
Co-Occurrence Analysis of Citrus Root Bacterial Microbiota under Citrus Greening Disease


Abstract
:1. Introduction
2. Results
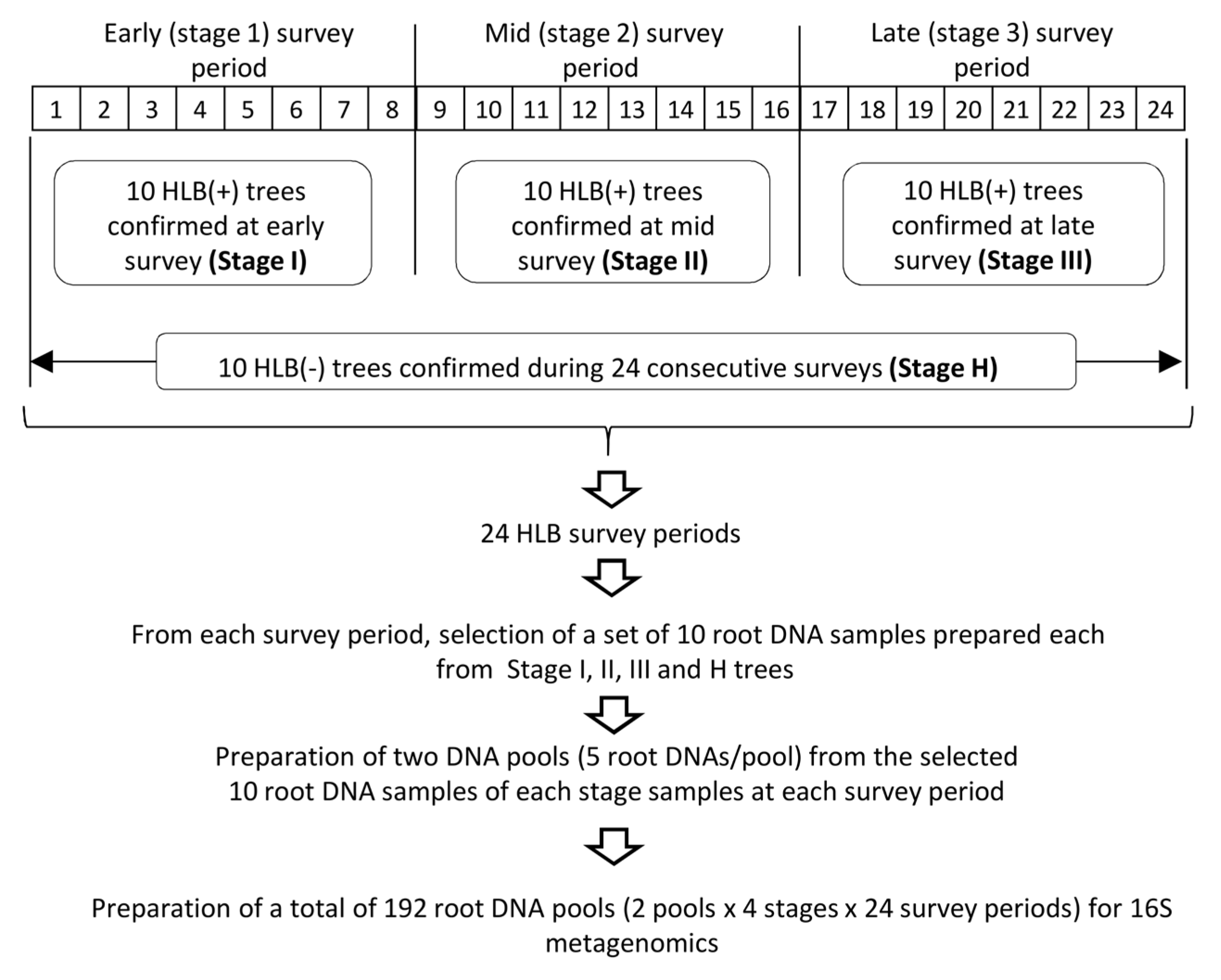
2.1. Sample Selection for 16S rDNA-Based Root Microbiome Analysis
2.2. 16S Metagenomics Sequencing Output and OTU Clustering
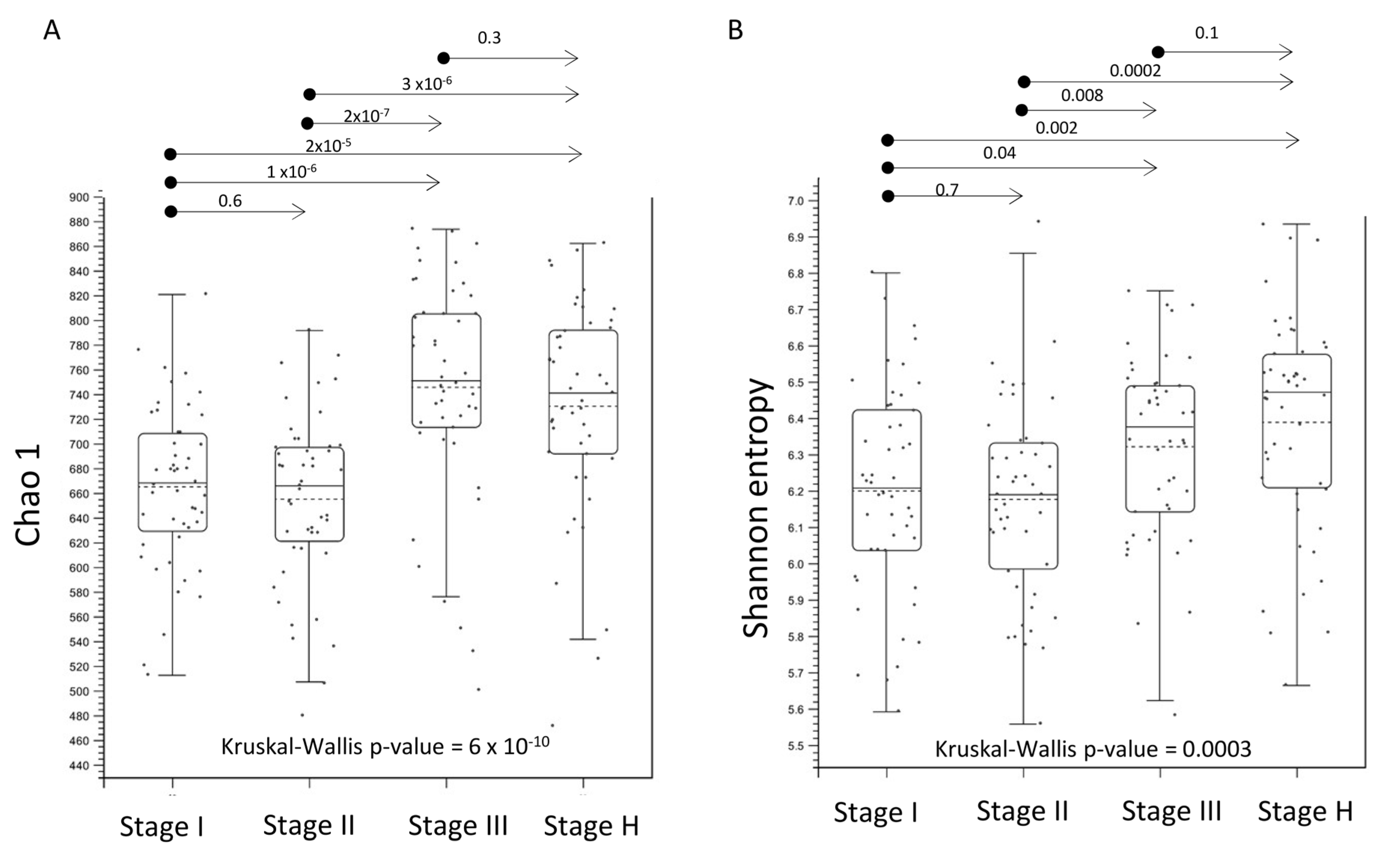
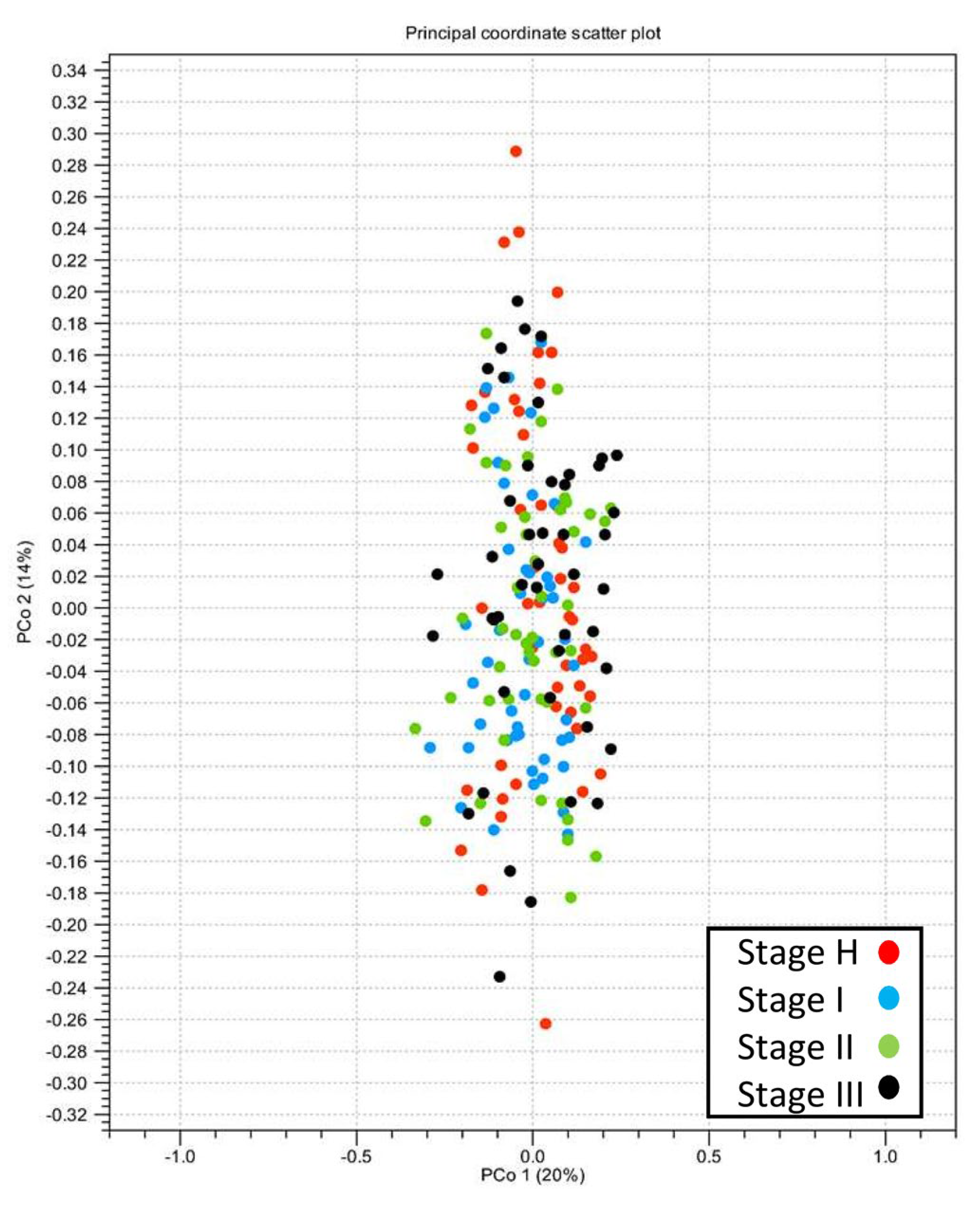
2.3. Alpha- and Beta-Diversity of Root Bacterial Microbiota of Trees with Different HLB Disease Status
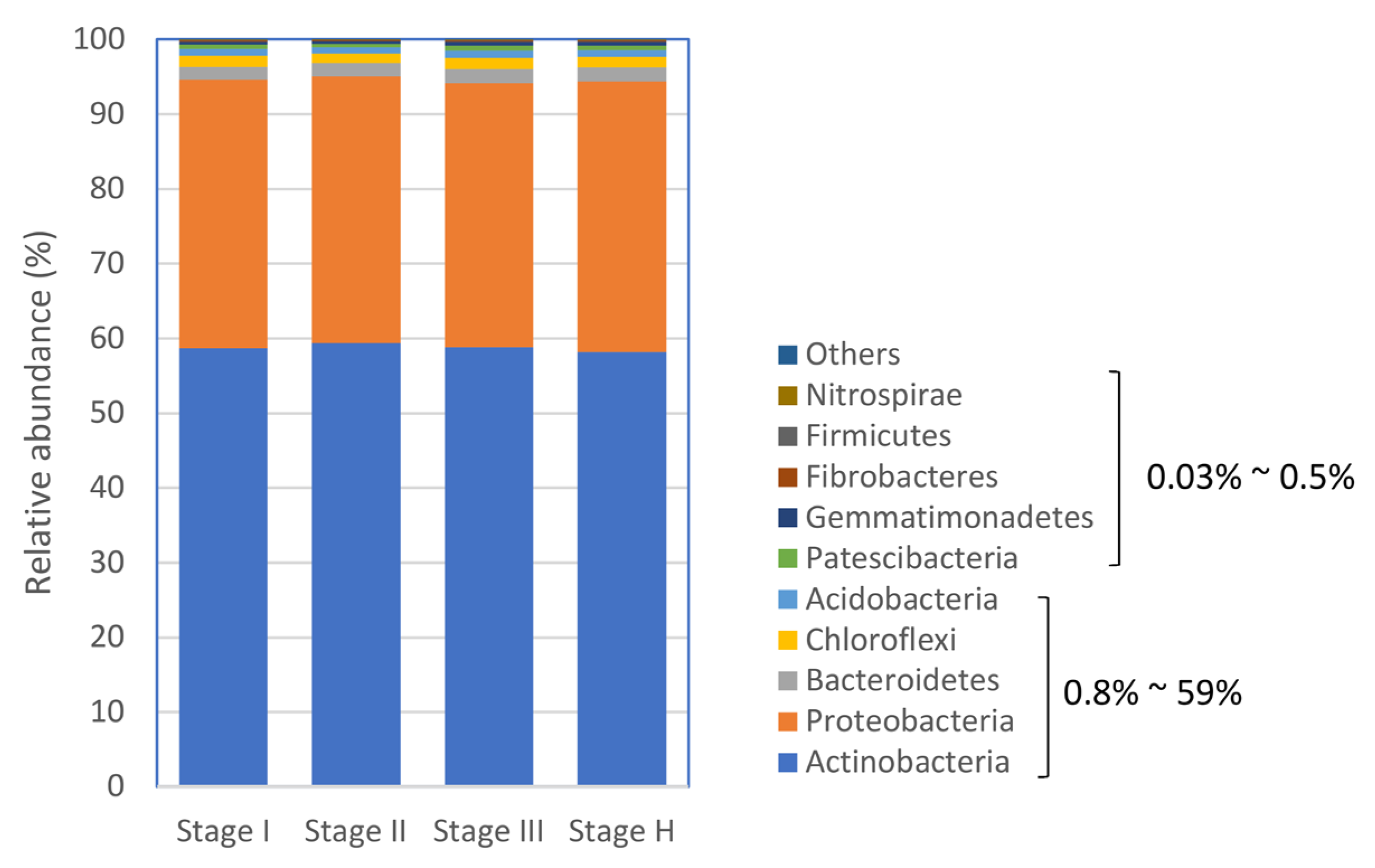
2.4. The Composition of Citrus Root Bacterial Microbiota
2.5. The Temporal Dynamics of Citrus Root Microbiota in the Trees with Different HLB Status
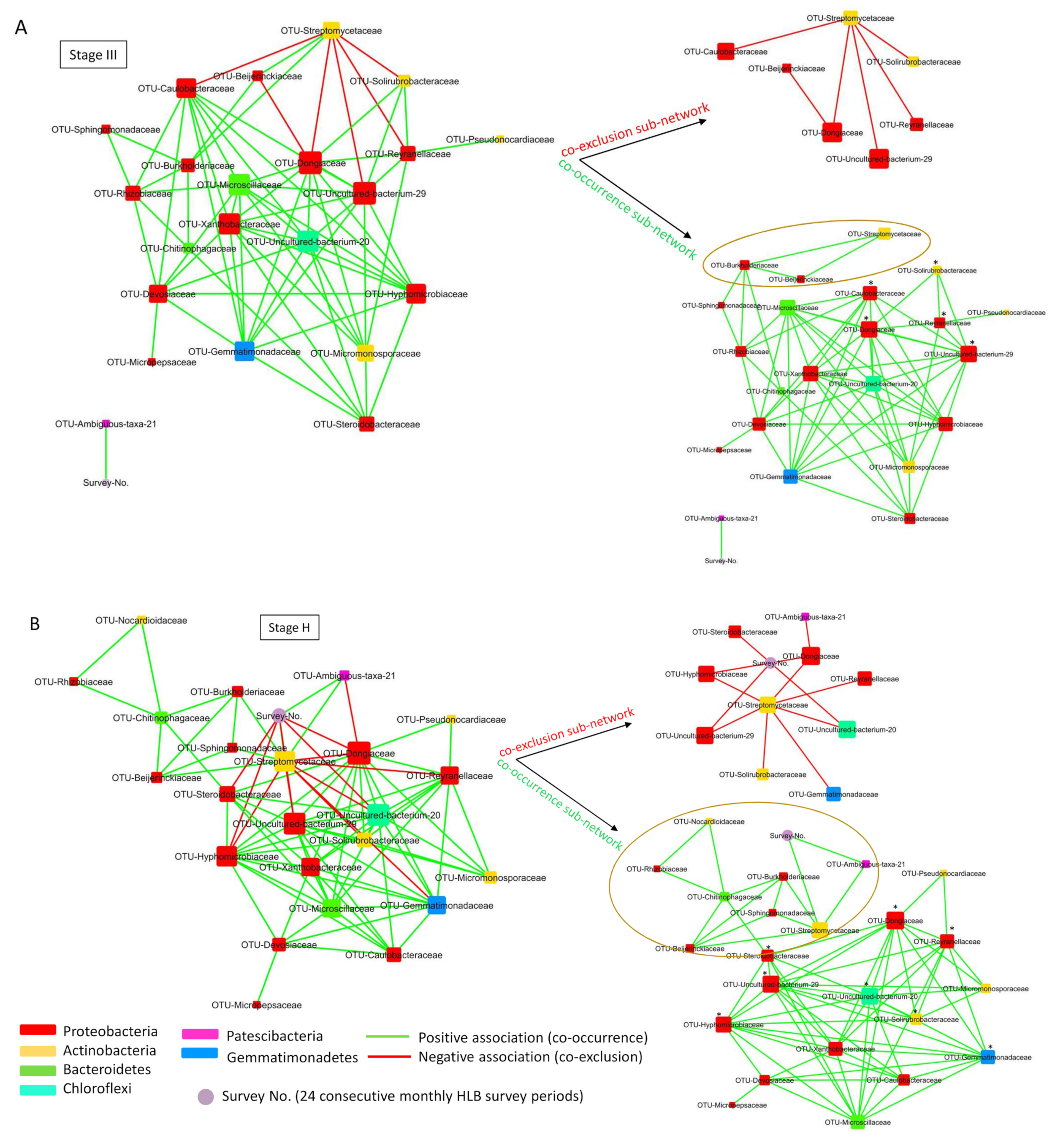
2.6. Co-Occurrence Network Analysis of Root Bacterial Microbiota of Trees with Different HLB Status
2.7. Root Bacterial Subnetwork Involving Streptomycetaceae Affected by HLB
3. Discussion
4. Materials and Methods
4.1. Root DNA Samples
4.2. 16S Metagenomics Data Analysis
4.3. Microbial Co-Occurrence Network Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jagoueix, S.; Bove, J.-M.; Garnier, M. The Phloem-Limited Bacterium of Greening Disease of Citrus Is a Member of the Subdivision of the Proteobacteria. Int. J. Syst. Bacteriol. 1994, 44, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Planet, P.; Jagoueix, S.; Bove, J.M.; Garnier, M. Detection and Characterization of the African Citrus Greening Liberobacter by Amplification, Cloning, and Sequencing of the rplKAJL-rpoBC Operon. Curr. Microbiol. 1995, 30, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Texeira, D.C.; Ayres, J.; Kitajima, E.W.; Danet, L.; Jagoueix-Eveillard, S.; Saillard, C.; Bové, J.M. First Report of a Huanglongbing-Like Disease of Citrus in Sao Paulo State, Brazil and Association of a New Liberibacter Species, “Candidatus Liberibacter Americanus”, with the Disease. Plant Dis. 2005, 89, 107. [Google Scholar] [CrossRef] [PubMed]
- Bové, J.M. Huanglongbing: A Destructive, Newly-Emerging, Century-Old Disease of Citrus. J. Plant Pathol. 2006, 88, 7–37. [Google Scholar]
- Halbert, S. The Discovery of Huanglongbing in Florida. In Proceedings of the Second International Citrus Canker and Huanglongbing Research Workshop, Orlando, FL, USA, 7–11 November 2005. H-3. [Google Scholar]
- Kunta, M.; Setamou, M.; Skaria, M.; Li, W.; Nakhla, M.K.; da Graça, J.V. First Report of Citrus Huanglongbing in Texas. Phytopathology 2012, 102, S4.66. [Google Scholar]
- Kumagai, L.B.; LeVesque, C.S.; Blomquist, C.L.; Madishetty, K.; Guo, Y.; Woods, P.W.; Rooney-Latham, S.; Rascoe, J.; Gallindo, T.; Schnabel, D.; et al. First Report of Candidatus Liberibacter Asiaticus Associated with Citrus Huanglongbing in California. Plant Dis. 2013, 97, 283. [Google Scholar] [CrossRef]
- da Graça, J.V. Citrus Greening Disease. Annu. Rev. Phytopathol. 1991, 29, 109–136. [Google Scholar] [CrossRef]
- Li, S.; Wu, F.; Duan, Y.; Singerman, A.; Guan, Z. Citrus Greening: Management Strategies and Their Economic Impact. Horts 2020, 55, 604–612. [Google Scholar] [CrossRef]
- Riera, N.; Handique, U.; Zhang, Y.; Dewdney, M.M.; Wang, N. Characterization of Antimicrobial-Producing Beneficial Bacteria Isolated from Huanglongbing Escape Citrus Trees. Front. Microbiol. 2017, 8, 2415. [Google Scholar] [CrossRef]
- Li, X.; Ruan, H.; Zhou, C.; Meng, X.; Chen, W. Controlling Citrus Huanglongbing: Green Sustainable Development Route Is the Future. Front. Plant Sci. 2021, 12, 760481. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, J.; Riera, N.; Jin, T.; Li, J.; Wang, N. Huanglongbing Impairs the Rhizosphere-to-Rhizoplane Enrichment Process of the Citrus Root-Associated Microbiome. Microbiome 2017, 5, 97. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Duan, Y.; Wang, N. Huanglongbing, a Systemic Disease, Restructures the Bacterial Community Associated with Citrus Roots. Appl. Env. Microbiol. 2010, 76, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.-H.; Veliz Vallejos, D.F.; Nicotra, A.B.; Mathesius, U. The Impact of Beneficial Plant-Associated Microbes on Plant Phenotypic Plasticity. J. Chem. Ecol. 2013, 39, 826–839. [Google Scholar] [CrossRef]
- Xu, L.; Naylor, D.; Dong, Z.; Simmons, T.; Pierroz, G.; Hixson, K.K.; Kim, Y.-M.; Zink, E.M.; Engbrecht, K.M.; Wang, Y.; et al. Drought Delays Development of the Sorghum Root Microbiome and Enriches for Monoderm Bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, 18. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Brettell, L.E.; Qiu, Z.; Singh, B.K. Microbiome-Mediated Stress Resistance in Plants. Trends Plant Sci. 2020, 25, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Monohon, S.J.; Manter, D.K.; Vivanco, J.M. Conditioned Soils Reveal Plant-Selected Microbial Communities That Impact Plant Drought Response. Sci. Rep. 2021, 11, 21153. [Google Scholar] [CrossRef]
- Santos-Medellín, C.; Liechty, Z.; Edwards, J.; Nguyen, B.; Huang, B.; Weimer, B.C.; Sundaresan, V. Prolonged Drought Imparts Lasting Compositional Changes to the Rice Root Microbiome. Nat. Plants 2021, 7, 1065–1077. [Google Scholar] [CrossRef]
- Hou, S.; Wolinska, K.W.; Hacquard, S. Microbiota-Root-Shoot-Environment Axis and Stress Tolerance in Plants. Curr. Opin. Plant Biol. 2021, 62, 102028. [Google Scholar] [CrossRef]
- Hong, Y.; Zhou, Q.; Hao, Y.; Huang, A.C. Crafting the Plant Root Metabolome for Improved Microbe-assisted Stress Resilience. New Phytol. 2022, 234, 1945–1950. [Google Scholar] [CrossRef]
- Bai, B.; Liu, W.; Qiu, X.; Zhang, J.; Zhang, J.; Bai, Y. The Root Microbiome: Community Assembly and Its Contributions to Plant Fitness. JIPB 2022, 64, 230–243. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–Microbiome Interactions: From Community Assembly to Plant Health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.-J.; Kong, H.G.; Choi, K.; Kwon, S.-K.; Song, J.Y.; Lee, J.; Lee, P.A.; Choi, S.Y.; Seo, M.; Lee, H.J.; et al. Rhizosphere Microbiome Structure Alters to Enable Wilt Resistance in Tomato. Nat. Biotechnol. 2018, 36, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Robert, C.A.M.; Cadot, S.; Zhang, X.; Ye, M.; Li, B.; Manzo, D.; Chervet, N.; Steinger, T.; Van Der Heijden, M.G.A.; et al. Root Exudate Metabolites Drive Plant-Soil Feedbacks on Growth and Defense by Shaping the Rhizosphere Microbiota. Nat. Commun. 2018, 9, 2738. [Google Scholar] [CrossRef] [PubMed]
- Carrión, V.J.; Perez-Jaramillo, J.; Cordovez, V.; Tracanna, V.; De Hollander, M.; Ruiz-Buck, D.; Mendes, L.W.; Van Ijcken, W.F.J.; Gomez-Exposito, R.; Elsayed, S.S.; et al. Pathogen-Induced Activation of Disease-Suppressive Functions in the Endophytic Root Microbiome. Science 2019, 366, 606–612. [Google Scholar] [CrossRef]
- French, E.; Kaplan, I.; Iyer-Pascuzzi, A.; Nakatsu, C.H.; Enders, L. Emerging Strategies for Precision Microbiome Management in Diverse Agroecosystems. Nat. Plants 2021, 7, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Toju, H.; Peay, K.G.; Yamamichi, M.; Narisawa, K.; Hiruma, K.; Naito, K.; Fukuda, S.; Ushio, M.; Nakaoka, S.; Onoda, Y.; et al. Core Microbiomes for Sustainable Agroecosystems. Nat. Plants 2018, 4, 247–257. [Google Scholar] [CrossRef]
- Lawson, C.E.; Harcombe, W.R.; Hatzenpichler, R.; Lindemann, S.R.; Löffler, F.E.; O’Malley, M.A.; García Martín, H.; Pfleger, B.F.; Raskin, L.; Venturelli, O.S.; et al. Common Principles and Best Practices for Engineering Microbiomes. Nat. Rev. Microbiol. 2019, 17, 725–741. [Google Scholar] [CrossRef]
- Jansson, J.K.; McClure, R.; Egbert, R.G. Soil Microbiome Engineering for Sustainability in a Changing Environment. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef]
- Ikeda, S.; Kaneko, T.; Okubo, T.; Rallos, L.E.E.; Eda, S.; Mitsui, H.; Sato, S.; Nakamura, Y.; Tabata, S.; Minamisawa, K. Development of a Bacterial Cell Enrichment Method and Its Application to the Community Analysis in Soybean Stems. Microb. Ecol. 2009, 58, 703–714. [Google Scholar] [CrossRef]
- Ikeda, S.; Okubo, T.; Anda, M.; Nakashita, H.; Yasuda, M.; Sato, S.; Kaneko, T.; Tabata, S.; Eda, S.; Momiyama, A.; et al. Community- and Genome-Based Views of Plant-Associated Bacteria: Plant–Bacterial Interactions in Soybean and Rice. Plant Cell Physiol. 2010, 51, 1398–1410. [Google Scholar] [CrossRef]
- Braswell, W.E.; Park, J.-W.; Stansly, P.A.; Kostyk, B.C.; Louzada, E.S.; da Graça, J.V.; Kunta, M. Root Samples Provide Early and Improved Detection of Candidatus Liberibacter Asiaticus in Citrus. Sci. Rep. 2020, 10, 16982. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.-G.; Zhang, X.-D.; Cao, M.; Liu, F.; Qian, Y.; Zhang, S.-W. Comparison of Methods for Picking the Operational Taxonomic Units From Amplicon Sequences. Front. Microbiol. 2021, 12, 644012. [Google Scholar] [CrossRef] [PubMed]
- Brundrett, M.C. Coevolution of Roots and Mycorrhizas of Land Plants. New Phytol. 2002, 154, 275–304. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-R.; Cho, G.; Jeon, C.-W.; Weller, D.M.; Thomashow, L.S.; Paulitz, T.C.; Kwak, Y.-S. A Mutualistic Interaction between Streptomyces Bacteria, Strawberry Plants and Pollinating Bees. Nat. Commun. 2019, 10, 4802. [Google Scholar] [CrossRef] [PubMed]
- Aleklett, K.; Hart, M.; Shade, A. The Microbial Ecology of Flowers: An Emerging Frontier in Phyllosphere Research. Botany 2014, 92, 253–266. [Google Scholar] [CrossRef]
- Jacoby, R.; Peukert, M.; Succurro, A.; Koprivova, A.; Kopriva, S. The Role of Soil Microorganisms in Plant Mineral Nutrition—Current Knowledge and Future Directions. Front. Plant Sci. 2017, 8, 1617. [Google Scholar] [CrossRef]
- Mendes, R.; Kruijt, M.; De Bruijn, I.; Dekkers, E.; Van Der Voort, M.; Schneider, J.H.M.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.H.M.; et al. Deciphering the Rhizosphere Microbiome for Disease-Suppressive Bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef]
- Cannon, A. The Root Microbiome: Techniques for Exploration and Agricultural Applications. BioTechniques 2023, 75, 1–6. [Google Scholar] [CrossRef]
- Brader, G.; Compant, S.; Vescio, K.; Mitter, B.; Trognitz, F.; Ma, L.-J.; Sessitsch, A. Ecology and Genomic Insights into Plant-Pathogenic and Plant-Nonpathogenic Endophytes. Annu. Rev. Phytopathol. 2017, 55, 61–83. [Google Scholar] [CrossRef]
- Thoms, D.; Liang, Y.; Haney, C.H. Maintaining Symbiotic Homeostasis: How Do Plants Engage With Beneficial Microorganisms While at the Same Time Restricting Pathogens? MPMI 2021, 34, 462–469. [Google Scholar] [CrossRef]
- Li, H.; Song, F.; Wu, X.; Deng, C.; Xu, Q.; Peng, S.; Pan, Z. Microbiome and Metagenome Analysis Reveals Huanglongbing Affects the Abundance of Citrus Rhizosphere Bacteria Associated with Resistance and Energy Metabolism. Horticulturae 2021, 7, 151. [Google Scholar] [CrossRef]
- Olanrewaju, O.S.; Babalola, O.O. Streptomyces: Implications and Interactions in Plant Growth Promotion. Appl. Microbiol. Biotechnol. 2019, 103, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Tarkka, M.T.; Lehr, N.A.; Hampp, R.; Schrey, S.D. Plant Behavior upon Contact with Streptomycetes. Plant Signal. Behav. 2008, 3, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Estrada-De Los Santos, P.; Bustillos-Cristales, R.; Caballero-Mellado, J. Burkholderia, a Genus Rich in Plant-Associated Nitrogen Fixers with Wide Environmental and Geographic Distribution. Appl. Environ. Microbiol. 2001, 67, 2790–2798. [Google Scholar] [CrossRef]
- Miyasaka, N.R.S.; Strauss Thuler, D.; Floh, E.I.S.; Handro, W.; Toledo, M.B.D.; Gagioti, S.M.; Barbosa, H.R. During Stationary Phase, Beijerinckia Derxii Shows Nitrogenase Activity Concomitant with the Release and Accumulation of Nitrogenated Substances. Microbiol. Res. 2003, 158, 309–315. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial Co-Occurrence Relationships in the Human Microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Reference Database Size (Silva 16S v132) | No. of OTUs Based on the Database | No. of de novo OTUs | Total No. of Predicted OTUs | No. of OTUs after Removing Low Abundance OTUs | No. of OTUs without Mitochondria and Chloroplast Sequences |
|---|---|---|---|---|---|---|
| Stage I | 17,222 | 3655 | 1496 | 5151 | 1598 | 1576 |
| Stage II | 3674 | 1430 | 5104 | 1562 | 1530 | |
| Stage III | 3897 | 2001 | 5898 | 1849 | 1816 | |
| Stage H | 3808 | 1975 | 5765 | 1809 | 1776 |
| Edge | Node | ||||||
|---|---|---|---|---|---|---|---|
| Stage | Total Number of Interactions (Edges) | No. of Positive Interaction (Co-Occurrence) | No. of Negative Interaction (Co-Exclusion) | Stage | Total Number of Nodes * | No. of Nodes with Positive Interaction (Positive Degree (Edge)) | Number of Nodes with Negative Interaction (Negative Degree (Edge)) |
| Stage I | 50 | 49 | 1 | Stage I | 21 | 20 | 2 |
| Stage II | 34 | 34 | - | Stage II | 22 | 22 | - |
| Stage III | 70 | 64 | 6 | Stage III | 23 | 23 | 7 |
| Stage H | 85 | 72 | 13 | Stage H | 24 | 24 | 11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.-W.; Braswell, W.E.; Kunta, M. Co-Occurrence Analysis of Citrus Root Bacterial Microbiota under Citrus Greening Disease. Plants 2024, 13, 80. https://doi.org/10.3390/plants13010080
Park J-W, Braswell WE, Kunta M. Co-Occurrence Analysis of Citrus Root Bacterial Microbiota under Citrus Greening Disease. Plants. 2024; 13(1):80. https://doi.org/10.3390/plants13010080
Chicago/Turabian StylePark, Jong-Won, W. Evan Braswell, and Madhurababu Kunta. 2024. "Co-Occurrence Analysis of Citrus Root Bacterial Microbiota under Citrus Greening Disease" Plants 13, no. 1: 80. https://doi.org/10.3390/plants13010080
APA StylePark, J. -W., Braswell, W. E., & Kunta, M. (2024). Co-Occurrence Analysis of Citrus Root Bacterial Microbiota under Citrus Greening Disease. Plants, 13(1), 80. https://doi.org/10.3390/plants13010080