


Transcriptome-Based Study on the Phylogeny and Hybridization of Marattialean Ferns (Marattiaceae)

 , ,
, ,
Abstract
:1. Introduction
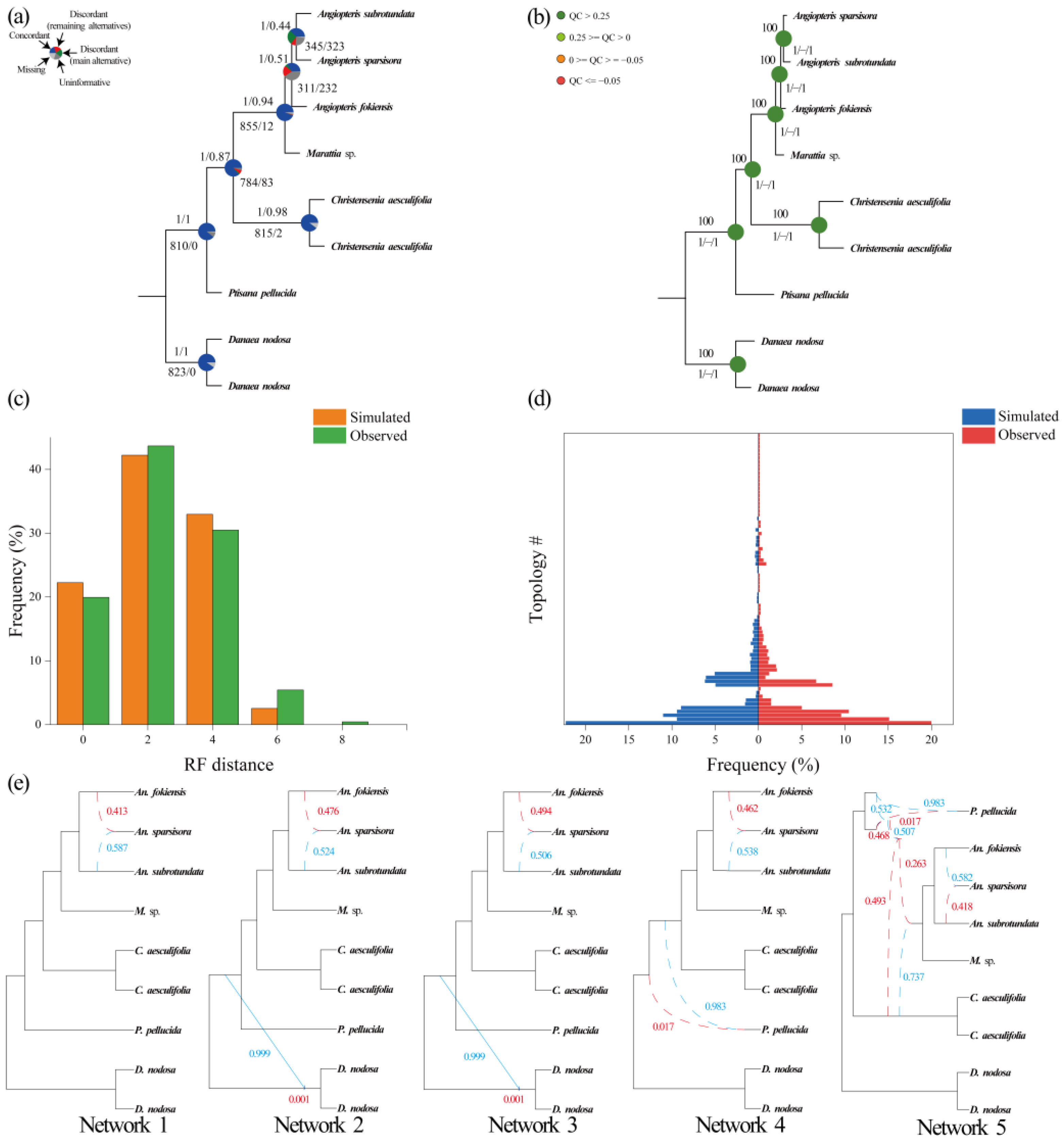
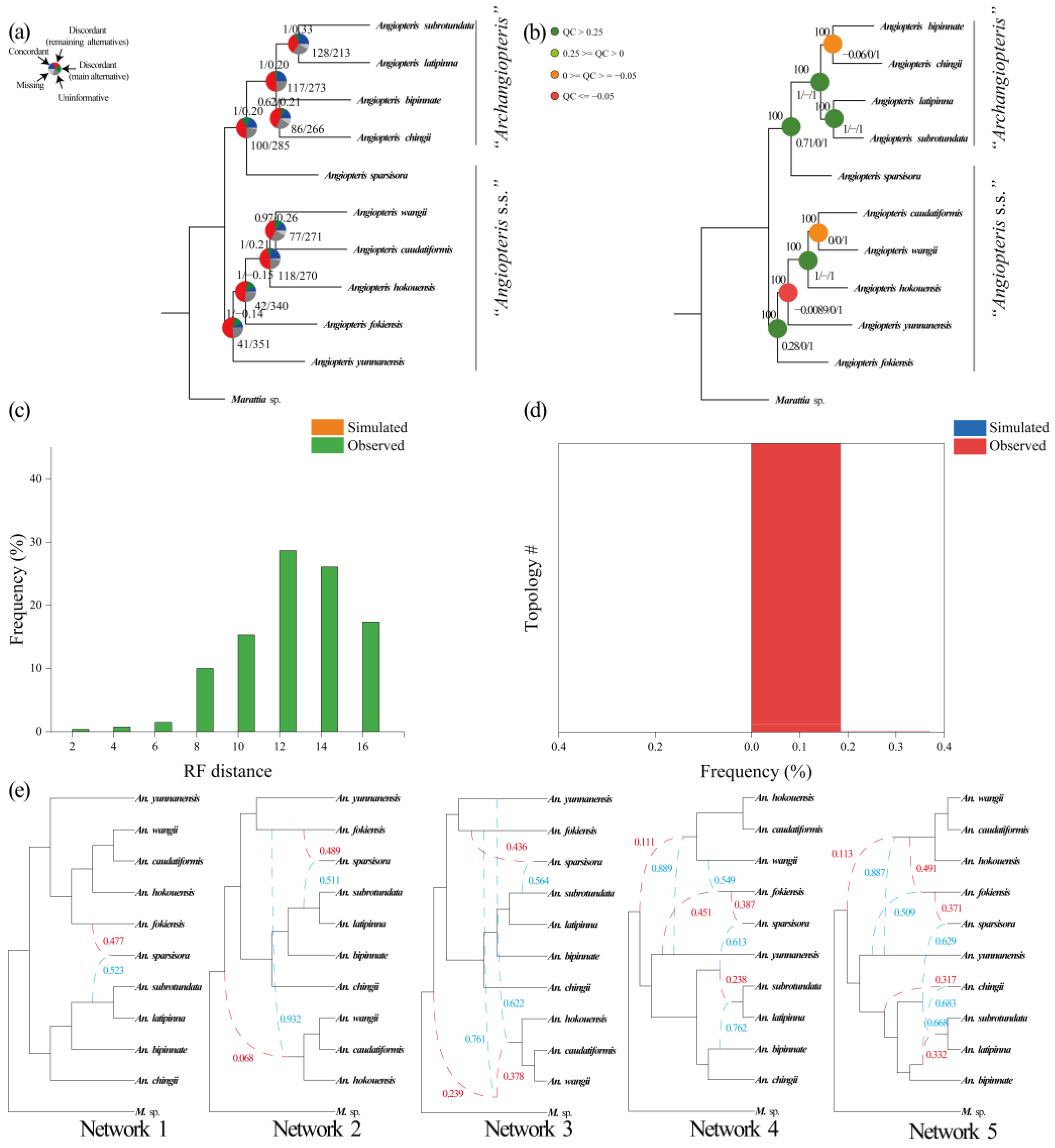
2. Results
2.1. Characteristics of Transcriptomes and Nuclear Gene Datasets
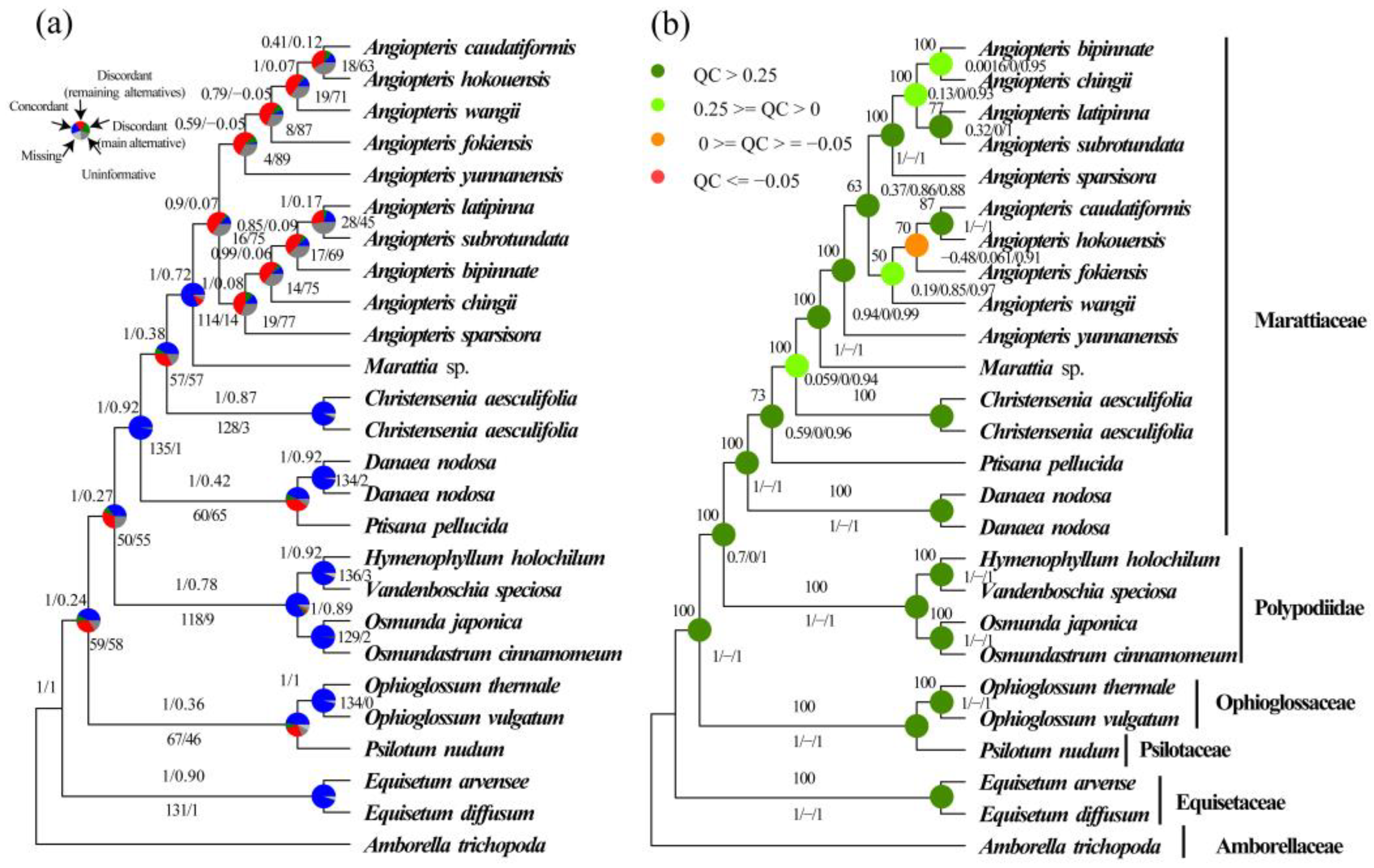
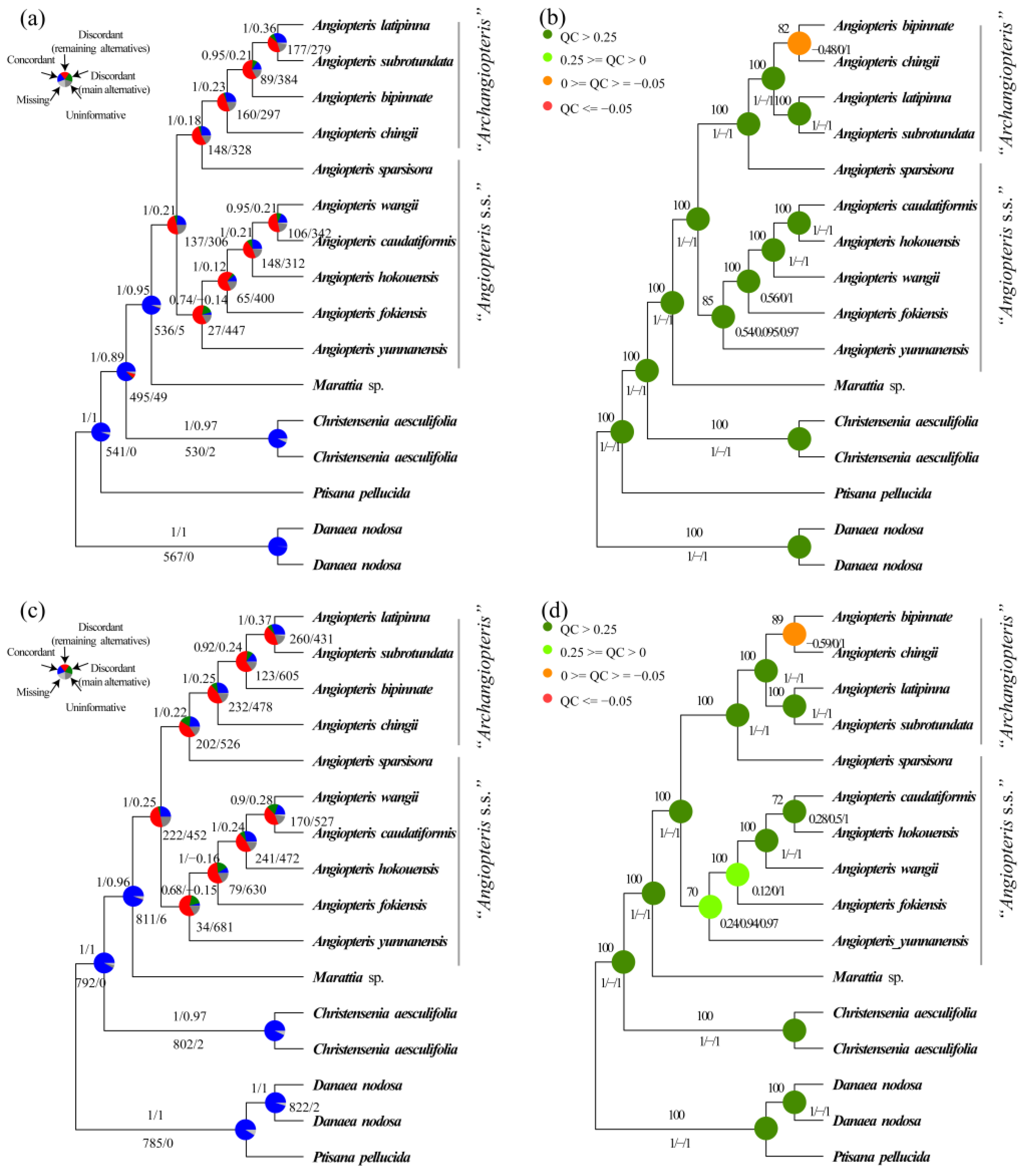
2.2. Phylogenetic Reconstruction Using Single-Copy Nuclear Genes
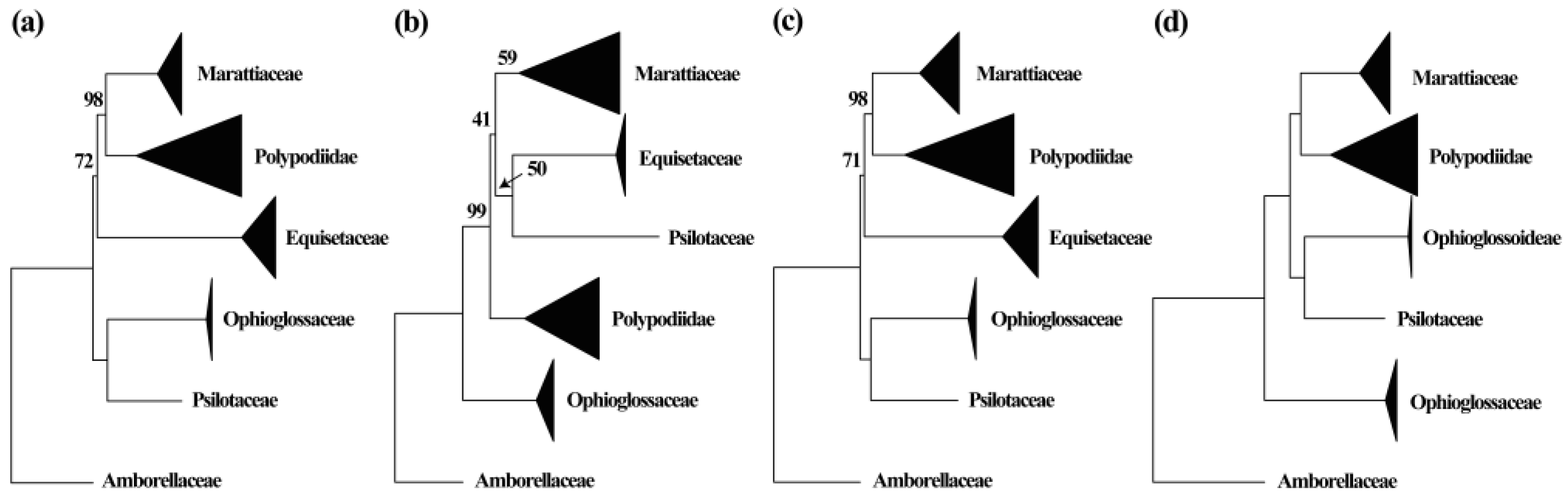
2.3. Phylogenetic Reconstruction of Organelle Genes
2.4. Coalescent Simulations
2.5. Network Analysis
2.6. Divergence Dating Estimation
3. Discussion
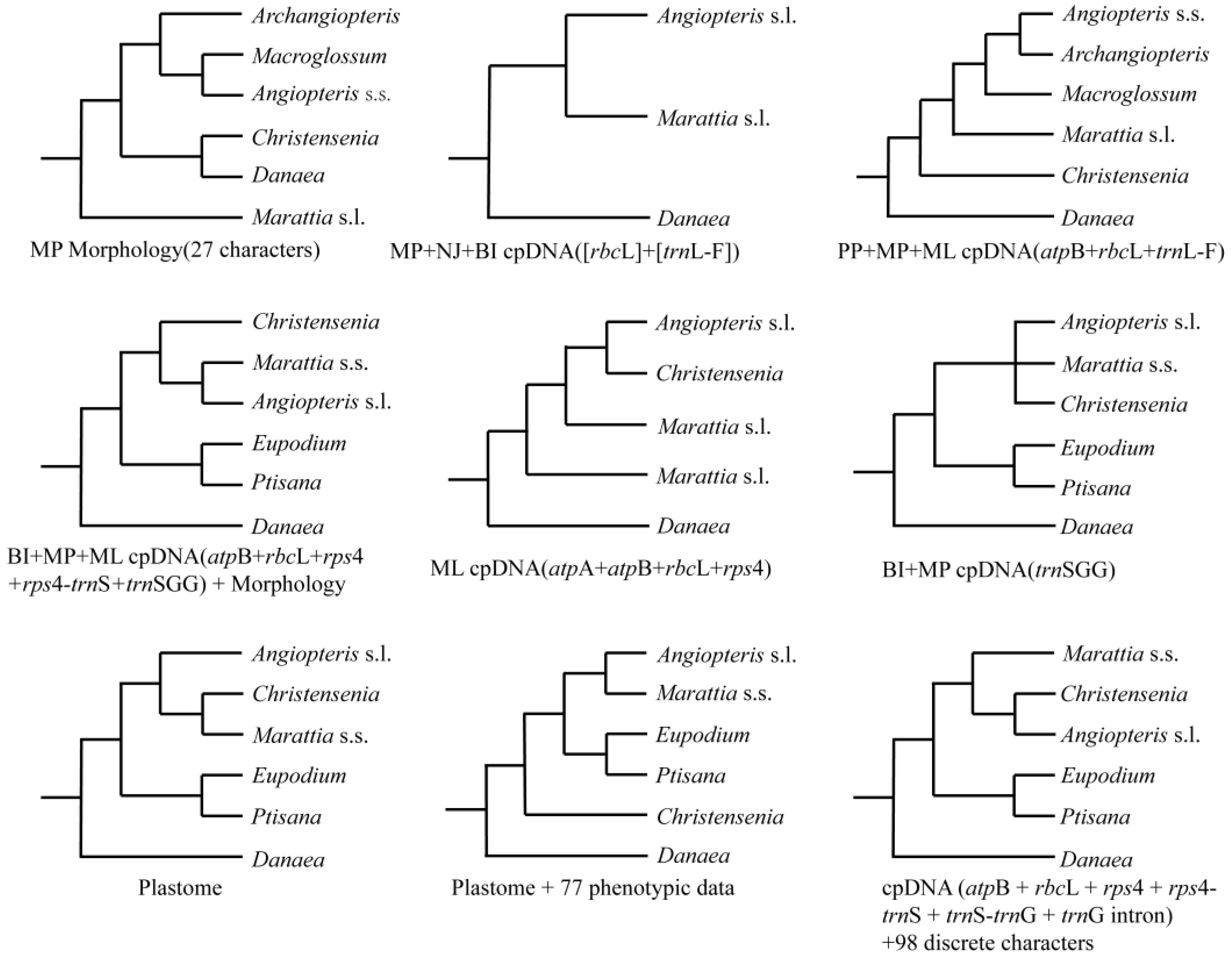
3.1. New Insights into Phylogeny of Marattiaceae
3.2. Resolution of Angiopteris s.l.
3.3. Phylogenetic Incongruence as Further Evidence for ILS and Hybridization
4. Materials and Methods
4.1. Taxon Sampling and RNA Sequencing
4.2. Assembly and Single-Copy Orthologue Identification
4.3. Nuclear Genes Dataset Generation and Phylogenetic Inference
4.4. Organelle Genome Assembly, Locus Extraction, and Phylogenetic Analysis
4.5. Concordance, ILS Simulation, and Hybridization Inference
4.6. Age Estimation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murdock, A.G. A taxonomic revision of the eusporangiate fern family Marattiaceae, with description of a new genus Ptisana. Taxon 2008, 57, 737–755. [Google Scholar] [CrossRef]
- Ppg, I. A community-derived classifification for extant lycophytes and ferns. J. Syst. Evol. 2016, 54, 563–603. [Google Scholar] [CrossRef]
- Mamay, S.H. Some American Carboniferous fern fructififications. Ann. Mo. Bot. Gard. 1950, 37, 409–476. [Google Scholar] [CrossRef]
- Millay, M.A.; Taylor, T.N. An unusual Botryopterid sporangial aggregation from the middle Pennsylvanian of north America. Am. J. Bot. 1980, 67, 758–773. [Google Scholar] [CrossRef]
- Stewart, W.N.; Rothwell, G.W. Paleobotany and the Evolution of Plants, 2nd ed.; Cambridge University Press: Cambridge, UK, 1993; pp. 1–405. [Google Scholar]
- Millay, M.A. A review of permineralized Euramerican Carboniferous tree ferns. Rev. Palaeobot. Palynol. 1997, 95, 191–209. [Google Scholar] [CrossRef]
- DiMichele, W.L.; Phillips, T.L. The ecology of Paleozoic ferns. Rev. Palaeobot. Palynol. 2002, 119, 143–159. [Google Scholar] [CrossRef]
- Taylor, T.N.; Taylor, E.L.; Krings, M. Paleobotany, the Biology and Evolution of Fossil Plants, 2nd ed.; Academic Press: Burlington, MA, USA, 2009. [Google Scholar]
- Rothwell, G.W.; Millay, M.A.; Stockey, R.A. Escapia gen. nov.: Morphological Evolution, Paleogeographic Diversifification, and the Environmental Distribution of Marattialean Ferns through Time. In Transformative Paleobotany: Papers to Commemorate the Life and Legacy of Thomas N. Taylor; Krings, M., Harper, C.J., Cúneo, N.R., Rothwell, G.W., Eds.; Elsevier: New York, NY, USA, 2018; pp. 271–360. [Google Scholar]
- Murdock, A.G. Phylogeny of marattioid ferns (Marattiaceae): Inferring a root in the absence of a closely related outgroup. Am. J. Bot. 2008, 95, 626–641. [Google Scholar] [CrossRef]
- Hill, C.R.; Wagner, R.H.; El-Khayal, A.A. Qasimia gen. nov., an early Marattia-like fern from the Permian of Saudi Arabia. Scripta. Geol. 1985, 79, 1–50. [Google Scholar]
- Hill, C.R. Jurassic Angiopteris (Marattiales) from north Yorkshire. Rev. Palaeobot. Palynol. 1987, 51, 65–93. [Google Scholar] [CrossRef]
- Lesnikowska, A.; Galtier, J. A reconsideration of four genera of permineralized Marattiales from the Stephanian and Autunian of France. Rev. Palaeobot. Palynol. 1991, 67, 141–152. [Google Scholar] [CrossRef]
- Delevoryas, T.; Taylor, T.N.; Taylor, E.L. A marattialean fern from the Triassic of Antarctica. Rev. Palaeobot. Palynol. 1992, 74, 101–107. [Google Scholar] [CrossRef]
- Liu, Z.-H.; Li, C.-S.; Hilton, J. Zhutheca Liu, Li et Hilton gen. nov., the fertile pinnules of Fascipteris densata Gu et Zhi and their significance in marattialean evolution. Rev. Palaeobot. Palynol. 2000, 109, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-H.; Li, C.-S.; Hilton, J. Fertile pinnules of Danaeites rigida Gu and Zhi (Marattiales) from the Upper Permian of south China. Bot. J. Linn. Soc. 2001, 136, 107–117. [Google Scholar] [CrossRef]
- Hu, S.; Taylor, D.W.; Brenner, G.J.; Basha, S.H. A new marsilealean fern species from the Early Cretaceous of Jordan. Palaeoworld 2008, 17, 235–245. [Google Scholar] [CrossRef]
- Escapa, I.H.; Bomflfleur, B.; Cuneo, N.R.; Scasso, R. A new marattiaceous fern from the Lower Jurassic of Patagonia (Argentina): The renaissance of Marattiopsis. J. Syst. Palaeontol. 2014, 13, 677–689. [Google Scholar] [CrossRef]
- Lehtonen, S.; Poczai, P.; Sablok, G.; Hyvonen, J.; Karger, D.N.; Flores, J. Exploring the phylogeny of the marattialean ferns. Cladistics 2020, 36, 569–593. [Google Scholar] [CrossRef]
- May, M.R.; Contreras, D.L.; Sundue, M.A.; Nagalingum, N.S.; Rothfels, C.J. Inferring the total-evidence timescale of Marattialean fern evolution in the face of model sensitivity. Syst. Biol. 2021, 70, 1232–1255. [Google Scholar] [CrossRef]
- Rai, H.S.; Graham, S.W. Utility of a large; multigene plastid data set in inferring higher-order relationships in ferns and relatives (monilophytes). Am. J. Bot. 2010, 97, 1444–1456. [Google Scholar] [CrossRef]
- Kuo, L.-Y.; Li, F.-W.; Chiou, W.-L.; Wang, C.-N. First insights into fern matK phylogeny. Mol. Phylogenet. Evol. 2011, 59, 556–566. [Google Scholar] [CrossRef]
- Lehtonen, S. Towards resolving the complete fern tree of life. PLoS ONE 2011, 6, e24851. [Google Scholar] [CrossRef] [Green Version]
- Grewe, F.; Guo, W.; Gubbels, E.A.; Hansen, A.K.; Mower, J.P. Complete plastid genomes from Ophioglossum californicum; Psilotum nudum; and Equisetum hyemale reveal an ancestral land plant genome structure and resolve the position of Equisetales among monilophytes. BMC Evol. Biol. 2013, 13, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.T.; Chung, M.G.; Kim, K.-J. Chloroplast genome evolution in early diverged leptosporangiate ferns. Mol. Cells. 2014, 37, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.-M.; Zhang, N.; Du, X.-Y.; Wen, J.; Li, D.-Z. Chloroplast phylogenomics resolves key relationships in ferns. J. Syst. Evol. 2015, 53, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Knie, N.; Fischer, S.; Grewe, F.; Polsakiewicz, M.; Knoop, V. Horsetails are the sister group to all other monilophytes and Marattiales are sister to leptosporangiate ferns. Mol. Phylogenet. Evol. 2015, 90, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Rothfels, C.J.; Johnson, A.K.; Hovenkamp, P.H.; Swofford, D.L.; Roskam, H.C.; Fraseer-Jenkins, C.R.; Windham, M.D.; Pryer, K. Natural hybridization between genera that diverged from each other approximately 60 million years ago. Am. Nat. 2015, 185, 433–442. [Google Scholar] [CrossRef]
- Testo, W.; Sundue, M. A 4000-species dataset provides new insight into the evolution of ferns. Mol. Phylogenet. Evol. 2016, 105, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Labiak, P.H.; Karol, K.G. Plastome sequences of an ancient fern lineage reveal remarkable changes in gene content and architecture. Am. J. Bot. 2017, 104, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Lehtonen, S.; Silvestro, D.; Karger, D.N.; Scotese, C.; Tuomisto, H.; Kessler, M.; Pena, C.; Wahlberg, N.; Antonelli, A. Environmentally driven extinction and opportunistic origination explain fern diversification patterns. Sci. Rep. 2017, 7, 4831. [Google Scholar] [CrossRef] [Green Version]
- Gitzendanner, M.A.; Soltis, P.S.; Wong, G.K.S.; Ruhfel, B.R.; Soltis, D.E. Plastid phylogenomic analysis of green plants: A billion years of evolutionary history. Am. J. Bot. 2018, 105, 291–301. [Google Scholar] [CrossRef]
- Kuo, L.-Y.; Qi, X.-P.; Ma, H.; Li, F.-W. Order-level fern plastome phylogenomics: New insights from Hymenophyllales. Am. J. Bot. 2018, 105, 1545–1555. [Google Scholar] [CrossRef]
- Qi, X.-P.; Kuo, L.-Y.; Guo, C.; Li, H.; Li, Z.-Y.; Qi, J.; Wang, L.-B.; Hu, Y.; Xiang, J.-Y.; Zhang, C.-F.; et al. A well-resolved fern nuclear phylogeny reveals the evolution history of numerous transcription factor families. Mol. Phylogenet. Evol. 2018, 127, 961–977. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, S.; Cardenas, G.G. Dynamism in plastome structure observed across the phylogenetic tree of ferns. Bot. J. Linn. Soc. 2019, 190, 229–241. [Google Scholar] [CrossRef]
- Du, X.-Y.; Kuo, L.-Y.; Zuo, Z.-Y.; Li, D.-Z.; Lu, J.M. Structural variation of plastomes provides key insight into the deep phylogeny of ferns. Front. Plant Sci. 2022, 13, 862772. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, J.A.; Kim, E.H.; Barbazuk, W.B.; Sessa, E.B. Phylotranscriptomics illuminates the placement of whole genome duplications and gene retention in ferns. Front. Plant Sci. 2022, 13, 882441. [Google Scholar] [CrossRef]
- Wickett, N.J.; Mirarab, S.; Nguyen, N.; Warnow, T.; Carpenter, E.; Matasci, N.; Ayyampalayam, S.; Barker, M.S.; Burleigh, J.G.; Gitzendanner, M.A.; et al. Phylotranscriptomic analysis of the origin and early diversifification of land plants. Proc. Natl. Acad. Sci. USA 2014, 111, E4859–E4868. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Jin, D.; Shu, J.-P.; Zhou, X.L.; Lei, M.; Wei, R.; Shang, H.; Wei, H.J.; Zhang, R.; Liu, L.; et al. Large-scale phylogenomic analysis resolves a backbone phylogeny in ferns. GigaScience 2018, 7, gix116. [Google Scholar] [CrossRef] [Green Version]
- One Thousand Plant Transcriptomes Initiative. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679–685. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.-L.; Li, L.; Wang, B.; Chen, Z.; Dombrovska, O.; Lee, J.; Kent, L.; Li, R.; Jobson, R.W.; Hendry, T.A.; et al. A nonflowering land plant phylogeny inferred from nucleotide sequences of seven chloroplast; mitochondrial; and nuclear genes. Int. J. Plant Sci. 2007, 68, 691–708. [Google Scholar] [CrossRef]
- He, Z.-R.; Christenhusz, M.J.M. Marattiaceae. In Flora of China; Wu, Z.Y., Raven, P.H., Hong, D.Y., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MI, USA, 2013; Volume 2–3, pp. 82–89. [Google Scholar]
- Yan, Y.-H.; Zhou, X.-L. Ferns of Hainan; China Forestry Publishing: Beijing, China, 2018; 45p. [Google Scholar]
- Wang, T.; Xiao, B.; Liu, E.-D.; Nguyen, K.S.; Duan, J.-Q.; Wang, K.-L.; Yan, Y.-H.; Xiang, J.-Y. Rediscovery of Angiopteris tonkinensis (Marattiaceae) after 100 years; and its revision. PhytoKeys 2020, 161, 1–9. [Google Scholar] [CrossRef]
- Hill, C.R.; Camus, J.M. Evolutionary cladistics of marattialean ferns. Bull. Br. Mus. Nat. Hist. 1986, 14, 219–300. [Google Scholar]
- Hill, C.R.; Camus, J.M. Pattern cladistics or evolutionary cladistics. Cladistics 1986, 2, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-H.; Hilton, J.; Li, C.-S. Review on the origin; evolution and phylogeny of Marattiales. Chinese Bull. Bot. 2000, 17, 39–52. [Google Scholar]
- Senterre, B.; Rouhan, G.; Fabre, I.; Morel, C.; Christenhusz, M.J.M. Revision of the fern family Marattiaceae in the Seychelles with two new species and a discussion of the African Ptisana fraxinea complex. Phytotaxa 2014, 158, 19–57. [Google Scholar] [CrossRef] [Green Version]
- Rothwell, G.W.; Millay, M.A.; Stockey, R.A. Resolving the overall pattern of marattialean fern phylogeny. Am. J. Bot. 2018, 105, 1304–1314. [Google Scholar] [CrossRef] [PubMed]
- Ching, R.-C. Systematic arrangements of families and genera of Chinese Pteridophytes with corresponding names in Chinese. Acta Phytotax. Sin. 1954, 3, 93–99. [Google Scholar]
- Ching, R.-C. A revision of the ferns genus Archangiopteris Christ & Giesenhagen. Acta Phytotax. Sin. 1958, 7, 212–224. [Google Scholar]
- Ching, R.-C. Flora of China; Science Press: Beijing, China, 1959; Volume II, pp. 330–345. [Google Scholar]
- Chu, W.-M.; He, Z.-R. Flora of Yunnan; Science Press: Beijing, China, 2006; Volume 20, pp. 117–128. [Google Scholar]
- Pichi-Sermolli, R.E.G. Tentamen Pteridophytorum genera in taxonomicum ordinem redigendi. Webbia 1977, 31, 315–512. [Google Scholar] [CrossRef]
- Smith, A.R.; Pryer, K.M.; Schuettpelz, E.; Korall, P.; Schneider, H.; Wolf, P. A classification for extant ferns. Taxon 2006, 55, 705–731. [Google Scholar] [CrossRef]
- Li, C.-X.; Lu, S.-G. Phylogeny and divergence of Chinese Angiopteridaceae based on chloroplast DNA sequence data (rbcL and trnL-F). Chin. Sci. Bull. 2007, 52, 91–97. [Google Scholar] [CrossRef]
- Ching, R.-C.; Wang, Z.-R. Angiopteris sparsisora Ching; sp. nov.—A putative bigeneric hybrid. Acta Phytotax. Sin. 1982, 20, 347–350. [Google Scholar]
- Wang, C.-Y.; Yang, B.; He, Z.-R.; Wang, H.-B. Analysis on the geographical distribution characteristics of Angiopteridaceae and its causes in Yungui Plateau and Hengduan Mountains; China. Plant Div. Res. 2012, 344, 317–325. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, G.-L.; Yan, Y.-H.; Sun, D.-K.; Shu, J.-P.; Duan, J.-Q.; Zhao, G.-H.; Zhang, G.-S.; Xiang, J.-Y.; Wang, K.-L. Angiopteris sugongii (Marattiaceae); a new diploid species with transitional morphology between Angiopteris (sen. str.) and Archangiopteris. Phytotaxa 2021, 516, 275–282. [Google Scholar] [CrossRef]
- Wu, W.H. A Study on the Natural Fern Hybrid–Archangiopteris Itoi Shieh; National Taiwan University: Taipei, Taiwan, 2002; 40p. [Google Scholar]
- Hsieh, T.-H.; Chang, Y.-H.; Chiou, W.-L.; Huang, Y.-M. Chromosome numbers of five species of the Marattiaceae in Taiwan. Taiwan. J. For. Sci. 2008, 234, 335–339. [Google Scholar]
- Christ, H.; Giesenhagen, K. Archangiopteris. Flora 1899, 89, 72–79. [Google Scholar]
- Christenhusz, M.J.M.; Tuomisto, H.; Metzgar, J.S.; Pryer, K.M. Evolutionary relationships within the neotropical; eusporangiate fern genus Danaea (Marattiaceae). Mol. Phylogenet. Evol. 2008, 46, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Y. The morphylogy of Archangiopteris Christ et Gies and its relationship with Angiopteris Hoffm. Acta. Bot. Sin. 1973, 15, 261–270. (In Chinese) [Google Scholar]
- Duarte, J.M.; Wall, P.K.; Edger, P.P.; Landherr, L.L.; Ma, H.; Pires, J.C.; Leebens-Mack, J.; Depamphilis, C. Identification of shared single copy nuclear genes in Arabidopsis; Populus; Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol. Biol. 2010, 10, 61. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Zeng, L.-P.; Shan, H.-Y.; Ma, H. Highly conserved low-copy nuclear genes as effective markers for phylogenetic analyses in angiosperms. New Phytol. 2012, 195, 923–937. [Google Scholar] [CrossRef]
- Zimmer, E.A.; Wen, J. Using nuclear gene data for plant phylogenetics: Progress and prospects. Mol. Phylogenet. Evol. 2012, 65, 774–785. [Google Scholar] [CrossRef]
- Li, Z.; de La Torre, A.R.; Sterck, L.; Cánovas, F.M.; Avila, C.; Merino, I.; Cabezas, J.A.; Cercas, F.M.; Ingvarsson, P.K.; de Peer, Y.V. Single-copy genes as molecular markers for phylogenomic studies in seed plants. Genome Biol. Evol. 2017, 9, 1130–1147. [Google Scholar] [CrossRef] [Green Version]
- Bakkali, M.; Martín-Blázquez, R.; Ruiz-Estévez, M.; Garrido-Ramos, M.A. De novo sporophyte transcriptome assembly and functional annotation in the endangered fern species Vandenboschia speciosa (Willd.) G. Kunkel. Genes 2021, 12, 1017. [Google Scholar] [CrossRef]
- Mai, U.; Mirarab, S. TreeShrink: Fast and accurate detection of outlier long branches in collections of phylogenetic trees. BMC Genom. 2018, 19, 4046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gastony, G.J.; Yatskievych, G. Maternal inheritance of the chloroplast and mitochondrial genomes in cheilanthoid ferns. Am. J. Bot. 1992, 79, 716–722. [Google Scholar] [CrossRef]
- Maddison, W.P. Gene trees in species trees. Syst. Biol. 1997, 46, 523–536. [Google Scholar] [CrossRef]
- Vogel, J.C.; Russell, S.J.; Rumsey, F.J.; Barrett, J.A.; Gibby, M. Evidence for the maternal transmission of chloroplast DNA in the genus Asplenium (Aspleniaceae; Pteridophyta). Botanica Acta 1998, 111, 247–249. [Google Scholar] [CrossRef]
- Guillon, J.M.; Raquin, C. Maternal inheritance of chloroplasts in the horsetail Equisetum variegatum (Schleich.). Curr. Genet. 2000, 37, 53–56. [Google Scholar] [CrossRef]
- Kuo, L.-Y.; Tang, T.-Y.; Li, F.-W. Organelle Genome Inheritance in Deparia Ferns (Athyriaceae; Aspleniineae; Polypodiales). Front. Plant Sci. 2018, 9, 486. [Google Scholar] [CrossRef] [Green Version]
- Olmstead, R.G.; Bedoya, A.M. Whole genomes: The holy grail. A commentary on: ‘Molecular phylogenomics of the tribe Shoreeae (Dipterocarpaceae) using whole plastidgenomes’. Ann. Bot. 2019, 123, iv–v. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.F.; Walker-Hale, N.; Vargas, O.M.; Larson, D.A.; Stull, G.W. Characterizing gene tree conflict in plastome inferred phylogenies. PeerJ 2019, 7, e7747. [Google Scholar] [CrossRef] [Green Version]
- Galtier, N.; Daubin, V. Dealing with incongruence in phylogenomic analyses. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 4023–4029. [Google Scholar] [CrossRef] [Green Version]
- Camus, J.M. Marattiaceae. In The Families and Genera of Vascular Plants: Pteridophytes and Gymnosperms; Kubitzki, K., Kramer, K.U., Green, P.S., Eds.; Springer: Berlin, Germany, 1990; Volume 1, pp. 174–180. [Google Scholar]
- Walker, T.G. A chromosome count for Macroglossum (Marattiales). Fern Gaz. 1981, 12, 161–163. [Google Scholar]
- Christenhusz, M.J.M. Evolutionary history and taxonomy of Neotropical marattioid ferns: Studies of an ancient lineage of plants. Ann. Univ. Turku. Ser. AII. 2007, 216, 1–78. [Google Scholar]
- Wang, K.; Lenstra, J.A.; Liu, L.; Hu, Q.-J.; Ma, T.; Qiu, Q.; Liu, J.-Q. Incomplete lineage sorting rather than hybridization explains the inconsistent phylogeny of the wisent. Commun. Biol. 2018, 1, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-L.; Zhang, Y.-J.; Ruhsam, M.; Milne, R.I.; Wang, Y.; Wu, D.-Y.; Jia, S.-Y.; Tao, T.-Z.; Mao, K.-S. Seeing through the hedge: Phylogenomics of Thuja (Cupressaceae) reveals prominent incomplete lineage sorting and ancient introgression for Tertiary relict flora. Cladistics 2021, 38, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.-X.; Sun, P.-C.; Wang, D.-S.; Wang, Z.-Y.; Yang, J.; Li, Y.; Mu, W.-J.; Xu, P.-R.; Wu, Y.; Dong, C.-C.; et al. The Chloranthus sessilifolius genome provides insight into early diversification of angiosperms. Nat. Commun. 2021, 12, 6929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, L.; Folk, R.A.; Zhao, J.-L.; Zamora, N.A.; Yang, S.-X.; Soltis, D.E.; Soltis, D.E.; Gao, L.-M.; Peng, H.; et al. Phylotranscriptomics of Theaceae: Generic-level relationships; reticulation and whole-genome duplication. Ann. Bot. 2022, 129, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.-B.; Ren, C.; Kwak, M.; Hodel, R.G.; Xu, C.; He, J.; Zhou, W.-B.; Huang, C.-H.; Ma, H.; Qian, G.-Z.; et al. Phylogenomic conflict analyses in the apple genus Malus s.l. reveal widespread hybridization and allopolyploidy driving diversification; with insights into the complex biogeographic history in the Northern Hemisphere. J. Integr. Plant Biol. 2022, 64, 1020–1043. [Google Scholar] [CrossRef]
- Degnan, J.H.; Rosenberg, N.A. Gene tree discordance; phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009, 24, 332–340. [Google Scholar] [CrossRef]
- Flouri, T.; Jiao, X.; Rannala, B.; Yang, Z. Species tree inference with BPP using genomic sequences and the multispecies coalescent. Mol. Biol. Evol. 2018, 35, 2585–2593. [Google Scholar] [CrossRef] [Green Version]
- Warren, H.; Wagner, J. Reticulate Evolution in the Appalachian Aspleniums. Evolution 1953, 8, 103–118. [Google Scholar]
- Barrington, D.S.; Haufler, C.H.; Werth, C.R. Hybridization; reticulation; and species concepts in the ferns. Am. Fern J. 1989, 79, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Wood, T.E.; Takebayashi, N.; Barker, M.S.; Mayrose, I.; Greenspoon, P.B.; Rieseberg, L.H. The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. USA 2009, 106, 13875–13879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitney, K.D.; Ahern, J.R.; Cambell, L.G.; Albert, L.; King, M.S. Patterns of hybridization in plants. Perspect. Plant Ecol. Evol. Syst. 2010, 12, 175–182. [Google Scholar] [CrossRef]
- Sessa, E.B.; Zimmer, E.A.; Givnish, T.J. Unraveling reticulate evolution in north american Dryopteris (Dryopteridaceae). BMC Evol. Biol. 2012, 12, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigel, E.M. Genetic and genomic aspects of hybridization in ferns. J. Syst. Evol. 2016, 54, 638–655. [Google Scholar] [CrossRef]
- Christenhusz, M.J.M. Revision of the Neotropical fern genus Eupodium (Marattiaceae). Kew Bull. 2010, 65, 115–121. [Google Scholar] [CrossRef]
- Chen, S.-F.; Zhou, Y.-Q.; Chen, Y.-R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, 2. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clusteringand comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Cosentino, S.; Iwasaki, W. SonicParanoid: Fast accurate and easy orthology inference. Bioinformatics 2019, 35, 149–151. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K. MAFFT: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.-X.; Wang, G.-T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Res. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Morales-Briones, D.F.; Kadereit, G.; Tefarikis, D.T.; Moore, M.J.; Smith, S.A.; Brockington, S.F.; Timoneda, A.; Yim, W.C.; Cushman, J.C.; Yang, Y. Disentangling sources of gene tree discordance in phylogenomic datasets: Testing ancient hybridizations in Amaranthaceae s.l. Syst. Biol. 2020, 70, 219–235. [Google Scholar] [CrossRef]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Haeseler, A.V.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayyari, E.; Mirarab, S. Fast coalescent-based computation of local branch support from quartet frequencies. Mol. Biol. Evol. 2016, 33, 1654–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salichos, L.; Stamatakis, A.; Rokas, A. Novel information theory-based measures for quantifying incongruence among phylogenetic trees. Mol. Biol. Evol. 2014, 31, 1261–1271. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.A.; Moore, M.J.; Brown, J.W.; Yang, Y. Analysis of phylogenomic datasets reveals conflict; concordance; and gene duplications with examples from animals and plants. BMC Evol. Biol. 2015, 15, 150. [Google Scholar] [CrossRef] [Green Version]
- Pease, J.B.; Brown, J.W.; Walker, J.F.; Hinchliff, C.E.; Smith, S.A. Quartet Sampling distinguishes lack of support from conflicting support in the green plant tree of life. Am. J. Bot. 2018, 105, 385–403. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, J.; Holder, M.T. DendroPy: A Python library for phylogenetic computing. Bioinformatics 2010, 26, 1569–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Than, C.; Ruths, D.; Nakhleh, L. PhyloNet: A software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinform. 2008, 9, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Dong, J.; Liu, K.-J.; Nakhleh, L. Maximum likelihood inference of reticulate evolutionary histories. Proc. Natl. Acad. Sci. USA 2014, 111, 16448–16453. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Degnan, J.H.; Nakhleh, L. The probability of a gene tree topology within a phylogenetic network with applications to hybridization detection. PLoS Genet. 2012, 8, e1002660. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Scornavacca, C. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 2012, 61, 1061–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.A.; O’Meara, B.C. treePL: Divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics 2012, 28, 2689–2690. [Google Scholar] [CrossRef] [Green Version]
- Lundblad, A.B. Studies in the Rhaeto-Liassie Flora of Sweden; 1. Pteridophyta; Pteridospermae and Cycadophyta from the mining district of NW Scania. K. Sven. Vetensk. Handl. Fjärde Ser. 1950, 1, 1–82. [Google Scholar]
- Harris, T.M. The Yorkshire Jurassic Flora. I. Thalophyta-Pteridophyta. Trustees of the British Museum (Natural History); India. Nat. Hist. 1961. [Google Scholar] [CrossRef]
- Maurin, J.L.K. An empirical guide for producing a dated phylogeny with treePL in a maximum likelihood framework. arXiv 2020, arXiv:2008.07054v2. [Google Scholar]
- Rambaut, A. FigTree-Version 1.4.3; A Graphical Viewer of Phylogenetic Tree. 2017. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 5 May 2023).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Genera (PPG I) | Species | Voucher Data | Locations or Sample Provider | Citation |
|---|---|---|---|---|---|
| Marattiaceae | Angiopteris | An. caudatiformis Hieronymus | AN_CA (PYU) | Yunnan, China, | This study |
| Marattiaceae | Angiopteris | An. fokiensis Hieronymus | AN_FO (PYU) | Yunnan, China | This study |
| Marattiaceae | Angiopteris | An. hokouensis Ching | AN_HO (PYU) | Yunnan, China | This study |
| Marattiaceae | Angiopteris | An. sparsisora Ching | AN_SP (PYU) | Yunnan, China | This study |
| Marattiaceae | Angiopteris | An. wangii Ching | AN_WA (PYU) | Yunnan, China | This study |
| Marattiaceae | Angiopteris | An. yunnanensis Hieronymus | AN_YN (PYU) | Yunnan, China | This study |
| Marattiaceae | Angiopteris | * An. bipinnata (Ching) J.M. Camus (=Ar. bipinnata Ching) | AN_BP (PYU) | Yunnan, China | This study |
| Marattiaceae | Angiopteris | * An. latipinna Z.R. He, W.M. Chu & Christenhusz (=Ar. henryi Christ & Giesenhagen) | AN_LA (PYU) | Yunnan, China | This study |
| Marattiaceae | Angiopteris | * An. chingii J. M. Camus (=Ar. hokouensis Ching) | AN_CH (PYU) | Yunnan, China | This study |
| Marattiaceae | Angiopteris | * An. subrotundata (Ching) Z.R. He & Christenhusz (=Ar. subrotundata Ching) | AN_SU (PYU) | Yunnan, China | This study |
| Marattiaceae | Christensenia | Christensenia aesculifolia (Blume) Maxon | CH_AE (PYU) | Yunnan, China | This study |
| Marattiaceae | Christensenia | Christensenia aesculifolia (Blume) Maxon | K024247 (KBCC) | Dr. Ceceilia Koo of Botanic Conservation Center | [34] |
| Marattiaceae | Danaea | Danaea nodosa (L.) Sm. | Vasco 879 (NY) | Sample Provider: Alejandra Vasco | 1 kp., 2019 |
| Marattiaceae | Danaea | Danaea nodosa (L.) Sm. | K034856(KBCC) | Dr. Ceceilia Koo of Botanic Conservation Center | [34] |
| Marattiaceae | Marattia | Marattia sp. | NYBG1295/78-A (NYBG) | Sample Provider: D. W. Stevenson | 1 kp., 2019 |
| Marattiaceae | Ptisana | Ptisana pellucida (C.Presl) Murdock | K013208(FUS) | Dr. Ceceilia Koo of Botanic Conservation Center | [34] |
| Equisetaceae | Equisetum | Equisetum arvense L. | xp705 (FUS) | Zhejiang, China | [34] |
| Equisetaceae | Equisetum | Equisetum diffusum D.Don | RS-107 (CSH) | Sichuan, China | [39] |
| Psilotaceae | Psilotum | Psilotum nudum (L.) P.Beauv. | xp703 (FUS) | Guangxi, China | [34] |
| Ophioglossaceae | Ophioglossum | Ophioglossum thermale Kom. | xp746 (FUS) | Shanghai, China | [34] |
| Ophioglossaceae | Ophioglossum | Ophioglossum vulgatum L. | RS-84 (CSH) | Guangdong, China | [39] |
| Osmundaceae | Osmundastrum | Osmundastrum cinnamomeum (L.) C.Presl | Unknown (NYBG) | Sample Provider: D. W. Stevenson | 1 kp., 2019 |
| Osmundaceae | Osmunda | Osmunda japonica Thunb. | RS38 (CSH) | Shanghai, China | [39] |
| Hymenophyllaceae | Hymenophyllum | Hymenophyllum holochilum (Bosch) C.Chr. | Kuo4290 (TAFI) | Taiwan, China | [34] |
| Hymenophyllaceae | Vandenboschia | Vandenboschia speciosa (Willd.) G.Kunkel | Unknown | Alcornocales Natural Park (Cádiz, Spain): Valdeinfierno (VDI). | [69] |
| Amborellaceae | Amborella | Amborella trichopoda Baill. | Unknown | Unknown | Unknown |
| Dataset | 26-O-aa | 26-O-cds | 26-S-aa | 26-S-cds | 16-O-aa | 16-O-cds | 16-S-aa | 16-S-cds | Family-O-aa | Family-O-cds |
|---|---|---|---|---|---|---|---|---|---|---|
| Number of Taxon | 26 | 26 | 26 | 26 | 16 | 16 | 16 | 16 | 11 | 11 |
| Number of Gene | 65 | 80 | 147 | 162 | 474 | 595 | 782 | 947 | 749 | 834 |
| Number of Sites | 23,136 | 78,828 | 62,321 | 194,713 | 212,344 | 679,941 | 381,413 | 1,189,191 | 351,399 | 1,093,200 |
| Number of Informative sites | 11,278 | 43,172 | 29,032 | 103,057 | 32,577 | 83,068 | 55,719 | 137,762 | 148,631 | 523,878 |
| Number of Invariable sites | 8888 | 26,923 | 24,912 | 69,474 | 157,262 | 541,874 | 285,182 | 955,281 | 144,968 | 416,330 |
| Sequence Length | 23,136 | 78,828 | 62,321 | 194,713 | 212,344 | 679,941 | 381,413 | 1,189,191 | 351,399 | 1,093,200 |
| Pairwise Identity (%) | 71.10 | 70.70 | 69.50 | 69.20 | 81.30 | 83.70 | 78.90 | 81.30 | 62.40 | 62.50 |
| Identical Sites (%) | 14.40 | 12.60 | 12.10 | 10.70 | 27.10 | 32.00 | 25.40 | 30.20 | 27.90 | 24.60 |
| Average GC content (%) | − | 46.60 | − | 46.40 | − | 47.30 | − | 46.90 | − | 45.50 |
| Max Sequence Length (bp) | 22,918 | 77,973 | 60,898 | 190,133 | 207,882 | 665,631 | 367,727 | 1,147,431 | 344,823 | 1,072,395 |
| Min Sequence Length (bp) | 11,416 | 38,560 | 25,970 | 84,138 | 93,695 | 323,922 | 159,932 | 542,583 | 296,857 | 917,580 |
| Mean coverage (%) | 25.98 | 26.00 | 25.99 | 25.99 | 16.00 | 16.00 | 16.00 | 16.00 | 11.00 | 11.00 |
| Dataset | Family-S-aa | Family-S-cds | Genus-O-aa | Genus-O-cds | Genus-S-aa | Genus-S-cds | Species-O-aa | Species-O-cds | Species-S-aa | Species-S-cds |
| Number of Taxon | 11 | 11 | 9 | 9 | 9 | 9 | 11 | 11 | 11 | 11 |
| Number of Gene | 1293 | 1397 | 700 | 911 | 1105 | 1389 | 541 | 1058 | 616 | 1216 |
| Number of Sites | 678,528 | 2,102,052 | 314,325 | 1,032,691 | 532,963 | 1,702,007 | 244,990 | 967,773 | 269,150 | 1,094,673 |
| Number of Informative sites | 289,393 | 1,012,805 | 45,580 | 118,504 | 73,153 | 186,245 | 4850 | 10,908 | 5510 | 12,700 |
| Number of Invariable sites | 273,670 | 785,106 | 235,281 | 830,260 | 401,729 | 1,375,289 | 232,965 | 939,305 | 254,861 | 1,060,028 |
| Sequence Length | 678,528 | 2,102,052 | 314,325 | 1,032,691 | 532,963 | 1,702,007 | 244,990 | 967,773 | 269,150 | 1,094,673 |
| Pairwise Identity (%) | 60.70 | 61.00 | 71.80 | 75.50 | 69.90 | 73.60 | 84.90 | 89.10 | 84.60 | 89.20 |
| Identical Sites (%) | 25.90 | 22.80 | 27.30 | 33.10 | 26.50 | 31.70 | 38.80 | 54.50 | 42.80 | 58.00 |
| Average GC content (%) | − | 45.30 | − | 46.90 | − | 46.60 | − | 47.50 | − | 47.50 |
| Max Sequence Length (bp) | 661,831 | 2,050,271 | 305,614 | 1,005,724 | 508,729 | 1,631,249 | 239,958 | 952,194 | 261,892 | 1,073,154 |
| Min Sequence Length (bp) | 575,170 | 1,774,443 | 133,950 | 488,227 | 221,802 | 779,205 | 111,063 | 578,586 | 136,543 | 702,030 |
| Mean coverage (%) | 11.00 | 11.00 | 9.00 | 9.00 | 9.00 | 9.00 | 11.00 | 11.00 | 11.00 | 11.00 |
| Total Log Probability | Family-O-aa | Family-O-cds | Family-S-aa | Family-S-cds | Genus-O-aa | Genus-O-cds |
| Network 1 | −6190.5703 | −3501.058081 | −11295.45488 | −5950.690602 | −744.3201955 | −457.4195646 |
| Network 2 | −6113.875518 | −3227.002002 | −11199.63915 | −5872.642211 | −743.1046667 | −487.2788227 |
| Network 3 | −6231.19481 | −3287.813375 | −11385.39823 | −5950.690602 | −720.0360015 | −487.2788227 |
| Network 4 | −5643.849581 | −3323.11141 | −10901.25444 | −5158.20478 | −743.3484675 | −452.0974283 |
| Network 5 | −6165.723153 | −3484.299045 | −11033.73158 | −5699.184902 | −712.7726379 | −510.6857333 |
| Total log probability | Genus-S-aa | Genus-S-cds | Species-O-aa | Species-O-cds | Species-S-aa | Species-S-cds |
| Network 1 | −1357.70562 | −772.4172772 | −247.1150431 | −199.7265792 | −328.9024602 | −267.86003727524957 |
| Network 2 | −1358.256057 | −772.0158611 | −249.0607795 | −213.7213554 | −332.0709943 | −267.86003727524945 |
| Network 3 | −1358.256057 | −773.8058876 | −247.117924 | −220.5996779 | −331.5036725 | −272.798912 |
| Network 4 | −1377.690453 | −808.015031 | −249.0611516 | −199.8006603 | −331.5036725 | −268.2104459 |
| Network 5 | −1389.124744 | −808.015031 | −249.0611364 | −226.4899819 | −328.9071446 | −300.2662086 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, J.; Zhou, X.; Fang, S.; Zhu, Z.; Li, Y.; Yu, H.; He, Z. Transcriptome-Based Study on the Phylogeny and Hybridization of Marattialean Ferns (Marattiaceae). Plants 2023, 12, 2237. https://doi.org/10.3390/plants12122237
Zhao J, Zhou X, Fang S, Zhu Z, Li Y, Yu H, He Z. Transcriptome-Based Study on the Phylogeny and Hybridization of Marattialean Ferns (Marattiaceae). Plants. 2023; 12(12):2237. https://doi.org/10.3390/plants12122237
Chicago/Turabian StyleZhao, Jing, Xinmao Zhou, Shaoli Fang, Zhangming Zhu, Yuxin Li, Hong Yu, and Zhaorong He. 2023. "Transcriptome-Based Study on the Phylogeny and Hybridization of Marattialean Ferns (Marattiaceae)" Plants 12, no. 12: 2237. https://doi.org/10.3390/plants12122237
APA StyleZhao, J., Zhou, X., Fang, S., Zhu, Z., Li, Y., Yu, H., & He, Z. (2023). Transcriptome-Based Study on the Phylogeny and Hybridization of Marattialean Ferns (Marattiaceae). Plants, 12(12), 2237. https://doi.org/10.3390/plants12122237