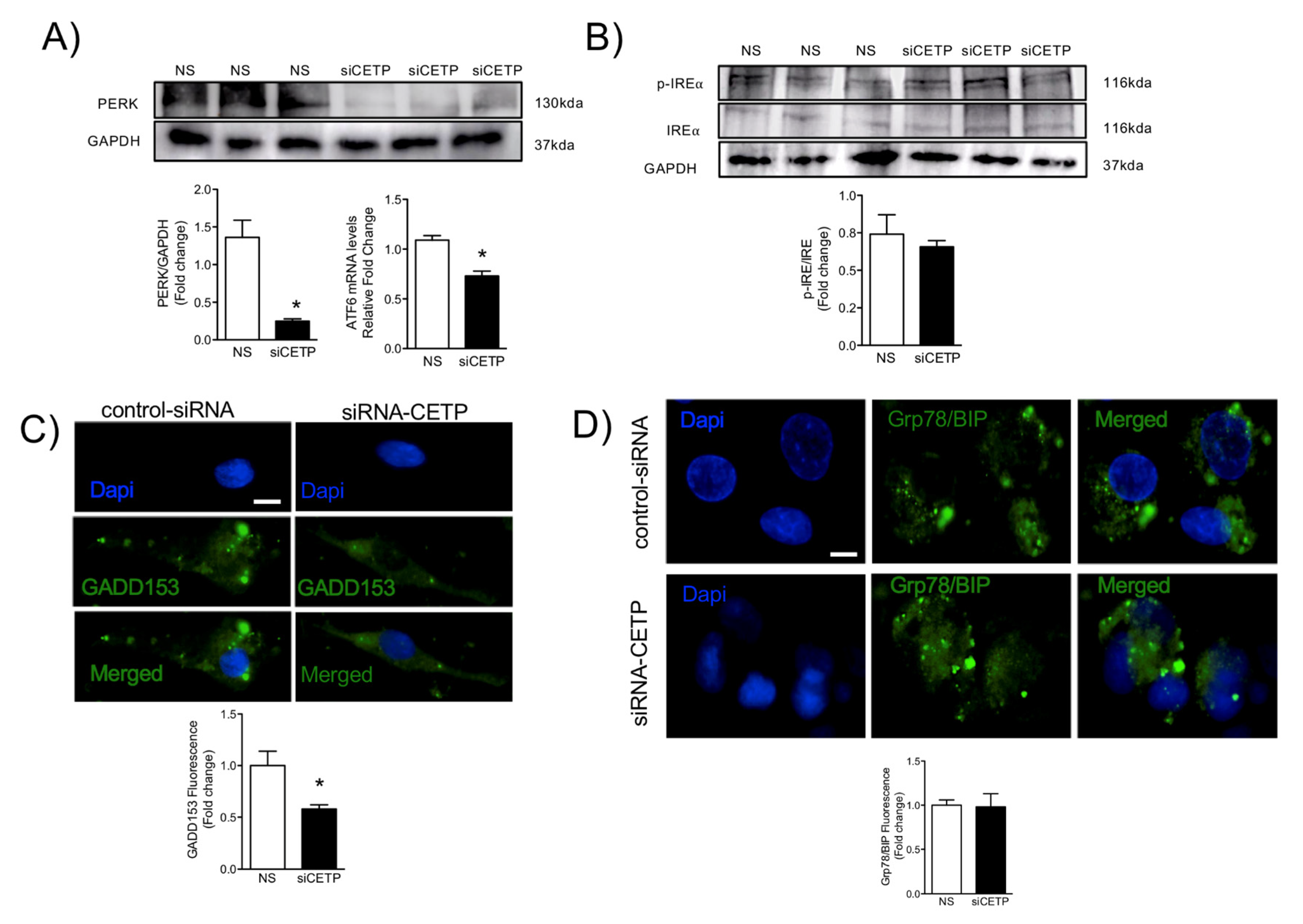
3.4. CETP Inhibition Downregulates Endoplasmic Reticulum Stress
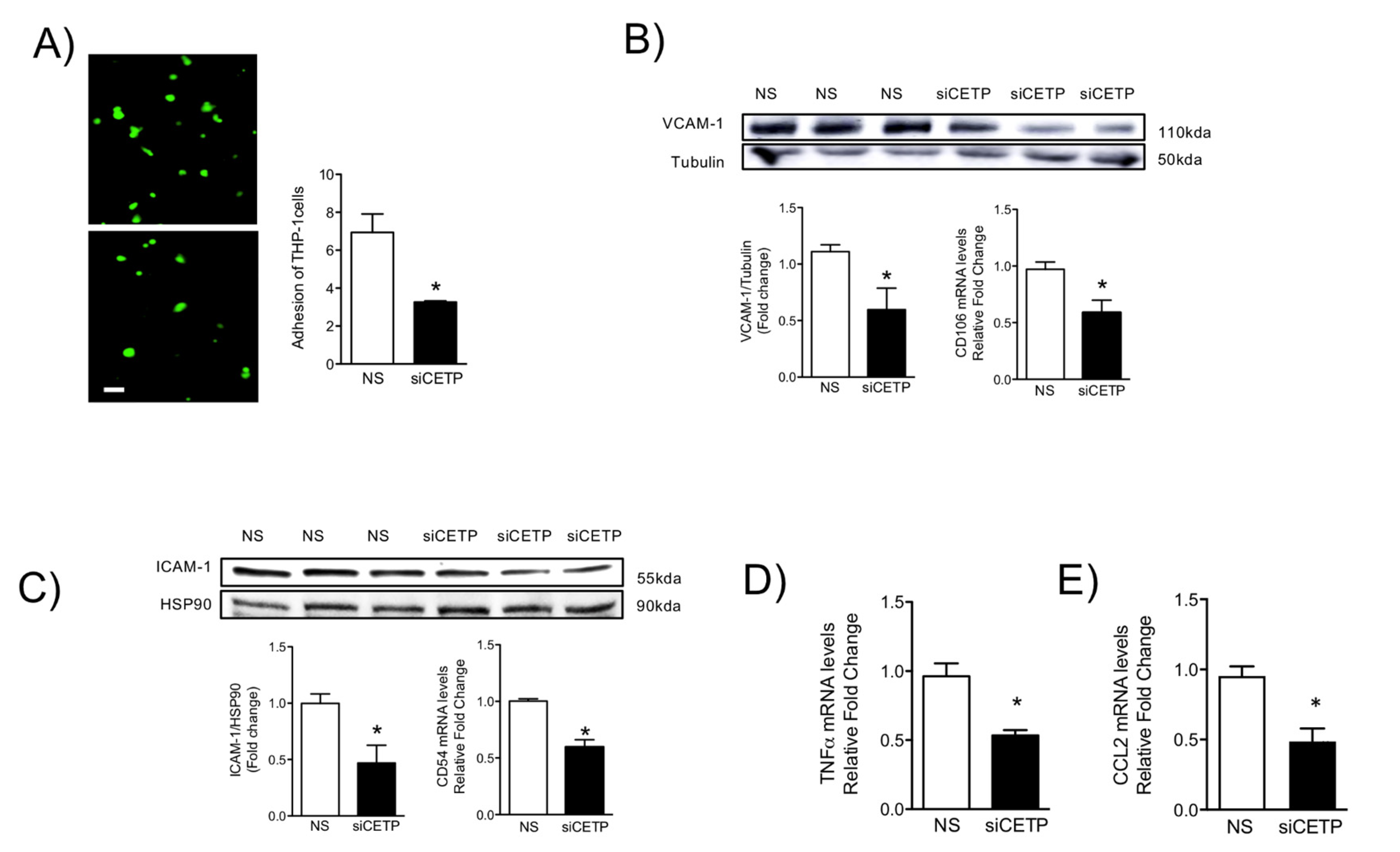
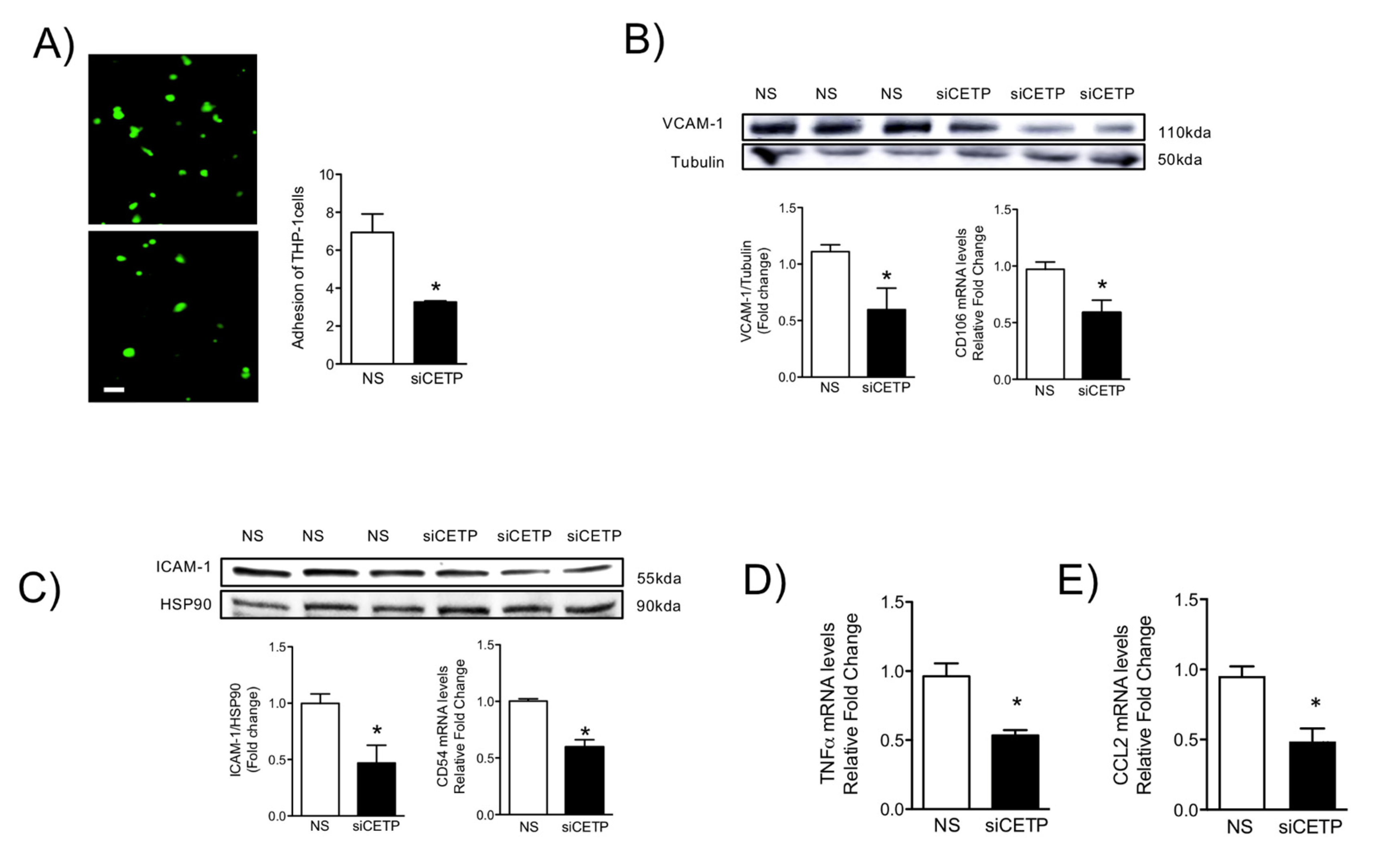
3.5. Removal of CETP Reduces Cell Adhesion Molecules in Endothelial Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Ference, B.A.; Cannon, C.P.; Landmesser, U.; Luscher, T.F.; Catapano, A.L.; Ray, K.K. Reduction of low density lipopro-tein-cholesterol and cardiovascular events with proprotein convertase subtilisin-kexin type 9 (pcsk9) inhibitors and statins: An analysis of fourier, spire, and the cholesterol treatment trialists collaboration. Eur. Heart J. 2018, 39, 2540–2545. [Google Scholar] [CrossRef] [Green Version]
- Lieb, W.; Enserro, D.M.; Larson, M.G.; Vasan, R.S. Residual cardiovascular risk in individuals on lipid-lowering treatment: Quan-tifying absolute and relative risk in the community. Open Heart 2018, 5, e000722. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Targeting residual cardiovascular risk: Raising high-density lipoprotein cholesterol levels. Heart 2008, 94, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.; Harrington, R. Cholesterol, Cardiovascular Risk, Statins, PCSK9 Inhibitors, and the Future of LDL-C Lowering. JAMA 2016, 316, 1967–1968. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.J.; Le Goff, W.; Guerin, M.; Kontush, A. Cholesteryl ester transfer protein: At the heart of the action of li-pid-modulating therapy with statins, fibrates, niacin, and cholesteryl ester transfer protein inhibitors. Eur. Heart J. 2010, 31, 149–164. [Google Scholar] [CrossRef] [Green Version]
- Kontush, A. HDL-mediated mechanisms of protection in cardiovascular disease. Cardiovasc. Res. 2014, 103, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Mineo, C.; Deguchi, H.; Griffin, J.H.; Shaul, P.W. Endothelial and Antithrombotic Actions of HDL. Circ. Res. 2006, 98, 1352–1364. [Google Scholar] [CrossRef] [Green Version]
- Armitage, J.; Holmes, M.V.; Preiss, D. Cholesteryl ester transfer protein inhibition for preventing cardiovascular events: Jacc review topic of the week. J. Am. Coll. Cardiol. 2019, 73, 477–487. [Google Scholar] [CrossRef]
- Shrestha, S.; Wu, B.J.; Guiney, L.; Barter, P.J.; Rye, K. Cholesteryl ester transfer protein and its inhibitors. J. Lipid Res. 2018, 59, 772–783. [Google Scholar] [CrossRef] [Green Version]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.P.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.-C.; Waters, D.D.; et al. Effects of Torcetrapib in Patients at High Risk for Coronary Events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [Green Version]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.; Gibson, C.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.K.; Kallend, D.; Leiter, L.A.; et al. Effects of Dalcetrapib in Patients with a Recent Acute Coronary Syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [Green Version]
- Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; Sammons, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R. HDL in Morbidity and Mortality: A 40+ Year Perspective. Clin. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Berti, J.; Amaral, M.E.C.D.; Boschero, A.; Nunes, V.S.; Harada, L.; Castilho, L.; Oliveira, H.C. Thyroid hormone increases plasma cholesteryl ester transfer protein activity and plasma high-density lipoprotein removal rate in transgenic mice. Metabolism 2001, 50, 530–536. [Google Scholar] [CrossRef]
- Davel, A.P.; Ceravolo, G.S.; Wenceslau, C.F.; Carvalho, M.H.C.; Brum, P.C.; Rossoni, L.V. Increased vascular contractility and oxida-tive stress in beta(2)-adrenoceptor knockout mice: The role of nadph oxidase. J. Vasc. Res. 2012, 49, 342–352. [Google Scholar] [CrossRef]
- Dulce, R.A.; Yiginer, O.; Gonzalez, D.R.; Goss, G.; Feng, N.; Zheng, M.; Hare, J.M. Hydralazine and Organic Nitrates Restore Impaired Excitation-Contraction Coupling by Reducing Calcium Leak Associated with Nitroso-Redox Imbalance. J. Biol. Chem. 2013, 288, 6522–6533. [Google Scholar] [CrossRef] [Green Version]
- Hissin, P.J.; Hilf, R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]
- Alom-Ruiz, S.P.; Anilkumar, N.; Shah, A.M. Reactive Oxygen Species and Endothelial Activation. Antioxid. Redox Signal. 2008, 10, 1089–1100. [Google Scholar] [CrossRef]
- Pober, J.S.; Lapierre, L.A.; Stolpen, A.H.; Brock, T.A.; Springer, T.A.; Fiers, W.; Bevilacqua, M.P.; Mendrick, D.L.; Gimbrone, M.A., Jr. Acti-vation of cultured human endothelial cells by recombinant lymphotoxin: Comparison with tumor necrosis factor and in-terleukin 1 species. J. Immunol. 1987, 138, 3319–3324. [Google Scholar]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arter. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.A.; Deanfield, J.; Ganz, P.; Hamburg, N.M.; Luscher, T.F.; Shechter, M.; Taddei, S.; et al. The assessment of endothelial function: From research into clinical practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.D.; Hryniewicz, K.; Hriljac, I.; Balidemaj, K.; Dimayuga, C.; Hudaihed, A.; Yasskiy, A. Vascular Endothelial Dysfunction and Mortality Risk in Patients With Chronic Heart Failure. Circulation 2005, 111, 310–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Premer, C.; Kanelidis, A.J.; Hare, J.M.; Schulman, I.H. Rethinking Endothelial Dysfunction as a Crucial Target in Fighting Heart Failure. In Mayo Clinic Proceedings: Innovations, Quality & Outcomes; Elsevier BV: Amsterdam, The Netherlands, 2019; Volume 3, pp. 1–13. [Google Scholar]
- Rosenson, R.S.; Brewer, H.B.; Ansell, B.J.; Barter, P.J.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef]
- Harder, C.; Lau, P.; Meng, A.; Whitman, S.C.; McPherson, R. Cholesteryl Ester Transfer Protein (CETP) Expression Protects Against Diet Induced Atherosclerosis in SR-BI Deficient Mice. Arter. Thromb. Vasc. Biol. 2007, 27, 858–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomaraschi, M.; Ossoli, A.; Pozzi, S.; Nilsson, P.; Cefalù, A.B.; Averna, M.; Kuivenhoven, J.A.; Hovingh, G.K.; Veglia, F.; Franceschini, G.; et al. eNOS Activation by HDL Is Impaired in Genetic CETP Deficiency. PLoS ONE 2014, 9, e95925. [Google Scholar] [CrossRef] [PubMed]
- Kurano, M.; Hara, M.; Ikeda, H.; Tsukamoto, K.; Yatomi, Y. Involvement of CETP (Cholesteryl Ester Transfer Protein) in the Shift of Sphingosine-1-Phosphate Among Lipoproteins and in the Modulation of its Functions. Arter. Thromb. Vasc. Biol. 2017, 37, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Zimmet, J.M.; Hare, J.M. Nitroso-redox interactions in the cardiovascular system. Circulation 2006, 114, 1531–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hare, J.M. Nitroso-redox balance in the cardiovascular system. N. Engl. J. Med. 2004, 351, 2112–2214. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Li, H.; Förstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharmacol. 2013, 13, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Benna, J.; Dang, P.M.; Gougerot-Pocidalo, M.A.; Marie, J.C.; Braut-Boucher, F. P47phox, the phagocyte nadph oxidase/nox2 or-ganizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 2009, 41, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kröller-Schön, S.; Steven, S.; Schulz, E.; Münzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017, 174, 1670–1689. [Google Scholar] [CrossRef] [Green Version]
- Battson, M.L.; Lee, D.M.; Gentile, C.L. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am. J. Physiol. Circ. Physiol. 2017, 312, H355–H367. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2003, 11, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Lira, M.E.; Paciga, S.A.; Lloyd, D.B.; Thompson, J.F.; Loomis, A.K. Expression of CETP and of splice variants induces the same level of ER stress despite secretion efficiency differences. J. Lipid Res. 2008, 49, 1955–1962. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Harrison, D.G. Endothelial Dysfunction in Cardiovascular Diseases: The Role of Oxidant Stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [Green Version]
- Hordijk, P.L. Endothelial signalling events during leukocyte transmigration. FEBS J. 2006, 273, 4408–4415. [Google Scholar] [CrossRef]
- Izem, L.; Morton, R.E. Possible Role for Intracellular Cholesteryl Ester Transfer Protein in Adipocyte Lipid Metabolism and Storage. J. Biol. Chem. 2007, 282, 21856–21865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferri, N.; Corsini, A.; Sirtori, C.R.; Ruscica, M. Present therapeutic role of cholesteryl ester transfer protein inhibitors. Pharmacol. Res. 2018, 128, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.C.F.; Raposo, H.F. Cholesteryl Ester Transfer Protein and Lipid Metabolism and Cardiovascular Diseases. Advances Exp. Med. Biol. 2020, 1276, 15–25. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wanschel, A.C.B.A.; Guizoni, D.M.; Lorza-Gil, E.; Salerno, A.G.; Paiva, A.A.; Dorighello, G.G.; Davel, A.P.; Balkan, W.; Hare, J.M.; Oliveira, H.C.F. The Presence of Cholesteryl Ester Transfer Protein (CETP) in Endothelial Cells Generates Vascular Oxidative Stress and Endothelial Dysfunction. Biomolecules 2021, 11, 69. https://doi.org/10.3390/biom11010069
Wanschel ACBA, Guizoni DM, Lorza-Gil E, Salerno AG, Paiva AA, Dorighello GG, Davel AP, Balkan W, Hare JM, Oliveira HCF. The Presence of Cholesteryl Ester Transfer Protein (CETP) in Endothelial Cells Generates Vascular Oxidative Stress and Endothelial Dysfunction. Biomolecules. 2021; 11(1):69. https://doi.org/10.3390/biom11010069
Chicago/Turabian StyleWanschel, Amarylis C. B. A., Daniele M. Guizoni, Estela Lorza-Gil, Alessandro G. Salerno, Adriene A. Paiva, Gabriel G. Dorighello, Ana Paula Davel, Wayne Balkan, Joshua M. Hare, and Helena C. F. Oliveira. 2021. "The Presence of Cholesteryl Ester Transfer Protein (CETP) in Endothelial Cells Generates Vascular Oxidative Stress and Endothelial Dysfunction" Biomolecules 11, no. 1: 69. https://doi.org/10.3390/biom11010069
APA StyleWanschel, A. C. B. A., Guizoni, D. M., Lorza-Gil, E., Salerno, A. G., Paiva, A. A., Dorighello, G. G., Davel, A. P., Balkan, W., Hare, J. M., & Oliveira, H. C. F. (2021). The Presence of Cholesteryl Ester Transfer Protein (CETP) in Endothelial Cells Generates Vascular Oxidative Stress and Endothelial Dysfunction. Biomolecules, 11(1), 69. https://doi.org/10.3390/biom11010069