
Potential Roles of the GRF Transcription Factors in Sorghum Internodes during Post-Reproductive Stages

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
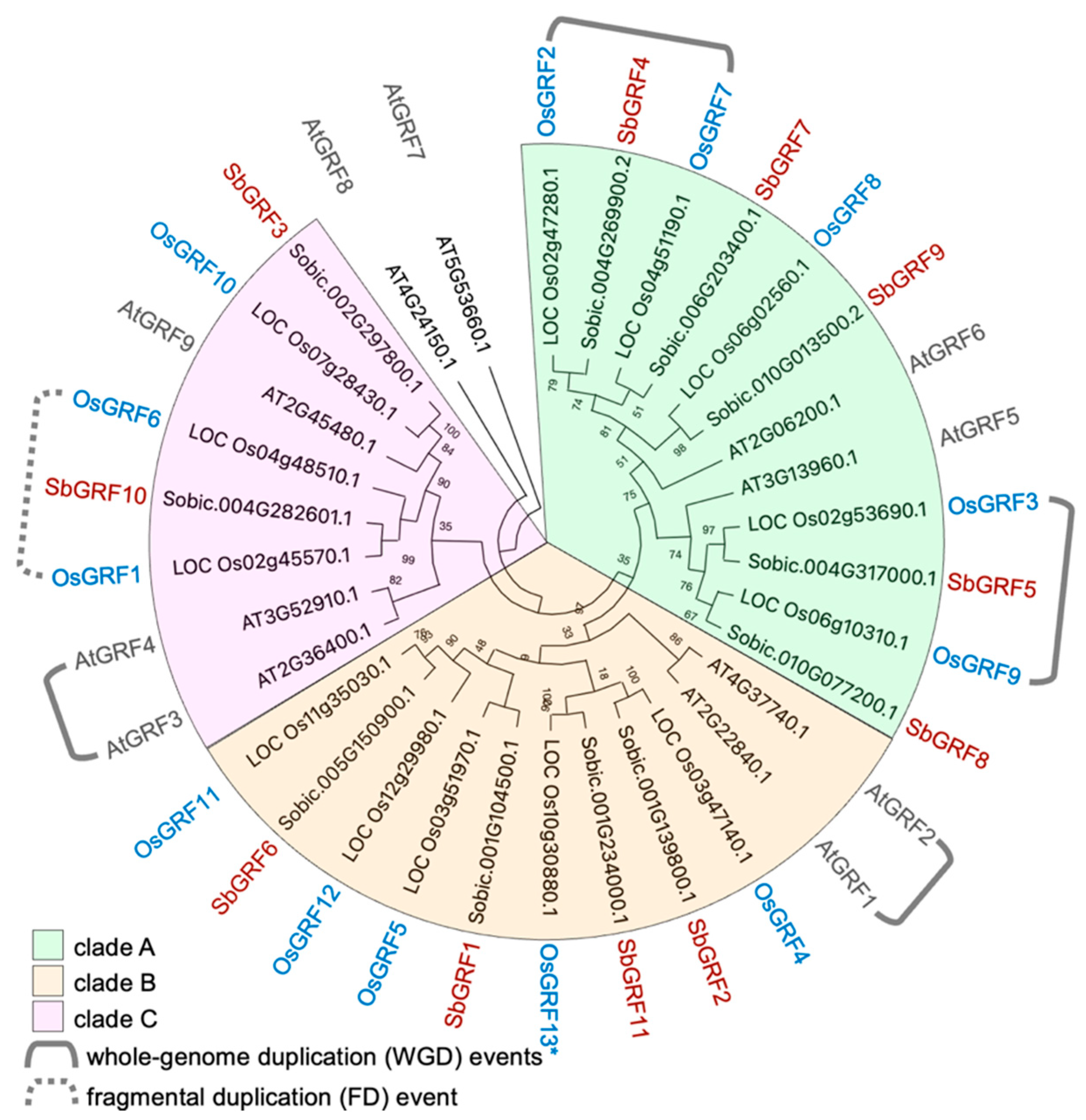
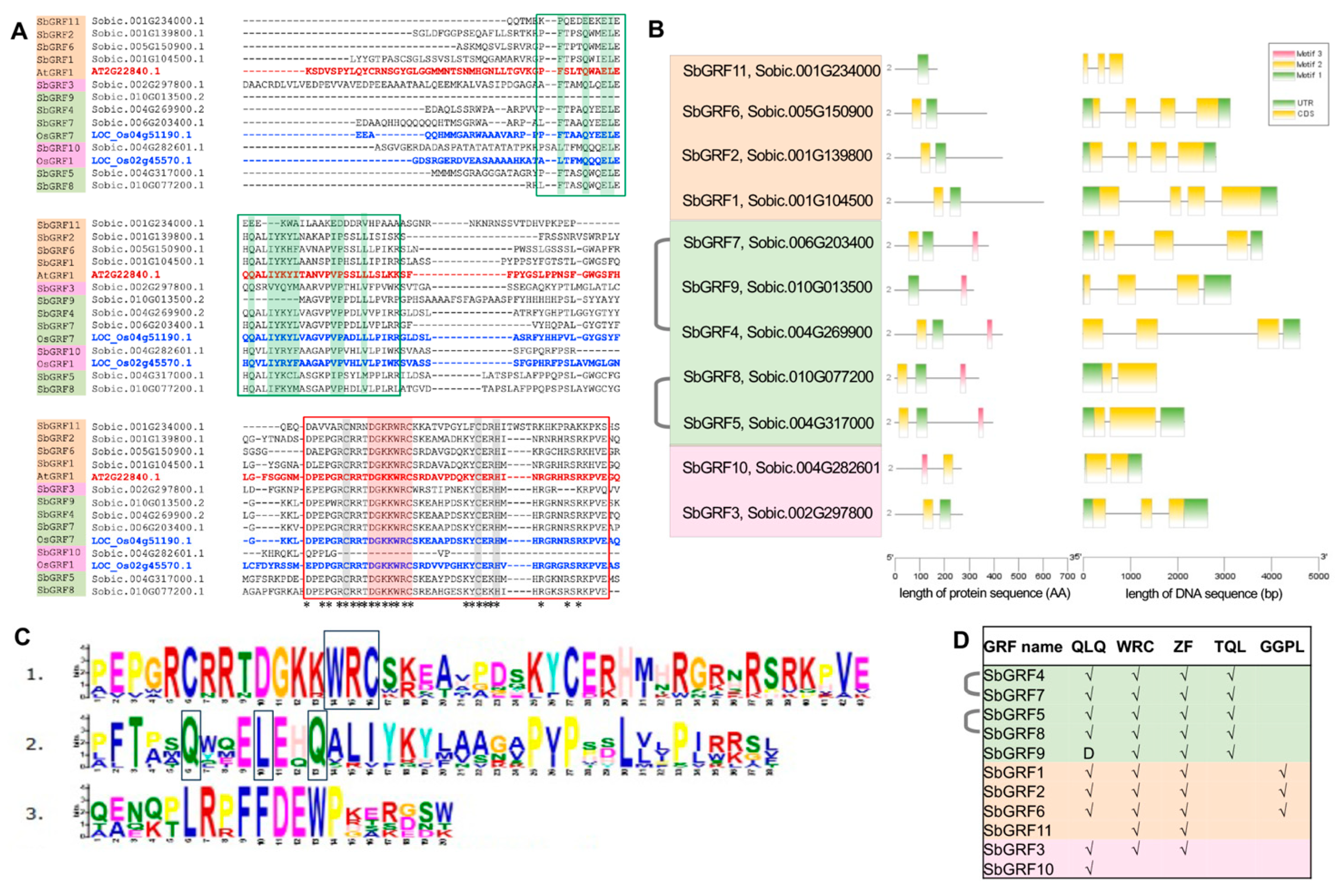
2.1. SbGRF Family Is Expanded by Whole-Genome Duplication
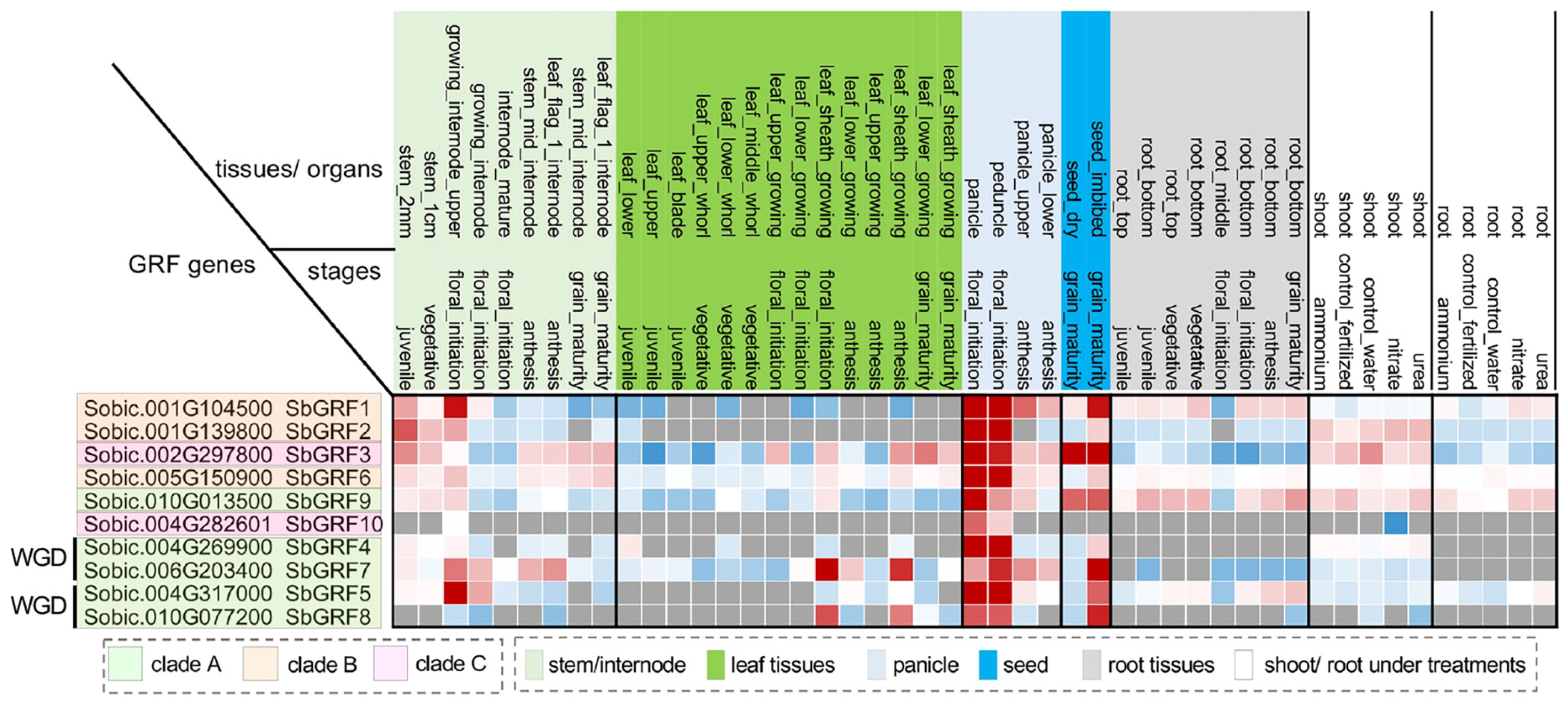
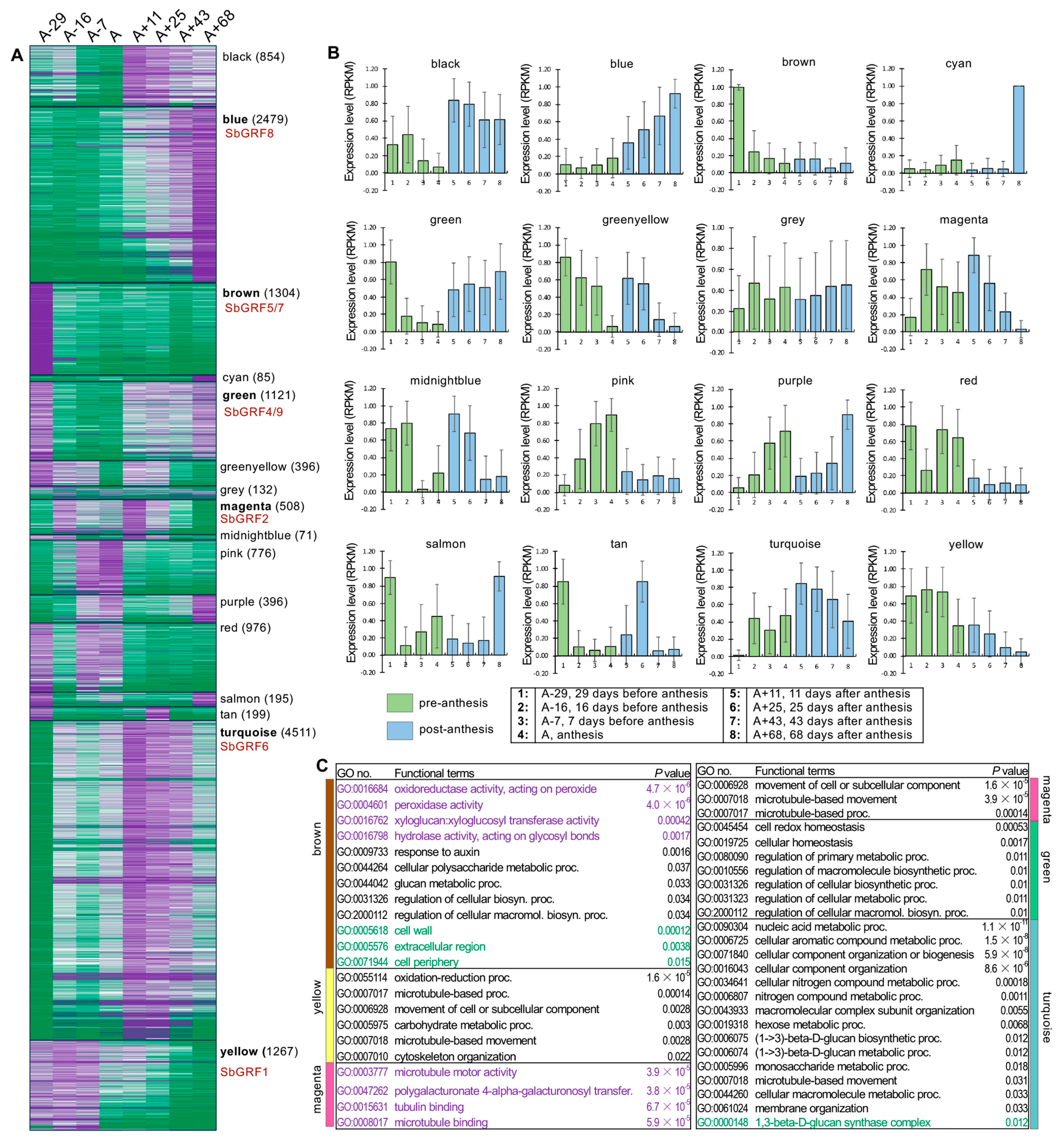
2.2. Expression Analysis Suggests Functional Divergence of SbGRFs
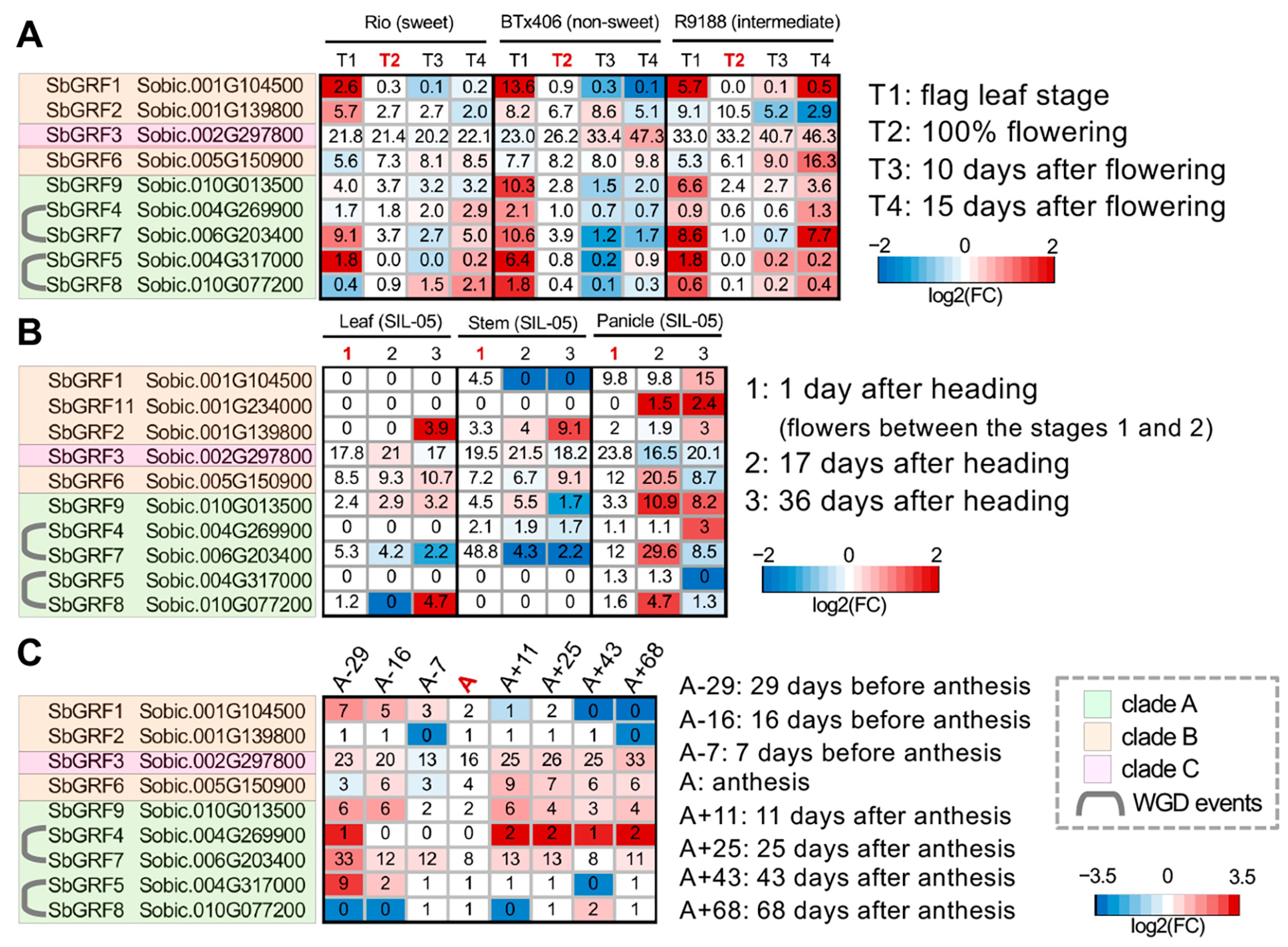
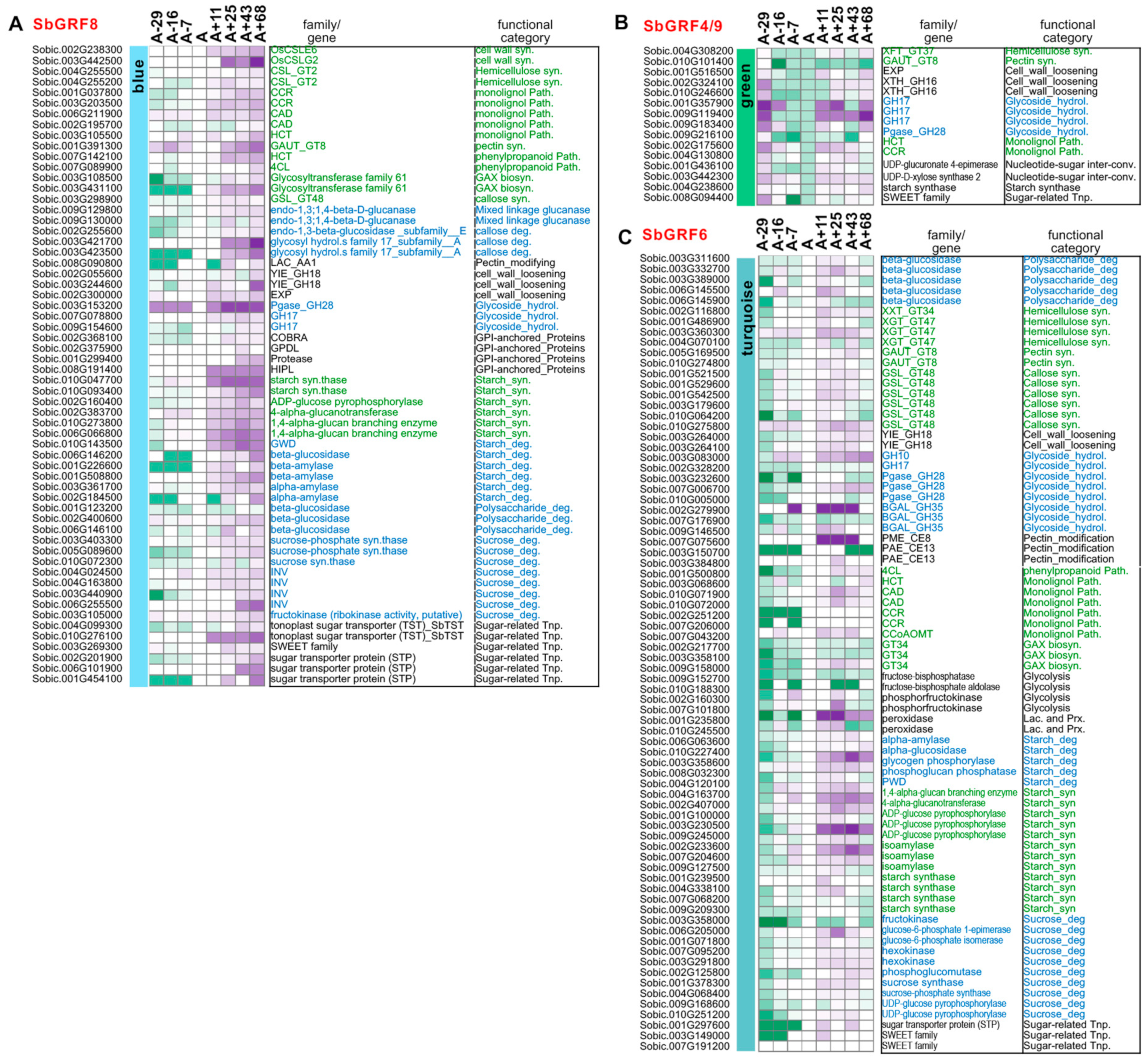
2.3. Analyses of SbGRF Expression Patterns in Sorghum Internodes Identifies Their Roles in Internode Elongation and Metabolism
3. Materials and Methods
3.1. Genome-Wide Identification and Sequence Analysis of Sorghum GRFs
3.2. Phylogenetic Analysis Sorghum GRFs
3.3. Gene Expression Analysis of SbGRFs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, J.H. Biological roles and an evolutionary sketch of the GRF-GIF transcriptional complex in plants. BMB. Rep. 2019, 52, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Choi, D.; Kende, H. The AtGRF family of putative transcription factors is involved in leaf and cotyledon growth in Arabidopsis. Plant. J. 2003, 36, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hua, W.; Yang, H.L.; Zhan, G.M.; Li, R.J.; Deng, L.B.; Wang, X.F.; Liu, G.H.; Wang, H.Z. The BnGRF2 gene (GRF2- like gene from Brassica napus) enhances seed oil production through regulating cell number and plant photosynthesis. J. Exp. Bot. 2012, 63, 3727–3740. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.; Bian, H.; Zha, Y.; Li, F.; Sun, Y.; Bai, B.; Chen, Z.; Wang, J.; Zhu, M.; Han, N. MiR396a-mediated basic helix-loop- helix transcription factor bHLH74 repression acts as a regulator for root growth in Arabidopsis seedlings. Plant Cell Physiol. 2014, 6, 1343–1353. [Google Scholar] [CrossRef]
- Pajoro, A.; Madrigal, P.; Muiño, J.M.; Matus, J.T.; Jin, J.; Mecchia, M.A.; Debernardi, J.M.; Palatnik, J.F.; Balazadeh, S.; Arif, M.; et al. Dynamics of chromatin accessibility and gene regulation by MADS-domain transcription factors in flower development. Genome Biol. 2014, 15, R41. [Google Scholar] [CrossRef]
- Chen, Y.; Dan, Z.; Gao, F.; Chen, P.; Fan, F.; Li, S. Rice growth-regulating factor7 modulates plant architecture through regulating GA and indole-3-acetic acid metabolism. Plant Physiol. 2020, 184, 393–406. [Google Scholar] [CrossRef]
- Liu, H.; Tian, X.; Li, Y.; Wu, C.; Zheng, C. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 2008, 14, 836–843. [Google Scholar] [CrossRef]
- Hewezi, T.; Baum, T.J. Complex feedback regulations govern the expression of miRNA396 and its GRF target genes. Plant Signaling. Behav. 2012, 7, 749–751. [Google Scholar] [CrossRef]
- Kim, J.S.; Mizoi, J.; Kidokoro, S.; Maruyama, K.; Nakajima, J.; Nakashima, K.; Mitsuda, N.; Takiguchi, Y.; Ohme-Takagi, M.; Kondou, Y. Arabidopsis growth-regulating factor7 functions as a transcriptional repressor of abscisic acid- and osmotic stress-responsive genes, including DREB2A. Plant Cell 2012, 24, 3393–3405. [Google Scholar] [CrossRef]
- Liu, J.; Rice, J.H.; Chen, N.; Baum, T.J.; Hewezi, T. Synchronization of developmental processes and defense signaling by growth regulating transcription factors. PloS ONE 2014, 9, e98477. [Google Scholar] [CrossRef]
- Sakuma, Y.; Maruyama, K.; Osakabe, Y.; Qin, F.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Functional analysis of an Arabidopsis transcription factor, DREB2A, involved in drought-responsive gene expression. Plant Cell 2016, 18, 1292–1309. [Google Scholar] [CrossRef]
- Kim, J.H.; Kende, H. A transcriptional coactivator, AtGIF1, is involved in regulating leaf growth and morphology in Arabidopsis. Proc. Natl. Acad. Sci. USA 2004, 101, 13374–13379. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Ko, J.H.; Lee, S.; Lee, Y.; Pak, J.-H.; Kim, J.H. The Arabidopsis grf-interacting factor gene family performs an overlapping function in determining organ size as well as multiple developmental properties. Plant Physiol. 2009, 151, 655–668. [Google Scholar] [CrossRef]
- Lee, B.H.; Wynn, A.N.; Franks, R.G.; Hwang, Y.; Lim, J.; Kim, J.H. The Arabidopsis thaliana grf-interacting factor gene family plays an essential role in control of male and female reproductive development. Dev. Biol. 2014, 386, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Omidbakhsfard, M.A.; Proost, S.; Fujikura, U.; Mueller-Roeber, B. Growth-regulating factors (GRFs): A small transcription factor family with important functions in plant biology. Mol. Plant 2015, 8, 998–1010. [Google Scholar] [CrossRef] [PubMed]
- Ferela, A.; Debernadi, J.M.; Rosatti, S.; Liebsch, D.; Schommer, C.; Palatnik, J.F. Interplay among ZF-HD and GRF transcription factors during Arabidopsis leaf development. Plant Physiol. 2023, 191, 1789–1802. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, B.H. Growth-regulating factor4 of Arabidopsis thaliana is required for development of leaves, cotyledons, and shoot apical meristem. J. Plant Biol. 2006, 49, 463–468. [Google Scholar] [CrossRef]
- Liu, D.; Song, Y.; Chen, Z.; Yu, D. Ectopic expression of miR396 suppresses GRF target gene expression and alters leaf growth in Arabidopsis. Physiol. Plant. 2009, 136, 223–236. [Google Scholar] [CrossRef]
- Liebsch, D.; Palatnik, J. MicroRNA miR396, GRF transcription factors and GIF co-regulators: A conserved plant growth regulatory module with potential for breeding and biotechnology. Curr. Opin. Plant Biol. 2020, 53, 31–42. [Google Scholar] [CrossRef]
- van der Knaap, E.; Kim, J.H.; Kende, H. A novel gibberellin-induced gene from rice and its potential regulatory role in stem growth. Plant Physiol. 2000, 122, 695–704. [Google Scholar] [CrossRef]
- Choi, D.; Kim, J.H.; Kende, H. Whole genome analysis of the OsGRF gene family encoding plant-specific putative transcription activators in Rice (Oryza sativa L.). Plant Cell Physiol. 2004, 45, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tian, Y.; Wu, K.; Ye, Y.; Yu, J.; Zhang, J.; Liu, Q.; Hu, M.; Li, H.; Tong, Y.; et al. Modulating plant growth-metabolism coordination for sustainable agriculture. Nature 2018, 560, 595–600. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, H.; Guo, S.; Wang, B.; Li, Z.; Chong, K.; Xu, Y. OsmiR396d affects gibberellin and brassinosteroid signaling to regulate plant architecture in rice. Plant Physiol. 2018, 176, 946–959. [Google Scholar] [CrossRef]
- Tsukaya, H. GRFs provide the key. Nat. Plant 2015, 2, 15210. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guo, P.; Wang, J.; Xu, Z. Growth-regulating factors: Conserved and divergent roles in plant growth and development and potential value for crop improvement. Plant J. 2023, 113, 1122–1145. [Google Scholar] [CrossRef]
- Lazzara, F.E.; Rodriguez, R.E.; Palatnik, J.F. Molecular mechanisms regulating GROWTH-REGULATING FACTORS activity in plant growth, development, and environmental responses. J. Exp. Bot. 2024, 75, 4360–4372. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cheng, M.; Wei, X.; Wang, R.; Fan, F.; Wang, Z.; Tian, Z.; Li, S.; Yuan, H. Comprehensive evolutionary analysis of growth-regulating factor gene family revealing the potential molecular basis under multiple hormonal stress in Gramineae crops. Front. Plant Sci. 2023, 14, 1174955. [Google Scholar] [CrossRef]
- Qin, L.; Chen, H.; Wu, Q.; Wang, X. Identification and exploration of the GRF and GIF families in maize and foxtail millet. Physiol. Mol. Biol. Plants 2022, 28, 1717–1735. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, X.; Wang, J.; Niu, J.; Du, R.; Ji, G.; Zhu, L.; Zhang, J.; Lv, P.; Cao, J. Systematical characterization of GRF gene family in sorghum, and their potential functions in aphid resistance. Gene 2022, 836, 146669. [Google Scholar] [CrossRef]
- Li, S.; Gao, F.; Xie, K.; Zeng, X.; Cao, Y.; Zeng, J.; He, Z.; Ren, Y.; Li, W.; Deng, Q.; et al. The OsmiR396c-OsGRF4-OsGIF1 regulatory module determines grain size and yield in rice. Plant Biotechnol. J. 2016, 14, 2134–2146. [Google Scholar] [CrossRef]
- Liu, H.; Guo, S.; Xu, Y.; Li, C.; Zhang, Z.; Zhang, D.; Xu, S.; Zhang, C.; Chong, K. OsmiR396d-Regulated OsGRFs Function in Floral Organogenesis in Rice through Binding to Their Targets OsJMJ706 and OsCR4. Plant Physiol. 2014, 165, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Cheng, M.; Wang, R.; Wang, Z.; Fan, F.; Wang, W.; Si, F.; Gao, F.; Li, S. miR396b/GRF6 module contributes to salt tolerance in rice. Plant Biotechnol. J. 2024, 22, 2079–2092. [Google Scholar] [CrossRef]
- Bowers, J.E.; Chapman, B.A.; Rong, J.; Paterson, A.H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 2003, 422, 433–438. [Google Scholar] [CrossRef]
- Paterson, A.H.; Bowers, J.E.; Chapman, B.A. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. USA 2004, 101, 9903–9908. [Google Scholar] [CrossRef]
- Wu, Y.; Feng, J.; Zhang, Q.; Wang, Y.; Guan, Y.; Wang, R.; Shi, F.; Zeng, F.; Wang, Y.; Chen, M.; et al. Integrative gene duplication and genome-wide analysis as an approach to facilitate Wheat reverse genetics: An example in the TaCIPK family. J. Adv. Res. 2024, 61, 19–33. [Google Scholar] [CrossRef]
- Calvino, M.; Messing, J. Sweet sorghum as a model system for bioenergy crops. Curr. Opin. Biotechnol. 2012, 23, 323–329. [Google Scholar] [CrossRef]
- McKinley, B.; Rooney, W.; Wilkerson, C.; Mullet, J. Dynamics of biomass partitioning, stem gene expression, cell wall biosynthesis, and sucrose accumulation during development of Sorghum bicolor. Plant J. 2016, 88, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, W.; Feng, Y.; Tu, M.; Wittich, P.E.; Bate, N.J.; Messing, J. Transcriptome and metabolome reveal distinct carbon allocation patterns during internode sugar accumulation in different Sorghum genotypes. Plant Biotechnol. J. 2019, 17, 472–487. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tu, M.; Feng, Y.; Wang, W.; Messing, J. Common metabolic networks contribute to carbon sink strength of Sorghum internodes: Implications for bioenergy improvement. Biotechnol. Biofuels 2019, 12, 274. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Tang, H.; Zhang, Q.; Hua, X.; Ma, X.; Zhu, F.; Jones, T.; Zhu, X.; Bowers, J.; et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef]
- Wang, B.; Hou, M.; Shi, J.; Ku, L.; Song, W.; Li, C.; Ning, Q.; Li, X.; Li, C.; Zhao, B.; et al. De novo genome assembly and analyses of 12 founder inbred lines provide insights into maize heterosis. Nat. Genet. 2023, 55, 312–323. [Google Scholar] [CrossRef]
- Yang, L.; Zhou, Q.; Sheng, X.; Chen, X.; Hua, Y.; Lin, S.; Luo, Q.; Yu, B.; Shao, T.; Wu, Y.; et al. Harnessing the genetic basis of sorghum biomass-related traits to facilitate bioenergy applications. Int. J. Mol. Sci. 2023, 24, 14549. [Google Scholar] [CrossRef]
- Wang, T.; Wang, B.; Hua, X.; Tang, H.; Zhang, Z.; Gao, R.; Qi, Y.; Zhang, Q.; Wang, G.; Yu, Z.; et al. A complete gap-free diploid genome in Saccharum complex and the genomic footprints of evolution in the highly polyploid Saccharum genus. Nat. Plants 2023, 9, 554–571. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zeng, J.; Du, C.; Tang, Q.; Hua, Y.; Chen, M.; Yang, G.; Tu, M.; He, G.; Li, Y.; et al. Divergent roles of the auxin response factors in lemongrass (Cymbopogon flexuosus (Nees ex Steud.) W. Watson) during plant growth. Int. J. Mol. Sci. 2024, 25, 8154. [Google Scholar] [CrossRef]
- Mizuno, H.; Kasuga, S.; Kawahigashi, H. The sorghum SWEET gene family: Stem sucrose accumulation as revealed through transcriptome profiling. Biotechnol. Biofuels 2016, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- McKinley, B.; Casto, A.; Rooney, W.; Mullet, J. Developmental dynamics of stem starch accumulation in Sorghum Bicolor. Plant Direct 2018, 2, e00074. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.M.; Joel, O.; McKinley, B.; Weers, B.; Fabich, H.T.; Evetts, N.; Cornadi, M.S.; Altobelli, S.A.; Marshall-Colon, A.; Mullet, J. Bioenergy sorghum stem growth regulation: Intercalary meristem localization, development, and gene regulatory network analysis. Plant J. 2022, 112, 476–492. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ngu, D.W.; Carvalho, D.; Liang, Z.; Qiu, Y.; Roston, R.L.; Schnable, J.C. Differentially regulated orthologs in sorghum and the subgenomes of maize. Plant Cell 2017, 29, 1938–1951. [Google Scholar] [CrossRef]
- RaventoÂs, D.; Skriver, K.; Schlein, M.; Karnahl, K.; Rogers, S.W.; Rogers, J.C.; Mundy, J. HRT, a novel zinc finger, transcriptional repressor from barley. J. Biol. Chem. 1998, 273, 23313–23320. [Google Scholar] [CrossRef]
- Sood, R.; Zehnder, J.L.; Druzin, M.L.; Brown, P.O. Gene expression patterns in human placenta. Proc. Natl. Acad. Sci. USA 2006, 103, 5478–5483. [Google Scholar] [CrossRef]
- McCormick, R.F.; Truong, F.K.; Sreedasyam, A.; Jenkins, J.; Shu, S.; Sims, D.; Kennedy, M.; Amirebrahimi, M.; Weers, B.D.; McKinley, B.; et al. The Sorghum Bicolor reference genome: Improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J. 2018, 93, 338–354. [Google Scholar] [CrossRef] [PubMed]
- Sang, J.; Zou, D.; Wang, Z.; Wang, F.; Zhang, Y.; Xia, L.; Li, Z.; Ma, L.; Li, M.; Xu, B. IC4R-2.0: Rice genome reannotation using massive RNA-seq data. Genom. Proteom. Bioinf. 2020, 18, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.M.; Thu, S.W.; Balasubramanian, V.K.; Cobos, C.J.; Disasa, T.; Mendu, V. Identification, characterization, and expression analysis of cell wall related genes in Sorghum bicolor (L.) moench, a food, fodder, and biofuel crop. Front. Plant Sci. 2016, 7, 1287. [Google Scholar] [CrossRef]
- Li, M.; Xiong, G.; Li, R.; Cui, J.; Tang, D.; Zhang, B.; Pauly, M.; Cheng, Z.; Zhou, Y. Rice cellulose synthase-like D4 is essential for normal cell-wall biosynthesis and plant growth. Plant J. 2009, 60, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Liepman, A.H.; Cavalier, D.M. The cellulose synthase-like a and cellulose synthase-like C families: Recent advances and future perspectives. Front. Plant Sci. 2012, 3, 109. [Google Scholar] [CrossRef] [PubMed]
- Verhertbruggen, Y.; Yin, L.; Oikawa, A.; Scheller, H.V. Mannan synthase activity in the CSLD family. Plant Signal. Behav. 2011, 6, 1620–1623. [Google Scholar] [CrossRef]
- Nishitani, K.; Vissenberg, K. Roles of the XTH protein family in the expanding cell. In The Expanding Cell. Plant Cell Monographs; Verbelen, J.-P., Vissenberg, K., Eds.; Springer: Berlin, Heidelberg, 2007; pp. 89–116. [Google Scholar] [CrossRef]
- Park, Y.B.; Cosgrove, D.J. Xyloglucan and its interactions with other components of the growing cell wall. Plant Cell Physiol. 2015, 56, 180–194. [Google Scholar] [CrossRef]
- MacMillan, C.P.; Mansfield, S.D.; Stachurski, Z.H.; Rvans, R.; Southerton, S.G. Fasciclin-like arabinogalactan proteins: Specialization for stem biomechanics and cell wall architecture in Arabidopsis and Eucalyptus. Plant J. 2010, 62, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jin, Y.; Wang, C.; Bei, L.; Chunmei, J.; Zhencang, S.; Zhiping, Z.; Fanjing, K.; Honxia, Z. Fasciclin-like arabinogalactan proteins, PtFLAs, play important roles in GA-mediated tension wood formation in populus. Sci. Rep. 2017, 7, 6182. [Google Scholar] [CrossRef]
- Ma, Y.; Shafee, T.; Mudiyanselage, A.M.; Ratcliffe, J.; MacMillan, C.P.; Mansfield, S.D.; Bacic, A.; Johnson, K.L. Distinct functions of fasciclin-like arabinogalactan proteins relate to domain structure. Plant Physiol. 2023, 192, 119–132. [Google Scholar] [CrossRef]
- Hatfield, R.D.; Wilson, J.R.; Mertens, D.R. Composition of cell walls isolated from cell types of grain sorghum stems. J. Sci. Food. Agr. 1999, 79, 891–899. [Google Scholar] [CrossRef]
- Penning, B.W.; Hunter, C.T., 3rd; Tayengwa, R.; Eveland, A.L.; Dugard, C.K.; Olek, A.T.; Vermerris, W.; Koch, K.E.; McCarty, D.R.; Davis, M.F.; et al. Genetic resources for maize cell wall biology. Plant Physiol. 2009, 151, 1703–1728. [Google Scholar] [CrossRef]
- Carpita, N.C. Structure and biogenesis of the cell walls of grasses. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 445–476. [Google Scholar] [CrossRef]
- Mitchell, R.A.; Dupree, P.; Shewry, P.R. A novel bioinformatics approach identifies candidate genes for the synthesis and feruloylation of arabinoxylan. Plant Physiol. 2007, 144, 43–53. [Google Scholar] [CrossRef]
- Jun, S.Y.; Sattler, S.A.; Cortez, G.S.; Vermerris, W.; Sattler, S.E.; Kang, C. Biochemi-cal and structural analysis of substrate specificity of a phenylalanine ammonia-lyase. Plant Physiol. 2018, 176, 1452–1468. [Google Scholar] [CrossRef]
- Chen, X.Y.; Kim, J.Y. Callose synthesis in higher plants. Plant Signal. Behav. 2009, 4, 489–492. [Google Scholar] [CrossRef]
- Vega-Sanchez, M.E.; Verhertbruggen, Y.; Christensen, U.; Chen, X.; Sharma, V.; Varanasi, P.; Jobling, S.A.; Talbot, M.; White, R.G.; Joo, M. Loss of cellulose synthase-like f6 function affects mixed-linkage glucan deposition, cell wall mechanical properties, and defense responses in vegetative tissues of rice. Plant Physiol. 2012, 159, 56–69. [Google Scholar] [CrossRef]
- Vega-Sanchez, M.E.; Verhertbruggen, Y.; Scheller, H.V.; Ronald, P.C. Abundance of mixed linkage glucan in mature tissues and secondary cell walls of grasses. Plant Signal. Behav. 2013, 8, e23143. [Google Scholar] [CrossRef]
- Burton, R.A.; Fincher, G.B. Current challenges in cell wall biology in the cereals and grasses. Front. Plant Sci. 2012, 3, 130. [Google Scholar] [CrossRef]
- Ermawar, R.A.; Collins, H.M.; Byrt, C.S.; Betts, N.S.; Henderson, M.; Shirley, N.J.; Schwerdt, J.; Lahnstein, J.; Fincher, G.B.; Burton, R.A. Distribution, structure and biosynthetic gene families of (1,3;1,4)-beta-glucan in Sorghum bicolor. J. Integr. Plant Biol. 2015, 57, 429–445. [Google Scholar] [CrossRef]
- Purashotham, P.; Ho, R.; Yu, L.; Fincher, G.B.; Bulone, V.; Zimmer, J. Mechanism of mixed-linkage glucan biosynthesis by barley cellulose synthase–like CslF6 (1,3;1,4)-β-glucan synthase. Sci. Adv. 2022, 8, eadd1596. [Google Scholar] [CrossRef]
- Gupta, C.; Pereira, A. Recent advances in gene function prediction using context-specific coexpression networks in plants. F100 Res. 2019, 8, 153. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Jiang, Y.; Chen, T.; Li, H.; Fu, M.; Wang, Y.; Xu, Y.; Li, Y.; Zhou, Z.; Jia, L.; et al. New Data and New Features Open Access of the FunRiceGenes (Functionally Characterized Rice Genes) Database: 2021 Update. Rice 2022, 15, 23. [Google Scholar] [CrossRef]
- Dadras, A.; Furst-Janson, J.M.R.; Darienko, T.; Krone, D.; Scholz, P.; Sun, S.; Herrfurth, C.; Rieseberg, T.P.; Irisarri, I.; Steinkamp, R.; et al. Environmental gradients reveal stress hubs pre-dating plant terrestrialization. Nat. Plants 2023, 9, 1419–1438. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Xie, X.; Zhang, Y.; Yu, X.; He, G.; Li, Y.; Yang, G. Evolution of the DEHYDRATION-RESPONSIVE ELEMENT-BINDING PROTEIN subfamily in green plants. Plant Physiol. 2022, 190, 421–440. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. Tbtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE v5 enables improved estimates of phylogenetic tree confidence by ensemble bootstrapping. bioRxiv 2021, 06.20.449169. [Google Scholar] [CrossRef]
- Madeira, F.; Madhusoodanan, N.; Lee, J.; Eusebi, A.; Niewielska, A.; Tivey, A.R.N.; Lopez, R.; Butcher, S. The EMBL-EBI job dispatcher sequence analysis tools framework in 2024. Nucleic Acids Res. 2024, 52, W521–W525. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Salse, J.; Bolot, S.; Throude, M.; Jouffe, V.; Piegu, B.; Quraishi, U.M.; Calcagno, T.; Cooke, R.; Delseny, M.; Feuillet, C. Identification and characterization of shared duplications between Rice and Wheat provide new insight into grass genome evolution. Plant Cell 2008, 20, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mehta, S.; Messing, J. A new high-throughput assay for determining soluble sugar in Sorghum internode-extracted juice. Planta 2018, 248, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinf. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. Fast R functions for robust correlations and hierarchical clustering. J. Stat. Softw. 2012, 46, i11. Available online: http://www.ncbi.nlm.nih.gov/pmc/articles/pmc3465711/ (accessed on 19 August 2024). [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 1, 100141. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, M.; Li, Z.; Zhu, Y.; Wang, P.; Jia, H.; Wang, G.; Zhou, Q.; Hua, Y.; Yang, L.; Xiao, J.; et al. Potential Roles of the GRF Transcription Factors in Sorghum Internodes during Post-Reproductive Stages. Plants 2024, 13, 2352. https://doi.org/10.3390/plants13172352
Tu M, Li Z, Zhu Y, Wang P, Jia H, Wang G, Zhou Q, Hua Y, Yang L, Xiao J, et al. Potential Roles of the GRF Transcription Factors in Sorghum Internodes during Post-Reproductive Stages. Plants. 2024; 13(17):2352. https://doi.org/10.3390/plants13172352
Chicago/Turabian StyleTu, Min, Zhuang Li, Yuanlin Zhu, Peng Wang, Hongbin Jia, Guoli Wang, Qin Zhou, Yuqing Hua, Lin Yang, Jiangrong Xiao, and et al. 2024. "Potential Roles of the GRF Transcription Factors in Sorghum Internodes during Post-Reproductive Stages" Plants 13, no. 17: 2352. https://doi.org/10.3390/plants13172352