
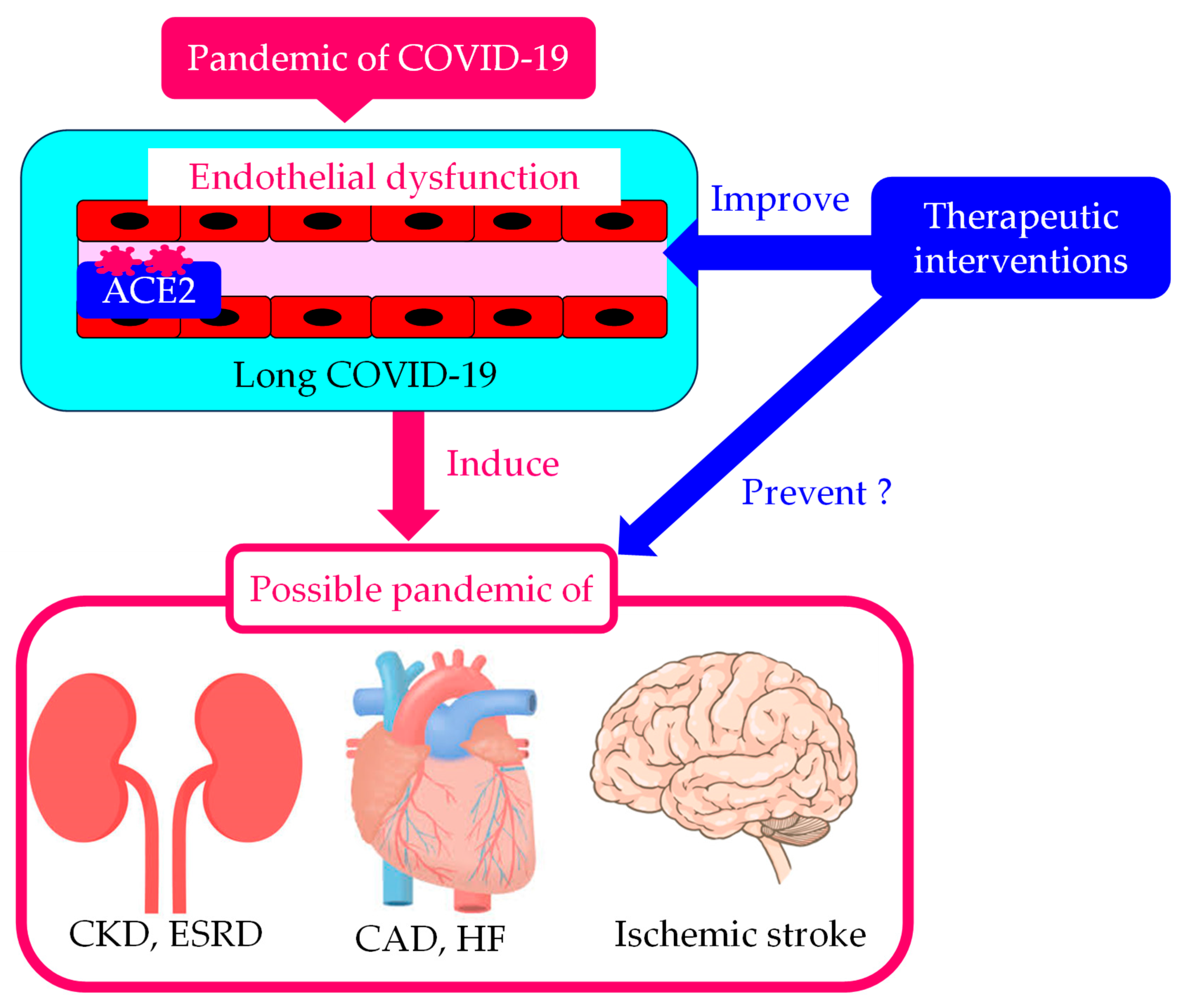
The Significance of Endothelial Dysfunction in Long COVID-19 for the Possible Future Pandemic of Chronic Kidney Disease and Cardiovascular Disease

 , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Association of Pre-Existing Endothelial Dysfunction with the Development of Severe COVID-19
3. A Significant Association of Endothelial Dysfunction with the Development of Long COVID-19
4. The Biomarkers for Endothelial Dysfunction in Patients with Long COVID-19 as Risk Factors for ASCVD
4.1. NADPH Oxidase (NOX) 2 (NOX2), IL-6, and Monocyte Chemoattractant Protein-1 (MCP-1)
4.1.1. The Association of NOX2, IL-6, and MCP-1 with Long COVID-19
4.1.2. The Association of NOX2, IL-6, and MCP-1 with ASCVD
4.2. VWF, Factor VIII, and ADAMST-13
4.2.1. The Association of VWF, Factor VIII, and ADAMST-13 with Long COVID-19
4.2.2. The Association of VWF, Factor VIII, and ADAMST-13 with ASCVD
4.3. Tissue Plasminogen Activator (tPA) and PAI-1
4.3.1. The Association of tPA and PAI-1 with Long COVID-19
4.3.2. The Association of tPA and PAI-1 with ASCVD
4.4. Neutrophil Extracellular Traps (NETs)
4.4.1. The Association of NETs with Long COVID-19
4.4.2. The Association of NETs with ASCVD
4.5. Selectins
4.5.1. The Association of Selectins with Long COVID-19
4.5.2. The Association of Selectins with ASCVD
5. The Association of Long COVID-19 with Atherosclerotic Risk Factors
5.1. Diabetes
5.2. Hypertension
5.3. Dyslipidemia
6. The Association of Long COVID-19 with Chronic Kidney Disease (CKD) and Cardiovascular Diseases
6.1. CKD
6.2. HF
6.3. CAD
6.4. Ischemic Stroke
7. Possible Therapeutic Interventions for Long COVID-19 Considering Endothelial Dysfunction as the Therapeutic Target
7.1. NO
7.2. Antioxidants
7.2.1. Hydrogen-Rich Water
7.2.2. L-Arginine with Vitamin C
7.2.3. Coenzyme Q10 and Alpha Lipoic Acid
7.3. Statins
7.4. N-3 PUFAs
7.5. SGLT2is
8. Limitations of This Review
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lancet, T. Facing up to long COVID. Lancet 2020, 396, 1861. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Wadman, M.; Couzin-Frankel, J.; Kaiser, J.; Matacic, C. A rampage through the body. Science 2020, 368, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Morrone, M.C.; Patrono, C.; Santoro, M.G.; Schiaffino, S.; Remuzzi, G.; Bussolati, G. Long Covid: Where we stand and challenges ahead. Cell Death Differ. 2022, 29, 1891–1900. [Google Scholar] [CrossRef]
- Yelin, D.; Wirtheim, E.; Vetter, P.; Kalil, A.C.; Bruchfeld, J.; Runold, M.; Guaraldi, G.; Mussini, C.; Gudiol, C.; Pujol, M.; et al. Long-term consequences of COVID-19: Research needs. Lancet Infect. Dis. 2020, 20, 1115–1117. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. COVID-19: What do we know about “long covid”? BMJ 2020, 370, m2815. [Google Scholar] [CrossRef]
- Su, S.; Cui, H.; Wang, T.; Shen, X.; Ma, C. Pain: A potential new label of COVID-19. Brain Behav. Immun. 2020, 87, 159–160. [Google Scholar] [CrossRef]
- Hoshijima, H.; Mihara, T.; Seki, H.; Hyuga, S.; Kuratani, N.; Shiga, T. Incidence of long-term post-acute sequelae of SARS-CoV-2 infection related to pain and other symptoms: A systematic review and meta-analysis. PLoS ONE 2023, 18, e0250909. [Google Scholar] [CrossRef]
- Yanai, H. A Significance of High Prevalence of Diabetes and Hypertension in Severe COVID-19 Patients. J. Clin. Med. Res. 2020, 12, 389–392. [Google Scholar] [CrossRef]
- Busetto, L.; Bettini, S.; Fabris, R.; Serra, R.; Pra, C.D.; Maffei, P.; Rossato, M.; Fioretto, P.; Vettor, R. Obesity and COVID-19: An Italian Snapshot. Obesity 2020, 28, 1600–1605. [Google Scholar] [CrossRef] [PubMed]
- Higham, A.; Singh, D. Increased ACE2 Expression in Bronchial Epithelium of COPD Patients who are Overweight. Obesity 2020, 28, 1586–1589. [Google Scholar] [CrossRef] [PubMed]
- Yudkin, J.S. Abnormalities of coagulation and fibrinolysis in insulin resistance. Evidence for a common antecedent? Diabetes Care 1999, 22 (Suppl. S3), C25–C30. [Google Scholar] [PubMed]
- Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H. Significance of Endothelial Dysfunction Amelioration for Sodium-Glucose Cotransporter 2 Inhibitor-Induced Improvements in Heart Failure and Chronic Kidney Disease in Diabetic Patients. Metabolites 2023, 13, 736. [Google Scholar] [CrossRef] [PubMed]
- Sarafidis, P.A.; Bakris, G.L. Review: Insulin and endothelin: An interplay contributing to hypertension development? J. Clin. Endocrinol. Metab. 2007, 92, 379–385. [Google Scholar] [CrossRef]
- Yanai, H. Adiposity is the Crucial Enhancer of COVID-19. Cardiol. Res. 2020, 11, 353–354. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 patients in intensive care unit: A report of thromboelastography findings and other parameters of hemostasis. J. Thromb. Haemost. 2020, 18, 1738–1742. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H. Metabolic Syndrome and COVID-19. Cardiol. Res. 2020, 11, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Moutchia, J.; Pokharel, P.; Kerri, A.; McGaw, K.; Uchai, S.; Nji, M.; Goodman, M. Clinical laboratory parameters associated with severe or critical novel coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. PLoS ONE 2020, 15, e0239802. [Google Scholar] [CrossRef]
- Georgieva, E.; Ananiev, J.; Yovchev, Y.; Arabadzhiev, G.; Abrashev, H.; Abrasheva, D.; Atanasov, V.; Kostandieva, R.; Mitev, M.; Petkova-Parlapanska, K.; et al. COVID-19 Complications: Oxidative Stress, Inflammation, and Mitochondrial and Endothelial Dysfunction. Int. J. Mol. Sci. 2023, 24, 14876. [Google Scholar] [CrossRef] [PubMed]
- Charfeddine, S.; Ibn Hadj Amor, H.; Jdidi, J.; Torjmen, S.; Kraiem, S.; Hammami, R.; Bahloul, A.; Kallel, N.; Moussa, N.; Touil, I.; et al. Long COVID 19 Syndrome: Is It Related to Microcirculation and Endothelial Dysfunction? Insights From TUN-EndCOV Study. Front. Cardiovasc. Med. 2021, 8, 745758. [Google Scholar] [CrossRef]
- Oikonomou, E.; Souvaliotis, N.; Lampsas, S.; Siasos, G.; Poulakou, G.; Theofilis, P.; Papaioannou, T.G.; Haidich, A.B.; Tsaousi, G.; Ntousopoulos, V.; et al. Endothelial dysfunction in acute and long standing COVID-19: A prospective cohort study. Vasc. Pharmacol. 2022, 144, 106975. [Google Scholar] [CrossRef]
- Nicolai, L.; Kaiser, R.; Stark, K. Thromboinflammation in long COVID-the elusive key to postinfection sequelae? J. Thromb. Haemost. 2023, 21, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, H.; Townsend, L.; Morrin, H.; Ahmad, A.; Comerford, C.; Karampini, E.; Englert, H.; Byrne, M.; Bergin, C.; O’Sullivan, J.M.; et al. Persistent endotheliopathy in the pathogenesis of long COVID syndrome. J. Thromb. Haemost. 2021, 19, 2546–2553. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Liberale, L.; Portincasa, P.; Khalil, M.; Galerati, I.; Farella, I.; Noto, A.; JohnBritto, S.; Moriero, M.; Michelauz, C.; et al. Neutrophil degranulation, endothelial and metabolic dysfunction in unvaccinated long COVID patients. Eur. J. Clin. Investig. 2024, 54, e14155. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Sanduzzi Zamparelli, S.; Mosella, M.; Formisano, R.; Molino, A.; Spedicato, G.A.; Papa, A.; Motta, A.; Di Minno, M.N.D.; Maniscalco, M. Clinical assessment of endothelial function in convalescent COVID-19 patients: A meta-analysis with meta-regressions. Ann. Med. 2022, 54, 3234–3249. [Google Scholar] [CrossRef]
- Martins-Gonçalves, R.; Hottz, E.D.; Bozza, P.T. Acute to post-acute COVID-19 thromboinflammation persistence: Mechanisms and potential consequences. Curr. Res. Immunol. 2023, 4, 100058. [Google Scholar] [CrossRef]
- Youn, J.Y.; Zhang, Y.; Wu, Y.; Cannesson, M.; Cai, H. Therapeutic application of estrogen for COVID-19: Attenuation of SARS-CoV-2 spike protein and IL-6 stimulated, ACE2-dependent NOX2 activation, ROS production and MCP-1 upregulation in endothelial cells. Redox. Biol. 2021, 46, 102099. [Google Scholar] [CrossRef]
- Yin, J.X.; Agbana, Y.L.; Sun, Z.S.; Fei, S.W.; Zhao, H.Q.; Zhou, X.N.; Chen, J.H.; Kassegne, K. Increased interleukin-6 is associated with long COVID-19: A systematic review and meta-analysis. Infect. Dis. Poverty 2023, 12, 43. [Google Scholar] [CrossRef]
- Alfadda, A.A.; Rafiullah, M.; Alkhowaiter, M.; Alotaibi, N.; Alzahrani, M.; Binkhamis, K.; Siddiqui, K.; Youssef, A.; Altalhi, H.; Almaghlouth, I.; et al. Clinical and biochemical characteristics of people experiencing post-coronavirus disease 2019-related symptoms: A prospective follow-up investigation. Front. Med. 2022, 9, 1067082. [Google Scholar] [CrossRef]
- Cai, H.; Griendlingm, K.K.; Harrison, D.G. The vascular NAD (P) H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol. Sci. 2003, 24, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, X.L.; Zhao, C.R.; Pan, C.L.; Zhang, Z. Interleukin-6 as a Predictor of the Risk of Cardiovascular Disease: A Meta-Analysis of Prospective Epidemiological Studies. Immunol. Investig. 2018, 47, 689–699. [Google Scholar] [CrossRef]
- Lin, J.; Kakkar, V.; Lu, X. Impact of MCP-1 in atherosclerosis. Curr. Pharm. Des. 2014, 20, 4580–4588. [Google Scholar] [CrossRef]
- Georgakis, M.K.; Gill, D.; Rannikmäe, K.; Traylor, M.; Anderson, C.D.; Lee, J.M.; Kamatani, Y.; Hopewell, J.C.; Worrall, B.B.; Bernhagen, J.; et al. Genetically Determined Levels of Circulating Cytokines and Risk of Stroke. Impact of MCP-1 in atherosclerosis. Circulation 2019, 139, 256–268. [Google Scholar] [CrossRef]
- Ward, S.E.; Curley, G.F.; Lavin, M.; Fogarty, H.; Karampini, E.; McEvoy, N.L.; Clarke, J.; Boylan, M.; Alalqam, R.; Worrall, A.P.; et al. Von Willebrand factor propeptide in severe coronavirus disease 2019 (COVID-19): Evidence of acute and sustained endothelial cell activation. Br. J. Haematol. 2021, 192, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, A.; Pranata, R.; Lim, M.A.; Akbara, M.R.; Martha, J.W. Endotheliopathy marked by high von Willebrand factor (vWF) antigen in COVID-19 is associated with poor outcome: A systematic review and meta-analysis. Int. J. Infect. Dis. 2022, 117, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Terraube, V.; O’Donnell, J.S.; Jenkins, P.V. Factor VIII and von Willebrand factor interaction: Biological, clinical and therapeutic importance. Haemophilia 2010, 16, 3–13. [Google Scholar] [CrossRef]
- Zheng, X.L. ADAMTS13 and von willebrand factor in thrombotic thrombocytopenic purpura. Annu. Rev. Med. 2015, 66, 211–225. [Google Scholar] [CrossRef]
- Prasannan, N.; Heightman, M.; Hillman, T.; Wall, E.; Bell, R.; Kessler, A.; Neave, L.; Doyle, A.; Devaraj, A.; Singh, D.; et al. Impaired exercise capacity in post-COVID-19 syndrome: The role of VWF-ADAMTS13 axis. Blood. Adv. 2022, 6, 4041–4048. [Google Scholar] [CrossRef]
- Mancini, I.; Baronciani, L.; Artoni, A.; Colpani, P.; Biganzoli, M.; Cozzi, G.; Novembrino, C.; Boscolo Anzoletti, M.; De Zan, V.; Pagliari, M.T.; et al. The ADAMTS13-von Willebrand factor axis in COVID-19 patients. J. Thromb. Haemost. 2021, 19, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Feng, Y.; Jia, Y.; Zhang, X.; Li, L.; Bai, X.; Jiao, L. Prognostic value of von willebrand factor and ADAMTS13 in patients with COVID-19: A systematic review and meta-analysis. Thromb. Res. 2022, 218, 83–98. [Google Scholar] [CrossRef]
- Mei, Z.W.; van Wijk Xander, M.R.; Pham, H.P.; Marin, M.J. Role of von willebrand factor in COVID-19 associated coagulopathy. J. Appl. Lab. Med. 2021, 6, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Wang, X.; Fan, M.; Zhao, J.; Lin, L.; Liu, J. Plasma levels of von Willebrand factor in type 2 diabetes patients with and without cardiovascular diseases: A meta-analysis. Diabetes Metab. Res. Rev. 2020, 36, e3193. [Google Scholar] [CrossRef]
- Whincup, P.H.; Danesh, J.; Walker, M.; Lennon, L.; Thomson, A.; Appleby, P.; Rumley, A.; Lowe, G.D. von Willebrand factor and coronary heart disease: Prospective study and meta-analysis. Eur. Heart. J. 2002, 23, 1764–1770. [Google Scholar] [CrossRef]
- Sabater-Lleal, M.; Huffman, J.E.; de Vries, P.S.; Marten, J.; Mastrangelo, M.A.; Song, C.; Pankratz, N.; Ward-Caviness, C.K.; Yanek, L.R.; Trompet, S.; et al. Genome-Wide Association Transethnic Meta-Analyses Identifies Novel Associations Regulating Coagulation Factor VIII and von Willebrand Factor Plasma Levels. Circulation 2019, 139, 620–635. [Google Scholar] [CrossRef]
- Sonneveld, M.A.; de Maat, M.P.; Leebeek, F.W. von Willebrand factor and ADAMTS13 in arterial thrombosis: A systematic review and meta-analysis. Blood. Rev. 2014, 28, 167–178. [Google Scholar] [CrossRef]
- Maino, A.; Siegerink, B.; Lotta, L.A.; Crawley, J.T.; le Cessie, S.; Leebeek, F.W.; Lane, D.A.; Lowe, G.D.; Peyvandi, F.; Rosendaal, F.R. Plasma ADAMTS-13 levels and the risk of myocardial infarction: An individual patient data meta-analysis. J. Thromb. Haemost. 2015, 13, 1396–1404. [Google Scholar] [CrossRef]
- Medcalf, R.L.; Keragala, C.B. The fibrinolytic system: Mysteries and opportunities. Hemasphere 2021, 5, e570. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, K.; Zieger, B. Endothelial cells and coagulation. Cell Tissue Res. 2022, 387, 391–398. [Google Scholar] [CrossRef]
- Zuo, Y.; Warnock, M.; Harbaugh, A.; Yalavarthi, S.; Gockman, K.; Zuo, M.; Madison, J.A.; Knight, J.S.; Kanthi, Y.; Lawrence, D.A. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Sci. Rep. 2021, 11, 1580. [Google Scholar] [CrossRef] [PubMed]
- Nougier, C.; Benoit, R.; Simon, M.; Desmurs-Clavel, H.; Marcotte, G.; Argaud, L.; David, J.S.; Bonnet, A.; Negrier, C.; Dargaud, Y. Hypofibrinolytic state and high thrombin generation may play a major role in SARS-CoV2 associated thrombosis. J. Thromb. Haemost. 2020, 18, 2215–2219. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Garcia, D.; Miltiades, A.; Yim, P.; Parsons, S.; Elisman, K.; Mansouri, M.T.; Wagener, G.; Harrison, N.L. Plasma biomarkers associated with survival and thrombosis in hospitalized COVID-19 patients. Int. J. Hematol. 2022, 116, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, E.; Barion, B.G.; da Rocha, T.R.F.; Di Giacomo, G.; Ho, Y.L.; Rothschild, C.; Fatobene, G.; de Carvalho Moraes, B.D.G.; Stefanello, B.; Villaça, P.R.; et al. Persistent hypofibrinolysis in severe COVID-19 associated with elevated fibrinolysis inhibitors activity. J. Thromb. Thrombolysis 2024, 57, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Jarahzadeh, M.H.; Dastgheib, S.A.; Seifi-Shalamzari, N.; Raee-Ezzabadi, A.; Sadeghizadeh-Yazdi, J.; Akbarian, E.; Neamatzadeh, H. Association of PAI-1 rs1799889 Polymorphism with Susceptibility to Ischemic Stroke: A Huge Meta-Analysis based on 44 Studies. Acta. Medica 2020, 63, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Nikolopoulos, G.K.; Bagos, P.G.; Tsangaris, I.; Tsiara, C.G.; Kopterides, P.; Vaiopoulos, A.; Kapsimali, V.; Bonovas, S.; Tsantes, A.E. The association between plasminogen activator inhibitor type 1 (PAI-1) levels, PAI-1 4G/5G polymorphism, and myocardial infarction: A Mendelian randomization meta-analysis. Clin. Chem. Lab. Med. 2014, 52, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y. Plasminogen activator inhibitor-1 4G/5G gene polymorphism and coronary artery disease in the Chinese Han population: A meta-analysis. PLoS ONE 2012, 7, e33511. [Google Scholar] [CrossRef] [PubMed]
- Jung, R.G.; Motazedian, P.; Ramirez, F.D.; Simard, T.; Di Santo, P.; Visintini, S.; Faraz, M.A.; Labinaz, A.; Jung, Y.; Hibbert, B. Association between plasminogen activator inhibitor-1 and cardiovascular events: A systematic review and meta-analysis. Thromb. J. 2018, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Urban, C.F.; Ermert, D.; Schmid, M.; Abu-Abed, U.; Goosmann, C.; Nacken, W.; Brinkmann, V.; Jungblut, P.R.; Zychlinsky, A. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009, 5, e1000639. [Google Scholar] [CrossRef]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Matsuyama, S.; Shirato, K.; Maejima, M.; Fukushi, S.; Morikawa, S.; Taguchi, F. Entry from the cell surface of severe acute respiratory syndrome coronavirus with cleaved S protein as revealed by pseudotype virus bearing cleaved S protein. J. Virol. 2008, 82, 11985–11991. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef] [PubMed]
- Pramitasuri, T.I.; Laksmidewi, A.A.A.P.; Putra, I.B.K.; Dalimartha, F.A. Neutrophil Extracellular Traps in Coronavirus Disease-19-Associated Ischemic Stroke: A Novel Avenue in Neuroscience. Exp. Neurobiol. 2021, 30, 1–12. [Google Scholar] [CrossRef]
- Krinsky, N.; Sizikov, S.; Nissim, S.; Dror, A.; Sas, A.; Prinz, H.; Pri-Or, E.; Perek, S.; Raz-Pasteur, A.; Lejbkowicz, I.; et al. NETosis induction reflects COVID-19 severity and long COVID: Insights from a 2-center patient cohort study in Israel. J. Thromb. Haemost. 2023, 21, 2569–2584. [Google Scholar] [CrossRef]
- Shafqat, A.; Omer, M.H.; Albalkhi, I.; Alabdul Razzak, G.; Abdulkader, H.; Abdul Rab, S.; Sabbah, B.N.; Alkattan, K.; Yaqinuddin, A. Neutrophil extracellular traps and long COVID. Front. Immunol. 2023, 14, 1254310. [Google Scholar] [CrossRef]
- Megens, R.T.; Vijayan, S.; Lievens, D.; Doring, Y.; van Zandvoort, M.A.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 2012, 107, 597–598. [Google Scholar] [CrossRef]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef]
- Soehnlein, O.; Zernecke, A.; Eriksson, E.E.; Rothfuchs, A.G.; Pham, C.T.; Herwald, H.; Bidzhekov, K.; Rottenberg, M.E.; Weber, C.; Lindbom, L. Neutrophil secretion products pave the way for inflammatory monocytes. Blood 2008, 112, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Tangeten, C.; Zouaoui Boudjeltia, K.; Delporte, C.; Van Antwerpen, P.; Korpak, K. Unexpected Role of MPO-Oxidized LDLs in Atherosclerosis: In between Inflammation and Its Resolution. Antioxidants 2022, 11, 874. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Larragoiti, N.; Cano-Mendez, A.; Jimenez-Vega, Y.; Trujillo, M.; Guzman-Cancino, P.; Ambriz-Murillo, Y.; Viveros-Sandoval, M.E. Inflammatory and Prothrombotic Biomarkers Contribute to the Persistence of Sequelae in Recovered COVID-19 Patients. Int. J. Mol. Sci. 2023, 24, 17468. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Chen, A.; Delahunty, M.D.; Moore, K.L.; Watson, S.R.; McEver, R.P.; Tedder, T.F. L-selectin binds to P-selectin glycoprotein ligand-1 on leukocytes: Interactions between the lectin, epidermal growth factor, and consensus repeat domains of the selectins determine ligand binding specificity. J. Immunol. 1996, 157, 3995–4004. [Google Scholar] [CrossRef]
- Ding, G.; Wang, J.; Liu, K.; Huang, B.; Deng, W.; He, T. Association of E-Selectin gene rs5361 polymorphism with ischemic stroke susceptibility: A systematic review and Meta-analysis. Int. J. Neurosci. 2021, 131, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lou, Y.; Lu, L.; Liu, Y.; Chen, Q.; Chen, X.; Jin, W. Heterogeneous effect of two selectin gene polymorphisms on coronary artery disease risk: A meta-analysis. PLoS ONE 2014, 9, e88152. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, J.; Du, X.; Song, M.; Jia, C.; Liu, H. Association of A561C and G98T polymorphisms in E-selectin gene with coronary artery disease: A meta-analysis. PLoS ONE 2013, 8, e79301. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.; Yang, M.; Sun, M.; Pan, B.; Wang, Q.; Wang, J.; Tian, J.; Ding, G.; Yang, K.; Song, X.; et al. Risk of incident diabetes after COVID-19 infection: A systematic review and meta-analysis. Metabolism 2022, 137, 155330. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Pal, R.; Dutta, S. Risk of incident diabetes post-COVID-19: A systematic review and meta-analysis. Prim. Care Diabetes 2022, 16, 591–593. [Google Scholar] [CrossRef]
- Zuin, M.; Rigatelli, G.; Bilato, C.; Pasquetto, G.; Mazza, A. Risk of Incident New-Onset Arterial Hypertension after COVID-19 Recovery: A Systematic Review and Meta-analysis. High Blood Press. Cardiovasc. Prev. 2023, 30, 227–233. [Google Scholar] [CrossRef]
- Xu, E.; Xie, Y.; Al-Aly, Z. Risks and burdens of incident dyslipidaemia in long COVID: A cohort study. Lancet Diabetes Endocrinol. 2023, 11, 120–128. [Google Scholar] [CrossRef]
- Yilmaz, M.I.; Saglam, M.; Caglar, K.; Cakir, E.; Sonmez, A.; Ozgurtas, T.; Aydin, A.; Eyileten, T.; Ozcan, O.; Acikel, C.; et al. The determinants of endothelial dysfunction in CKD: Oxidative stress and asymmetric dimethylarginine. Am. J. Kidney Dis. 2006, 47, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.C.; Imig, J.D.; Kalantar-Zadeh, K.; Gupta, A. Kidney in the net of acute and long-haul coronavirus disease 2019: A potential role for lipid mediators in causing renal injury and fibrosis. Curr. Opin. Nephrol. Hypertens. 2022, 31, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Teng, L.; Song, X.; Zhang, M.; Han, Y.; Chang, G.; Chang, W.; Shen, Z. The pattern of cytokines expression and dynamic changes of renal function at 6 months in patients with Omicron COVID-19. J. Med. Virol. 2023, 95, e28477. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Yuan, S.; Wei, J.; Xia, S.; Huang, Y.; Chen, X.; Han, Y.; Li, Z.; Xiao, Y.; Peng, F.; et al. Clinical and pathological features of omicron variant of SARS-CoV-2-associated kidney injury. J. Med. Virol. 2023, 95, e29196. [Google Scholar] [CrossRef] [PubMed]
- Atiquzzaman, M.; Thompson, J.R.; Shao, S.; Djurdjev, O.; Bevilacqua, M.; Wong, M.M.Y.; Levin, A.; Birks, P.C. Long-term effect of COVID-19 infection on kidney function among COVID-19 patients followed in post-COVID-19 recovery clinics in British Columbia, Canada. Nephrol. Dial. Transplant. 2023, 38, 2816–2825. [Google Scholar] [CrossRef] [PubMed]
- Lam, I.C.H.; Wong, C.K.H.; Zhang, R.; Chui, C.S.L.; Lai, F.T.T.; Li, X.; Chan, E.W.Y.; Luo, H.; Zhang, Q.; Man, K.K.C.; et al. Long-term post-acute sequelae of COVID-19 infection: A retrospective, multi-database cohort study in Hong Kong and the UK. EClinicalMedicine 2023, 60, 102000. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Cao, B. Severe COVID-19 and chronic kidney disease: Bidirectional mendelian randomization study. Virol. J. 2024, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Taneska, A.C.; Rambabova-Bushljetik, I.; Markovska, Z.S.; Milenkova, M.; Vasileva, A.S.; Zafirova, B.; Pushevski, V.; Severova, G.; Trajceska, L.; Spasovski, G. Predictive Admission Risk Factors, Clinical Features and Kidney Outcomes in COVID-19 Hospitalised Patients with Acute Kidney Injury. Prilozi 2023, 44, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Babel, N.; Hugo, C.; Westhoff, T.H. Vaccination in patients with kidney failure: Lessons from COVID-19. Nat. Rev. Nephrol. 2022, 18, 708–723. [Google Scholar] [CrossRef]
- Stai, S.; Lioulios, G.; Christodoulou, M.; Kasimatis, E.; Fylaktou, A.; Stangou, M. COVID-19 Infection and Response to Vaccination in Chronic Kidney Disease and Renal Transplantation: A Brief Presentation. Life 2022, 12, 1358. [Google Scholar] [CrossRef] [PubMed]
- Tsai, E.J.; Čiháková, D.; Tucker, N.R. Cell-Specific Mechanisms in the Heart of COVID-19 Patients. Circ. Res. 2023, 132, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Honchar, O.; Ashcheulova, T. Short-term echocardiographic follow-up after hospitalization for COVID-19: A focus on early post-acute changes. Front. Cardiovasc. Med. 2023, 10, 1250656. [Google Scholar] [CrossRef] [PubMed]
- Zuin, M.; Rigatelli, G.; Roncon, L.; Pasquetto, G.; Bilato, C. Risk of incident heart failure after COVID-19 recovery: A systematic review and meta-analysis. Heart. Fail. Rev. 2023, 28, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, P.U.; Udupa, H.; Ks, J.; Babu, S.K.N.; Jain, N.; Das, R.; Upadhyai, P. The impact of COVID-19 on pulmonary, neurological, and cardiac outcomes: Evidence from a Mendelian randomization study. Front. Public Health 2023, 11, 1303183. [Google Scholar] [CrossRef]
- Zuin, M.; Rigatelli, G.; Battisti, V.; Costola, G.; Roncon, L.; Bilato, C. Increased risk of acute myocardial infarction after COVID-19 recovery: A systematic review and meta-analysis. Int. J. Cardiol. 2023, 372, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Zuin, M.; Mazzitelli, M.; Rigatelli, G.; Bilato, C.; Cattelan, A.M. Risk of ischemic stroke in patients recovered from COVID-19 infection: A systematic review and meta-analysis. Eur. Stroke J. 2023, 8, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ming, X.F. Recent advances in understanding endothelial dysfunction in atherosclerosis. Clin. Med. Res. 2006, 4, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Bauer, V.; Sotnikova, R. Nitric oxide-the endothelium-derived relaxing factor and its role in endothelial functions. Gen. Physiol. Biophys. 2010, 29, 319–340. [Google Scholar] [CrossRef]
- Stankevicius, E.; Kevelaitis, E.; Vainorius, E.; Simonsen, U. Role of nitric oxide and other endothelium-derived factors. Medicina 2003, 39, 333–341. [Google Scholar]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Anti-inflammatory Action of Statins in Cardiovascular Disease: The Role of Inflammasome and Toll-Like Receptor Pathways. Clin. Rev. Allergy Immunol. 2021, 60, 175–199. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Bakogiannis, C.; Leeson, P.; Guzik, T.J.; Zhang, M.H.; Tousoulis, D.; Antonopoulos, A.S.; Demosthenous, M.; Marinou, K.; Hale, A.; et al. Rapid, direct effects of statin treatment on arterial redox state and nitric oxide bioavailability in human atherosclerosis via tetrahydrobiopterin-mediated endothelial nitric oxide synthase coupling. Circulation 2011, 124, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Sola, S.; Mir, M.Q.; Lerakis, S.; Tandon, N.; Khan, B.V. Atorvastatin improves left ventricular systolic function and serum markers of inflammation in nonischemic heart failure. J. Am. Coll. Cardiol. 2006, 47, 332–337. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Jiang, H.; Sun, A.; Wang, Y.; Zou, Y.; Ge, J.; Chen, H. Effects of statin therapy on inflammatory markers in chronic heart failure: A meta-analysis of randomized controlled trials. Arch. Med. Res. 2010, 41, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Gubari, M.I.M. Effect of omega-3 fatty acid supplementation on markers of inflammation and endothelial function in patients with chronic heart disease: A systematic review and meta-analysis. Cell. Mol. Biol 2024, 70, 171–177. [Google Scholar]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H. Sodium-glucose cotransporter 2 inhibitors and death and heart failure in type 2 diabetes. Ann. Transl. Med. 2017, 5, 470. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Cherney, D.Z.I.; Zinman, B.; Inzucchi, S.E.; Koitka-Weber, A.; Mattheus, M.; von Eynatten, M.; Wanner, C. Effects of empagliflozin on the urinary albumin-to-creatinine ratio in patients with type 2 diabetes and established cardiovascular disease: An exploratory analysis from the EMPA-REG OUTCOME randomised, placebo-controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Shigiyama, F.; Kumashiro, N.; Miyagi, M.; Ikehara, K.; Kanda, E.; Uchino, H.; Hirose, T. Effectiveness of dapagliflozin on vascular endothelial function and glycemic control in patients with early-stage type 2 diabetes mellitus: DEFENCE study. Cardiovasc. Diabetol. 2017, 16, 84. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.J.; Sanchez, M.J.; Sanchez, R.A. Diabetic patients with essential hypertension treated with amlodipine: Blood pressure and arterial stiffness effects of canagliflozin or perindopril. J. Hypertens. 2019, 37, 636–642. [Google Scholar] [CrossRef]
- Sezai, A.; Sekino, H.; Unosawa, S.; Taoka, M.; Osaka, S.; Tanaka, M. Canagliflozin for Japanese patients with chronic heart failure and type II diabetes. Cardiovasc. Diabetol. 2019, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Pozdnyakova, D.D.; Bakhareva, T.A.; Baranova, I.A.; Selemir, V.D.; Chuchalin, A.G. Rehabilitation program of post-COVID-19 syndrome with the use of nitric oxide and molecular hydrogen. Ter. Arkh. 2024, 96, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Xie, Y.; Dong, G.; Yin, M.; Shang, Z.; Zhou, K.; Bao, D.; Zhou, J. The Effect of 14-Day Consumption of Hydrogen-Rich Water Alleviates Fatigue but Does Not Ameliorate Dyspnea in Long-COVID Patients: A Pilot, Single-Blind, and Randomized, Controlled Trial. Nutrients 2024, 16, 1529. [Google Scholar] [CrossRef]
- Wu, G.; Meininger, C.J.; McNeal, C.J.; Bazer, F.W.; Rhoads, J.M. Role of l-Arginine in nitric oxide synthesis and health in humans. Adv. Exp. Med. Biol. 2021, 1332, 167–187. [Google Scholar]
- Adebayo, A.; Varzideh, F.; Wilson, S.; Gambardella, J.; Eacobacci, M.; Jankauskas, S.S.; Donkor, K.; Kansakar, U.; Trimarco, V.; Mone, P.; et al. l-Arginine and COVID-19: An Update. Nutrients 2021, 13, 3951. [Google Scholar] [CrossRef]
- Bronte, V.; Zanovello, P. Regulation of immune responses by l-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef]
- Morelli, M.B.; Gambardella, J.; Castellanos, V.; Trimarco, V.; Santulli, G. Vitamin C and Cardiovascular Disease: An Update. Antioxidants 2020, 9, 1227. [Google Scholar] [CrossRef]
- Heller, R.; Munscher-Paulig, F.; Grabner, R.; Till, U. L-Ascorbic acid potentiates nitric oxide synthesis in endothelial cells. J. Biol. Chem. 1999, 274, 8254–8260. [Google Scholar] [CrossRef]
- Huang, A.; Vita, J.A.; Venema, R.C.; Keaney, J.F., Jr. Ascorbic acid enhances endothelial nitric-oxide synthase activity by increasing intracellular tetrahydrobiopterin. J. Biol. Chem. 2000, 275, 17399–17406. [Google Scholar] [CrossRef]
- Baker, T.A.; Milstien, S.; Katusic, Z.S. Effect of vitamin C on the availability of tetrahydrobiopterin in human endothelial cells. J. Cardiovasc. Pharmacol. 2001, 37, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Calvani, R.; Gervasoni, J.; Picca, A.; Ciciarello, F.; Galluzzo, V.; Coelho-Júnior, H.J.; Di Mario, C.; Gremese, E.; Lomuscio, S.; Paglionico, A.M.; et al. Effects of l-Arginine Plus Vitamin C Supplementation on l-Arginine Metabolism in Adults with Long COVID: Secondary Analysis of a Randomized Clinical Trial. Int. J. Mol. Sci. 2023, 24, 5078. [Google Scholar] [CrossRef]
- Izzo, R.; Trimarco, V.; Mone, P.; Aloè, T.; Capra Marzani, M.; Diana, A.; Fazio, G.; Mallardo, M.; Maniscalco, M.; Marazzi, G.; et al. Combining L-Arginine with vitamin C improves long-COVID symptoms: The LINCOLN Survey. Pharmacol. Res. 2022, 183, 106360. [Google Scholar] [CrossRef] [PubMed]
- Tosato, M.; Calvanim, R.; Picca, A.; Ciciarello, F.; Galluzzo, V.; Coelho-Júnior, H.J.; Di Giorgio, A.; Di Mario, C.; Gervasoni, J.; Gremese, E.; et al. Effects of l-Arginine Plus Vitamin C Supplementation on Physical Performance, Endothelial Function, and Persistent Fatigue in Adults with Long COVID: A Single-Blind Randomized Controlled Trial. Nutrients 2022, 14, 4984. [Google Scholar] [CrossRef]
- Barletta, M.A.; Marino, G.; Spagnolo, B.; Bianchi, F.P.; Falappone, P.C.F.; Spagnolo, L.; Gatti, P. Coenzyme Q10 + alpha lipoic acid for chronic COVID syndrome. Clin. Exp. Med. 2023, 23, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Sadeghdoust, M.; Aligolighasemabadi, F.; Dehesh, T.; Taefehshokr, N.; Sadeghdoust, A.; Kotfis, K.; Hashemiattar, A.; Ravandi, A.; Aligolighasemabadi, N.; Vakili, O.; et al. The Effects of Statins on Respiratory Symptoms and Pulmonary Fibrosis in COVID-19 Patients with Diabetes Mellitus: A Longitudinal Multicenter Study. Arch. Immunol. Ther. Exp. 2023, 71, 8. [Google Scholar] [CrossRef]
- Grote, K.; Schaefer, A.C.; Soufi, M.; Ruppert, V.; Linne, U.; Mukund Bhagwat, A.; Szymanski, W.; Graumann, J.; Gercke, Y.; Aldudak, S.; et al. Targeting the High-Density Lipoprotein Proteome for the Treatment of Post-Acute Sequelae of SARS-CoV-2. Int. J. Mol. Sci. 2024, 25, 4522. [Google Scholar] [CrossRef]
- Yang, C.P.; Chang, C.M.; Yang, C.C.; Pariante, C.M.; Su, K.P. Long COVID and long chain fatty acids (LCFAs): Psychoneuroimmunity implication of omega-3 LCFAs in delayed consequences of COVID-19. Brain. Behav. Immun. 2022, 103, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Mroueh, A.; Fakih, W.; Carmona, A.; Trimaille, A.; Matsushita, K.; Marchandot, B.; Qureshi, A.W.; Gong, D.S.; Auger, C.; Sattler, L.; et al. COVID-19 promotes endothelial dysfunction and thrombogenicity: Role of proinflammatory cytokines/SGLT2 prooxidant pathway. J. Thromb. Haemost. 2024, 22, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Talotta, R.; Robertson, E. Autoimmunity as the comet tail of COVID-19 pandemic. World J. Clin. Cases 2020, 8, 3621–3644. [Google Scholar] [CrossRef] [PubMed]
- Kozłowski, P.; Leszczyńska, A.; Ciepiela, O. Long COVID Definition, Symptoms, Risk Factors, Epidemiology and Autoimmunity: A Narrative Review. Am. J. Med. Open 2024, 11, 100068. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanai, H.; Adachi, H.; Hakoshima, M.; Katsuyama, H.; Sako, A. The Significance of Endothelial Dysfunction in Long COVID-19 for the Possible Future Pandemic of Chronic Kidney Disease and Cardiovascular Disease. Biomolecules 2024, 14, 965. https://doi.org/10.3390/biom14080965
Yanai H, Adachi H, Hakoshima M, Katsuyama H, Sako A. The Significance of Endothelial Dysfunction in Long COVID-19 for the Possible Future Pandemic of Chronic Kidney Disease and Cardiovascular Disease. Biomolecules. 2024; 14(8):965. https://doi.org/10.3390/biom14080965
Chicago/Turabian StyleYanai, Hidekatsu, Hiroki Adachi, Mariko Hakoshima, Hisayuki Katsuyama, and Akahito Sako. 2024. "The Significance of Endothelial Dysfunction in Long COVID-19 for the Possible Future Pandemic of Chronic Kidney Disease and Cardiovascular Disease" Biomolecules 14, no. 8: 965. https://doi.org/10.3390/biom14080965
APA StyleYanai, H., Adachi, H., Hakoshima, M., Katsuyama, H., & Sako, A. (2024). The Significance of Endothelial Dysfunction in Long COVID-19 for the Possible Future Pandemic of Chronic Kidney Disease and Cardiovascular Disease. Biomolecules, 14(8), 965. https://doi.org/10.3390/biom14080965