
Updated Gene Prediction of the Cucumber (9930) Genome through Manual Annotation


 , and
, and
Abstract
1. Introduction
2. Results
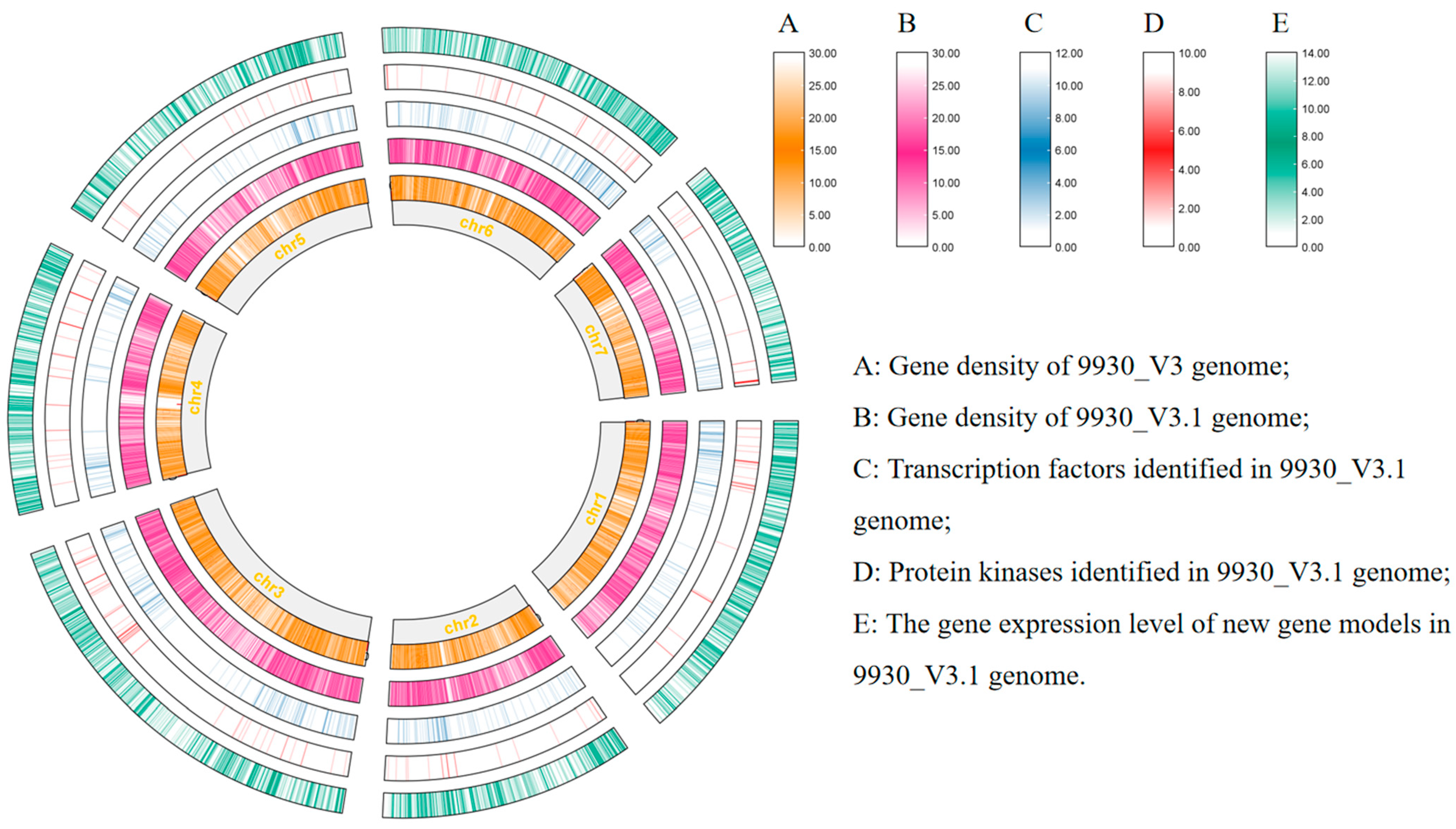
2.1. Reannotation of the Cucumber Genome through Manual Operation
2.2. Functional Annotation of Genes in the New Annotation File
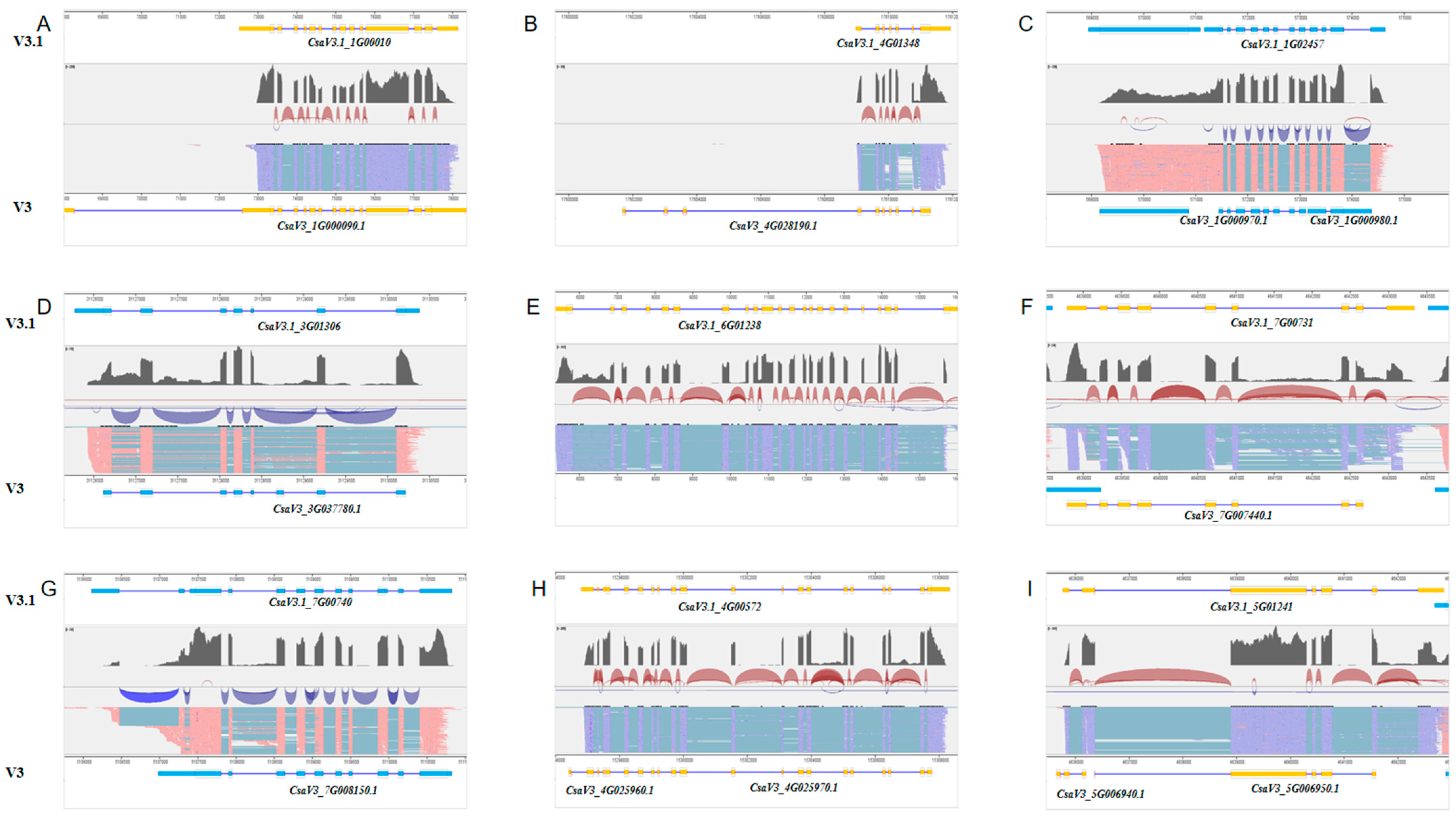
2.3. Identification of Novel Gene Features in the New Annotation File
2.4. Validation of Selected Novel Transcripts
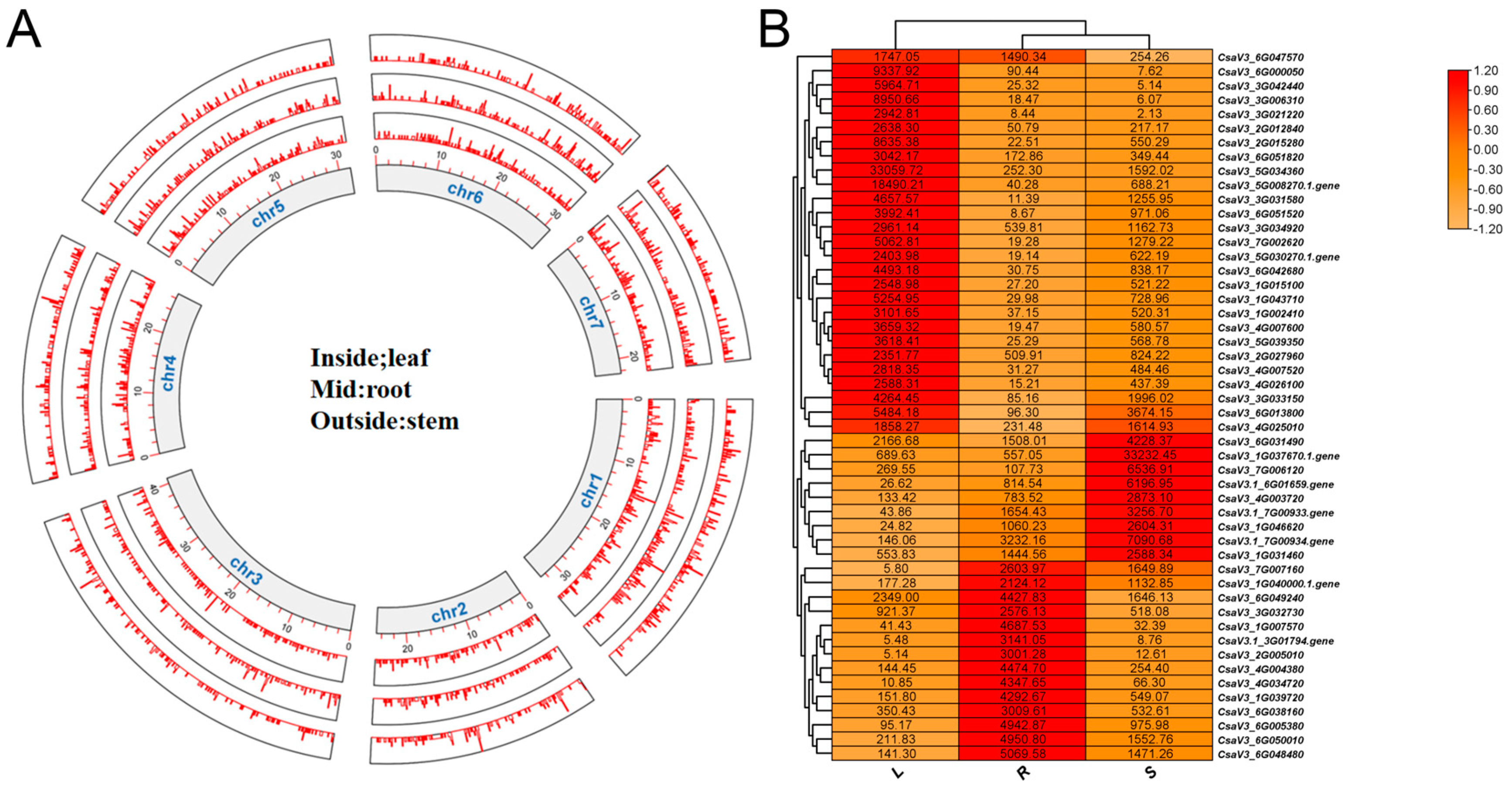
2.5. Expression Profiles of Genes in the Novel Annotation File Based on RNA-Seq Data
2.6. Transcripts with Diverse Expression Levels across Different Tissues
3. Discussion
3.1. Enhancing Functional Genomics Research into Cucumber through Accurate Gene Annotation
3.2. Gene Expression Profiles Provide Support for Gene Identification
4. Materials and Methods
4.1. Construction and Sequencing of RNA Libraries
4.2. Reannotation of the 9930_V3 Reference Genome
4.3. Validation of Novel Transcripts
4.4. Functional Annotation of Genes in the Novel Annotation File
4.5. Gene Expression Profiles in the Novel Annotation File
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haas, B.J.; Wortman, J.R.; Ronning, C.M.; Hannick, L.I.; Smith, R.K.; Maiti, R.; Chan, A.P.; Yu, C.; Farzad, M.; Wu, D.; et al. Complete Reannotation of the Arabidopsis Genome: Methods, Tools, Protocols and the Final Release. BMC Biol. 2005, 3, 7. [Google Scholar] [CrossRef]
- Li, Q.; Li, H.; Huang, W.; Xu, Y.; Zhou, Q.; Wang, S.; Ruan, J.; Huang, S.; Zhang, Z. A Chromosome-Scale Genome Assembly of Cucumber (Cucumis sativus L.). GigaScience 2019, 8, giz072. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Z.; Yan, P.; Huang, S.; Fei, Z.; Lin, K. RNA-Seq Improves Annotation of Protein-Coding Genes in the Cucumber Genome. BMC Genom. 2011, 12, 540. [Google Scholar] [CrossRef]
- Andorf, C.M.; Cannon, E.K.; Portwood, J.L.; Gardiner, J.M.; Harper, L.C.; Schaeffer, M.L.; Braun, B.L.; Campbell, D.A.; Vinnakota, A.G.; Sribalusu, V.V.; et al. MaizeGDB Update: New Tools, Data and Interface for the Maize Model Organism Database. Nucleic Acids Res. 2016, 44, D1195–D1201. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11: A Complete Reannotation of the Arabidopsis Thaliana Reference Genome. Plant J. Cell Mol. Biol. 2017, 89, 789–804. [Google Scholar] [CrossRef]
- Liu, H.; Wang, X.; Liu, S.; Huang, Y.; Guo, Y.-X.; Xie, W.-Z.; Liu, H.; Tahir Ul Qamar, M.; Xu, Q.; Chen, L.-L. Citrus Pan-Genome to Breeding Database (CPBD): A Comprehensive Genome Database for Citrus Breeding. Mol. Plant 2022, 15, 1503–1505. [Google Scholar] [CrossRef]
- Sang, J.; Zou, D.; Wang, Z.; Wang, F.; Zhang, Y.; Xia, L.; Li, Z.; Ma, L.; Li, M.; Xu, B.; et al. IC4R-2.0: Rice Genome Reannotation Using Massive RNA-Seq Data. Genom. Proteom. Bioinform. 2020, 18, 161–172. [Google Scholar] [CrossRef]
- Zhang, H.; Feng, B.; Wang, C.; Lian, X.; Wang, X.; Zheng, X.; Cheng, J.; Wang, W.; Zhang, L.; Ye, X.; et al. Manually Annotated Gene Prediction of the CN14 Peach Genome. Sci. Hortic. 2023, 321, 112242. [Google Scholar] [CrossRef]
- Pilkington, S.M.; Crowhurst, R.; Hilario, E.; Nardozza, S.; Fraser, L.; Peng, Y.; Gunaseelan, K.; Simpson, R.; Tahir, J.; Deroles, S.C.; et al. A Manually Annotated Actinidia chinensis Var. chinensis (Kiwifruit) Genome Highlights the Challenges Associated with Draft Genomes and Gene Prediction in Plants. BMC Genom. 2018, 19, 257. [Google Scholar] [CrossRef]
- Liang, B.; Zhou, Y.; Liu, T.; Wang, M.; Liu, Y.; Liu, Y.; Li, Y.; Zhu, G. Genome Reannotation of the Sweetpotato (Ipomoea batatas (L.) Lam.) Using Extensive Nanopore and Illumina-Based RNA-Seq Datasets. Trop. Plants 2024, 3, e008. [Google Scholar] [CrossRef]
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The Genome of the Cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Liu, X.; Shen, D.; Miao, H.; Xie, B.; Li, X.; Zeng, P.; Wang, S.; Shang, Y.; Gu, X.; et al. A Genomic Variation Map Provides Insights into the Genetic Basis of Cucumber Domestication and Diversity. Nat. Genet. 2013, 45, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Ma, Y.; Zhou, Y.; Zhang, H.; Duan, L.; Chen, H.; Zeng, J.; Zhou, Q.; Wang, S.; Gu, W.; et al. Plant Science. Biosynthesis, Regulation, and Domestication of Bitterness in Cucumber. Science 2014, 346, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant. 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.; Fabris, F.; Doherty, A.; Freitas, A.A.; de Magalhães, J.P. Ageing Transcriptome Meta-Analysis Reveals Similarities and Differences between Key Mammalian Tissues. Aging 2021, 13, 3313–3341. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Du, Y.; Long, Z.; Li, Y.; Kong, W.; Wang, H.; Wei, A.; Du, S.; Yang, R.; Li, J.; et al. Fine Mapping of a Novel QTL CsFSG1 for Fruit Skin Gloss in Cucumber (Cucumis sativus L.). Mol. Breed. New Strateg. Plant Improv. 2022, 42, 25. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lin, S.; Xiang, C.; Liu, W.; Zhang, X.; Wang, C.; Lu, X.; Liu, M.; Wang, T.; Liu, Z.; et al. CUCUME: An RNA Methylation Database Integrating Systemic mRNAs Signals, GWAS and QTL Genetic Regulation and Epigenetics in Different Tissues of Cucurbitaceae. Comput. Struct. Biotechnol. J. 2023, 21, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Mansfeld, B.N.; Tang, X.; Colle, M.; Chen, F.; Weng, Y.; Fei, Z.; Grumet, R. Identification of QTL Associated with Resistance to Phytophthora Fruit Rot in Cucumber (Cucumis sativus L.). Front. Plant Sci. 2023, 14, 1281755. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Nie, J.; Xiao, T.; Guo, C.; Lv, D.; Zhang, K.; He, H.-L.; Pan, J.; Cai, R.; Wang, G. CsPM5.2, a Phosphate Transporter Protein-like Gene, Promotes Powdery Mildew Resistance in Cucumber. Plant J. Cell Mol. Biol. 2024, 117, 1487–1502. [Google Scholar] [CrossRef]
- Zhang, R.-J.; Liu, B.; Song, S.-S.; Salah, R.; Song, C.-J.; Xia, S.-W.; Hao, Q.; Liu, Y.-J.; Li, Y.; Lai, Y.-S. Lipid-Related Domestication Accounts for the Extreme Cold Sensitivity of Semiwild and Tropic Xishuangbanna Cucumber (Cucumis sativus L. Var. Xishuangbannanesis). Int. J. Mol. Sci. 2024, 25, 79. [Google Scholar] [CrossRef]
- Yang, Y.; Dong, S.; Miao, H.; Liu, X.; Dai, Z.; Li, X.; Gu, X.; Zhang, S. Genome-Wide Association Studies Reveal Candidate Genes Related to Stem Diameter in Cucumber (Cucumis sativus L.). Genes 2022, 13, 1095. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Liu, X.; Han, J.; Miao, H.; Beckles, D.M.; Bai, Y.; Liu, X.; Guan, J.; Yang, R.; Gu, X.; et al. CsMLO8/11 Are Required for Full Susceptibility of Cucumber Stem to Powdery Mildew and Interact with CsCRK2 and CsRbohD. Hortic. Res. 2024, 11, uhad295. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, Z.; Wang, L.; Yan, S.; Cheng, Z.; Liu, X.; Han, L.; Chen, G.; Wang, S.; Song, W.; et al. The CsHEC1-CsOVATE Module Contributes to Fruit Neck Length Variation via Modulating Auxin Biosynthesis in Cucumber. Proc. Natl. Acad. Sci. USA 2022, 119, e2209717119. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wei, C.; Liu, Q.; Qu, W.; Qi, X.; Xu, Q.; Chen, X. The Major-Effect Quantitative Trait Locus Fnl7.1 Encodes a Late Embryogenesis Abundant Protein Associated with Fruit Neck Length in Cucumber. Plant Biotechnol. J. 2020, 18, 1598–1609. [Google Scholar] [CrossRef]
- Campbell, M.S.; Yandell, M. An Introduction to Genome Annotation. Curr. Protoc. Bioinforma. 2015, 52, 4.1.1–4.1.17. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pi, M.; Gao, Q.; Liu, Z.; Kang, C. Updated Annotation of the Wild Strawberry Fragaria Vesca V4 Genome. Hortic. Res. 2019, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.A.; Hermans, F.W.K.; Beenders, F.; Lou, L.; Vriezen, W.H.; Visser, R.G.F.; Bai, Y.; Schouten, H.J. Analysis of QTL DM4.1 for Downy Mildew Resistance in Cucumber Reveals Multiple subQTL: A Novel RLK as Candidate Gene for the Most Important subQTL. Front. Plant Sci. 2020, 11, 569876. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yuan, Z.; Zhang, H.; Li, W.; Shi, M.; Peng, Z.; Li, M.; Tian, J.; Deng, X.; Cheng, Y.; et al. Cit1,2RhaT and Two Novel CitdGlcTs Participate in Flavor-Related Flavonoid Metabolism during Citrus Fruit Development. J. Exp. Bot. 2019, 70, 2759–2771. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.-G.; Sun, C.-H.; Ma, Q.-J.; You, C.-X.; Cheng, L.; Hao, Y.-J. MdMYB1 Regulates Anthocyanin and Malate Accumulation by Directly Facilitating Their Transport into Vacuoles in Apples. Plant Physiol. 2016, 170, 1315–1330. [Google Scholar] [CrossRef]
- Ma, B.; Liao, L.; Fang, T.; Peng, Q.; Ogutu, C.; Zhou, H.; Ma, F.; Han, Y. A Ma10 Gene Encoding P-Type ATPase Is Involved in Fruit Organic Acid Accumulation in Apple. Plant Biotechnol. J. 2019, 17, 674–686. [Google Scholar] [CrossRef]
- Tian, Y.; Thrimawithana, A.; Ding, T.; Guo, J.; Gleave, A.; Chagné, D.; Ampomah-Dwamena, C.; Ireland, H.S.; Schaffer, R.J.; Luo, Z.; et al. Transposon Insertions Regulate Genome-Wide Allele-Specific Expression and Underpin Flower Colour Variations in Apple (Malus spp.). Plant Biotechnol. J. 2022, 20, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J.; Peng, Z.; Shi, M.; Liu, X.; Wen, H.; Jiang, Y.; Cheng, Y.; Xu, J.; Zhang, H. Integrated Transcriptomic and Metabolomic Analysis Reveals a Transcriptional Regulation Network for the Biosynthesis of Carotenoids and Flavonoids in “Cara Cara” Navel Orange. BMC Plant Biol. 2021, 21, 29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, M.; Wen, H.; Wang, Z.; Chen, J.; Fang, L.; Zhang, H.; Xie, Z.; Jiang, D.; Cheng, Y.; et al. Transcriptomic and Metabolomic Analyses Provide Insight into the Volatile Compounds of Citrus Leaves and Flowers. BMC Plant Biol. 2020, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Xuan, C.; Feng, M.; Li, X.; Hou, Y.; Wei, C.; Zhang, X. Genome-Wide Identification and Expression Analysis of Chitinase Genes in Watermelon under Abiotic Stimuli and Fusarium Oxysporum Infection. Int. J. Mol. Sci. 2024, 25, 638. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Fu, J.; Wang, L.; Fang, M.; Zhang, W.; Yang, M.; Yang, X.; Xu, Y.; Shi, L.; Ma, X.; et al. Diverse O-Methyltransferases Catalyze the Biosynthesis of Floral Benzenoids That Repel Aphids from the Flowers of Waterlily Nymphaea Prolifera. Hortic. Res. 2023, 10, uhad237. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Su, T.; Xin, X.; Li, P.; Wang, W.; Yu, Y.; Zhang, D.; Zhao, X.; Wang, J.; Sun, L.; et al. The Adaxial/Abaxial Patterning of Auxin and Auxin Gene in Leaf Veins Functions in Leafy Head Formation of Chinese Cabbage. Front. Plant Sci. 2022, 13, 918112. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-Level Expression Analysis of RNA-Seq Experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent Prioritization and Exploratory Visualization of Biological Functions for Gene Enrichment Analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Lohse, M.; Nagel, A.; Herter, T.; May, P.; Schroda, M.; Zrenner, R.; Tohge, T.; Fernie, A.R.; Stitt, M.; Usadel, B. Mercator: A Fast and Simple Web Server for Genome Scale Functional Annotation of Plant Sequence Data. Plant Cell Environ. 2014, 37, 1250–1258. [Google Scholar] [CrossRef]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-Wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liao, L.; Xu, S.; Ren, F.; Zhao, J.; Ogutu, C.; Wang, L.; Jiang, Q.; Han, Y. Two Amino Acid Changes in the R3 Repeat Cause Functional Divergence of Two Clustered MYB10 Genes in Peach. Plant Mol. Biol. 2018, 98, 169–183. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| V3 | V3.1 | |
|---|---|---|
| Number of genes | 24,317 | 24,145 |
| Mean mRNA length (bp) | 1716 | 1819 |
| Mean exon number | 5.22 | 5.27 |
| Mean CDS length (bp) | 1128 | 1131 |
| Genes with GO terms | 11,086 | 11,142 |
| Transcripts with KEGG terms | 20,167 | 20,317 |
| Complete BUSCOs | 95.4% | 96.9% |
| Complete and single-copy BUSCOs | 91.3% | 92.6% |
| Complete and duplicated BUSCOs | 4.1% | 4.3% |
| Fragmented BUSCOs | 1.5% | 0.7% |
| Missing BUSCOs | 3.1% | 2.2% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, W.; Xia, L.; Li, R.; Zhao, X.; Jin, D.; Wang, X.; Pei, Y.; Zhou, R.; Chen, J.; Yu, X. Updated Gene Prediction of the Cucumber (9930) Genome through Manual Annotation. Plants 2024, 13, 1604. https://doi.org/10.3390/plants13121604
Du W, Xia L, Li R, Zhao X, Jin D, Wang X, Pei Y, Zhou R, Chen J, Yu X. Updated Gene Prediction of the Cucumber (9930) Genome through Manual Annotation. Plants. 2024; 13(12):1604. https://doi.org/10.3390/plants13121604
Chicago/Turabian StyleDu, Weixuan, Lei Xia, Rui Li, Xiaokun Zhao, Danna Jin, Xiaoning Wang, Yun Pei, Rong Zhou, Jinfeng Chen, and Xiaqing Yu. 2024. "Updated Gene Prediction of the Cucumber (9930) Genome through Manual Annotation" Plants 13, no. 12: 1604. https://doi.org/10.3390/plants13121604
APA StyleDu, W., Xia, L., Li, R., Zhao, X., Jin, D., Wang, X., Pei, Y., Zhou, R., Chen, J., & Yu, X. (2024). Updated Gene Prediction of the Cucumber (9930) Genome through Manual Annotation. Plants, 13(12), 1604. https://doi.org/10.3390/plants13121604