
Multi-Omics Analyses Reveal Systemic Insights into Maize Vivipary

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
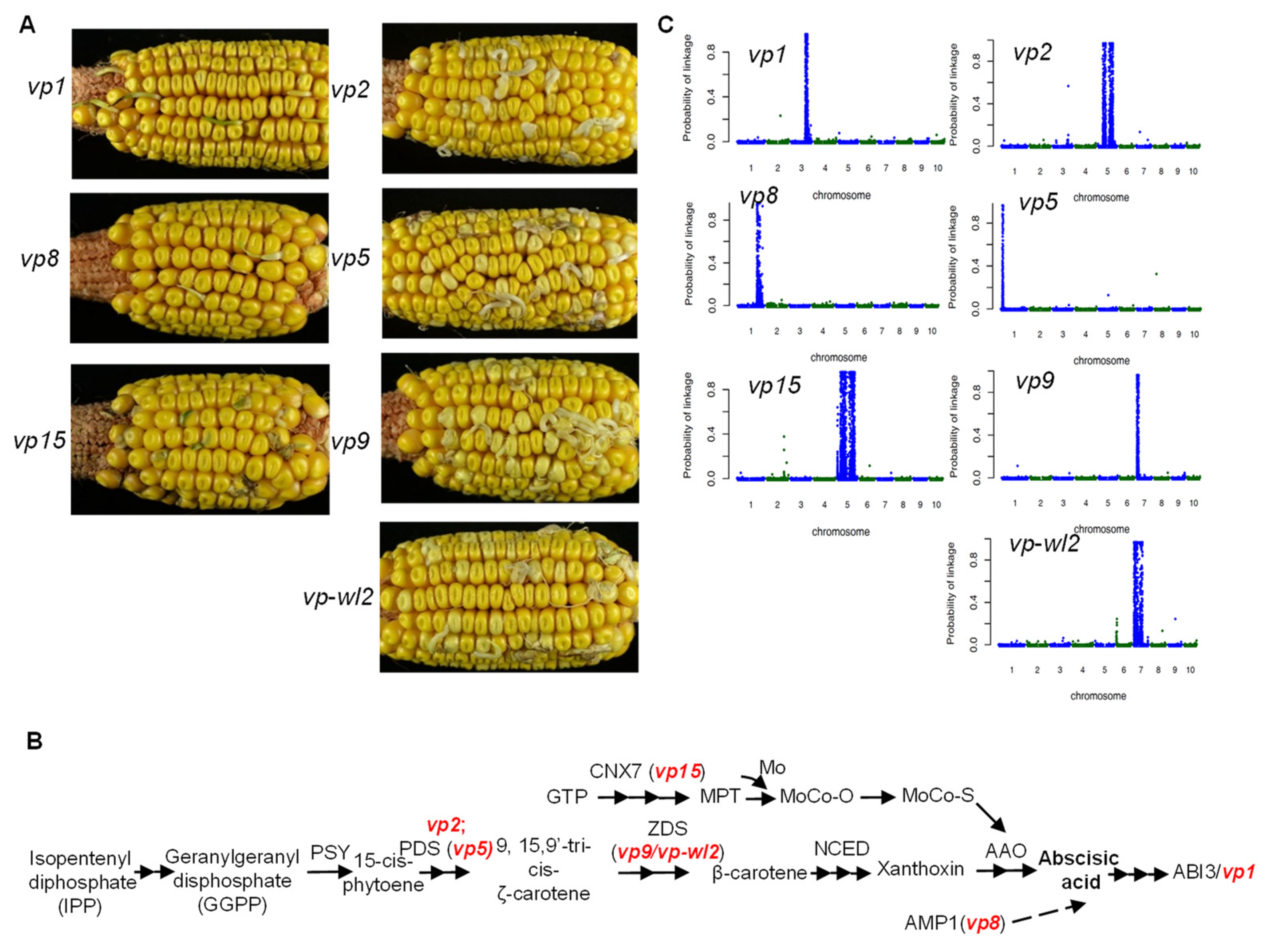
2.1. Phenotype of Vivipary Mutants
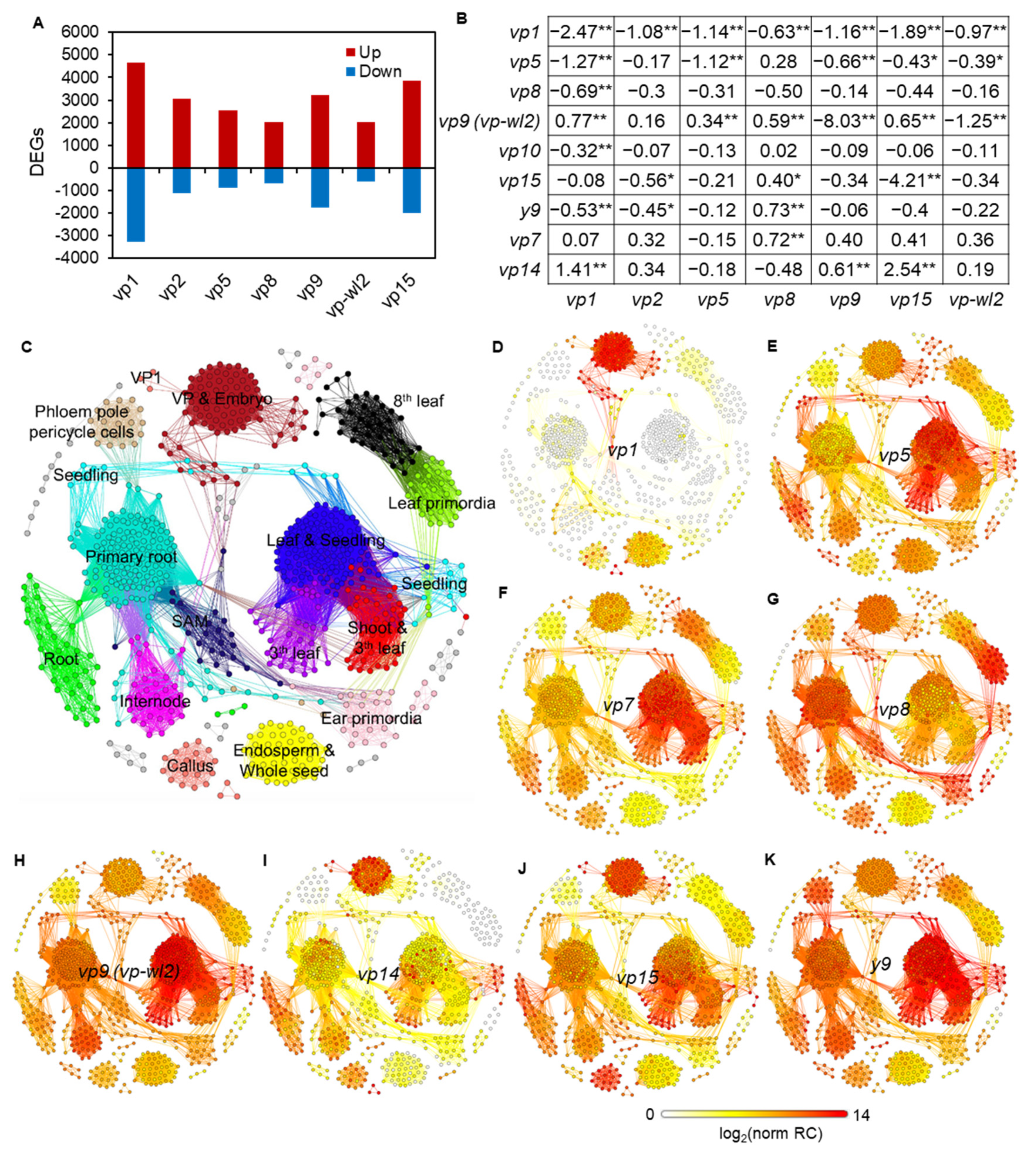
2.2. Common Biological Processes Affected in Vivipary Mutants
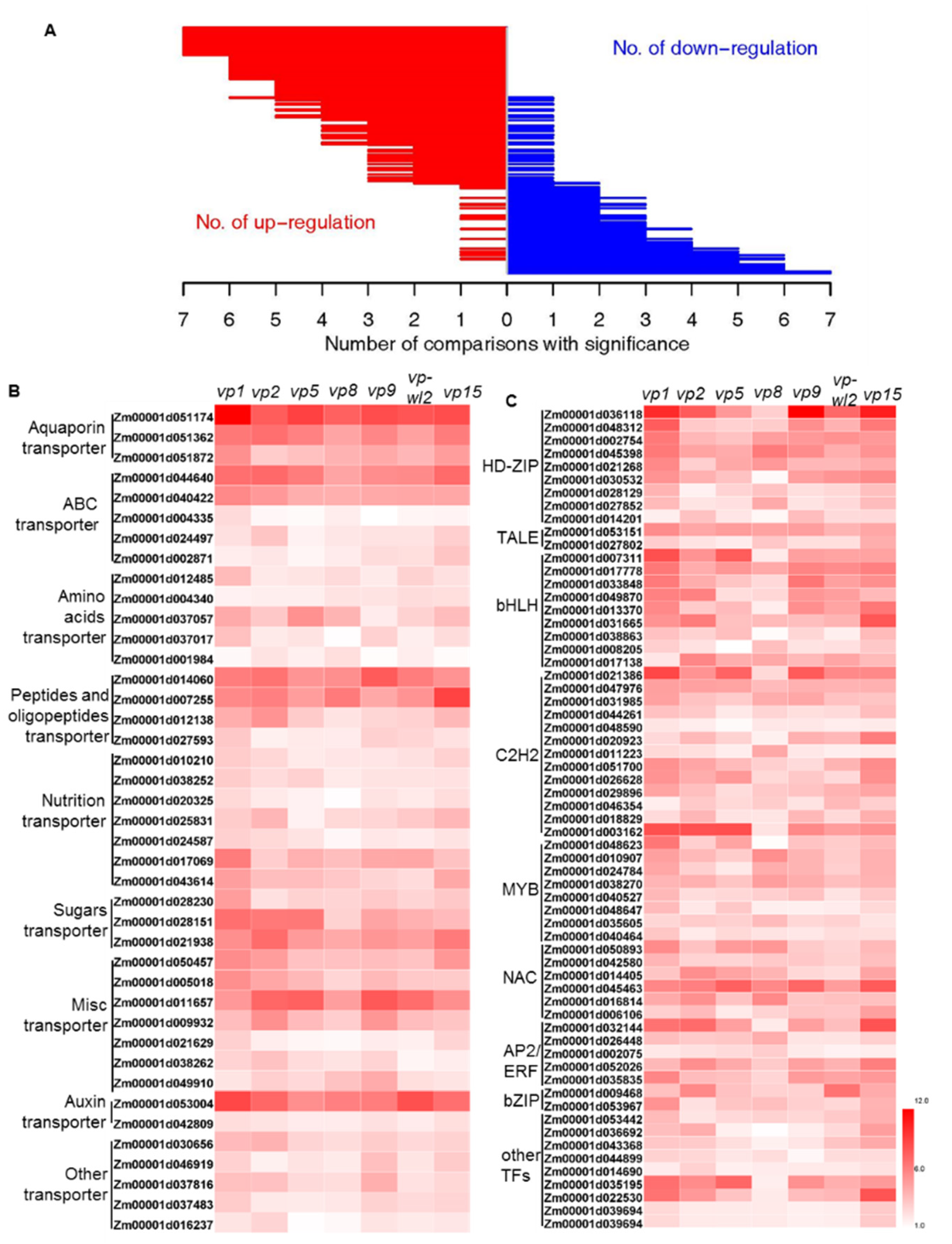
2.3. Transporters and Transcription Factors (TFs) with Altered Expression in Seven Vivipary Mutants
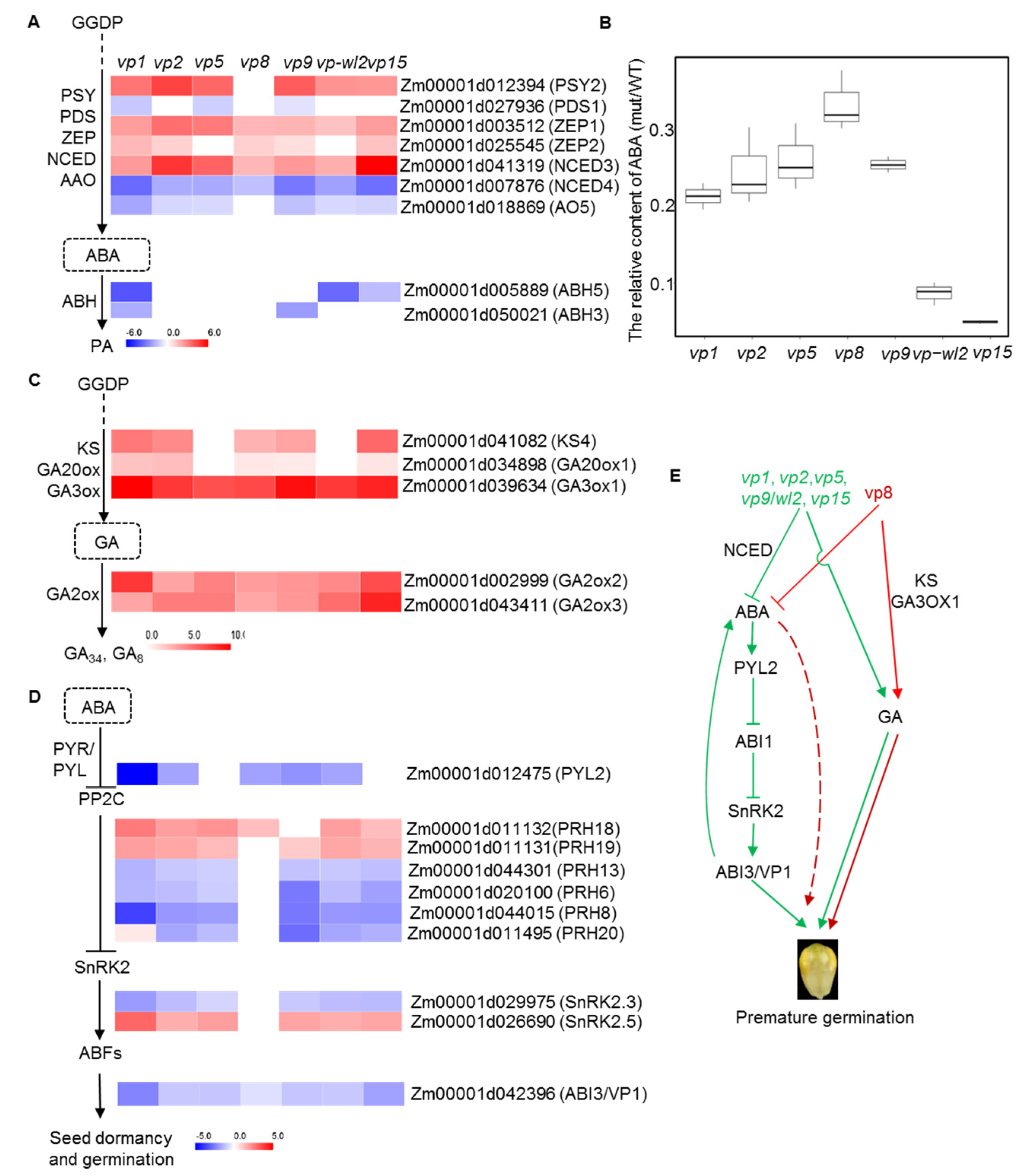
2.4. ABA- and GA-Related Genes Were Differentially Expressed in Vivipary Mutants
2.5. Vivipary Mutants Had Distinct Patterns of Metabolite Accumulation
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction and Sequencing
4.3. BSR-Seq Mapping
4.4. Gene Ontology Analysis
4.5. Analysis of ABA Content
4.6. WGCNA Network Construction and Module Identification
4.7. Metabolomic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fang, J.; Chai, C.; Qian, Q.; Li, C.; Tang, J.; Sun, L.; Huang, Z.; Guo, X.; Sun, C.; Liu, M.; et al. Mutations of genes in synthesis of the carotenoid precursors of ABA lead to pre-harvest sprouting and photo-oxidation in rice. Plant J. 2008, 54, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Xu, F.; Fang, J.; Gao, S.; Tang, J.; Fang, S.; Wang, H.; Tong, H.; Zhang, F.; Chu, J.; et al. Endosperm sugar accumulation caused by mutation of PHS8/ISA1 leads to pre-harvest sprouting in rice. Plant J. 2018, 95, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Burbidge, A.; Grieve, T.M.; Jackson, A.; Thompson, A.; McCarty, D.R.; Taylor, I.B. Characterization of the ABA-deficient tomato mutant notabilis and its relationship with maize Vp14. Plant J. 1999, 17, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Sagi, M.; Scazzocchio, C.; Fluhr, R. The absence of molybdenum cofactor sulfuration is the primary cause of the flacca phenotype in tomato plants. Plant J. 2002, 31, 305–317. [Google Scholar] [CrossRef]
- Harrison, E.; Burbidge, A.; Okyere, J.P.; Thompson, A.J.; Taylor, I.B. Identification of the tomato ABA-deficient mutant sitiens as a member of the ABA-aldehyde oxidase gene family using genetic and genomic analysis. Plant Growth Regul. 2011, 64, 301–309. [Google Scholar] [CrossRef]
- Robertson, D.S. The genetics of vivipary in maize. Genetics 1955, 40, 745–760. [Google Scholar] [CrossRef] [PubMed]
- McCarty, D.R.; Hattori, T.; Carson, C.B.; Vasil, V.; Lazar, M.; Vasil, I.K. The Viviparous-1 developmental gene of maize encodes a novel transcriptional activator. Cell 1991, 66, 895–905. [Google Scholar] [CrossRef]
- Hable, W.E.; Oishi, K.K.; Schumaker, K.S. Viviparous-5 encodes phytoene desaturase, an enzyme essential for abscisic acid (ABA) accumulation and seed development in maize. Mol. Gen. Genet. 1998, 257, 167–176. [Google Scholar] [CrossRef]
- Singh, M.; Lewis, P.E.; Hardeman, K.; Bai, L.; Rose, J.K.C.; Mazourek, M.; Chomet, P.; Brutnell, T.P. Activator mutagenesis of the Pink scutellum1/viviparous7 locus of maize. Plant Cell 2003, 15, 874–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Li, J.; Fan, K.; Du, Y.; Ren, Z.; Xu, J.; Zheng, J.; Liu, Y.; Fu, J.; Ren, D.; et al. Mutations in the maize zeta-carotene desaturase gene lead to viviparous kernel. PLoS ONE 2017, 12, e0174270. [Google Scholar] [CrossRef]
- Porch, T.G.; Tseung, C.W.; Schmelz, E.A.; Mark Settles, A. The maize Viviparous10/Viviparous13 locus encodes the Cnx1 gene required for molybdenum cofactor biosynthesis. Plant J. 2006, 45, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.H.; Tan, B.C.; Gage, D.A.; Zeevaart, J.A.D.; McCarty, D.R. Specific oxidative cleavage of carotenoids by VP14 of maize. Science 1997, 276, 1872–1874. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Mark Settles, A.; Tseung, C.W.; Li, Q.B.; Latshaw, S.; Wu, S.; Porch, T.G.; Schmelz, E.A.; James, M.G.; McCarty, D.R. The maize viviparous15 locus encodes the molybdopterin synthase small subunit. Plant J. 2006, 45, 264–274. [Google Scholar] [CrossRef]
- Li, F.; Murillo, C.; Wurtzel, E.T. Maize Y9 encodes a product essential for 15-cis-ζ-carotene isomerization. Plant Physiol. 2007, 144, 1181–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Li, G.J.; Bressan, R.A.; Song, C.P.; Zhu, J.K.; Zhao, Y. Abscisic acid dynamics, signaling, and functions in plants. J. Integr. Plant Biol. 2020, 62, 25–54. [Google Scholar] [CrossRef] [Green Version]
- Tuan, P.A.; Kumar, R.; Rehal, P.K.; Toora, P.K.; Ayele, B.T. Molecular mechanisms underlying abscisic acid/gibberellin balance in the control of seed dormancy and germination in cereals. Front. Plant Sci. 2018, 9, 668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucera, B.; Cohn, M.A.; Leubner-Metzger, G. Plant hormone interactions during seed dormancy release and germination. Seed Sci. Res. 2005, 15, 281–307. [Google Scholar] [CrossRef]
- Finkelstein, R.; Reeves, W.; Ariizumi, T.; Steber, C. Molecular aspects of seed dormancy. Annu. Rev. Plant Biol. 2008, 59, 387–415. [Google Scholar] [CrossRef] [Green Version]
- McCarty, D.R.; Carson, C.B.; Stinard, P.S.; Robertson, D.S. Molecular Analysis of viviparous-1: An Abscisic Acid-Insensitive Mutant of Maize. Plant Cell 1989, 1, 523. [Google Scholar] [CrossRef]
- Suzuki, M.; Kao, C.-Y.; Cocciolone, S.; McCarty, D.R. Maize VP1 complements Arabidopsis abi3 and confers a novel ABA/auxin interaction in roots. Plant J. 2001, 28, 409–418. [Google Scholar] [CrossRef]
- Tan, B.-C.; Cline, K.; McCarty, D.R. Localization and targeting of the VP14 epoxy-carotenoid dioxygenase to chloroplast membranes. Plant J. 2001, 27, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, G.; Mendel, R.R. Molybdenum cofactor biosynthesis and molybdenum enzymes. Annu. Rev. Plant Biol. 2006, 57, 623–647. [Google Scholar] [CrossRef]
- Nakai, Y.; Harada, A.; Hashiguchi, Y.; Nakai, M.; Hayashi, H. Arabidopsis molybdopterin biosynthesis protein Cnx5 collaborates with the ubiquitin-like protein urm11 in the thio-modification of tRNA. J. Biol. Chem. 2012, 287, 30874–30884. [Google Scholar] [CrossRef] [Green Version]
- Matthews, P.D.; Luo, R.B.; Wurtzel, E.T. Maize phytoene desaturase and ζ-carotene desaturase catalyse a poly-Z desaturation pathway: Implications for genetic engineering of carotenoid content among cereal crops. J. Exp. Bot. 2003, 54, 2215–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Yeh, C.T.; Tang, H.M.; Nettleton, D.; Schnable, P.S. Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS ONE 2012, 7, e36406. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Latshaw, S.; Sato, Y.; Settles, A.M.; Koch, K.E.; Hannah, L.C.; Kojima, M.; Sakakibara, H.; McCarty, D.R. The maize viviparous8 locus, encoding a putative ALTERED MERISTEM PROGRAM1-like peptidase, regulates abscisic acid accumulation and coordinates embryo and endosperm development. Plant Physiol. 2008, 146, 1193–1206. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.G.; Kim, E.J.; Song, J.Y.; Choi, S.B.; Cho, S.W.; Park, C.S.; Kang, C.S.; Park, Y.-Il. Peptide transporter2 (PTR2) enhances water uptake during early seed germination in Arabidopsis thaliana. Plant Mol. Biol. 2020, 102, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Rolland, F.; Baena-Gonzalez, E.; Sheen, J. Sugar sensing and signaling in plants: Conserved and novel mechanisms. Annu. Rev. Plant Biol. 2006, 57, 675–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, N.; Hirose, T.; Takahashi, S.; Ono, K.; Ishimaru, K.; Ohsugi, R. Molecular cloning and expression analysis of a gene for a sucrose transporter in maize (Zea mays L.). Plant cell Physiol. 1999, 40, 1072–1078. [Google Scholar] [CrossRef] [Green Version]
- Scofield, G.N.; Hirose, T.; Gaudron, J.A.; Upadhyaya, N.M.; Ohsugi, R.; Furbank, R.T. Antisense suppression of the rice sucrose transporter gene, OsSUT1, leads to impaired grain filling and germination but does not affect photosynthesis. Funct. Plant Biol. 2002, 29, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Jakoby, M.; Weisshaar, B.; Dröge-Laser, W.; Vicente-Carbajosa, J.; Tiedemann, J.; Kroj, T.; Parcy, F. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 2002, 7, 106–111. [Google Scholar] [CrossRef]
- Valdés, A.E.; Övernäs, E.; Johansson, H.; Rada-Iglesias, A.; Engström, P. The homeodomain-leucine zipper (HD-Zip) class I transcription factors ATHB7 and ATHB12 modulate abscisic acid signalling by regulating protein phosphatase 2C and abscisic acid receptor gene activities. Plant Mol. Biol. 2012, 80, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.Y.; Nam, K.H. RAV1 negatively regulates seed development by directly repressing MINI3 and IKU2 in Arabidopsis. Mol. Cells 2018, 41, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Nambara, E.; Marion-Poll, A. Abscisic acid biosynthesis and catabolism. Annu. Rev. Plant Biol. 2005, 56, 165–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, F.; Qanmber, G.; Li, F.; Wang, Z. Updated role of ABA in seed maturation, dormancy, and germination. J. Adv. Res. 2021. [Google Scholar] [CrossRef]
- Weiner, J.J.; Peterson, F.C.; Volkman, B.F.; Cutler, S.R. Structural and functional insights into core ABA signaling. Curr. Opin. Plant Biol. 2010, 13, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Matsumoto, M.; Hakomori, Y.; Takagi, H.; Shimada, H.; Sakamoto, A. The purine metabolite allantoin enhances abiotic stress tolerance through synergistic activation of abscisic acid metabolism. Plant, Cell Environ. 2014, 37, 1022–1036. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, R.; Stransky, H.; Koch, W. The amino acid permease AAP8 is important for early seed development in Arabidopsis thaliana. Planta 2007, 226, 805–813. [Google Scholar] [CrossRef]
- Tsay, Y.F.; Chiu, C.C.; Tsai, C.B.; Ho, C.H.; Hsu, P.K. Nitrate transporters and peptide transporters. FEBS Lett. 2007, 581, 2290–2300. [Google Scholar] [CrossRef] [Green Version]
- Stacey, G.; Koh, S.; Granger, C.; Becker, J.M. Peptide transport in plants. Trends Plant Sci. 2002, 7, 257–263. [Google Scholar] [CrossRef]
- Fait, A.; Angelovici, R.; Less, H.; Ohad, I.; Urbanczyk-Wochniak, E.; Fernie, A.R.; Galili, G. Arabidopsis seed development and germination is associated with temporally distinct metabolic switches. Plant Physiol. 2006, 142, 839–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glevarec, G.; Bouton, S.; Jaspard, E.; Riou, M.T.; Cliquet, J.B.; Suzuki, A.; Limami, A.M. Respective roles of the glutamine synthetase/glutamate synthase cycle and glutamate dehydrogenase in ammonium and amino acid metabolism during germination and post-germinative growth in the model legume Medicago truncatula. Planta 2004, 219, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Yang, P. Studies on the molecular mechanisms of seed germination. Proteomics 2015, 15, 1671–1679. [Google Scholar] [CrossRef]
- Galili, G.; Avin-Wittenberg, T.; Angelovici, R.; Fernie, A.R. The role of photosynthesis and amino acid metabolism in the energy status during seed development. Front. Plant Sci. 2014, 5, 447. [Google Scholar] [CrossRef]
- Tnani, H.; López-Ribera, I.; García-Muniz, N.; Vicient, C.M. ZmPTR1, a maize peptide transporter expressed in the epithelial cells of the scutellum during germination. Plant Sci. 2013, 207, 140–147. [Google Scholar] [CrossRef]
- Miranda, M.; Borisjuk, L.; Tewes, A.; Dietrich, D.; Rentsch, D.; Weber, H.; Wobus, U. Peptide and amino acid transporters are differentially regulated during seed development and germination in faba bean. Plant Physiol. 2003, 132, 1950–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghi, L.; Kang, J.; Ko, D.; Lee, Y.; Martinoia, E. The role of ABCG-type ABC transporters in phytohormone transport. Biochem. Soc. Trans. 2015, 43, 924–930. [Google Scholar] [CrossRef]
- Do, T.H.T.; Martinoia, E.; Lee, Y. Functions of ABC transporters in plant growth and development. Curr. Opin. Plant Biol. 2018, 41, 32–38. [Google Scholar] [CrossRef]
- Pawela, A.; Banasiak, J.; Biała, W.; Martinoia, E.; Jasiński, M. MtABCG20 is an ABA exporter influencing root morphology and seed germination of Medicago truncatula. Plant J. 2019, 98, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Hirose, T.; Imaizumi, N.; Scofield, G.N.; Furbank, R.T.; Ohsugi, R. cDNA cloning and tissue specific expression of a gene for sucrose transporter from rice (Oryza sativa L.). Plant Cell Physiol. 1997, 38, 1389–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsukura, C.; Saitoh, T.; Hirose, T.; Ohsugi, R.; Perata, P.; Yamaguchi, J. Sugar uptake and transport in rice embryo. Expression of companion cell-specific sucrose transporter (OsSUT1) induced by sugar and light. Plant Physiol. 2000, 124, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himmelbach, A.; Hoffmann, T.; Leube, M.; Höhener, B.; Grill, E. Homeodomain protein ATHB6 is a target of the protein phosphatase ABI1 and regulates hormone responses in Arabidopsis. EMBO J. 2002, 21, 3029–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elhiti, M.; Stasolla, C. Structure and function of homodomain-leucine zipper (HD-Zip) proteins. Plant Signal. Behav. 2009, 4, 86–88. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.Z.; Chen, Y.; Wang, C.; Kong, Y.H.; Wu, W.H.; Chen, Y.F. Arabidopsis RAV1 transcription factor, phosphorylated by SnRK2 kinases, regulates the expression of ABI3, ABI4, and ABI5 during seed germination and early seedling development. Plant J. 2014, 80, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Bai, X.; Wu, X.; Xiang, D.; Wan, Y.; Luo, Y.; Shi, X.; Li, Q.; Zhao, J.; Qin, P.; et al. Transcriptome profiling identifies transcription factors and key homologs involved in seed dormancy and germination regulation of Chenopodium quinoa. Plant Physiol. Biochem. 2020, 151, 443–456. [Google Scholar] [CrossRef]
- Okamoto, M.; Tatematsu, K.; Matsui, A.; Morosawa, T.; Ishida, J.; Tanaka, M.; Endo, T.A.; Mochizuki, Y.; Toyoda, T.; Kamiya, Y.; et al. Genome-wide analysis of endogenous abscisic acid-mediated transcription in dry and imbibed seeds of Arabidopsis using tiling arrays. Plant J. 2010, 62, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Koornneef, M.; Bentsink, L.; Hilhorst, H. Seed dormancy and germination. Curr. Opin. Plant Biol. 2002, 5, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Vallabhaneni, R.; Yu, J.; Rocheford, T.; Wurtzel, E.T. The maize phytoene synthase gene family: Overlapping roles for carotenogenesis in endosperm, photomorphogenesis, and thermal stress tolerance. Plant Physiol. 2008, 147, 1334–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, V.; North, H.; Frey, A.; Sotta, B.; Seo, M.; Okamoto, M.; Nambara, E.; Marion-Poll, A. Functional analysis of Arabidopsis NCED6 and NCED9 genes indicates that ABA synthesized in the endosperm is involved in the induction of seed dormancy. Plant J. 2006, 45, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Chinnusamy, V.; Rodrigues, A.; Rubio, S.; Antoni, R.; Park, S.Y.; Cutler, S.R.; Sheen, J.; Rodriguez, P.L.; Zhu, J.K. In vitro reconstitution of an abscisic acid signalling pathway. Nature 2009, 462, 660–664. [Google Scholar] [CrossRef] [Green Version]
- Parcy, F.; Valon, C.; Raynal, M.; Gaubier-Comella, P.; Delseny, M.; Giraudat, J. Regulation of gene expression programs during Arabidopsis seed development: Roles of the ABI3 locus and of endogenous abscisic acid. Plant Cell 1994, 6, 1567–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Ketterling, M.G.; Li, Q.B.; McCarty, D.R. Viviparous1 alters global gene expression patterns through regulation of abscisic acid signaling. Plant Physiol. 2003, 132, 1664–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Qu, B.; Li, H.P.; Zuo, D.Y.; Zhao, Z.X.; Liao, Y.C. A maize viviparous 1 gene increases seed dormancy and preharvest sprouting tolerance in transgenic wheat. J. Cereal Sci. 2012, 55, 166–173. [Google Scholar] [CrossRef]
- Kim, W.; Lee, Y.; Park, J.; Lee, N.; Choi, G. HONSU, a protein phosphatase 2C, regulates seed dormancy by inhibiting ABA signaling in Arabidopsis. Plant Cell Physiol. 2013, 54, 555–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, M.; Nambara, E.; Choi, G.; Yamaguchi, S. Interaction of light and hormone signals in germinating seeds. Plant Mol. Biol. 2009, 69, 463–472. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 15 August 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. AgriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhang, J.; Sun, M.; He, C.; Yu, K.; Zhao, B.; Li, R.; Li, J.; Yang, Z.; Wang, X.; et al. Multi-Omics Analyses Reveal Systemic Insights into Maize Vivipary. Plants 2021, 10, 2437. https://doi.org/10.3390/plants10112437
Wang Y, Zhang J, Sun M, He C, Yu K, Zhao B, Li R, Li J, Yang Z, Wang X, et al. Multi-Omics Analyses Reveal Systemic Insights into Maize Vivipary. Plants. 2021; 10(11):2437. https://doi.org/10.3390/plants10112437
Chicago/Turabian StyleWang, Yiru, Junli Zhang, Minghao Sun, Cheng He, Ke Yu, Bing Zhao, Rui Li, Jian Li, Zongying Yang, Xiao Wang, and et al. 2021. "Multi-Omics Analyses Reveal Systemic Insights into Maize Vivipary" Plants 10, no. 11: 2437. https://doi.org/10.3390/plants10112437