
A Novel Plant-Derived Biopesticide Mitigates Fusarium Root Rot of Angelica sinensis by Modulating the Rhizosphere Microbiome and Root Metabolome

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Assessment of Different Treatments on Biocontrol Efficiency of Angelica sinensis Root Rot
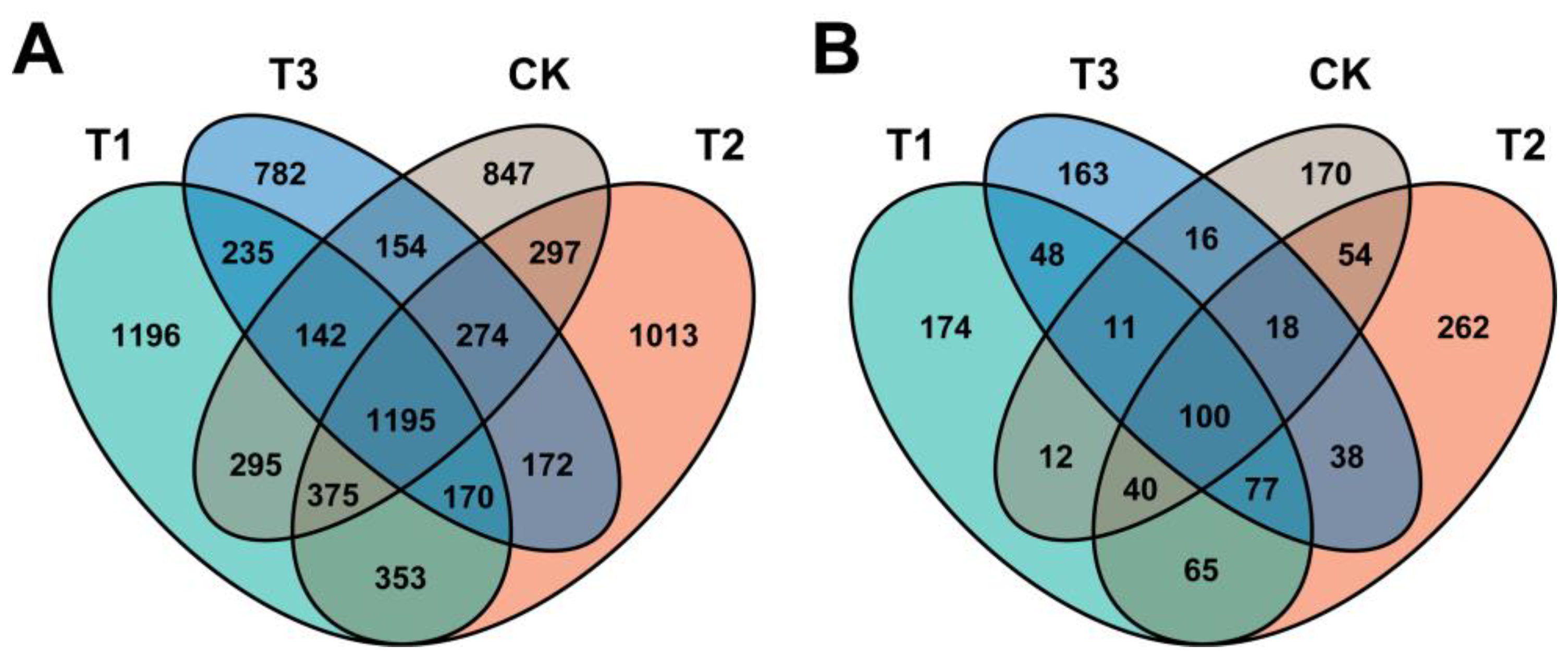
2.2. Effect of Different Treatments on the Angelica sinensis Rhizosphere Microbiome Assembly
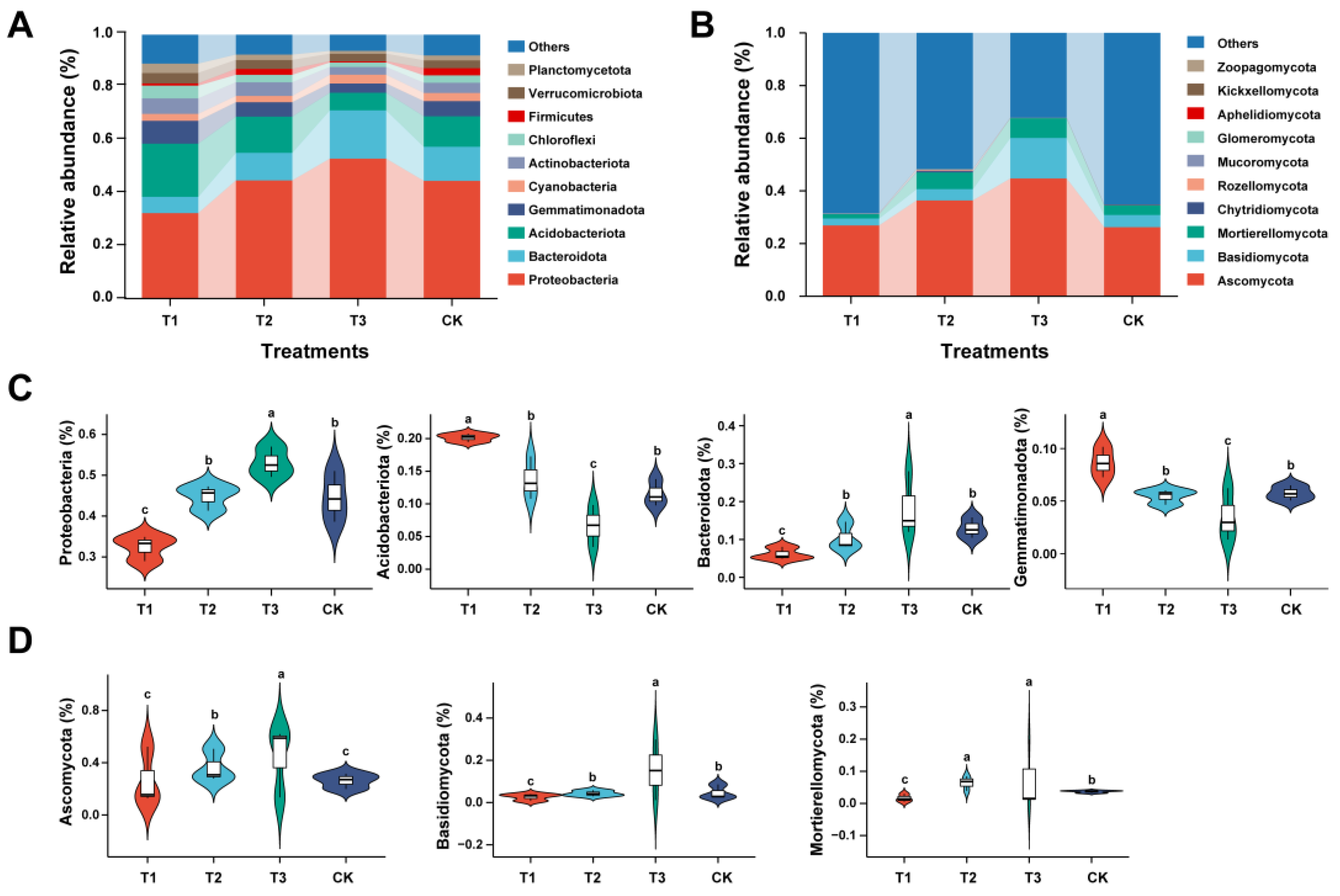
2.3. Analysis of Distribution and Abundance of Rhizosphere Microbial Communities at the Phyla Level
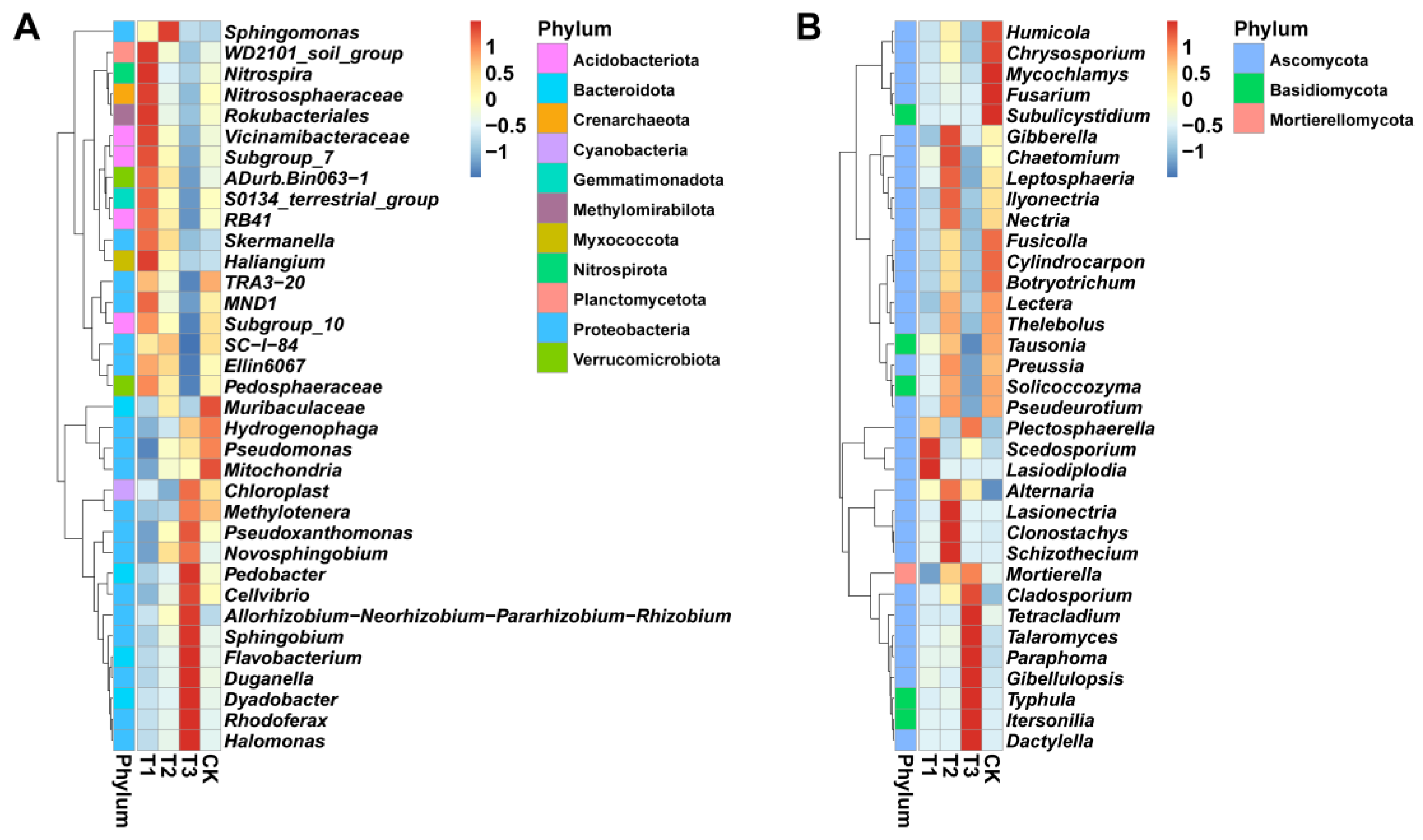
2.4. Relative Abundance Analysis of Rhizosphere Microbial Community Composition at the Genus Level
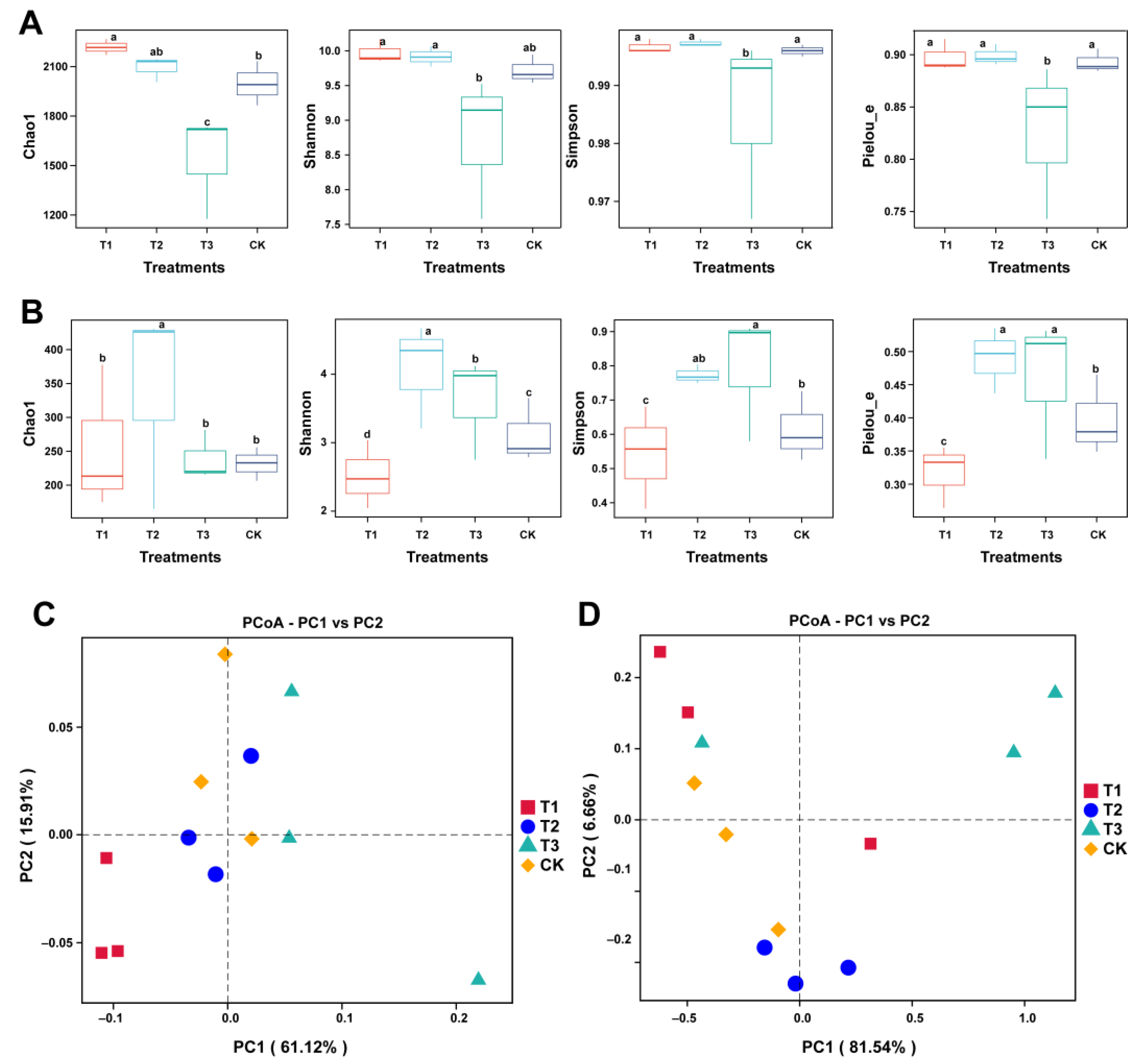
2.5. Assessment of Differences in Rhizosphere Microbial Community Diversity and Structure
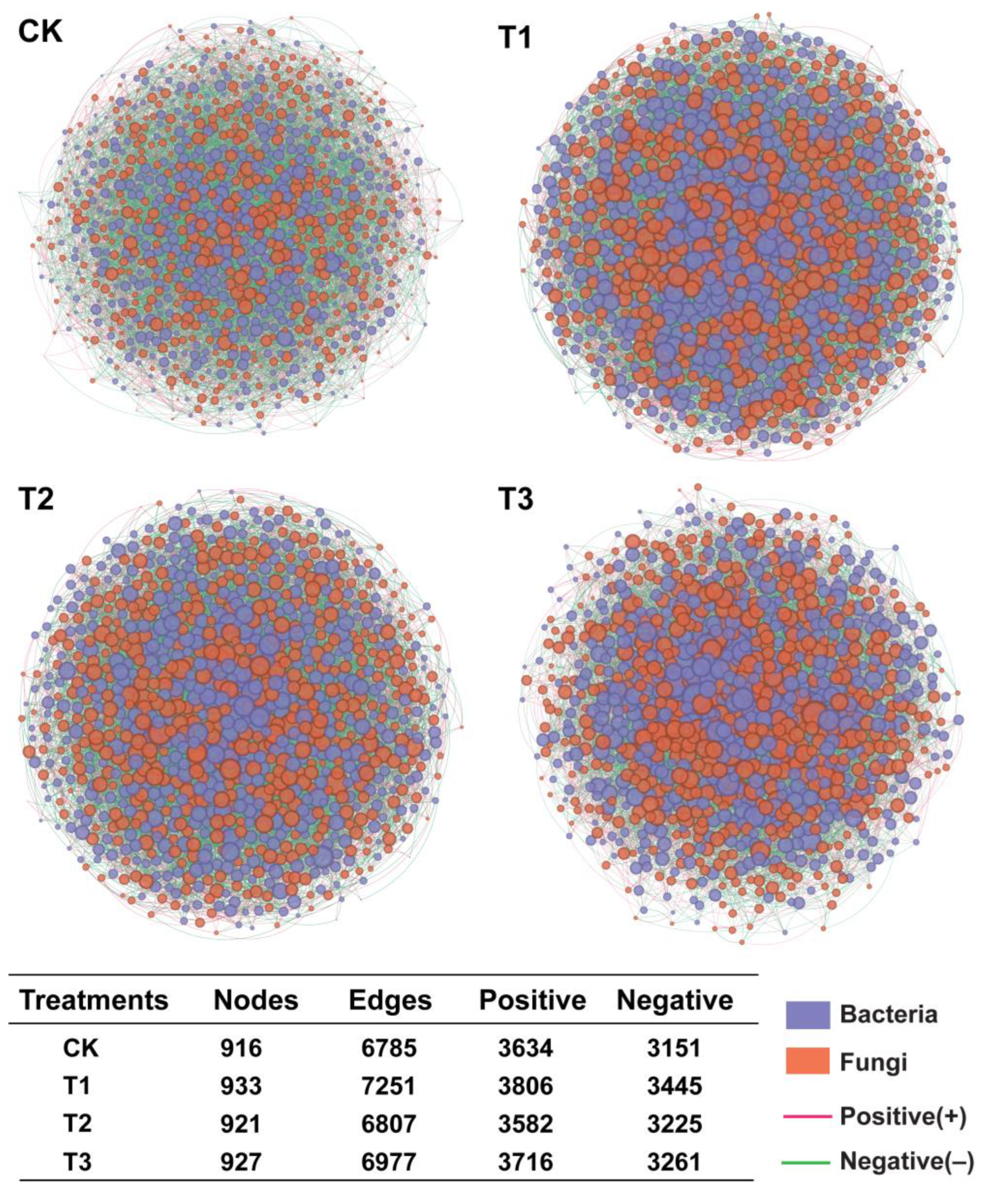
2.6. Characteristics of Angelica sinensis Intra-Kingdom Rhizosphere Microbial Co-Occurrence Network
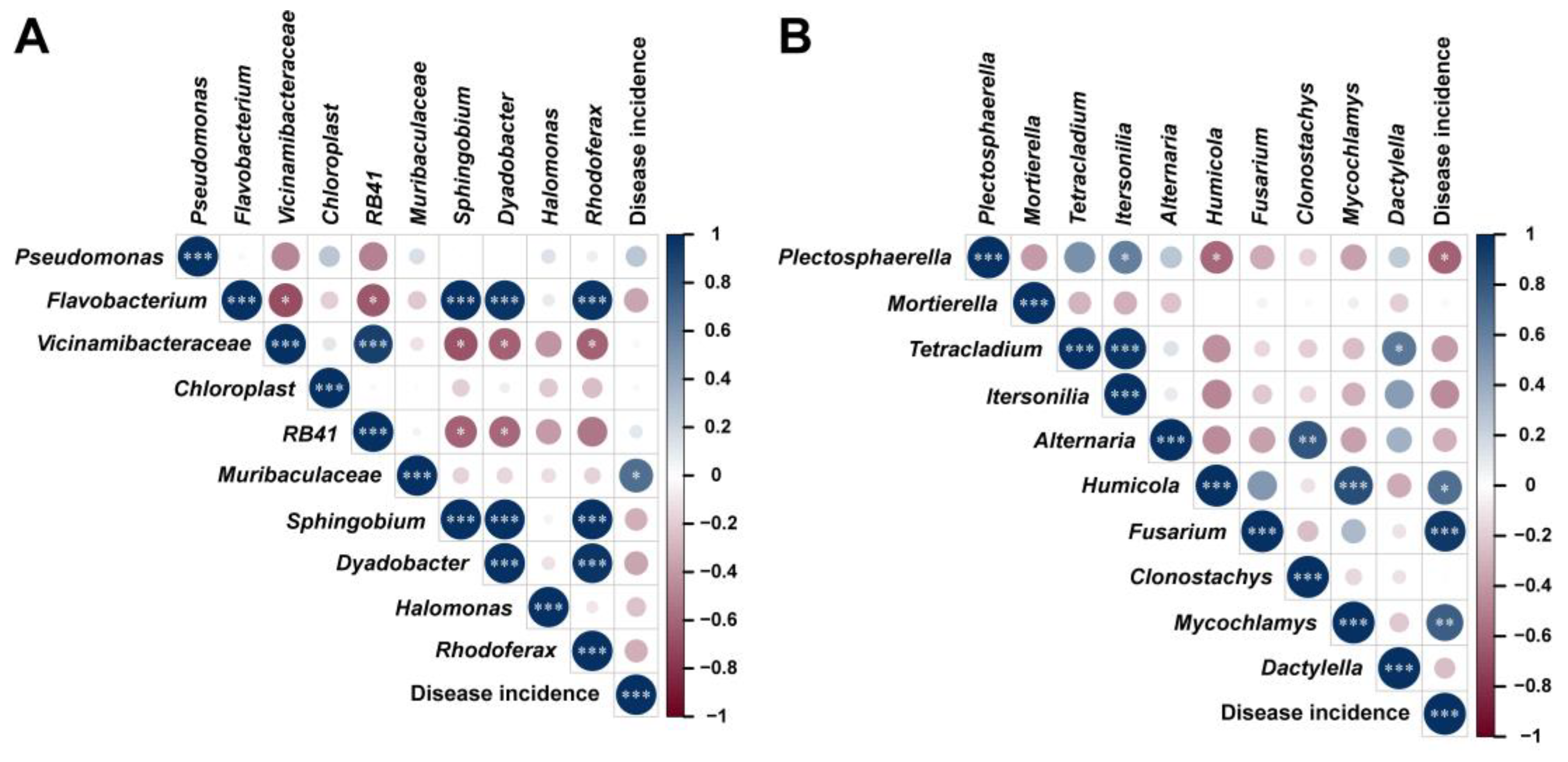
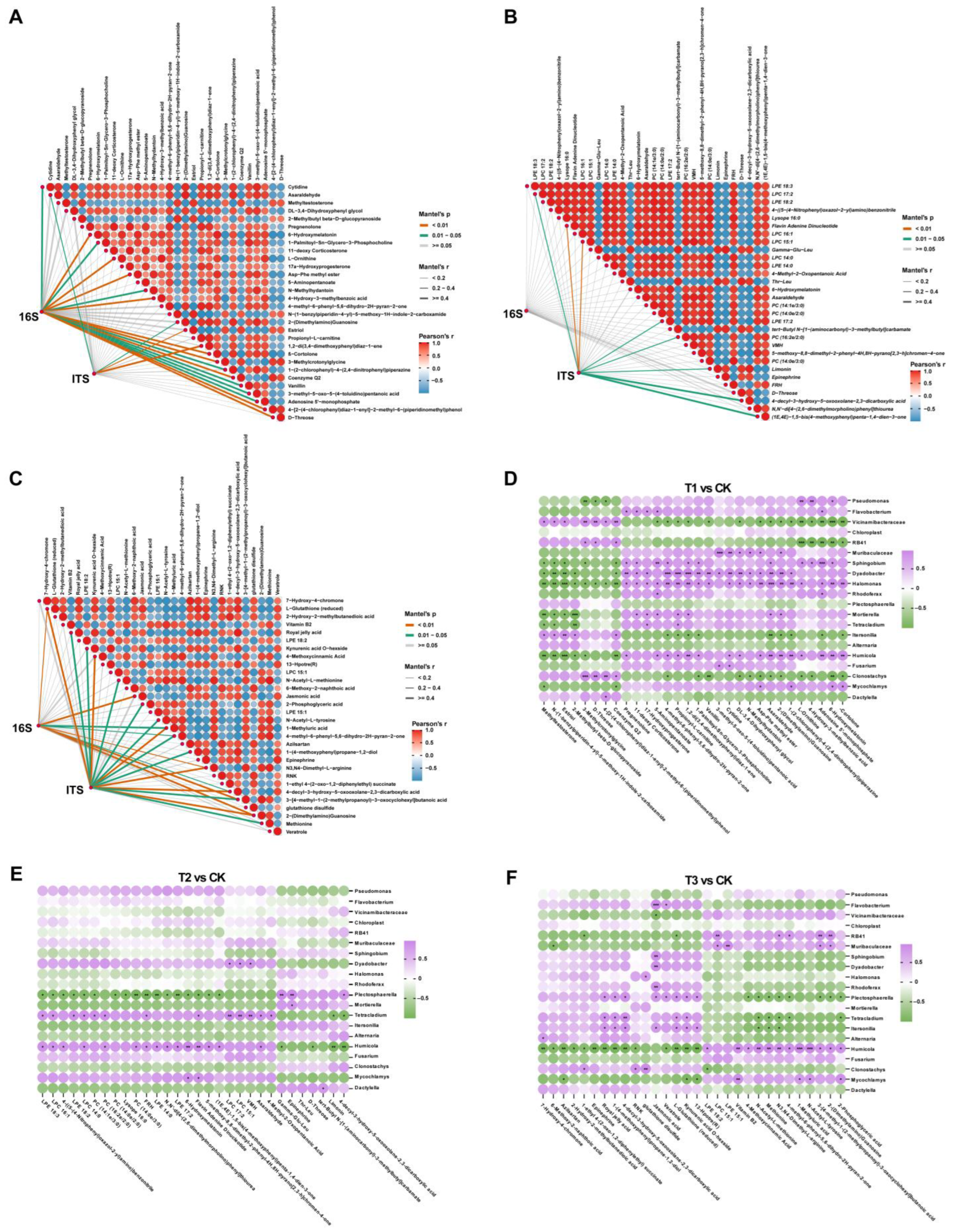
2.7. Correlation Analysis of Microbial Communities and Disease Incidence
2.8. Integrated Characteristics of Metabolomics of Angelica sinensis
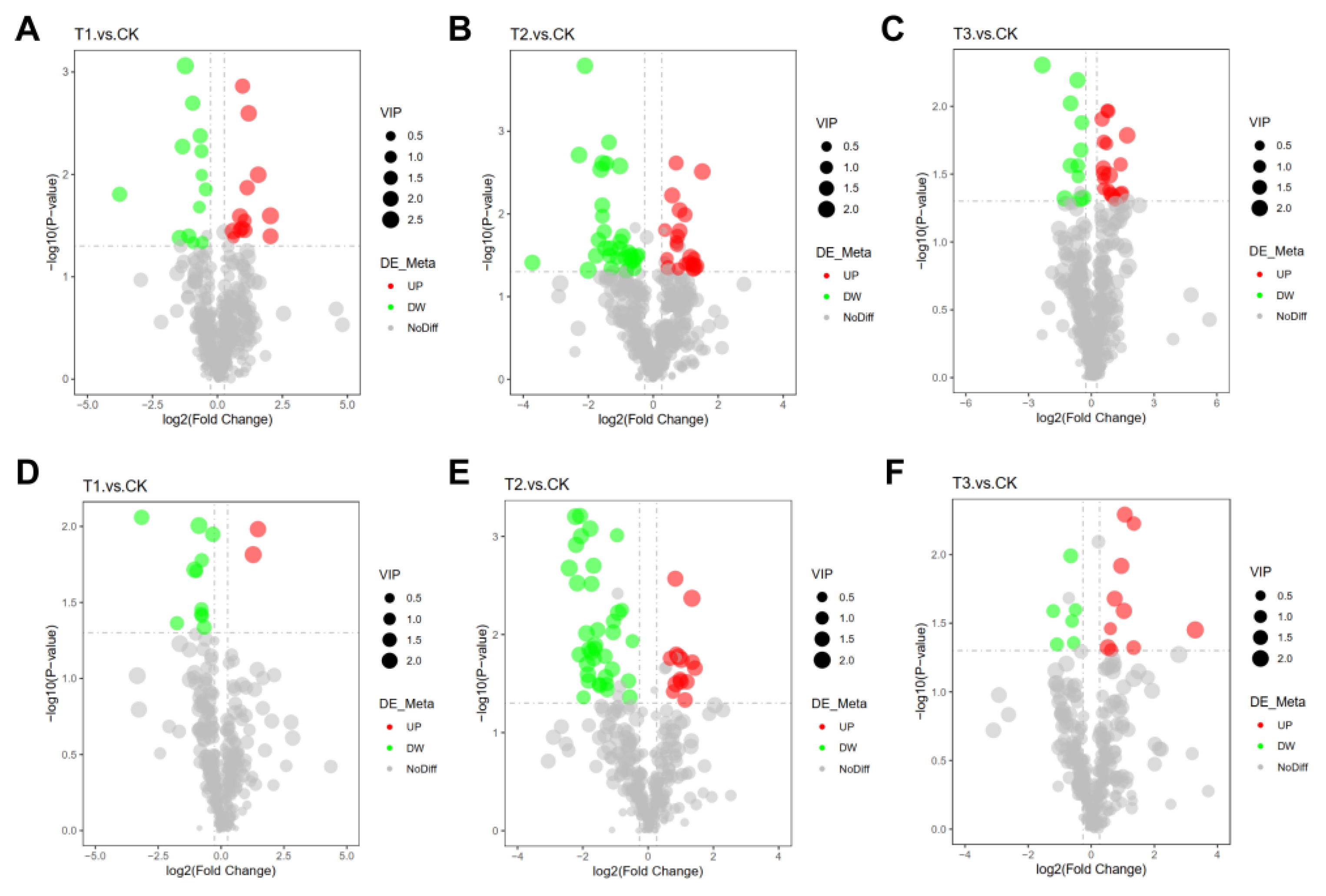
2.9. Assessment of Variations in Angelica sinensis Metabolites under Different Treatments
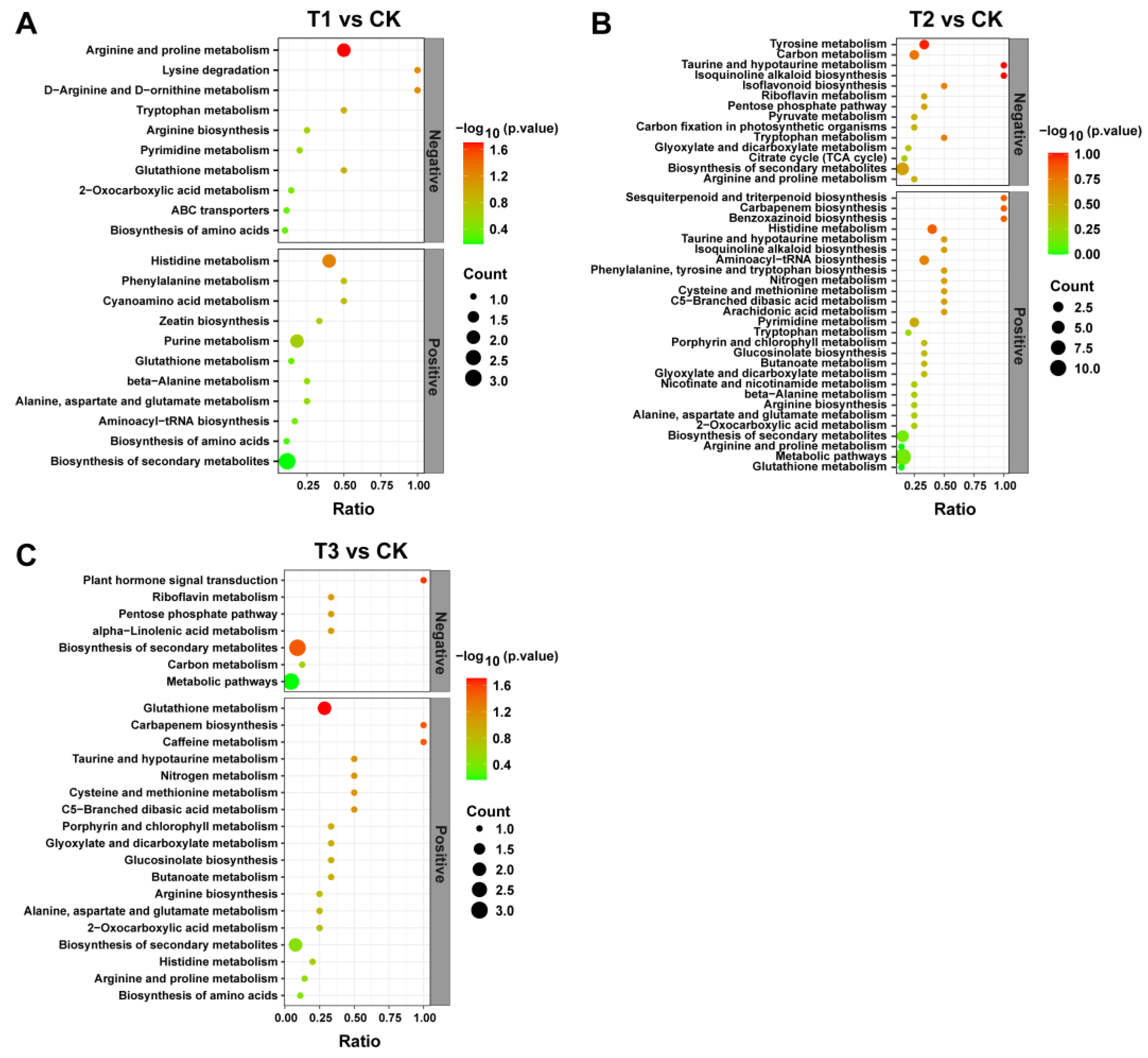
2.10. KEGG Enrichment Pathways Analysis for DEMs
2.11. Correlations between Differential Microorganisms and DEMs
3. Discussion
4. Materials and Methods
4.1. Site and Experimental Design Descriptions
4.2. Samples Collections and Disease Severity Analysis
4.3. Soil DNA Extraction, PCR Amplification, and Amplicon Sequencing
4.4. Microbiome Data Processing and Bioinformatics Analysis
4.5. Metabolites UHPLC-MS/MS Analysis
4.6. Data Processing and Metabolite Identification
4.7. Metabolomics Data Analysis
4.8. Correlations Analysis between Differential Microorganisms and DEMs and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, Y.; Chen, Y.; Zhang, Q.; Liu, B.-W.; Yang, L.; Xu, Y.-H.; Zhao, Y.-H. Overview of therapeutic potentiality of Angelica sinensis for ischemic stroke. Phytomedicine 2021, 90, 153652. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Wang, Y.; Su, W.; Zhou, X.; Lu, C. Angelica sinensis polysaccharide suppresses the Wnt/β-catenin-mediated malignant biological behaviors of breast cancer cells via the miR-3187-3p/PCDH10 axis. Biochem. Pharmacol. 2024, 225, 116295. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wu, Y.; Chen, P.; Bai, Y.; Liu, Y.; Jiang, Y.; Zhang, Y.; Yang, Z. Anti-platelet aggregation activities of different grades of Angelica sinensis and their therapeutic mechanisms in rats with blood deficiency: Insights from metabolomics and lipidomics analyses. Front. Pharmacol. 2024, 14, 1230861. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, W.; Wang, X.; Kou, Z.; Wang, Y.; Islam, R.; Zhang, J.; Liu, L.; Shen, T.; Tian, Y. Isolation and identification of antagonistic bacteria of Angelica root rot and their mechanism as biological control. Biol. Control 2023, 177, 105120. [Google Scholar] [CrossRef]
- Liu, Y.; Tian, Y.; Zhao, X.; Yue, L.; Uwaremwe, C.; Zhou, Q.; Wang, Y.; Zhang, Y.; Dun, Z.; Cui, Z. Identification of pathogenic Fusarium spp. responsible for root rot of Angelica sinensis and characterization of their biological enemies in Dingxi, China. Plant Dis. 2022, 106, 1898–1910. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, R.; Xie, Z.; Wang, Y.; Zhao, X.; Liu, Y.; Zhou, Q. Rapid visual detection of Japanese hornwort mosaic virus infecting Angelica sinensis by reverse transcription loop-mediated isothermal amplification. Ann. Appl. Biol. 2021, 178, 489–497. [Google Scholar] [CrossRef]
- Liu, Y.; Tian, Y.; Yue, L.; Constantine, U.; Zhao, X.; Zhou, Q.; Wang, Y.; Zhang, Y.; Chen, G.; Dun, Z. Effectively controlling Fusarium root rot disease of Angelica sinensis and enhancing soil fertility with a novel attapulgite-coated biocontrol agent. Appl. Soil Ecol. 2021, 168, 104121. [Google Scholar] [CrossRef]
- Uwaremwe, C.; Yue, L.; Liu, Y.; Tian, Y.; Zhao, X.; Wang, Y.; Xie, Z.; Zhang, Y.; Cui, Z.; Wang, R. Molecular identification and pathogenicity of Fusarium and Alternaria species associated with root rot disease of wolfberry in Gansu and Ningxia provinces, China. Plant Pathol. 2021, 70, 397–406. [Google Scholar] [CrossRef]
- Zhu, B.; Wu, J.; Ji, Q.; Wu, W.; Dong, S.; Yu, J.; Zhang, Q.; Qin, L. Diversity of rhizosphere and endophytic fungi in Atractylodes macrocephala during continuous cropping. PeerJ 2020, 8, e8905. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Duan, X.; Xu, W.; Ma, G.; Ma, W.; Qi, H. Root Rot of Angelica sinensis Caused by Clonostachys rosea and Fusarium acuminatum in China. Plant Dis. 2022, 106, 2264. [Google Scholar] [CrossRef]
- Farh, M.E.-A.; Kim, Y.-J.; Kim, Y.-J.; Yang, D.-C. Cylindrocarpon destructans/Ilyonectria radicicola-species complex: Causative agent of ginseng root-rot disease and rusty symptoms. J. Ginseng Res. 2018, 42, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xi, N.; Lang, D.; Zhou, L.; Zhang, Y.; Zhang, X. Potential biocontrol and plant growth promotion of an endophytic bacteria isolated from Glycyrrhiza uralensis seeds. Egypt. J. Biol. Pest Control 2022, 32, 55. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Z.; Yang, Y.; Ma, Q.; Zheng, Y.; Xu, C.; Gao, X.; Gao, W.; Huang, Z.; Liu, X. Characterization of Fusarium species causing soybean root rot in Heilongjiang, China, and mechanism underlying the differences in sensitivity to DMI fungicides. Pestic. Biochem. Physiol. 2024, 200, 105828. [Google Scholar] [CrossRef] [PubMed]
- Myresiotis, C.K.; Karaoglanidis, G.S.; Vryzas, Z.; Papadopoulou-Mourkidou, E. Evaluation of plant-growth-promoting rhizobacteria, acibenzolar-S-methyl and hymexazol for integrated control of Fusarium crown and root rot on tomato. Pest Manag. Sci. 2012, 68, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Naseri, B. Bean production and Fusarium root rot in diverse soil environments in Iran. J. Soil Sci. Plant Nutr. 2014, 14, 177–188. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, J.; Ahmed, W.; Wan, X.; Wei, L.; Ji, G. Exploiting the antibacterial mechanism of phenazine substances from Lysobacter antibioticus 13-6 against Xanthomonas oryzae pv. oryzicola. J. Microbiol. 2022, 60, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Yang, J.; Tan, Y.; Munir, S.; Liu, Q.; Zhang, J.; Ji, G.; Zhao, Z. Ralstonia solanacearum, a deadly pathogen: Revisiting the bacterial wilt biocontrol practices in tobacco and other Solanaceae. Rhizosphere 2022, 21, 100479. [Google Scholar] [CrossRef]
- Shen, T.; Wang, C.; Yang, H.; Deng, Z.; Wang, S.; Shen, B.; Shen, Q. Identification, solid-state fermentation and biocontrol effects of Streptomyces hygroscopicus B04 on strawberry root rot. Appl. Soil Ecol. 2016, 103, 36–43. [Google Scholar] [CrossRef]
- Kalantari, S.; Marefat, A.; Naseri, B.; Hemmati, R. Improvement of bean yield and Fusarium root rot biocontrol using mixtures of Bacillus, Pseudomonas and Rhizobium. Trop. Plant Pathol. 2018, 43, 499–505. [Google Scholar] [CrossRef]
- Zhang, J.; Wei, L.; Yang, J.; Ahmed, W.; Wang, Y.; Fu, L.; Ji, G. Probiotic consortia: Reshaping the rhizospheric microbiome and its role in suppressing root-rot disease of Panax notoginseng. Front. Microbiol. 2020, 11, 513301. [Google Scholar] [CrossRef]
- Zhang, J.; Ahmed, W.; Dai, Z.; Zhou, X.; He, Z.; Wei, L.; Ji, G. Microbial consortia: An engineering tool to suppress clubroot of Chinese cabbage by changing the rhizosphere bacterial community composition. Biology 2022, 11, 918. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Burke, D.J.; Medeiros, J.S.; Carrino-Kyker, S.R.; Burns, J.H. Phosphite indirectly mediates protection against root rot disease via altering soil fungal community in Rhododendron species. Plant Soil 2023, 491, 467–478. [Google Scholar] [CrossRef]
- Zhang, M.; Kong, Z.; Fu, H.; Shu, X.; Xue, Q.; Lai, H.; Guo, Q. Rhizosphere microbial ecological characteristics of strawberry root rot. Front. Microbiol. 2023, 14, 1286740. [Google Scholar] [CrossRef] [PubMed]
- Ling, N.; Wang, T.; Kuzyakov, Y. Rhizosphere bacteriome structure and functions. Nat. Commun. 2022, 13, 836. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Dai, Z.; Zhang, J.; Li, S.; Ahmed, A.; Munir, S.; Liu, Q.; Tan, Y.; Ji, G.; Zhao, Z. Plant-microbe interaction: Mining the impact of native Bacillus amyloliquefaciens WS-10 on tobacco bacterial wilt disease and rhizosphere microbial communities. Microbiol. Spectr. 2022, 10, e01471–e01422. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Ahmed, W.; Yang, J.; Yao, X.; Zhang, J.; Wei, L.; Ji, G. Seed coat treatment by plant-growth-promoting rhizobacteria Lysobacter antibioticus 13–6 enhances maize yield and changes rhizosphere bacterial communities. Biol. Fertil. Soils 2023, 59, 317–331. [Google Scholar] [CrossRef]
- Chaloner, T.M.; Gurr, S.J.; Bebber, D.P. Geometry and evolution of the ecological niche in plant-associated microbes. Nat. Commun. 2020, 11, 2955. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Li, Y.; Zhang, Z.; Cui, H.; Zhao, Q.; Liu, W.; Lu, T.; Qian, H. Effects of S-metolachlor on wheat (Triticum aestivum L.) seedling root exudates and the rhizosphere microbiome. J. Hazard. Mater. 2021, 411, 125137. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-M.; Feng, W.-M.; Yan, H.; Liu, P.; Zhou, G.-S.; Guo, S.; Yu, G.; Duan, J.-A. Explore the interaction between root metabolism and rhizosphere microbiota during the growth of Angelica sinensis. Front. Plant Sci. 2022, 13, 1005711. [Google Scholar] [CrossRef]
- Musilova, L.; Ridl, J.; Polivkova, M.; Macek, T.; Uhlik, O. Effects of secondary plant metabolites on microbial populations: Changes in community structure and metabolic activity in contaminated environments. Int. J. Mol. Sci. 2016, 17, 1205. [Google Scholar] [CrossRef]
- Wen, T.; Zhao, M.; Yuan, J.; Kowalchuk, G.A.; Shen, Q. Root exudates mediate plant defense against foliar pathogens by recruiting beneficial microbes. Soil Ecol. Lett. 2021, 3, 42–51. [Google Scholar] [CrossRef]
- Liu, H.; Li, J.; Carvalhais, L.C.; Percy, C.D.; Prakash Verma, J.; Schenk, P.M.; Singh, B.K. Evidence for the plant recruitment of beneficial microbes to suppress soil-borne pathogens. New Phytol. 2021, 229, 2873–2885. [Google Scholar] [CrossRef] [PubMed]
- Al-Khayri, J.M.; Rashmi, R.; Toppo, V.; Chole, P.B.; Banadka, A.; Sudheer, W.N.; Nagella, P.; Shehata, W.F.; Al-Mssallem, M.Q.; Alessa, F.M. Plant secondary metabolites: The weapons for biotic stress management. Metabolites 2023, 13, 716. [Google Scholar] [CrossRef] [PubMed]
- Yeshi, K.; Crayn, D.; Ritmejerytė, E.; Wangchuk, P. Plant secondary metabolites produced in response to abiotic stresses has potential application in pharmaceutical product development. Molecules 2022, 27, 313. [Google Scholar] [CrossRef] [PubMed]
- Carezzano, M.E.; Reyna, P.G.; Accotto, E.; Giordano, W.; Oliva, M.d.l.M.; Rodriguez Pardina, P.; Sabini, M.C. Plant-Derived Essential Oils and Aqueous Extract as Potential Ingredients for a Biopesticide: Phytotoxicity in Soybean and Activity against Soybean Mosaic Virus. Processes 2023, 11, 2265. [Google Scholar] [CrossRef]
- Thepbandit, W.; Athinuwat, D. Rhizosphere Microorganisms Supply Availability of Soil Nutrients and Induce Plant Defense. Microorganisms 2024, 12, 558. [Google Scholar] [CrossRef] [PubMed]
- Fatima, U.; Senthil-Kumar, M. Plant and pathogen nutrient acquisition strategies. Front. Plant Sci. 2015, 6, 162246. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Huang, X.; Zhang, J.; Cai, Z.; Jiang, K.; Chang, Y. Deciphering the relative importance of soil and plant traits on the development of rhizosphere microbial communities. Soil Biol. Biochem. 2020, 148, 107909. [Google Scholar] [CrossRef]
- Ginetti, B.; Uccello, A.; Bracalini, M.; Ragazzi, A.; Jung, T.; Moricca, S. Root rot and dieback of Pinus pinea caused by phytophthora Humicola in Tuscany, central Italy. Plant Dis. 2012, 96, 1694. [Google Scholar] [CrossRef]
- Menzies, J.G.; Ehret, D.L.; Koch, C.; Bogdanoff, C. Humicola fuscoatra infects tomato roots, but is not pathogenic. Eur. J. Plant Pathol. 1998, 104, 769–775. [Google Scholar] [CrossRef]
- Zhang, X.; Shao, J.; Chen, A.; Shang, C.; Hu, X.; Luo, S.; Lei, M.; Peng, L.; Zeng, Q. Effects of cadmium on calcium homeostasis in the white-rot fungus Phanerochaete chrysosporium. Ecotoxicol. Environ. Saf. 2018, 157, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ming, Z.; Li, H.; Yang, H.; Yu, B.; Wu, R.; Liu, X.; Bai, Y.; Yang, S.-T. Toxicity of graphene oxide to white rot fungus Phanerochaete chrysosporium. Chemosphere 2016, 151, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Vallance, J.; Déniel, F.; Floch, G.L.; Guérin-Dubrana, L.; Blancard, D.; Rey, P. Pathogenic and beneficial microorganisms in soilless cultures. Sustain. Agric. 2011, 2, 711–726. [Google Scholar]
- Tao, C.; Wang, Z.; Liu, S.; Lv, N.; Deng, X.; Xiong, W.; Shen, Z.; Zhang, N.; Geisen, S.; Li, R. Additive fungal interactions drive biocontrol of Fusarium wilt disease. New Phytol. 2023, 238, 1198–1214. [Google Scholar] [CrossRef] [PubMed]
- Shahid, M.; Khan, M.S. Ecotoxicological implications of residual pesticides to beneficial soil bacteria: A review. Pestic. Biochem. Physiol. 2022, 188, 105272. [Google Scholar] [CrossRef]
- Yang, H.; Li, J.; Xiao, Y.; Gu, Y.; Liu, H.; Liang, Y.; Liu, X.; Hu, J.; Meng, D.; Yin, H. An integrated insight into the relationship between soil microbial community and tobacco bacterial wilt disease. Front. Microbiol. 2017, 8, 2179. [Google Scholar] [CrossRef]
- Martins, S.J.; Pasche, J.; Silva, H.A.O.; Selten, G.; Savastano, N.; Abreu, L.M.; Bais, H.P.; Garrett, K.A.; Kraisitudomsook, N.; Pieterse, C.M. The use of synthetic microbial communities to improve plant health. Phytopathology® 2023, 113, 1369–1379. [Google Scholar] [CrossRef]
- Bai, Y.-C.; Li, B.-X.; Xu, C.-Y.; Raza, M.; Wang, Q.; Wang, Q.-Z.; Fu, Y.-N.; Hu, J.-Y.; Imoulan, A.; Hussain, M. Intercropping walnut and tea: Effects on soil nutrients, enzyme activity, and microbial communities. Front. Microbiol. 2022, 13, 852342. [Google Scholar] [CrossRef]
- Staley, C.; Ferrieri, A.P.; Tfaily, M.M.; Cui, Y.; Chu, R.K.; Wang, P.; Shaw, J.B.; Ansong, C.K.; Brewer, H.; Norbeck, A.D. Diurnal cycling of rhizosphere bacterial communities is associated with shifts in carbon metabolism. Microbiome 2017, 5, 65. [Google Scholar] [CrossRef]
- Liao, H.; Zheng, C.; Long, J.; Guzmán, I. Effects of biochar amendment on tomato rhizosphere bacterial communities and their utilization of plant-derived carbon in a calcareous soil. Geoderma 2021, 396, 115082. [Google Scholar] [CrossRef]
- Hu, Y.; Zhao, W.; Li, X.; Feng, J.; Li, C.; Yang, X.; Guo, Q.; Wang, L.; Chen, S.; Li, Y. Integrated biocontrol of tobacco bacterial wilt by antagonistic bacteria and marigold. Sci. Rep. 2021, 11, 16360. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Hu, J.; Yin, S.; Xu, Y.; Jousset, A.; Shen, Q.; Friman, V.-P. Ralstonia solanacearum pathogen disrupts bacterial rhizosphere microbiome during an invasion. Soil Biol. Biochem. 2018, 118, 8–17. [Google Scholar] [CrossRef]
- Bakker, P.A.; Doornbos, R.F.; Zamioudis, C.; Berendsen, R.L.; Pieterse, C.M. Induced systemic resistance and the rhizosphere microbiome. Plant Pathol. J. 2013, 29, 136. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Pommier, T.; Yin, Y.; Wang, J.; Gu, S.; Jousset, A.; Keuskamp, J.; Wang, H.; Wei, Z.; Xu, Y. Indirect reduction of Ralstonia solanacearum via pathogen helper inhibition. ISME J. 2022, 16, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, N.; Koss, M.J.; Greco, C.; Nickles, G.; Wiemann, P.; Keller, N.P. Secreted secondary metabolites reduce bacterial wilt severity of tomato in bacterial–fungal co-infections. Microorganisms 2021, 9, 2123. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Zarandi, M.; Saberi Riseh, R.; Tarkka, M.T. Actinobacteria as effective biocontrol agents against plant pathogens, an overview on their role in eliciting plant defense. Microorganisms 2022, 10, 1739. [Google Scholar] [CrossRef]
- Dubrovina, A.; Kiselev, K. Regulation of stilbene biosynthesis in plants. Planta 2017, 246, 597–623. [Google Scholar] [CrossRef] [PubMed]
- Divekar, P.A.; Narayana, S.; Divekar, B.A.; Kumar, R.; Gadratagi, B.G.; Ray, A.; Singh, A.K.; Rani, V.; Singh, V.; Singh, A.K. Plant secondary metabolites as defense tools against herbivores for sustainable crop protection. Int. J. Mol. Sci. 2022, 23, 2690. [Google Scholar] [CrossRef]
- Verma, N.; Shukla, S. Impact of various factors responsible for fluctuation in plant secondary metabolites. J. Appl. Res. Med. Aromat. Plants 2015, 2, 105–113. [Google Scholar] [CrossRef]
- Li, Q.; Chen, Y.; Gao, H.; Li, Z.; Qiu, D.; Hu, G. In situ analysis of volatile oil in Angelica sinensis roots by fluorescence imaging combined with mass spectrometry imaging. Talanta 2023, 255, 124253. [Google Scholar] [CrossRef]
- Kumar, N.; Kulsoom, M.; Shukla, V.; Kumar, D.; Priyanka; Kumar, S.; Tiwari, J.; Dwivedi, N. Profiling of heavy metal and pesticide residues in medicinal plants. Environ. Sci. Pollut. Res. 2018, 25, 29505–29510. [Google Scholar] [CrossRef]
- Jamwal, V.L.; Rather, I.A.; Ahmed, S.; Kumar, A.; Gandhi, S.G. Changing Rhizosphere Microbial Community and Metabolites with Developmental Stages of Coleus barbatus. Microorganisms 2023, 11, 705. [Google Scholar] [CrossRef]
- Bhuyan, B.; Debnath, S.; Pandey, P. The rhizosphere microbiome and its role in plant growth in stressed conditions. In Rhizosphere Microbes: Soil and Plant Functions; Springer: Berlin/Heidelberg, Germany, 2020; pp. 503–529. [Google Scholar]
- Dang, H.; Zhang, T.; Wang, Z.; Li, G.; Zhao, W.; Lv, X.; Zhuang, L. Succession of endophytic fungi and arbuscular mycorrhizal fungi associated with the growth of plant and their correlation with secondary metabolites in the roots of plants. BMC Plant Biol. 2021, 21, 165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ahmed, W.; Zhou, X.; Yao, B.; He, Z.; Qiu, Y.; Wei, F.; He, Y.; Wei, L.; Ji, G. Crop rotation with marigold promotes soil bacterial structure to assist in mitigating clubroot Incidence in Chinese Cabbage. Plants 2022, 11, 2295. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. Msystems 2016, 1, 10–1128. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, J.; Wei, X.; Huang, K.; Li, C.; Yang, G. Deciphering core microbiota in rhizosphere soil and roots of healthy and Rhizoctonia solani-infected potato plants from various locations. Front. Microbiol. 2024, 15, 1386417. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Kõljalg, U.; Larsson, K.H.; Abarenkov, K.; Nilsson, R.H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E. UNITE: A database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 2005, 166, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, X.; Zhang, Y.; Dai, Z.; He, Z.; Qiu, Y.; Alharbi, S.A.; Wei, F.; Wei, L.; Ahmed, W. Pre-soil fumigation with ammonium bicarbonate and lime modulates the rhizosphere microbiome to mitigate clubroot disease in Chinese cabbage. Front. Microbiol. 2024, 15, 1376579. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Deng, B.; Tian, S.; Guo, M.; Liu, H.; Zhao, X. Metabolic and transcriptomic analyses reveal different metabolite biosynthesis profiles between leaf buds and mature leaves in Ziziphus jujuba mill. Food Chem. 2021, 347, 129005. [Google Scholar] [CrossRef] [PubMed]
- Cooper, B.; Yang, R. An assessment of AcquireX and Compound Discoverer software 3.3 for non-targeted metabolomics. Sci. Rep. 2024, 14, 4841. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chen, S.; Wang, H.; Li, S.; Ma, C.; Wang, J.; Cui, L. Identification of secondary metabolites in Flammulina velutipes by UPLC-Q-Exactive-Orbitrap MS. J. Food Qual. 2021, 2021, 4103952. [Google Scholar] [CrossRef]
- Wen, B.; Mei, Z.; Zeng, C.; Liu, S. metaX: A flexible and comprehensive software for processing metabolomics data. BMC Bioinform. 2017, 18, 183. [Google Scholar] [CrossRef]
- Wei, L.; Yang, J.; Ahmed, W.; Xiong, X.; Liu, Q.; Huang, Q.; Ji, G. Unraveling the association between metabolic changes in inter-genus and intra-genus bacteria to mitigate clubroot disease of Chinese cabbage. Agronomy 2021, 11, 2424. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatments | Disease Index (%) | Disease Incidence (%) | Control Effect (%) |
|---|---|---|---|
| CK | 73.33 a | 92.00 a | ----- c |
| T1 | 19.67 c | 28.33 c | 73.17 a |
| T2 | 44.00 b | 47.83 b | 39.99 b |
| T3 | 18.00 c | 23.61 c | 75.45 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Ahmed, W.; Li, G.; He, Y.; Mohany, M.; Li, Z.; Shen, T. A Novel Plant-Derived Biopesticide Mitigates Fusarium Root Rot of Angelica sinensis by Modulating the Rhizosphere Microbiome and Root Metabolome. Plants 2024, 13, 2180. https://doi.org/10.3390/plants13162180
Liu Q, Ahmed W, Li G, He Y, Mohany M, Li Z, Shen T. A Novel Plant-Derived Biopesticide Mitigates Fusarium Root Rot of Angelica sinensis by Modulating the Rhizosphere Microbiome and Root Metabolome. Plants. 2024; 13(16):2180. https://doi.org/10.3390/plants13162180
Chicago/Turabian StyleLiu, Qi, Waqar Ahmed, Guoli Li, Yilin He, Mohamed Mohany, Zhaoyu Li, and Tong Shen. 2024. "A Novel Plant-Derived Biopesticide Mitigates Fusarium Root Rot of Angelica sinensis by Modulating the Rhizosphere Microbiome and Root Metabolome" Plants 13, no. 16: 2180. https://doi.org/10.3390/plants13162180