Abstract
MADS-box transcription factors (TFs) are involved in multiple plant development processes and are most known during the reproductive transition and floral organ development. Very few genes have been characterized in the genome of Humulus lupulus L. (Cannabaceae), an important crop for the pharmaceutical and beverage industries. The MADS-box family has not been studied in this species yet. We identified 65 MADS-box genes in the hop genome, of which 29 encode type-II TFs (27 of subgroup MIKCC and 2 MIKC*) and 36 type-I proteins (26 α, 9 β, and 1 γ). Type-II MADS-box genes evolved more complex architectures than type-I genes. Interestingly, we did not find FLOWERING LOCUS C (FLC) homologs, a transcription factor that acts as a floral repressor and is negatively regulated by cold. This result provides a molecular explanation for a previous work showing that vernalization is not a requirement for hop flowering, which has implications for its cultivation in the tropics. Analysis of gene ontology and expression profiling revealed genes potentially involved in the development of male and female floral structures based on the differential expression of ABC homeotic genes in each whorl of the flower. We identified a gene exclusively expressed in lupulin glands, suggesting a role in specialized metabolism in these structures. In toto, this work contributes to understanding the evolutionary history of MADS-box genes in hop, and provides perspectives on functional genetic studies, biotechnology, and crop breeding.
1. Introduction
MADS-box proteins are transcription factors (TFs) that interact with the promoters of their target genes through the binding to CArG-box cis-elements [1]. Phylogenetic data have classified MADS-box proteins into two groups: types I (e.g., SRF from human) and II (e.g., Mcm1 from yeast) [2]. A highly conserved sequence of about 60 amino acids called the MADS domain characterizes this family of TFs. In plants, MADS-box TFs have largely diversified and can be sub-classified into several clades. Type-I proteins are split into three groups: Mα, Mβ, and Mγ, whereas type-II proteins are classified into two groups: MIKC* and MIKCC [3]. MIKC represents the protein structure of type-II MADS-box TFs, which has the conserved MADS-box for DNA-binding closed to the N-terminus followed by an intervening domain (I), a keratin-like domain (K) for protein-protein interaction, and the variable C-terminal domain. MIKCC MADS-box proteins are sub-classified into 13 subfamilies, including the TM8 subfamily that is absent in Arabidopsis [4].
MADS-box TFs orchestrate multiple developmental programs in plants, most notably vegetative and reproductive development programs. More recently, a novel MADS-box TF in apple was implicated in regulating dormancy cycles in response to environmental cues [5]. MADS-box TFs are also involved in maintaining the spike morphology of barley under high-temperature stress [6], promoting bud break in ecodormant poplar [7], and controlling nitrogen fixation symbiosis in common beans [8]. Flowering transition is another process governed by MADS-box genes. In Arabidopsis, SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1) integrates multiple flowering signals derived from photoperiod, temperature, hormone, and age-related signals [9,10,11]. SOC1 interacts with AGAMOUS-like 24 (AGL24) and FRUITFULL (FUL) to promote flowering [12,13]. In addition, the transition from the vegetative to the reproductive phase in Arabidopsis is controlled by the MADS-box protein, SHORT VEGETATIVE PHASE (SVP), which is a repressor of flowering under short days [14] alike to what FLOWERING LOCUS C (FLC) does prior to vernalization [15,16]. FLC is a TF that acts as a floral repressor and is negatively regulated by cold periods or vernalization, being essential to synchronize flowering and winter [17,18]. FLC homologs were described in the three major eudicot lineages [19,20], including sugar-beet, apple, and coffee [19,21,22]. However, despite the role of FLC being described in the Brassicaceae [23] and more recently cereal crops and grasses [24,25], the extent to which the molecular mechanisms underlying vernalization have been conserved during the diversification of the angiosperms remains elusive.
Some of the most studied MADS-box transcription factors are involved in the development of floral organs in angiosperms [26]. Such genes are called homeotic, since their misexpression in a given whorl leads to the formation of a different floral organ [27,28,29]. This led to the formulation of the ABC model [30], which encompasses the combinatorial transcription of MADS-box TFs that elicit the developmental program of specific organs in each whorl of the flower. In this model, a class-A gene expressed alone in the first whorl leads to the formation of sepals, the co-expression of class A and B genes in the second whorl leads to the development of petals, the co-expression of class B and C genes in the third whorl elicits the formation of anthers, and finally, the expression of a C-class gene alone in the fourth whorl leads to the formation of carpels.
Further research extended the ABC model to an ABCDE model, where D-class genes expressed in the carpel lead to the formation of ovules, and E-class genes expressed in all whorls form tetramers with ABC TFs that coordinate the development of each whorl [1,31]. In Arabidopsis, the ABCDE model encompasses the A-class APETALA1 (AP1) [32]; the B-class APETALA3 (AP3) and PISTILLATA (PI) [33,34]; the C-class AGAMOUS (AG) [35]; the D-class AGAMOUS-like 1, 5, and 11 (AGL1, AGL5, and AGL11) [36]; and the E-class SEPALLATA 1,2,3,4 (SEP1, SEP2, SEP3, and SEP4) [37]. The ABCDE model has been conserved throughout angiosperm evolution. The genes encompassing each homeotic class have been determined in different species, from monocots, such as rice [38,39], wheat [40], and Easter lily [28]; to dicots, such as soybean [41,42], coffee [22], and the New Zealand endemic species, Clianthus maximus [43], among many others. Moreover, studies have also revealed the significance of type-I MADS-box transcription factors in plant reproduction [44].
Hop (Humulus lupulus L.) is a perennial crop that belongs to the Cannabaceae family. It blooms in short days once it develops a particular number of nodes. It has been phenotypically demonstrated that vernalization and dormancy do not influence flower yield and quality [45,46]. Moreover, hop is an economically important species because its cones (female inflorescences) are widely utilized in the pharmaceutical and beer industries [47,48,49]. However, the mechanisms involved in the reproductive phase transition and flower development remain poorly explored at the molecular genetics level. We carried out a genome-wide approach, identified 65 MADS-box genes in the hop genome, and further studied their phylogenetic relationships, genetic structure, and gene expression profiles using publicly available RNA-Seq data. This study revealed TFs that potentially coordinate critical aspects of plant development, phase transition, and glandular metabolism. Therefore, this work advances our understanding of the evolutionary history of the MADS-box TFs in hop and opens new avenues for functional genetic research and crop breeding toward expanding its production zones in the world, especially in the tropics.
2. Results
2.1. MADS-Box Genes Encoded in the Hop Genome and Gene Ontology Annotation
Using AUGUSTUS on RNA-Seq libraries, our bioinformatics pipeline identified 47 genes coding for proteins with canonical MADS-box domains. Moreover, the hop genome sequence has 69 genes annotated as coding for MADS-box proteins. The overlap of our results with the official hop genome annotation shows a set of 65 non-redundant genes (unigenes: HlMADS01 to HlMADS65), seven of which exclusively from our prediction, 23 exclusively in the hop genome annotation, and 35 represented in both sets. All seven novel genes identified in our de novo prediction pipeline were MIKCC-type proteins (HlMADS28-33 and HlMADS65). The encoded protein length ranged from 135 to 547 amino acid residues (aa), with an average of 249 aa; the molecular mass varied from 16 to 60 kDa, and the isoelectric point was between 4.55 and 10.25 (Table S1).
Gene Ontology (GO) analysis was performed on the 65 hop MADS-box proteins with the Blast2GO software (Table S3). All hop MADS-box proteins were classified into the three main categories (cellular component, molecular function, and biological process) and their subcategories (Figure S6). In this analysis, 48 proteins returned for cellular component and further split into six subgroups, being the ‘cell part, cell, and organelle’ subcategory the most over-represented subgroup (74%), which is a share even greater than that of Arabidopsis (56%), according to our analyses. In addition, 1.5% of the hop MADS-box proteins were annotated into the membrane subcategory, in contrast with 2.8% in Arabidopsis. On the other hand, all 65 hop MADS-box proteins were annotated in the subcategory binding of the category molecular function, a result identical to Arabidopsis. The second most represented molecular function subcategory in the hop MADS-box protein set was transcription regulator activity, with 75% of the proteins, in contrast with 87% for Arabidopsis. The subcategory catalytic activity did not contain any hop MADS-box proteins. Finally, the category biological process contained 51 hop MADS-box proteins (79%). The subcategories biological regulation, cellular processes, regulation of biological processes, and metabolic process were the most overrepresented (97.5%) in hop, followed by positive regulation of biological process subcategory (92.5%). All other subcategories were represented by less than 20% of the hop MADS-box protein set.
2.2. Phylogenetic Analyses Revealed Clades and Potential Functions of Hop MADS-Box Genes
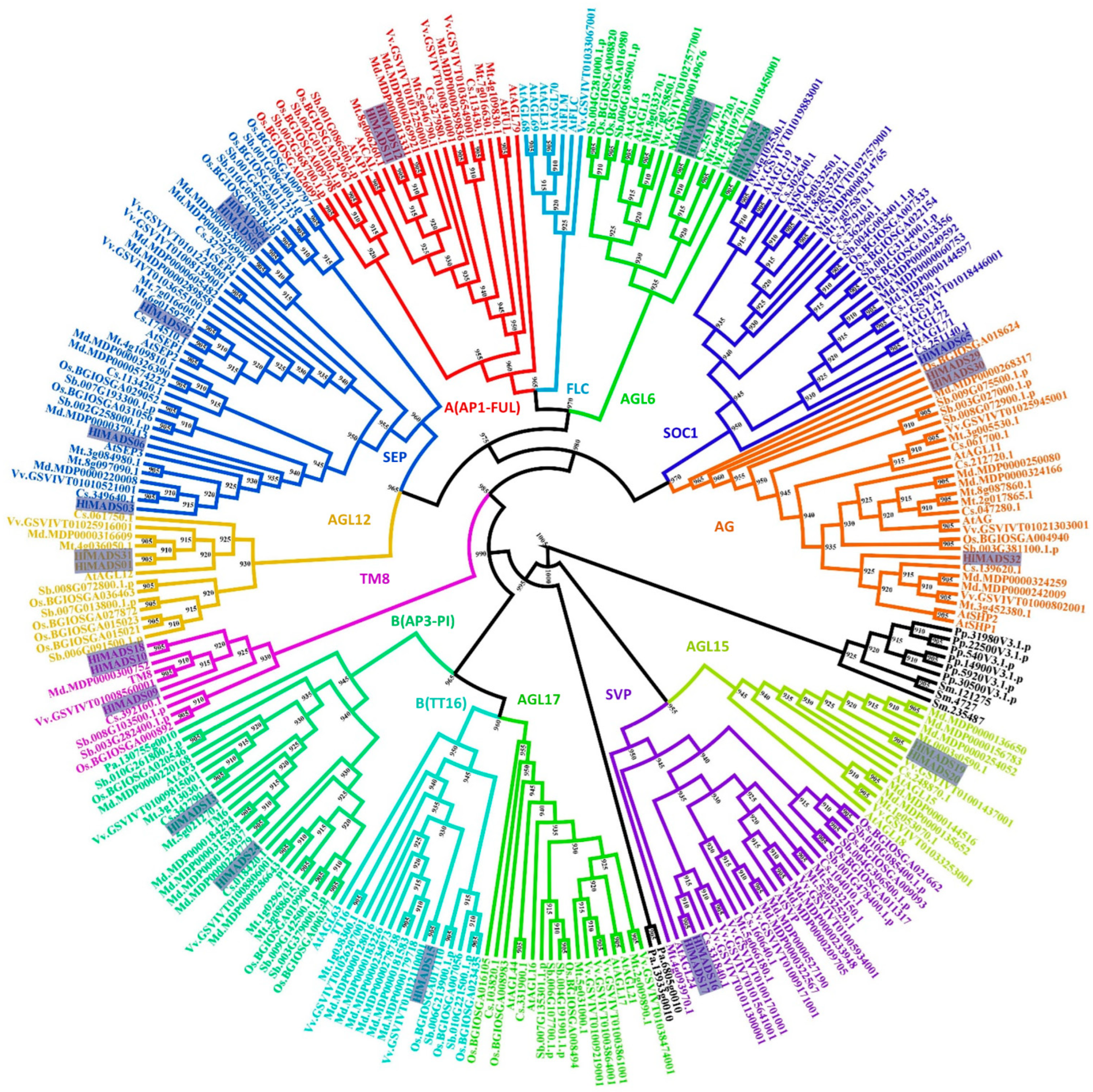
Protein domain analyses identified 29 type-II and 36 type-I MADS-box proteins encoded in the hop genome. Each set was submitted to separate phylogenetic analyses to further classify them into subfamilies (Figure 1, Figure 2 and Figure S1). Our results revealed five members in the SEP clade, two in the A clade (AP1-FUL), two in the B clade (AP3-PI), three in the C/D clade (AG), four in the AGL6, two in the AGL12, two in the AGL15, three in the TM8, one in the BS (TT16), two in the SVP, and one in the SOC1 clade (Figure 1, Table S1). Two type-II proteins were classified as MIKC* (Figure S1). Remarkably, the FLC and AGL17 subfamilies are not represented in the hop genome. The only member of the SOC1 subfamily (HlMADS65) was found when a tBLASTn was performed using the Arabidopsis SOC1 protein sequence as a query, and AUGUSTUS was run on the genome region identified in the output. HlMADS65 lies within the region annotated as an intron of 000453F.g47 (Figure S2). The same approach was used for other genes, resulting in no new sequences.
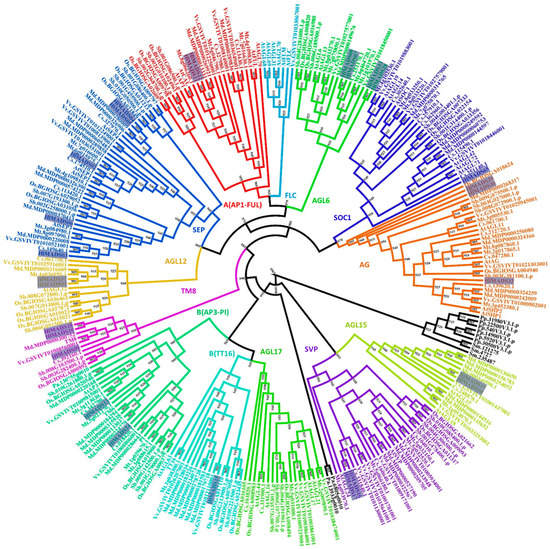
Figure 1.
Phylogenetic tree of MIKCc-type MADS-box proteins of Humulus lupulus ((Hl) (27 sequences, highlighted)), Arabidopsis thaliana ((At)(39)), Solanum lycopersicum (TM6 and TM8), Physcomitrella patens ((Pp) (6)), Selaginella moellendorffii ((Sm) (3)), Piceaabies ((Pa) (3)), Sorghum bicolor ((Sb) (32)), Oryza sativa subsp. indica ((Os) (32)), Cucumis sativus ((Cs) (26)), Malus domestica ((Md) (42)), Medicago truncatula ((Mt) (35)), and Vitis vinifera ((Vv) (33)). Sequences from Selaginella moellendorffii, Physcomitrella patens, and Piceaabies were used as outgroups (in black font).
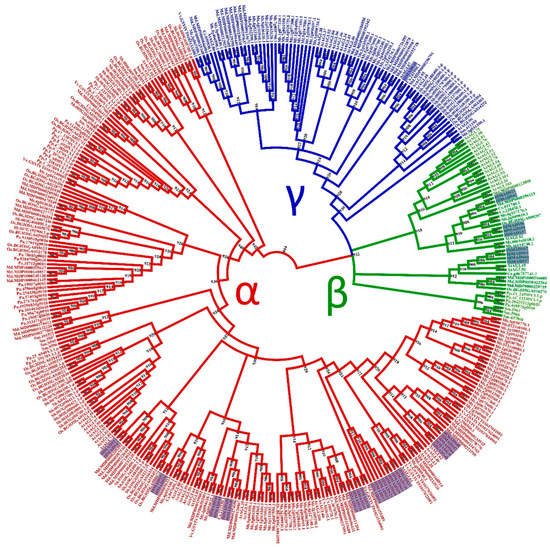
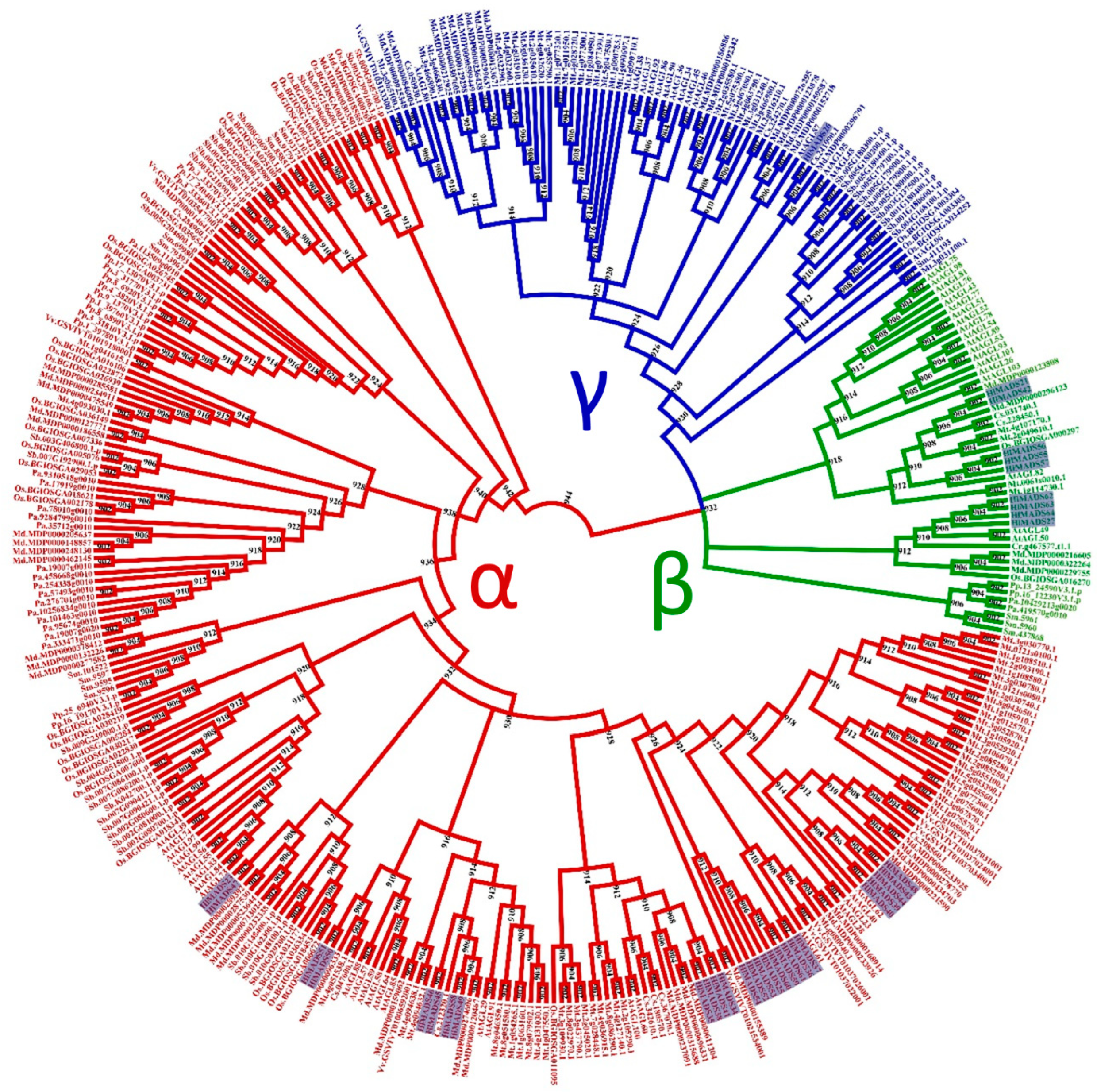
Figure 2.
Phylogenetic tree of I-type MADS-box proteins of Humulus lupulus ((Hl) (36 sequences, highlighted)), Arabidopsis thaliana ((At) (58)), Chlamydomonas reinhardtii ((Cr) (1)), Physcomitrella patens ((Pp) (16)), Selaginella moellendorffii ((Sm)(14)), Piceaabies ((Pa) (18)), Sorghum bicolor ((Sb) (38)), Oryza sativa subsp. indica ((Os) (36)), Cucumis sativus ((Cs) (12)), Malus domestica ((Md) (56)), Medicago truncatula ((Mt) (79)), and Vitis vinifera ((Vv) (10)).
For the type-I MADS-box proteins, the α-subfamily encompasses the most represented group, with 26 members, followed by the β-subfamily (nine members). In contrast, the γ-subfamily is represented by a single member (Figure 2).
2.3. Structural and Motif Analyses of Hop MADS-Box Genes
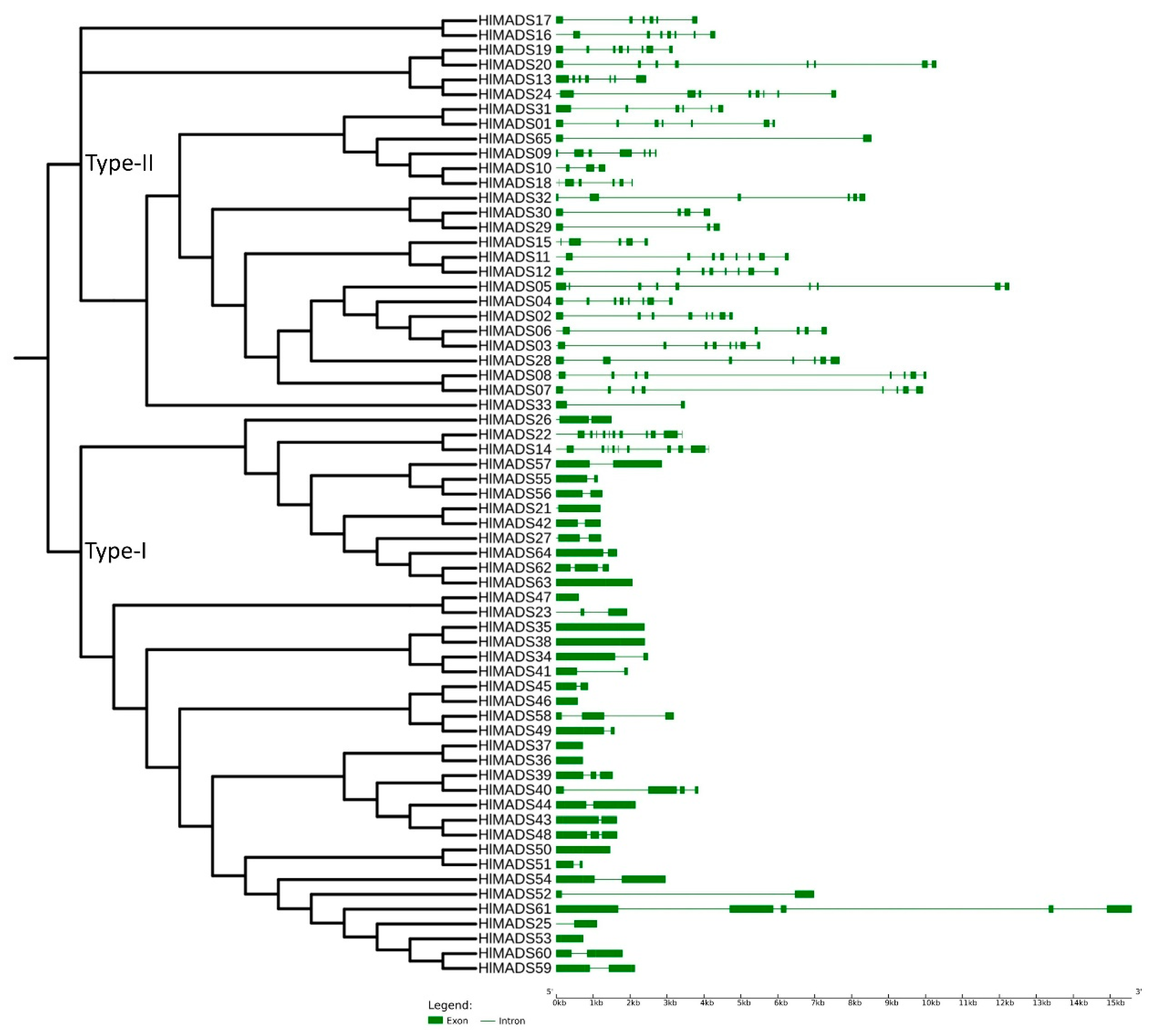
To confirm our phylogenetic relationships and gain further insights into gene functions, we explored the exon-intron architecture of MADS-box genes. The number of exons among the 29 type-II MADS-box genes varied from 2 (HlMADS33 and HLMADS65) to 13 (HlMADS22, the sole member of the MIKC* group). In hop, HlMADS05 is the longest type-II MADS-box gene (12.5 kb), with nine exons and eight introns. In the type-I group, the exon number varied from one to five. Overall, HlMADS61 was the longest gene (16 kb), whereas the shortest ones were HlMADS46 (584 bp) and HlMADS47 (613 bp), both with only one exon each (Figure 3), all falling in the type-I clade.
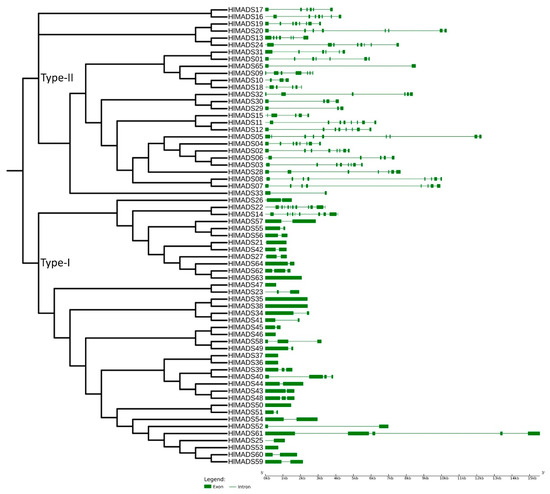
Figure 3.
Phylogenetic tree and structure of 65 hop MADS-box genes. Exons are represented by green solid boxes, and introns by green lines.
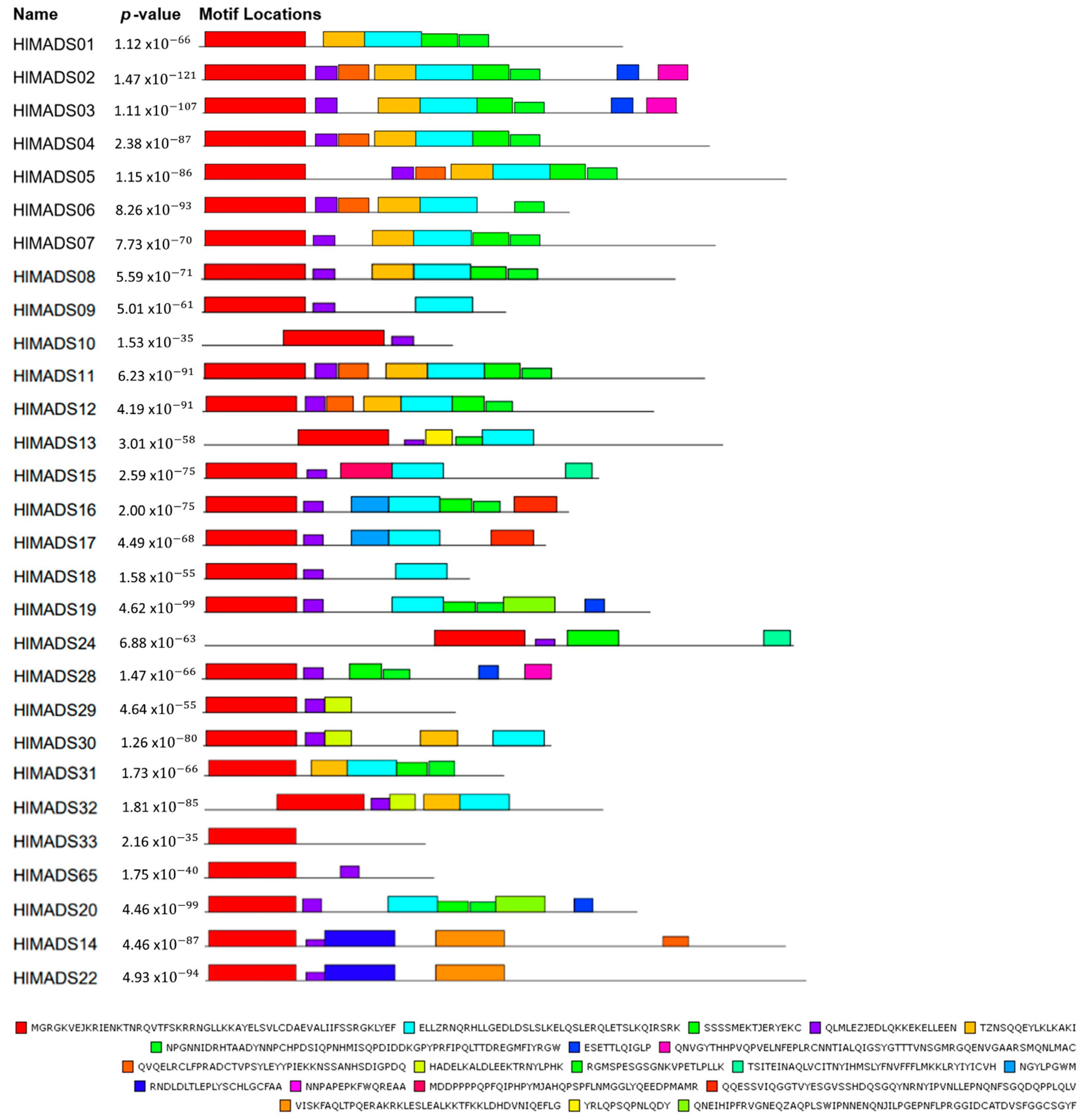
The MEME tool was used to visualize conserved protein domains. While all type-II proteins displayed the MADS-box domain in the tool output, only 21 proteins had the K-box domain, even though this motif is a characteristic feature of this group (Figure 4). However, the NCBI’s Conserved Domain Search tool resulted in 23 proteins with the K-box domain, consequently adding HlMADS28 and HlMADS29 to the list. Even though the MADS domain did not appear for HlMADS55, HlMADS56, and HlMADS57, in the MEME analyses, the domain presence was confirmed by NCBI and PFAM conserved domain inference tools. All I-type proteins displayed the MADS domain in MEME but at diverse locations of the peptide sequence (Figures S3 and S4).
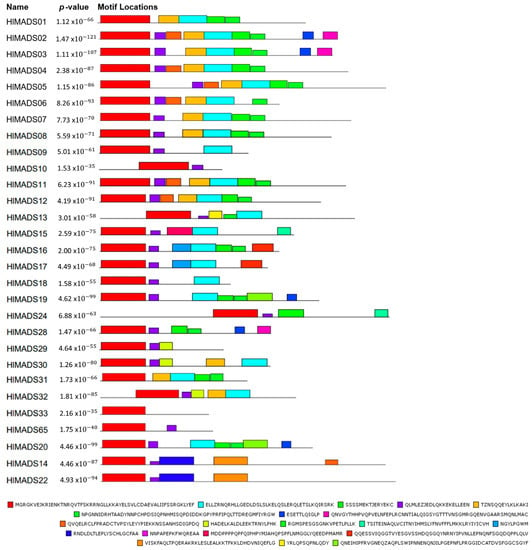
Figure 4.
Motif distribution of hop type-I MADS-box proteins. Protein sequences are represented by black lines, and the conserved motifs are represented by colored boxes with the MADS-box domain in red.
2.4. Transcriptional Profile of Hop MADS-Box Genes in Different Tissues
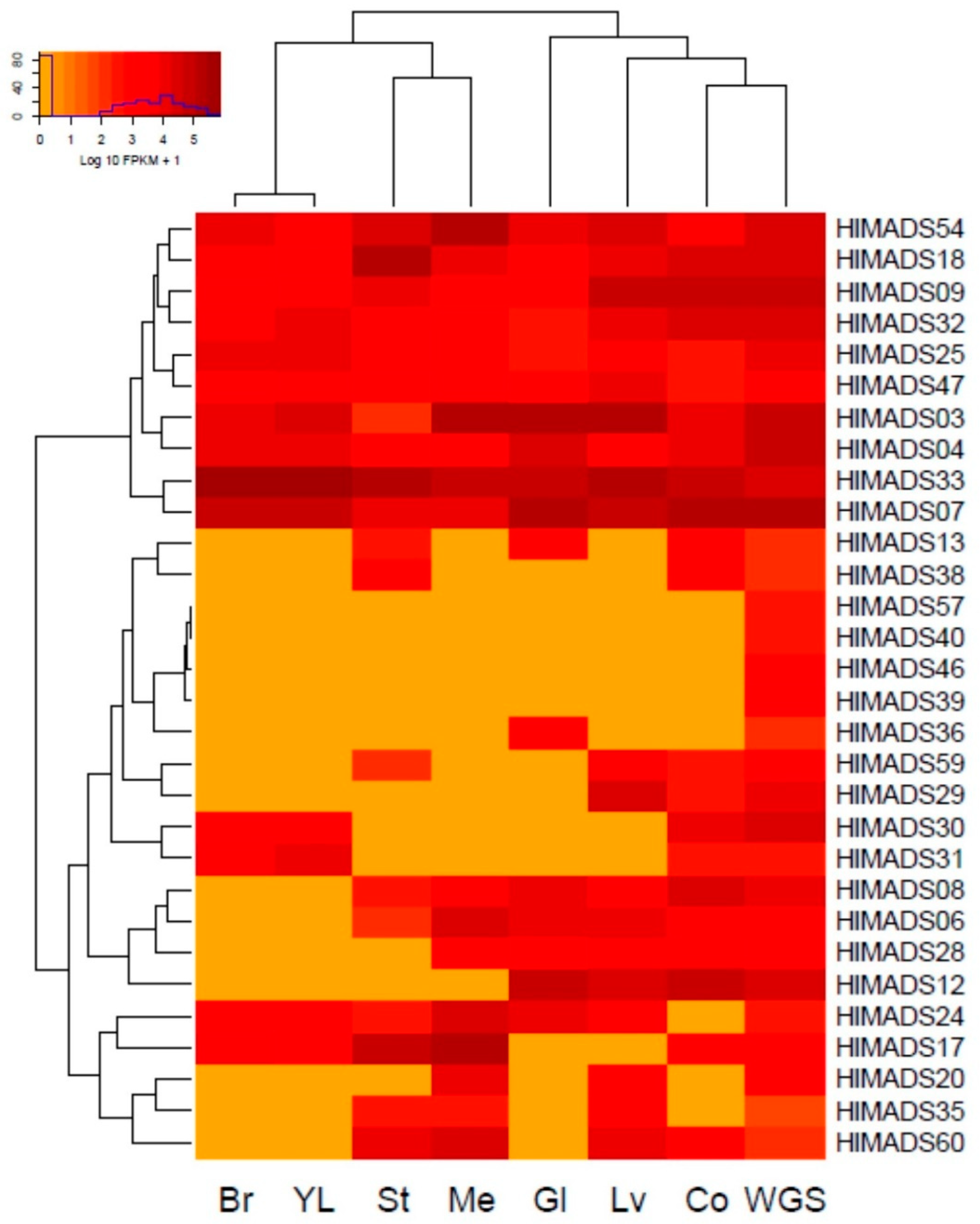
The expression profiling of MADS-box genes in different hop tissues was determined through the analysis of publicly available RNA-Seq data deposited in the NCBI-SRA database. Thirty genes were expressed in different samples (Figure 5), whereas 35 were not expressed. Of the expressed genes, 18 are of the MIKCC type, and 12 are type I. Some genes belonging to the same subfamily showed distinct expression patterns. For example, in the SEP subfamily, HlMADS03 and HlMADS04 were expressed in all samples analyzed, whereas HlMADS06 was expressed only in the stem, meristem, glands, mature leaves, and cones without glands. Interestingly, young leaves and bracts showed an identical expression pattern for the MADS-box genes. Moreover, HlMADS17, HlMADS30, and HlMADS31 (respective members of the SVP, C/D(AG), and AGL12 clades) were expressed in young leaves compared with the mature organ, indicating a possible function during leaf development and expansion.
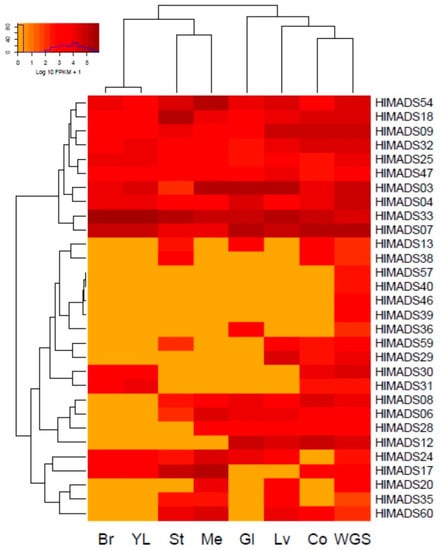
Figure 5.
Expression profiling of hop MADS-box genes in RNA-Seq libraries: bracts (Br), young leaves (YL), stem (St), vegetative meristem (Me), glands (Gl), mature leaves (Lv), cones without glands (Co), and a mélange of plant tissues collected throughout the whole growing season (WGS).
We identified two gene groups regarding the expression profile (Figure 5). The first encompassed constitutive genes: two SEP (HlMADS03 and HlMADS04), two TM8 (HlMADS09 and HlMADS18), two AGL6-like (HlMADS07 and HlMADS33), one of the C/D (AG) subfamily (HlMADS32), and three type-I α (HlMADS25, HlMADS47, and HlMADS54). The second group included 20 genes (9 type-I and 11 MIKCc) that showed variable expression among the samples. The B (AP3-PI) subfamily member, HlMADS13, was expressed in the stem, glands, and cones, whereas four type-I genes (HlMADS57, HlMADS40, HlMADS46, and HlMADS39) were expressed only in the whole plant sample. Interestingly, a type-I α gene, HlMADS36, was expressed only in the glands. On the other hand, two C/D(AG) subfamily genes (HlMADS29 and HlMADS30) are in this group, with HlMADS29 being expressed in mature leaves and cones, while HlMADS30 expressed in the bracts and young leaves, similar to HlMADS31. Meanwhile, HlMADS06 (SEP) and HlMADS08 (AGL6) did not express in bracts or young leaves; HlMADS28 (AGL6) expressed in the meristem, glands, mature leaf, and cones; HlMADS12 [A(AP1-FUL)] expressed in glands, leaf, and cones; HlMADS24 [B(AP3-PI)] expressed in all samples but cones without glands. In turn, HlMADS17 (SVP) was expressed in all samples but glands and mature leaves, whereas HlMADS20 was expressed only in the meristem and mature leaves. Lastly, HlMADS35 and HlMADS60 (type-I α-subfamily) expressed in the stem, meristem, and mature leaves, while HlMADS60 expressed in cones, and HlMADS35 did not.
3. Discussion
Many MADS-box proteins function as master regulatory transcription factors controlling critical aspects of plant development and agricultural traits. The genomic characterization of this gene family has been carried out for several plant species, such as Arabidopsis (107 genes: [50]), rice (75 genes: [51]), grapevine (74 genes: [52]), Pyrus (75 genes: [53]), to name a few. This work identified and provided the transcriptional profiling of 65 MADS-box genes in the hop (H. lupulus L.) genome. First, we carried out a de novo gene annotation of the hop genome and combined it with the official one available on the HopBase platform [54]. This pipeline identified seven novel MADS-box genes (HlMADS28-33 and HlMADS65) in the hop genome with reads from RNAseq libraries aligned on these genes (Figures S2 and S7). Additionally, it was possible to find some genes (i.e., HlMADS01 and HLMADS02) which were only identified when AUGUSTUS was run with the UTR parameters turned off. This finding shows that de novo gene prediction outputs, as well as the official functional genome annotation, must be interpreted with caution.
In our phylogenetic analyses, 27 MADS-box proteins were classified into 11 clades based on their relationships with the Arabidopsis MIKCC-type and TM8 subfamilies. We discovered that the AGL17 subfamily is not represented in the hop genome, similarly to pears [53]. Neither the FLC subfamily is represented in the hop genome, suggesting that the species lacks the vernalization route completely. Accordingly, it has been demonstrated that hop does not require vernalization to trigger flowering [45], but instead, the process must involve other induction routes, such as photoperiod and age [46].
According to the photoperiod route in Arabidopsis, SOC1 integrates photoperiod signals to promote flowering under long-day conditions [55]. It was possible that in hop, a functional homolog was similarly involved in flowering induction. However, no SOC1 ortholog was identified in our gene prediction pipelines. Notwithstanding, the HopBase genome annotation includes a gene (000453F.g47) that contains only the K-box domain characteristic of the SOC subfamily. That may be the reason our BlastP analysis using the MADS-box consensus sequence as the query did not identify this gene in the hop proteome. However, the MADS domain for this gene was recognized by AUGUSTUS in the first annotated intron of 000453F.g47, where RNA-Seq reads aligned (Figure S2). This gene displays two exons and encodes a protein with the MADS domain but without the K-box, suggesting that it was incorrectly annotated or may undergo intron retention, which is the most predominant mechanism of alternative splicing in plants [56]. Moreover, two SVP subfamily genes were identified in the hop genome, with HlMADS17 expressed in young leaves only, indicating potential participation in leaf development and expansion. Finally, two AGL15 subfamily genes (HlMADS19 and HlMADS20) were identified, with the latter expressed in mature leaves and the meristem. In hop, these genes could act as repressors of flowering transition since members of this subfamily act as flowering repressors in Arabidopsis [57] and promote the expression of miR156, a bona fide marker of plant juvenility [58].
Our phylogenetic study also identified two AP1-FUL subfamily genes (HlMADS11 and HlMADS12) in the hop genome. In Arabidopsis, AP1 determines floral meristem identity and, later, also in petal and sepal development [32,59]. The expression of HlMADS12 in hop cones suggests a possible participation in determining floral organ identity (Figure 5). Even though female flowers of hop (cones or strobiles) entirely lack the perianth (sepals and petals), male flowers have sepals [60], thus showing that the A function of the ABC model of flower development is present in hop. Another hypothesis for the lack of perianth structures in female hop flowers could be a lack of the E-class (SEP subfamily) function. However, this subfamily is the most represented within the MIKCC clade in this species, with five genes expressed in different tissues (Figure 5). The repression mechanism of the A- and B-functions in the first two whorls during cone development remain to be ascertained. Figure 1 shows the B-class HlMADS13 was closest to AP3 while HlMADS24 was closest to PI. These genes showed distinct gene structures, with HlMADS24 being longer than HlMADS13, containing an extra exon (Figure 3), and lacking the K-box (Figure 4). When comparing GO terms between HlMADS13 and HlMADS24 (Table S3), both displayed the same terms, results which are comparable for the Arabidopsis AP3 and PI. Therefore, it is plausible that the B-function in hop is conserved during flower development.
Dioecious reproduction is a common feature found in the Cannabaceae, including Cannabis and Humulus. The organ positions in male (staminate) and female (pistillate) hop flowers deviate from the prevalent four-whorl scheme (sepals-petals-stamens-carpels) observed in many angiosperm groups [60]. After the shoot meristem undergoes flowering transition, the formation of floral organs is initiated. Male and female inflorescence meristems are distinguishable at the anatomical level very early on [60], when the program of floral organ developmental fate has already been decided. According to the classic ABC model, also known as the ABCDE model, the expression of class A and C genes are mutually exclusive in the floral meristem [30]. Three C/D-class genes were identified in the hop genome (Figure 1). The presence of sepals in the first of the two whorls of the male flower suggests a partial presence of the A-class function. In contrast, the development of stamens in the second whorl reveals the expression of B and C class genes and repression of the class A gene function. On the other hand, in female flowers (cones), the formation of a rudimentary perianth (without the development of sepals or petals) indicates an absence of ABC gene expression. In contrast, the development of two carpels in its second whorl indicates the exclusive expression of the C function. Finally, HlMADS29 probably carries out the D-class function because it is expressed in the cones (Figure 5) and its gene annotation is associated with ovule development (GO:0048481). A more refined definition of gene expression within the developing flower and functional analyses to define the role of ABCDE genes are warranted to better understand the genetics of floral organ development in this species.
The 36 type-I MADS-box genes identified in the hop genome were classified into three subfamilies, α (26 genes), β (9), and γ (1). These genes were more structurally diverse but contained fewer exons than the type-II genes (Figure 3). Previous research reported similar results in other species [50,53,61]. The amino acid sequence in this group was also more diverse than in the type-II group, and the MADS-box was not detected in some analyses for three members of the β-subfamily (HlMADS55, HlMADS56, and HlMADS57), although they were present when manually inspected (Figure S4). The MADS motif is somewhat divergent in these three proteins and required a higher sensitivity from the sequence analysis tool. According to the gene expression profile, 12 type-I MADS-box were expressed in the RNA-Seq libraries analyzed, with HlMADS57 the only member of the β-subfamily to be expressed. Interestingly, HlMADS36 was exclusively expressed in all three gland samples analyzed (Figure S5), which allowed us to hypothesize that it may coordinate the biosynthesis of resin and specialized metabolites [62] or participate in gland development. Therefore, it is important to further verify the exact timing expression domain and function of HlMADS36. Moreover, HlMADS36 is associated with a GO term (0045944) involved in multiple processes related to transcriptional induction of genes related to the metabolism of organic compounds. Overall, our findings provide perspectives on functional analyses and breeding of hop.
4. Materials and Methods
4.1. Gene Prediction
H. lupulus L. gene prediction was performed using AUGUSTUS version 3.3.3 [63]. RNA-seq libraries retrieved from the NCBI’s SRA database guided the proper identification of exon-intron gene boundaries (Table S2; accessed on 7 January 2020). After quality evaluation with FastQC, the libraries were processed with Trimmomatic v.0.39 [64] to remove adapter sequences and fragments with poor overall Phred quality. High-quality libraries were then aligned to the H. lupulus L. masked genome sequence from the HopBase platform [54] (accessed on 7 January 2020) using HISAT2 v.2.1 [65]. During the training phase to establish AUGUSTUS metaparameters for the species, RNA-seq libraries from glands, leaf, cones without glands, and meristem (SRR575195, SRR10589377, SRR575201, SRR10320794, respectively) were assembled using Trinity v.2.11.0 [66]. Candidate coding regions were identified with TransDecoder v.5.5.0 [47]. Subsequently, protein sequences were generated and utilized to train AUGUSTUS according to Alternate protocol 1 [67]. The training was also enriched with EST and UTR coordinates utilizing coding sequences from Trandecoder and the PASA pipeline [68]. Sorted BAM files were used to generate exon (with Bam2wig) and intron hints. Finally, the trained metaparameters were fed into AUGUSTUS for gene prediction.
4.2. Identification of MADS-Box Genes in the Hop Genome
We used the Basic Local Alignment Search Tool BLAST v.2.11.0 [69] to scan the hop proteome searching for MADS-box proteins. A conserved domain sequence from Serum Response Factor (SRF) retrieved from the Pfam database (http://pfam.xfam.org/, accessed on 14 May 2021) was used as a query in BlastP against proteins obtained previously in the gene prediction step. In parallel, BlastP was carried out against the hop proteome retrieved from the HopBase platform. We only considered MADS-box proteins with sequences presenting a conserved domain with all three Pfam, SMART, and NCBI-BlastP analyses. Redundant proteins reported on the same locus were combined after manually curating genomic loci with IGV. Putative MADS-box protein sequences with less than 100 amino acid residues were re-submitted to AUGUSTUS, with UTR parameters turned off, until new sequences were no longer retrieved. Each non-redundant, putative MADS-box protein sequence identified in these analyses was named HlMADS01 to 65. Their physicochemical properties (length of amino acid sequence, molecular weight, and isoelectric point) were determined with the ExPASy Proteomics tool (https://web.expasy.org/protparam/, accessed on 14 May 2021).
4.3. Phylogenetic Analysis
MADS-box protein sequences retrieved from species spanning the plant kingdom (Chlamydomonas reinhardtii, Physcomitrella patens, Selaginella moellendorffii, Piceaabies, Sorghum bicolor, Oryza sativa, Cucumis sativus, Malus domestica, Medicago truncatula, and Vitis vinifera) were retrieved from the PlantTFDB database v.5.0 [70] along with those from Arabidopsis thaliana and Solanum lycopersicum retrieved from the NCBI database, along with those from H. lupulus identified above, were used in our phylogenetic analysis. For sequence types I, MIKCc, and MIKC*, multiple sequence alignment jobs were performed separately with MAFFT v.7.475 [71]. The alignment quality was evaluated with GUIDANCE 2 v.2.02 [72]. Both steps used default parameters. Phylogenetic trees were inferred with PHYLIP v.3.696 [73] with 1000 bootstrap replicates, using the Jones-Taylor-Thornton matrix and neighbor-joining method [74]. Finally, the tree was visualized with FigTree, and the hop MADS-box proteins were classified into subgroups according to the Arabidopsis MADS-box subfamilies [50] plus the subfamily TM8 first reported in tomato [4]. When a subfamily was absent in a first search, tBLASTn was performed using the protein sequence from Arabidopsis of that subfamily as the query.
4.4. MADS-Box Gene Structure and Conserved Protein Motif Analyses in Hop
The exon-intron structures of MADS-box genes were identified with the Gene Structure Display Server GSDS2.0 [75] using the GFF files generated from our gene prediction as well as the annotation available at the HopBase. The MEME suite online analysis tool [76] was used to identify putative motifs of hop MADS-box proteins with the following parameters: maximum of 20 motifs to be identified and motif width between 6 and 60. In this case, we used sequences from Chlamydomonas reinhardtii, Physcomitrella patens, Selaginella moellendorffii, Picea abies, Sorghum bicolor, Oryza sativa, Cucumis sativus, Malus domestica, Medicago truncatula, and Vitis vinifera, as background normalization. Finally, the conserved motifs obtained were verified with PFAM, SMART, and NCBI conserved domain inference tools.
4.5. Gene Ontology (GO) Annotation of the Hop MADS-Box Genes
The MADS-box genes of hop were annotated into each of the three categories of Gene Ontology (GO: biological process, molecular function, and cellular component) using the Blast2GO software [77], and the results were visualized with WEGO [78].
4.6. Expression Analysis of MADS-Box Genes in Hop Tissues
Gene transcription profiling of hop MADS-box genes was generated with NCBI-SRA RNA-seq libraries of meristems (SRR10320793), stems (SRR10320795), leaves (SRR575205), young leaves (ERR2040411), cones without glands (SRR575201), bracts (SRR10541757), glands (SRR575193), and a sample of the whole plant during the growing season (SRR4242068). The data were aligned to the hop masked genome sequence with STAR v.2.7.7 [79] using default parameters. The number of aligned reads was quantified with the htseq-count function in HTseq v.0.11.5 [80] assuming no strand specificity. The quantified reads were normalized as FPKM (Fragments Per Kilobase of transcript per Million mapped reads) with the edgeR package. Finally, a heatmap of MADS-box gene expression was generated in R v.3.6.3 using the gplots package. To corroborate these results, we used Samtoolsto filter all reads uniquely mapped to the genome. Subsequently, each locus was visualized with IGV, and the alignments for selected genes are reported in Figure S7.
5. Conclusions
In this work, we identified 65 MADS-box genes in the hop genome, with 36 being of type I and 29 genes of type II. Phylogenetic analyses showed that 27 type-II MADS-box genes belonged to 12 subfamilies, while two genes were of type MIKC*. Meanwhile, type-I MADS-box genes were classified in α-subfamily (26 members), β-subfamily (nine members), and γ-subfamily (a single member). The gene structure of type-I genes was less complex than that of type II genes, with fewer exons, even though the longest MADS-box gene was of type I. Some MIKCC-type MADS-box proteins did not display the K-box domain. Members of the FLC subfamily were not found in the hop genome. The only SOC1 subfamily member in the hop genome may undergo alternative splicing with intron retention. Genes of the ABCDE model of flower development were expressed in cones. One gene, a member of the α-subfamily, was found exclusively expressed in lupulin glands, with potential implications for specialized metabolism. Thus, this work contributes to understanding the evolutionary history of MADS-box in hop and provides perspectives on functional analysis and crop breeding.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/plants11091237/s1, Table S1: MADS-box genes identified in the hop genome. Table S2: RNA-seq libraries used to guide AUGUSTUS gene prediction and transcriptional profiling. Table S3: Gene Ontology terms of the hop MADS-box gene family. Figure S1: Phylogenetic tree of MIKC*-type MADS-box proteins of H. lupulus (2, in red font), Arabidopsis (6, in black font). Figure S2: RNA-Seq reads aligned on the gene 000453F.g47. Figure S3: Motif distribution of type-I MADS-box proteins in hop. Protein sequences are represented by black lines, and the conserved motifs are represented by colored boxes. Figure S4: Motif distribution of hop β-subfamily MADS-box proteins. Protein sequences are represented by black lines, and the conserved motifs are represented by colored boxes. (A) Logo of MADS-box domain. (B): Motif distribution of the hop β-subfamily MADS-box proteins. (C) Amino acid residues within MADS-box domains. Figure S5: Expression profile of the hop MADS-box genes in three RNA-Seq libraries from cones, leaves, and glands. Figure S6: Gene Ontology classification of MADS-box genes in hop and Arabidopsis. Figure S7: RNAseq reads that are uniquely mapped on six novel MADS-box genes (HlMADS28-33).
Author Contributions
R.R.d.O. and A.C.-J. conceptualized the project. R.M.G. conducted the analyses of the data. T.H.C.R. supported the bioinformatics analyses. T.H.C.R., R.R.d.O. and A.C.-J. supervised the project. R.M.G., R.R.d.O., V.A.B. and A.C.-J. discussed data analyses and results. R.M.G. wrote the first version of the manuscript. R.R.d.O., V.A.B. and A.C.-J. revised and contributed to the writing of the manuscript’s final version. All authors have read and agreed to the published version of the manuscript.
Funding
Student scholarship was funded by CNPq (grant 149043/2019-8). The Laboratory of Plant Molecular Physiology (LFMP) is partially supported by the Instituto Brasileiro de Ciência e Tecnologia do Café (INCT/Café), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Main data supporting the findings of this study are available in the manuscript and online supplementary materials. The raw data used for the analyses and figures are available upon request to A.C.-J.
Acknowledgments
The authors thank the Federal University of Lavras (UFLA/Brazil) and the members of the Laboratory of Plant Molecular Physiology (LFMP, UFLA/Brazil) for structural support of the experiments and data analyses; Léo Rufato (Universidade do Estado de Santa Catarina, UDESC) is thanked for the corrections and suggestions provided in a preliminary version of the manuscript; the Instituto Brasileiro de Ciência e Tecnologia do Café (INCT/Café), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) are acknowledged for financial support.
Conflicts of Interest
The authors declare no conflict of interest and that the funders had no role in the design of the study, data collection, analyses, interpretation, writing of the manuscript, or in the decision to publish the work.
References
- Riechmann, J.L.; Krizek, B.A.; Meyerowitz, E.M. Dimerization Specificity of Arabidopsis MADS Domain Homeotic Proteins APETALA1, APETALA3, PISTILLATA, and AGAMOUS. Proc. Natl. Acad. Sci. USA 1996, 93, 4793–4798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A.; Theißen, G. The Major Clades of MADS-Box Genes and Their Role in the Development and Evolution of Flowering Plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Smaczniak, C.; Immink, R.G.H.; Angenent, G.C.; Kaufmann, K. Developmental and Evolutionary Diversity of Plant MADS-Domain Factors: Insights from Recent Studies. Development 2012, 139, 3081–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daminato, M.; Masiero, S.; Resentini, F.; Lovisetto, A.; Casadoro, G. Characterization of TM8, a MADS-Box Gene Expressed in Tomato Flowers. BMC Plant Biol. 2014, 14, 319. [Google Scholar] [CrossRef] [Green Version]
- da Silveira Falavigna, V.; Severing, E.; Lai, X.; Estevan, J.; Farrera, I.; Hugouvieux, V.; Revers, L.F.; Zubieta, C.; Coupland, G.; Costes, E.; et al. Unraveling the Role of MADS Transcription Factor Complexes in Apple Tree Dormancy. New Phytol. 2021, 232, 2071–2088. [Google Scholar] [CrossRef]
- Li, G.; Kuijer, H.N.J.; Yang, X.; Liu, H.; Shen, C.; Shi, J.; Betts, N.; Tucker, M.R.; Liang, W.; Waugh, R.; et al. MADS1 Maintains Barley Spike Morphology at High Ambient Temperatures. Nat. Plants 2021, 7, 1093–1107. [Google Scholar] [CrossRef]
- Gómez-Soto, D.; Ramos-Sánchez, J.M.; Alique, D.; Conde, D.; Triozzi, P.M.; Perales, M.; Allona, I. Overexpression of a SOC1-Related Gene Promotes Bud Break in Ecodormant Poplars. Front. Plant Sci. 2021, 12, 670497. [Google Scholar] [CrossRef]
- Ayra, L.; Reyero-Saavedra, M.d.R.; Isidra-Arellano, M.C.; Lozano, L.; Ramírez, M.; Leija, A.; Fuentes, S.-I.; Girard, L.; Valdés-López, O.; Hernández, G. Control of the Rhizobia Nitrogen-Fixing Symbiosis by Common Bean MADS-Domain/AGL Transcription Factors. Front. Plant Sci. 2021, 12, 1061. [Google Scholar] [CrossRef]
- Hyun, Y.; Richter, R.; Vincent, C.; Martinez-Gallegos, R.; Porri, A.; Coupland, G. Multi-Layered Regulation of SPL15 and Cooperation with SOC1 Integrate Endogenous Flowering Pathways at the Arabidopsis Shoot Meristem. Dev. Cell 2016, 37, 254–266. [Google Scholar] [CrossRef]
- Immink, R.G.H.; Posé, D.; Ferrario, S.; Ott, F.; Kaufmann, K.; Valentim, F.L.; de Folter, S.; van der Wal, F.; van Dijk, A.D.J.; Schmid, M.; et al. Characterization of SOC1′s Central Role in Flowering by the Identification of Its Upstream and Downstream Regulators. Plant Physiol. 2012, 160, 433–449. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Lee, I. Regulation and Function of SOC1, a Flowering Pathway Integrator. J. Exp. Bot. 2010, 61, 2247–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Chen, H.; Er, H.L.; Soo, H.M.; Kumar, P.P.; Han, J.-H.; Liou, Y.C.; Yu, H. Direct Interaction of AGL24 and SOC1 Integrates Flowering Signals in Arabidopsis. Dev. Camb. Engl. 2008, 135, 1481–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torti, S.; Fornara, F. AGL24 Acts in Concert with SOC1 and FUL during Arabidopsis Floral Transition. Plant Signal. Behav. 2012, 7, 1251–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, U.; Höhmann, S.; Nettesheim, K.; Wisman, E.; Saedler, H.; Huijser, P. Molecular Cloning of SVP: A Negative Regulator of the Floral Transition in Arabidopsis. Plant J. 2000, 21, 351–360. [Google Scholar] [CrossRef]
- Michaels, S.D.; Amasino, R.M. Flowering Locus C Encodes a Novel MADS Domain Protein That Acts as a Repressor of Flowering. Plant Cell 1999, 11, 949–956. [Google Scholar] [CrossRef] [Green Version]
- Mateos, J.L.; Madrigal, P.; Tsuda, K.; Rawat, V.; Richter, R.; Romera-Branchat, M.; Fornara, F.; Schneeberger, K.; Krajewski, P.; Coupland, G. Combinatorial Activities of Short Vegetative Phase and Flowering Locus C Define Distinct Modes of Flowering Regulation in Arabidopsis. Genome Biol. 2015, 16, 31. [Google Scholar] [CrossRef] [Green Version]
- Madrid, E.; Chandler, J.W.; Coupland, G. Gene Regulatory Networks Controlled by FLOWERING LOCUS C That Confer Variation in Seasonal Flowering and Life History. J. Exp. Bot. 2021, 72, 4–14. [Google Scholar] [CrossRef]
- Kim, D.-H.; Doyle, M.R.; Sung, S.; Amasino, R.M. Vernalization: Winter and the Timing of Flowering in Plants. Annu. Rev. Cell Dev. Biol. 2009, 25, 277–299. [Google Scholar] [CrossRef] [Green Version]
- Reeves, P.A.; He, Y.; Schmitz, R.J.; Amasino, R.M.; Panella, L.W.; Richards, C.M. Evolutionary Conservation of the FLOWERING LOCUS C-Mediated Vernalization Response: Evidence From the Sugar Beet (Beta Vulgaris). Genetics 2007, 176, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Ruelens, P.; de Maagd, R.A.; Proost, S.; Theißen, G.; Geuten, K.; Kaufmann, K. Flowering Locus C in Monocots and the Tandem Origin of Angiosperm-Specific MADS-Box Genes. Nat. Commun. 2013, 4, 2280. [Google Scholar] [CrossRef] [Green Version]
- Kagaya, H.; Ito, N.; Shibuya, T.; Komori, S.; Kato, K.; Kanayama, Y. Characterization of FLOWERING LOCUS C Homologs in Apple as a Model for Fruit Trees. Int. J. Mol. Sci. 2020, 21, 4562. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, R.R.; Cesarino, I.; Mazzafera, P.; Dornelas, M.C. Flower Development in Coffea arabica L.: New Insights into MADS-Box Genes. Plant Reprod. 2014, 27, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, A.; Lloyd, A.; Hepworth, J.; Tudor, E.H.; Jones, D.M.; Woodhouse, S.; Bilham, L.; Chinoy, C.; Williams, K.; Corke, F.; et al. Total FLC Transcript Dynamics from Divergent Paralogue Expression Explains Flowering Diversity in Brassica Napus. New Phytol. 2021, 229, 3534–3548. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.; Geuten, K. The Role of Flowering Locus C Relatives in Cereals. Front. Plant Sci. 2020, 11, 617340. [Google Scholar] [CrossRef]
- Sharma, N.; Ruelens, P.; D’hauw, M.; Maggen, T.; Dochy, N.; Torfs, S.; Kaufmann, K.; Rohde, A.; Geuten, K. A Flowering Locus C Homolog Is a Vernalization-Regulated Repressor in Brachypodium and Is Cold Regulated in Wheat. Plant Physiol. 2017, 173, 1301–1315. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; dePamphilis, C. The ABCs of Floral Evolution. Cell 2000, 101, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Bowman, J.L.; Smyth, D.R.; Meyerowitz, E.M. Genes Directing Flower Development in Arabidopsis. Plant Cell 1989, 1, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Benedito, V.A.; Visser, P.B.; van Tuyl, J.M.; Angenent, G.C.; de Vries, S.C.; Krens, F.A. Ectopic Expression of LLAG1, an Agamous Homologue from Lily (Lilium longiflorum Thunb.) Causes Floral Homeotic Modifications in Arabidopsis. J. Exp. Bot. 2004, 55, 1391–1399. [Google Scholar] [CrossRef] [Green Version]
- Thomson, B.; Wellmer, F. Chapter Eight—Molecular Regulation of Flower Development. In Current Topics in Developmental Biology; Grossniklaus, U., Ed.; Plant Development and Evolution; Academic Press: Cambridge, MA, USA, 2019; Volume 131, pp. 185–210. [Google Scholar]
- Coen, E.S.; Meyerowitz, E.M. The War of the Whorls: Genetic Interactions Controlling Flower Development. Nature 1991, 353, 31–37. [Google Scholar] [CrossRef]
- Theißen, G.; Saedler, H. Floral Quartets. Nature 2001, 409, 469–471. [Google Scholar] [CrossRef]
- Irish, V.F.; Sussex, I.M. Function of the Apetala-1 Gene during Arabidopsis Floral Development. Plant Cell 1990, 2, 741–753. [Google Scholar] [PubMed] [Green Version]
- Goto, K.; Meyerowitz, E.M. Function and Regulation of the Arabidopsis Floral Homeotic Gene Pistillata. Genes Dev. 1994, 8, 1548–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, T.; Brockman, L.L.; Meyerowitz, E.M. The Homeotic Gene APETALA3 of Arabidopsis Thaliana Encodes a MADS Box and Is Expressed in Petals and Stamens. Cell 1992, 68, 683–697. [Google Scholar] [CrossRef]
- Yanofsky, M.F.; Ma, H.; Bowman, J.L.; Drews, G.N.; Feldmann, K.A.; Meyerowitz, E.M. The Protein Encoded by the Arabidopsis Homeotic Gene Agamous Resembles Transcription Factors. Nature 1990, 346, 35–39. [Google Scholar] [CrossRef]
- Savidge, B.; Rounsley, S.D.; Yanofsky, M.F. Temporal Relationship between the Transcription of Two Arabidopsis MADS Box Genes and the Floral Organ Identity Genes. Plant Cell 1995, 7, 721–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelaz, S.; Ditta, G.S.; Baumann, E.; Wisman, E.; Yanofsky, M.F. B and C Floral Organ Identity Functions Require SEPALLATA MADS-Box Genes. Nature 2000, 405, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Kyozuka, J.; Kobayashi, T.; Morita, M.; Shimamoto, K. Spatially and Temporally Regulated Expression of Rice MADS Box Genes with Similarity to Arabidopsis Class A, B and C Genes. Plant Cell Physiol. 2000, 41, 710–718. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Shi, X.; Lin, X.; Liu, Y.; Chong, K.; Theißen, G.; Meng, Z. The ABCs of Flower Development: Mutational Analysis of AP1/FUL-like Genes in Rice Provides Evidence for a Homeotic (A)-Function in Grasses. Plant J. 2017, 89, 310–324. [Google Scholar] [CrossRef]
- Kuijer, H.N.J.; Shirley, N.J.; Khor, S.F.; Shi, J.; Schwerdt, J.; Zhang, D.; Li, G.; Burton, R.A. Transcript Profiling of MIKCc MADS-Box Genes Reveals Conserved and Novel Roles in Barley Inflorescence Development. Front. Plant Sci. 2021, 12, 1834. [Google Scholar] [CrossRef]
- Chi, Y.; Huang, F.; Liu, H.; Yang, S.; Yu, D. An APETALA1-like Gene of Soybean Regulates Flowering Time and Specifies Floral Organs. J. Plant Physiol. 2011, 168, 2251–2259. [Google Scholar] [CrossRef]
- Huang, F.; Xu, G.; Chi, Y.; Liu, H.; Xue, Q.; Zhao, T.; Gai, J.; Yu, D. A Soybean MADS-Box Protein Modulates Floral Organ Numbers, Petal Identity and Sterility. BMC Plant Biol. 2014, 14, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Clemens, J.; Jameson, P.E. Expression of Floral Identity Genes in Clianthus Maximus during Mass Inflorescence Abortion and Floral Development. Ann. Bot. 2011, 107, 1501–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masiero, S.; Colombo, L.; Grini, P.E.; Schnittger, A.; Kater, M.M. The Emerging Importance of Type I MADS Box Transcription Factors for Plant Reproduction. Plant Cell 2011, 23, 865–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauerle, W.L. Disentangling Photoperiod from Hop Vernalization and Dormancy for Global Production and Speed Breeding. Sci. Rep. 2019, 9, 16003. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.G.; Schwabe, W.W. Factors Controlling Flowering in the Hop (Humulus lupulus L.). Ann. Bot. 1969, 33, 781–793. [Google Scholar] [CrossRef]
- Benkherouf, A.Y.; Logrén, N.; Somborac, T.; Kortesniemi, M.; Soini, S.L.; Yang, B.; Salo-Ahen, O.M.H.; Laaksonen, O.; Uusi-Oukari, M. Hops Compounds Modulatory Effects and 6-Prenylnaringenin Dual Mode of Action on GABAA Receptors. Eur. J. Pharmacol. 2020, 873, 172962. [Google Scholar] [CrossRef]
- Jiang, C.-H.; Sun, T.-L.; Xiang, D.-X.; Wei, S.-S.; Li, W.-Q. Anticancer Activity and Mechanism of Xanthohumol: A Prenylated Flavonoid From Hops (Humulus lupulus L.). Front. Pharmacol. 2018, 9, 530. [Google Scholar] [CrossRef]
- Yamashita, M.; Fukizawa, S.; Nonaka, Y. Hop-Derived Prenylflavonoid Isoxanthohumol Suppresses Insulin Resistance by Changing the Intestinal Microbiota and Suppressing Chronic Inflammation in High Fat Diet-Fed Mice. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1537–1547. [Google Scholar] [CrossRef]
- Parenicová, L.; de Folter, S.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B.; et al. Molecular and Phylogenetic Analyses of the Complete MADS-Box Transcription Factor Family in Arabidopsis: New Openings to the MADS World. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-Box Gene Family in Rice: Genome-Wide Identification, Organization and Expression Profiling during Reproductive Development and Stress. BMC Genom. 2007, 8, 242. [Google Scholar] [CrossRef] [Green Version]
- Grimplet, J.; Martínez-Zapater, J.M.; Carmona, M.J. Structural and Functional Annotation of the MADS-Box Transcription Factor Family in Grapevine. BMC Genom. 2016, 17, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, D.; Cao, Y.; Chen, T.; Abdullah, M.; Jin, Q.; Fan, H.; Lin, Y.; Cai, Y. Evolution and Functional Divergence of MADS-Box Genes in Pyrus. Sci. Rep. 2019, 9, 1266. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.T.; Sudarsanam, R.; Henning, J.; Hendrix, D. HopBase: A Unified Resource for Humulus Genomics. Database 2017, 2017, bax009. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.-H.; Ju, Y.; Seo, P.J.; Lee, J.-H.; Park, C.-M. The SOC1-SPL Module Integrates Photoperiod and Gibberellic Acid Signals to Control Flowering Time in Arabidopsis. Plant J. 2012, 69, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Ullah, F.; Hamilton, M.; Reddy, A.S.N.; Ben-Hur, A. Exploring the Relationship between Intron Retention and Chromatin Accessibility in Plants. BMC Genom. 2018, 19, 21. [Google Scholar] [CrossRef] [Green Version]
- Adamczyk, B.J.; Lehti-Shiu, M.D.; Fernandez, D.E. The MADS Domain Factors AGL15 and AGL18 Act Redundantly as Repressors of the Floral Transition in Arabidopsis. Plant J. Cell Mol. Biol. 2007, 50, 1007–1019. [Google Scholar] [CrossRef]
- Serivichyaswat, P.; Ryu, H.-S.; Kim, W.; Kim, S.; Chung, K.S.; Kim, J.J.; Ahn, J.H. Expression of the Floral Repressor MiRNA156 Is Positively Regulated by the AGAMOUS-like Proteins AGL15 and AGL18. Mol. Cells 2015, 38, 259–266. [Google Scholar] [CrossRef]
- Gustafson-Brown, C.; Savidge, B.; Yanofsky, M.F. Regulation of the Arabidopsis Floral Homeotic Gene APETALA1. Cell 1994, 76, 131–143. [Google Scholar] [CrossRef]
- Shephard, H.L.; Parker, J.S.; Darby, P.; Ainsworth, C.C. Sexual Development and Sex Chromosomes in Hop. New Phytol. 2000, 148, 397–411. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, J.; Feng, C.; Liu, M.; Wang, J.; Hu, Y. Genome-Wide Identification, Characterization of the MADS-Box Gene Family in Chinese Jujube and Their Involvement in Flower Development. Sci. Rep. 2017, 7, 1025. [Google Scholar] [CrossRef]
- Okada, Y.; Ito, K. Cloning and Analysis of Valerophenone Synthase Gene Expressed Specifically in Lupulin Gland of Hop (Humulus lupulus L.). Biosci. Biotechnol. Biochem. 2001, 65, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using Native and Syntenically Mapped CDNA Alignments to Improve de Novo Gene Finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-Level Expression Analysis of RNA-Seq Experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De Novo Transcript Sequence Reconstruction from RNA-Seq Using the Trinity Platform for Reference Generation and Analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Hoff, K.J.; Stanke, M. Predicting Genes in Single Genomes with AUGUSTUS. Curr. Protoc. Bioinform. 2019, 65, e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D.; et al. Improving the Arabidopsis Genome Annotation Using Maximal Transcript Alignment Assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Zhang, H.; Kong, L.; Gao, G.; Luo, J. PlantTFDB 3.0: A Portal for the Functional and Evolutionary Study of Plant Transcription Factors. Nucleic Acids Res. 2014, 42, D1182–D1187. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Sela, I.; Ashkenazy, H.; Katoh, K.; Pupko, T. GUIDANCE2: Accurate Detection of Unreliable Alignment Regions Accounting for the Uncertainty of Multiple Parameters. Nucleic Acids Res. 2015, 43, W7–W14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. PHYLIP—Phylogeny Inference Package (Version 3.2). Cladistics 1989, 5, 164–166. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An Upgraded Gene Feature Visualization Server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A Universal Tool for Annotation, Visualization and Analysis in Functional Genomics Research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; et al. WEGO 2.0: A Web Tool for Analyzing and Plotting GO Annotations, 2018 Update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data. bioRxiv 2014. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).