
Multi-Omics Analyses Uncover the Mechanism Underlying Polyploidization-Enhanced Steviol Glycosides Biosynthesis in Stevia rebaudiana

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
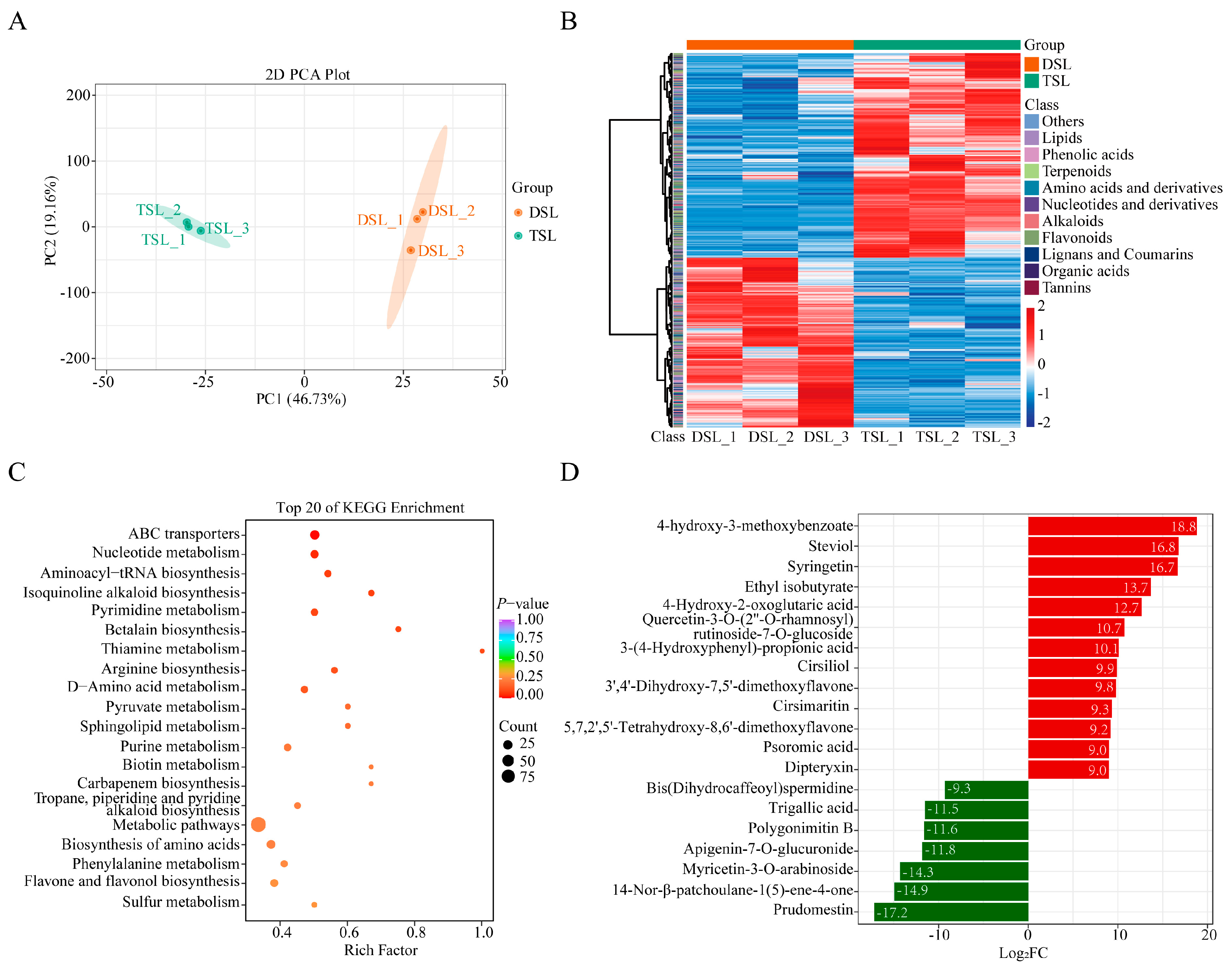
2.1. Differentially Accumulated Metabolites (DAMs) in Stevia Post-Polyploidization
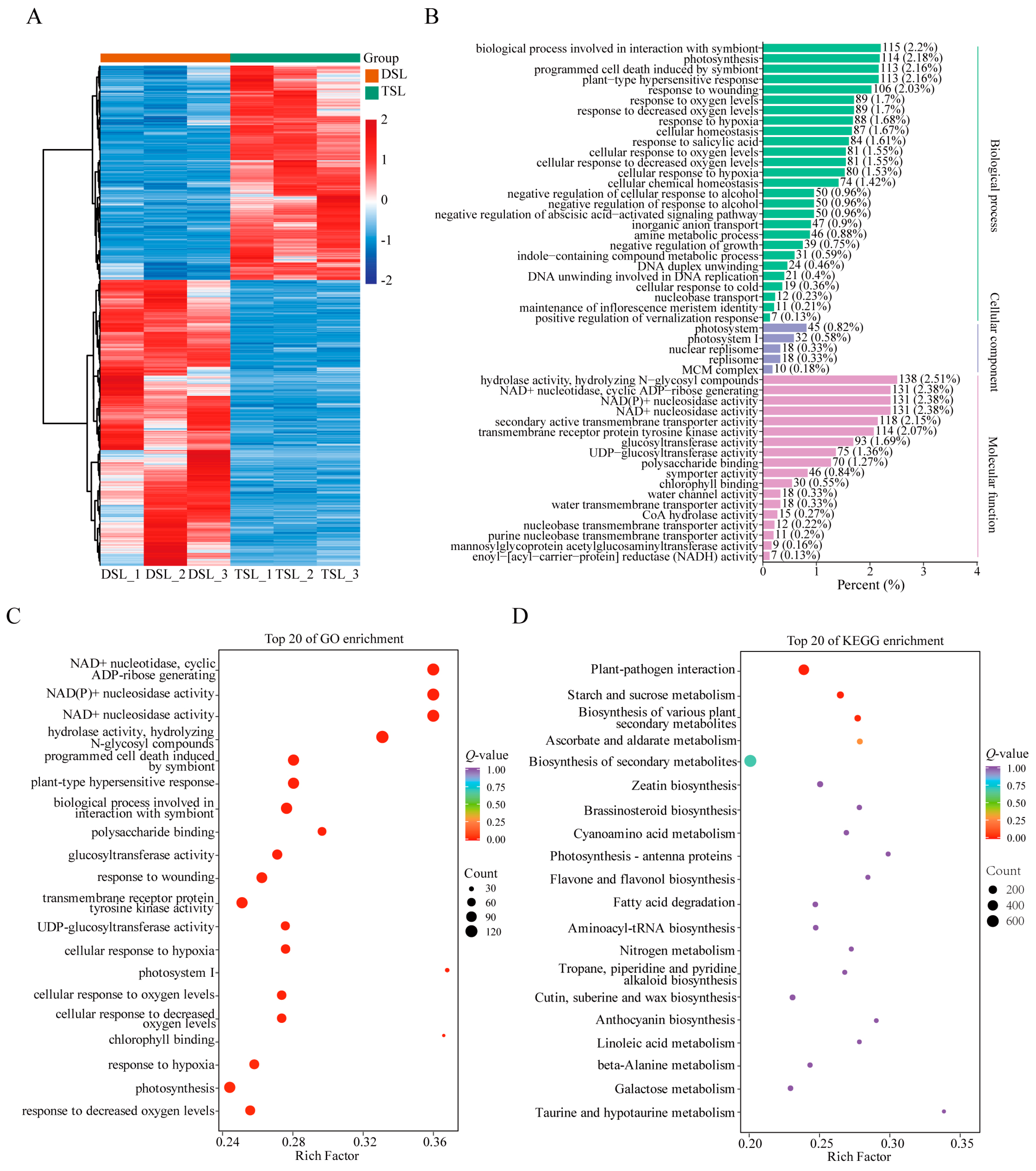
2.2. Differentially Expressed Genes (DEGs) in Stevia Post-Polyploidization
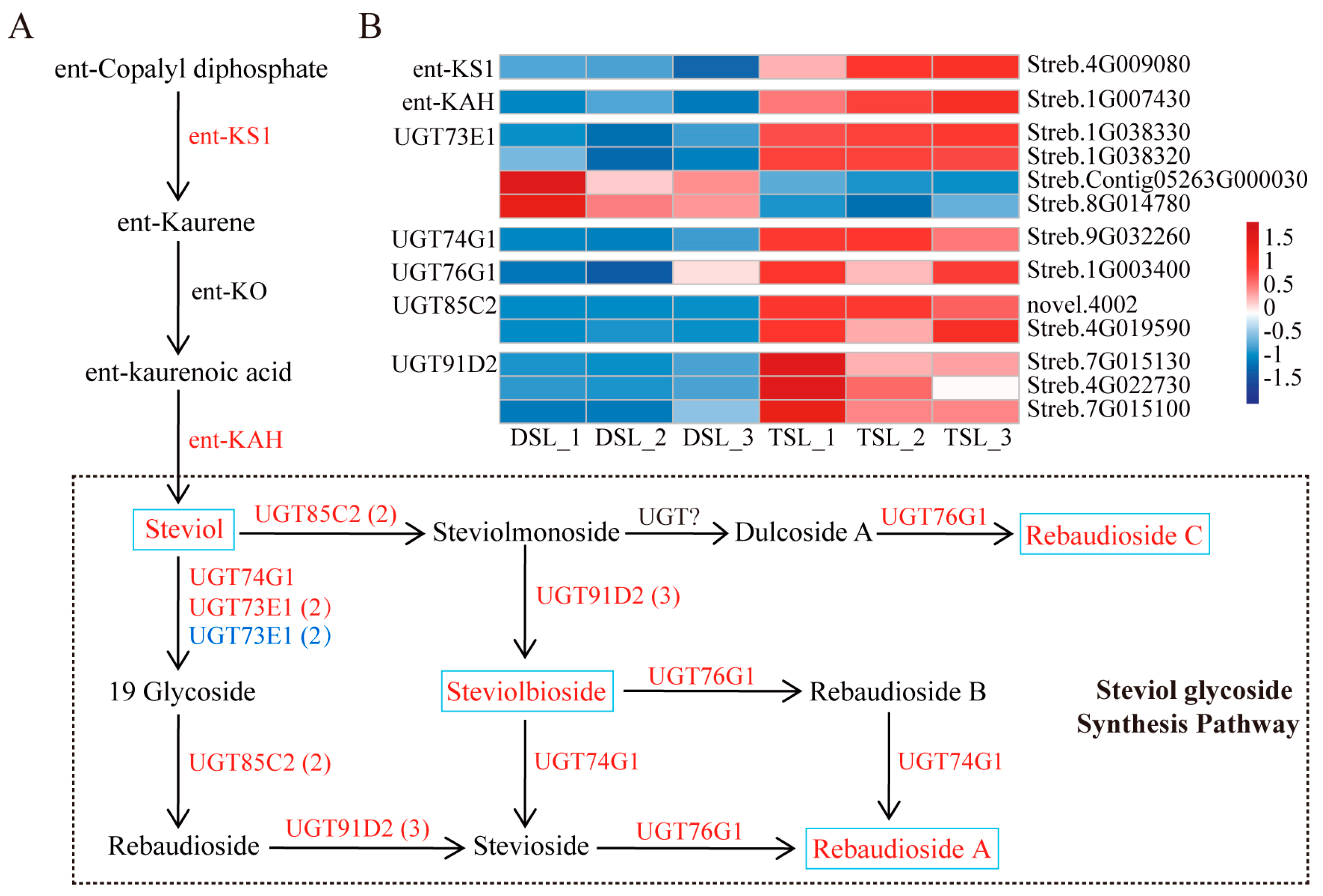
2.3. Integrative Transcriptome and Metabolome Analysis Revealed the Enhanced SG Biosynthesis in Stevia Post-Polyploidization
2.4. Taxonomic Features of the Rhizosphere Microbes of Diploid and Autotetraploid Stevia
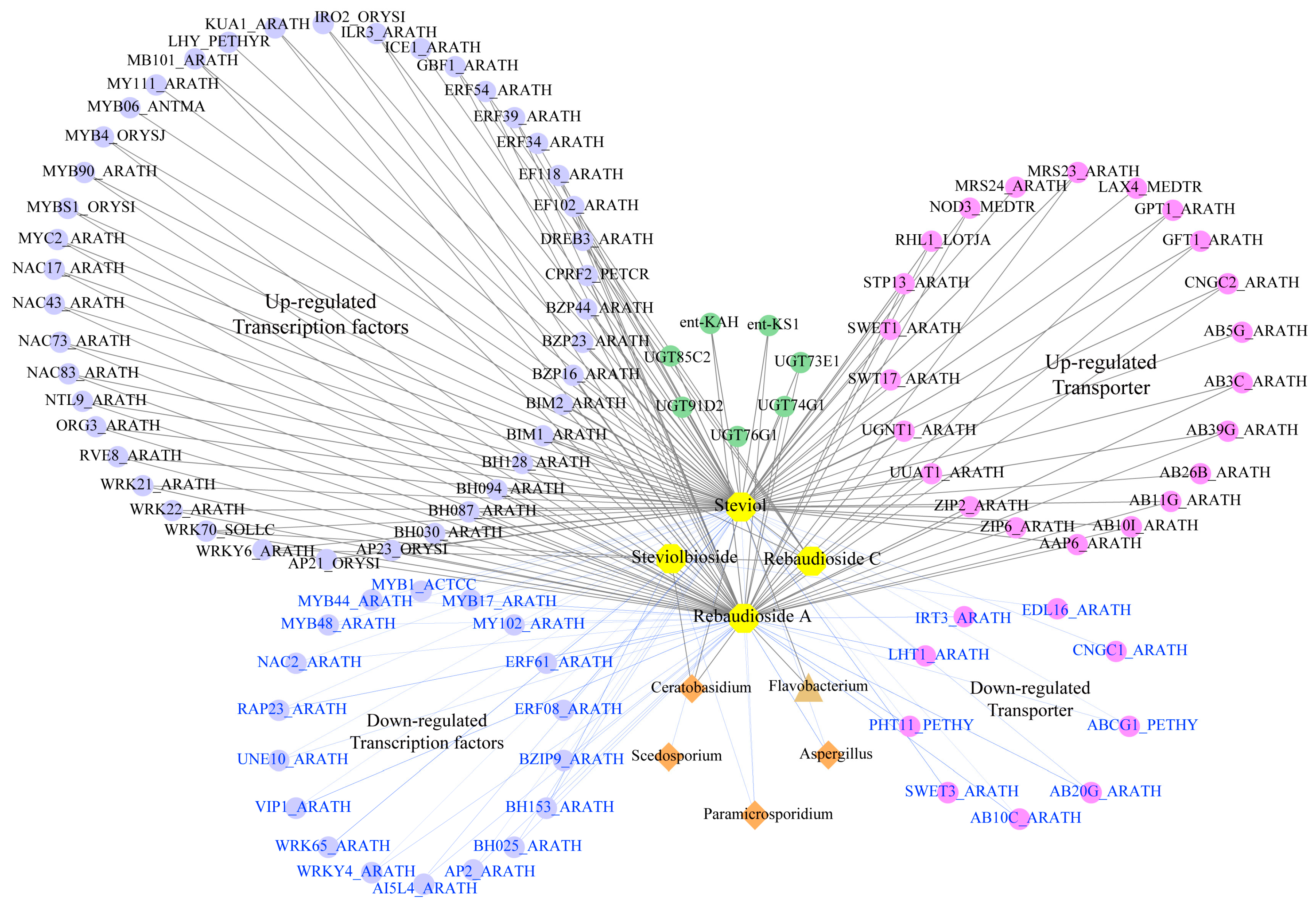
2.5. Multi-Omics Network Analysis of the Microbe–Gene–Metabolite Associations
3. Discussion
3.1. Steviol Glycosides Biosynthesis Mechanism Elucidated through Integrated Transcriptome and Metabolome Analysis
3.2. Optimized Rhizosphere Microbiome Contributes to SG Biosynthesis in Autotetraploid Stevia
4. Materials and Methods
4.1. Plant Materials
4.2. Total RNA Extraction, Library Preparation, and Transcriptome Sequencing
4.3. Transcriptome Assembly, Differential Expression, and Functional Enrichment Analysis
4.4. qRT-PCR Analysis
4.5. Metabolite Measurements and Analysis
4.6. Rhizosphere Microbial DNA Extraction and Amplicon Sequencing
4.7. Microbial Data Analysis
4.8. Multi-Omics Integrative Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yadav, S.K.; Guleria, P. Steviol glycosides from stevia:biosynthesis pathway review and their application in foods and medicine. Crit. Rev. Food Sci. Nutr. 2012, 52, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Ali, H.; Khan, H.; Begam, A.; Khan, S.; Ali, S.S.; Ahmad, N.; Fazal, H.; Ali, M.; Hano, C.; et al. Effect of gibberellic acid on production of biomass, polyphenolics and steviol glycosides in adventitious root cultures of Stevia rebaudiana (Bert.). Plants 2020, 9, 420. [Google Scholar] [CrossRef] [PubMed]
- Lemus-Mondaca, R.; Vega-Gálvez, A.; Zura-Bravo, L.; Ah-Hen, K. Stevia rebaudiana Bertoni, source of a high-potency natural sweetener: A comprehensive review on the biochemical, nutritional and functional aspects. Food Chem. 2012, 132, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A. Potential medicinal effects and applications of stevia constituents. Phytochem. Rev. 2022, 21, 161–178. [Google Scholar] [CrossRef]
- Tavarini, S.; Passera, B.; Martini, A.; Avio, L.; Sbrana, C.; Giovannetti, M.; Angelini, L.G. Plant growth, steviol glycosides and nutrient uptake as affected by arbuscular mycorrhizal fungi and phosphorous fertilization in Stevia rebaudiana Bert. Ind. Crops Prod. 2018, 111, 899–907. [Google Scholar] [CrossRef]
- Li, Y.P.; Qiu, Y.; Xu, X.; Luo, M. Genome-wide identification of SrbHLH transcription factors highlights its potential role in rebaudioside A (RA) biosynthesis in Stevia rebaudiana. BMC Plant Biol. 2023, 23, 352. [Google Scholar] [CrossRef]
- Libik-Konieczny, M.; Capecka, E.; Tuleja, M.; Konieczny, R. Synthesis and production of steviol glycosides: Recent research trends and perspectives. Appl. Microbiol. Biotechnol. 2021, 105, 3883–3900. [Google Scholar] [CrossRef]
- Sun, Y.M.; Guo, J.J.; Ruan, Y.; Zhang, T.; Fernie, A.R.; Yuan, H.Y. The recruitment of specific rhizospheric bacteria facilitates Stevia rebaudiana salvation under nitrogen and/or water deficit stresses. Ind. Crops Prod. 2022, 187, 115434. [Google Scholar] [CrossRef]
- Andrade, M.; Lucho, S.R.; Amaral, M.N.D.; Braga, E.J.B.; Ribeiro, P.R.; Castro, R.D.D. Long-day photoperiodic requirements for steviol glycosides and gibberellins biosynthesis and bio-sweetener levels optimization in Stevia rebaudiana Bertoni. Ind. Crops Prod. 2023, 204, 117363. [Google Scholar] [CrossRef]
- Xu, X.Y.; Yuan, H.Y.; Yu, X.Q.; Huang, S.Z.; Sun, Y.M.; Zhang, T.; Liu, Q.Q.; Tong, H.Y.; Zhang, Y.X.; Wang, Y.J.; et al. The chromosome-level Stevia genome provides insights into steviol glycoside biosynthesis. Hort. Res. 2021, 8, 129. [Google Scholar] [CrossRef]
- Nasrullah, N.; Ahmad, J.; Saifi, M.; Shah, I.G.; Nissar, U.; Quadri, S.N.; Ashrafi, K.; Abdin, M.Z. Enhancement of diterpenoid steviol glycosides by co-overexpressing SrKO and SrUGT76G1 genes in Stevia rebaudiana Bertoni. PLoS ONE 2023, 18, e0260085. [Google Scholar] [CrossRef] [PubMed]
- Almario, J.; Kyselková, M.; Kopecky, J.; Ságová-Marecková, M.; Muller, D.; Grundmann, G.L.; Moënne-Loccoz, Y. Assessment of the relationship between geologic origin of soil, rhizobacterial community composition and soil receptivity to tobacco black root rot in Savoie region (France). Plant Soil 2013, 371, 397–408. [Google Scholar] [CrossRef]
- Zhao, Y.S.; Sun, C.Y.; Wang, S.Z.; Zhang, M.L.; Li, Y.L.; Xue, Q.H.; Guo, Q.; Lai, H.X. Widely targeted metabolomic, transcriptomic, and metagenomic profiling reveal microbe-plant-metabolic reprogramming patterns mediated by Streptomyces pactum Act12 enhance the fruit quality of Capsicum annuum L. Food Res. Int. 2023, 166, 112587. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Langridge, H.; Straathof, A.L.; Muhamadali, H.; Hollywood, K.A.; Goodacre, R.; de Vries, F.T. Root functional traits explain root exudation rate and composition across a range of grassland species. J. Ecol. 2022, 110, 21–33. [Google Scholar] [CrossRef]
- Jain, A.; Chakraborty, J.; Das, S. Underlying mechanism of plant-microbe crosstalk in shaping microbial ecology of the rhizosphere. Acta Physiol. Plant. 2020, 42, 8. [Google Scholar] [CrossRef]
- Vafadar, F.; Amooaghaie, R.; Otroshy, M. Effects of plant-growth-promoting rhizobacteria and arbuscular mycorrhizal fungus on plant growth, stevioside, NPK, and chlorophyll content of Stevia rebaudiana. J. Plant Interact. 2014, 9, 128–136. [Google Scholar] [CrossRef]
- Mamta; Rahi, P.; Pathania, V.; Gulati, A.; Singh, B.; Bhanwra, R.K.; Tewari, R. Stimulatory effect of phosphate-solubilizing bacteria on plant growth, stevioside and rebaudioside-A contents of Stevia rebaudiana Bertoni. Appl. Soil Ecol. 2010, 46, 222–229. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; van Themaat, E.V.L.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Buckler, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef]
- Zhong, C.F.; Chen, C.Y.; Gao, X.; Tan, C.Y.; Bai, H.; Ning, K. Multi-omics profiling reveals comprehensive microbe-plant-metabolite regulation patterns for medicinal plant Glycyrrhiza uralensis Fisch. Plant Biotechnol. J. 2022, 20, 1874–1887. [Google Scholar] [CrossRef]
- Saravi, H.B.; Gholami, A.; Pirdashti, H.; Firouzabadi, M.B.; Asghari, H.; Yaghoubian, Y. Improvement of salt tolerance in Stevia rebaudiana by co-application of endophytic fungi and exogenous spermidine. Ind. Crops Prod. 2022, 177, 114443. [Google Scholar] [CrossRef]
- Zhang, H.; He, Q.; Xing, L.; Wang, R.; Wang, Y.; Liu, Y.; Zhou, Q.; Li, X.; Jia, Z.; Liu, Z.; et al. The haplotype-resolved genome assembly of autotetraploid rhubarb Rheum officinale provides insights into its genome evolution and massive accumulation of anthraquinones. Plant Commun. 2023, 5, 100677. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Zhu, Y.X. Plant polyploidy and evolution. J. Integr. Plant Biol. 2019, 61, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.X.; Li, Y.Q.; Bai, M.; Deng, Y.; Liang, G.X.; Wu, H. A comparative study of the dynamic accumulation of polyphenol components and the changes in their antioxidant activities in diploid and tetraploid Lonicera japonica. Plant Physiol. Biochem. 2017, 112, 87–96. [Google Scholar] [CrossRef]
- Li, M.J.; Xu, G.Y.; Xia, X.J.; Wang, M.L.; Yin, X.M.; Zhang, B.; Zhang, X.; Cui, Y.C. Deciphering the physiological and molecular mechanisms for copper tolerance in autotetraploid Arabidopsis. Plant Cell Rep. 2017, 36, 1585–1597. [Google Scholar] [CrossRef]
- Park, C.H.; Park, Y.E.; Yeo, H.J.; Yoon, J.S.; Park, S.Y.; Kim, J.K.; Park, S.U. Comparative analysis of secondary metabolites and metabolic profiling between diploid and tetraploid Morus alba L. J. Agric. Food Chem. 2021, 69, 1300–1307. [Google Scholar] [CrossRef]
- Bonnin, M.; Favreau, B.; Soriano, A.; Leonhardt, N.; Oustric, J.; Lourkisti, R.; Ollitrault, P.; Morillon, R.; Berti, L.; Santini, J. Insight into physiological and biochemical determinants of salt stress tolerance in tetraploid Citrus. Antioxidants 2023, 12, 640. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Tan, M.P.; Zhang, Y.Y.; Jia, Y.; Zhu, S.X.; Wang, J.; Zhao, J.J.; Liao, Y.Y.; Xiang, Z.X. Integrative analyses of targeted metabolome and transcriptome of Isatidis Radix autotetraploids highlighted key polyploidization-responsive regulators. BMC Genom. 2021, 22, 670. [Google Scholar] [CrossRef]
- Yoo, M.J.; Koh, J.; Boatwright, J.L.; Soltis, D.E.; Soltis, P.S.; Barbazuk, W.B.; Chen, S.X. Investigation of regulatory divergence between homoeologs in the recently formed allopolyploids, Tragopogon mirus and T. miscellus (Asteraceae). Plant J. 2023, 117, 1191–1205. [Google Scholar] [CrossRef]
- Xu, T.T.; Liu, Z.; Zhan, D.J.; Pang, Z.W.; Zhang, S.W.; Li, C.H.; Kang, X.Y.; Yang, J. Integrated transcriptomic and metabolomic analysis reveals the effects of polyploidization on the lignin content and metabolic pathway in Eucalyptus. Biotechnol. Biofuels Bioprod. 2023, 16, 117. [Google Scholar] [CrossRef]
- Ahmad, M.A.; Chaudhary, S.; Deng, X.; Cheema, M.; Javed, R. Nano-stevia interaction: Past, present, and future. Plant Physiol. Biochem. 2023, 201, 107807. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Hernandez, C.; Feregrino-Perez, A.A.; Perez-Ramirez, I.; Ocampo-Velazquez, R.V.; Rico-García, E.; Torres-Pacheco, I.; Guevara-Gonzalez, R.G. Controlled elicitation increases steviol glycosides (SGs) content and gene expression-associated to biosynthesis of SGs in Stevia rebaudiana B. cv. Morita II. Ind. Crops Prod. 2019, 139, 111479. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, P.Y.; Wang, W.X.; Jia, X.C.; Zhu, L.P.; Yin, H. Oligosaccharides increased both leaf biomass and steviol glycosides content of Stevia rebaudiana. Plant Physiol. Biochem. 2023, 202, 107937. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, G.; Yang, F.; Liang, Y.; Gao, Q.; Xiang, C.; Li, X.; Yang, R.; Zhang, G.; Jiang, H.; et al. Multilayered regulation of secondary metabolism in medicinal plants. Mol. Hortic. 2023, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Zha, W.J.; Li, W.; Wang, J.Y.; You, A.Q. Advances in the biosynthesis of terpenoids and their ecological functions in plant resistance. Int. J. Mol. Sci. 2023, 24, 11561. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, L.; Liu, X.; Xu, S.; Sun, X.; Zhang, Z.; Guo, H.; Shao, Q. The improvement of kinsenoside in wild-imitated cultivation Anoectochilus roxburghii associated with endophytic community. Ind. Crops Prod. 2023, 208, 117896. [Google Scholar] [CrossRef]
- Semwal, P.; Misra, S.; Misra, A.; Kar, S.; Majhi, B.; Mishra, S.K.; Srivastava, S.; Chauhan, P.S. Endophytic Bacillus strains enhance biomass and bioactive metabolites of Gloriosa superba. Ind. Crops Prod. 2023, 204, 117296. [Google Scholar] [CrossRef]
- Elsayed, A.; Abdelsattar, A.M.; Heikal, Y.M.; El-Esawi, M.A. Synergistic effects of Azospirillum brasilense and Bacillus cereus on plant growth, biochemical attributes and molecular genetic regulation of steviol glycosides biosynthetic genes in Stevia rebaudiana. Plant Physiol. Biochem. 2022, 189, 24–34. [Google Scholar] [CrossRef]
- Fox, D.T.; Soltis, D.E.; Soltis, P.S.; Ashman, T.L.; Van de Peer, Y. Polyploidy: A biological force from cells to ecosystems. Trends Cell Biol. 2020, 30, 688–694. [Google Scholar] [CrossRef]
- Anneberg, T.J.; Turcotte, M.M.; Ashman, T.L. Plant neopolyploidy and genetic background differentiate the microbiome of duckweed across a variety of natural freshwater sources. Mol. Ecol. 2023, 32, 5849–5863. [Google Scholar] [CrossRef]
- Lidbury, I.; Borsetto, C.; Murphy, A.R.J.; Bottrill, A.; Jones, A.M.E.; Bending, G.D.; Hammond, J.P.; Chen, Y.; Wellington, E.M.H.; Scanlan, D.J. Niche-adaptation in plant-associated Bacteroidetes favours specialisation in organic phosphorus mineralisation. ISME J. 2021, 15, 1040–1055. [Google Scholar] [CrossRef]
- Kraut-Cohen, J.; Shapiro, O.H.; Dror, B.; Cytryn, E. Pectin Induced Colony Expansion of soil-derived Flavobacterium strains. Front. Microbiol. 2021, 12, 651891. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.Y.; Xu, J.; An, X.L.; Yang, J.; Wang, Y.; Dou, M.J.; Wang, M.G.; Huang, J.; Fu, Y.J. Insight of endophytic fungi promoting the growth and development of woody plants. Crit. Rev. Biotechnol. 2023, 44, 78–99. [Google Scholar] [CrossRef] [PubMed]
- Jasim, B.; Anisha, C.; Rohini, S.; Kurian, J.M.; Jyothis, M.; Radhakrishnan, E.K. Phenazine carboxylic acid production and rhizome protective effect of endophytic Pseudomonas aeruginosa isolated from Zingiber officinale. World J. Microbiol. Biotechnol. 2014, 30, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Muazzam, K.A.A.R.; Darah, I. The evaluation of antibacterial activity of fungal endophyte Ceratobasidium ramicola IBRLCM127 colonizing in rhizomes of medicinal plant, Curcuma mangga Valeton & Zijp. IOP Conf. Ser. Earth Environ. Sci. 2020, 596, 012083. [Google Scholar]
- Zhang, Y.; Li, Y.Y.; Chen, X.M.; Meng, Z.X.; Guo, S.X. Combined metabolome and transcriptome analyses reveal the effects of mycorrhizal fungus Ceratobasidium sp. AR2 on the flavonoid accumulation in Anoectochilus roxburghii during different growth stages. Int. J. Mol. Sci. 2020, 21, 564. [Google Scholar] [CrossRef]
- Xiang, Z.X.; Tang, X.L.; Liu, W.H.; Song, C.N. A comparative morphological and transcriptomic study on autotetraploid Stevia rebaudiana (Bertoni) and its diploid. Plant Physiol. Biochem. 2019, 143, 154–164. [Google Scholar] [CrossRef]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Meth. 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Seijo, S.; Lozano, J.J.; Alonso, C.; Reverter, E.; Miquel, R.; Abraldes, J.G.; Martinez-Chantar, M.L.; Garcia-Criado, A.; Berzigotti, A.; Castro, A.; et al. Metabolomics Discloses Potential Biomarkers for the Noninvasive Diagnosis of Idiopathic Portal Hypertension. Am. J. Gastroenterol. 2013, 108, 926–932. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, Y.C.; Wang, R.M.; Feng, K.; Di, J.J.; Feng, T.T.; Cao, F.L. Transcriptome and physiological analysis highlights the hormone, phenylpropanoid, and photosynthesis effects on early somatic embryogenesis in Ginkgo biloba. Ind. Crops Prod. 2023, 203, 117176. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Ge, X.N.; Zhong, Q.P.; Tan, X.J.; Wang, J.F.; Cao, L.Q.; Zhou, Y.C.; Zou, Y.L.; Yuan, Y.Q.; Wan, X.R.; Yan, C.; et al. Integrated multi-omics analysis to elucidate the role of shikimic acid and phenethylamine in the effect of scions on rootstocks of Camellia oleifera. Ind. Crops Prod. 2023, 203, 117222. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Zheng, L.L.; Zheng, Y.; Xue, S.; Zhang, J.X.; Huang, P.; Zhao, Y.H.; Hao, X.C.; He, Z.K.; Hu, Z.B.; et al. Insight into the assembly of root-associated microbiome in the medicinal plant Polygonum cuspidatum. Ind. Crops Prod. 2020, 145, 112163. [Google Scholar] [CrossRef]
- Hou, Q.C.; Bai, X.Y.; Li, W.C.; Gao, X.; Zhang, F.M.; Sun, Z.H.; Zhang, H.P. Design of primers for evaluation of lactic acid bacteria populations in complex biological samples. Front. Microbiol. 2018, 9, 2045. [Google Scholar] [CrossRef]
- Kumari, P.; Shanker, K.; Singh, A. Insight into Andrographis paniculata associated bacterial endomicrobiome and assessment of culturable bacterial endophytes for enhancement of industrially important andrographolide content. Ind. Crops Prod. 2023, 200, 116840. [Google Scholar] [CrossRef]
- Li, S.Z.; Deng, Y.; Wang, Z.J.; Zhang, Z.J.; Kong, X.; Zhou, W.J.; Yi, Y.Y.; Qu, Y.Y. Exploring the accuracy of amplicon-based internal transcribed spacer markers for a fungal community. Mol. Ecol. Resour. 2020, 20, 170–184. [Google Scholar] [CrossRef]
- Lawley, B.; Tannock, G.W. Analysis of 16S rRNA gene amplicon sequences using the QIIME software package. In: Seymour GJ, Cullinan MP, Heng NCK, editors. Oral Biology: Molecular Techniques and Applications, 2nd Edition. Methods Mol. Biol. 2017, 1537, 153–163. [Google Scholar]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Log2FC | VIP |
|---|---|---|
| Terpenoids | ||
| 3β,21β,24-trihydroxyserrat-14-en-29-oic acid-3β(4-hydroxy-3-methoxybenzoate) | 18.8 | 1.5 |
| Steviol | 16.8 | 1.5 |
| Ent-kaurenal | 1.9 | 1.3 |
| Rebaudioside A | 0.9 | 1.4 |
| Steviolbioside | 0.8 | 1.0 |
| Rebaudioside C | 0.6 | 0.9 |
| Phenolic acids | ||
| 3-(4-hydroxyphenyl)-propionic acid | 10.1 | 1.5 |
| (S)-2-phenyloxirane | 2.4 | 1.4 |
| 1-O-feruloyl-3-O-p-coumaroylglycerol | 3.1 | 1.3 |
| 5-O-caffeoylshikimic acid | 2.1 | 1.4 |
| Galloyl methyl gallate | 2.0 | 1.2 |
| 3-O-feruloylquinic acid | 1.82 | 1.37 |
| Lipids | ||
| 9,10-dihydroxy-12,13-epoxyoctadecanoic acid | 4.4 | 1.4 |
| Dihydrosphingosine-1-phosphate | 3.4 | 1.3 |
| 3-dehydrosphinganine | 2.3 | 1.3 |
| Phytosphingosine | 2.1 | 1.3 |
| LysoPC 18:1 | 1.6 | 1.3 |
| LysoPE 18:0 | 1.7 | 1.2 |
| 1-(9Z-octadecenoyl)-sn-glycero-3-phosphocholine | 1.7 | 1.2 |
| LysoPE 16:1 | 1.6 | 1.1 |
| LysoPE 16:1 | 2.0 | 1.1 |
| Flavonoids | ||
| Syringetin | 16.7 | 1.5 |
| Quercetin-3-O-(2″-O-rhamnosyl) rutinoside-7-O-glucoside | 10.7 | 1.4 |
| Cirsiliol | 9.9 | 1.4 |
| Cirsimaritin | 9.3 | 1.5 |
| 3′,4′-dihydroxy-7,5′-dimethoxyflavone | 9.8 | 1.4 |
| Quercetin-3,3′-dimethyl ether | 8.1 | 1.4 |
| 5,7,2′,5′-tetrahydroxy-8,6′-dimethoxyflavone | 9.2 | 1.4 |
| 3,7-dimethylquercetagetin-(3′,4′,5,6-Tetrahydroxy-3,7-dimethoxyflavone) | 8.8 | 1.4 |
| Rhamnetin; 3,5,3′,4′-tetrahydroxy-7-methoxyflavone | 8.0 | 1.5 |
| Alkaloids | ||
| Tryptamine | 4.9 | 1.4 |
| N-caffeoylputrescine | 3.6 | 1.4 |
| Retronecine | 2.7 | 1.4 |
| 2-phenylethylamine | 2.7 | 1.4 |
| Vitamin | ||
| Retinol | 1.4 | 1.4 |
| Nicotinamide | 1.0 | 1.4 |
| Riboflavin | 2.0 | 1.4 |
| Biotin | 2.2 | 1.4 |
| Others | ||
| 2-phenylethanol | 2.2 | 1.4 |
| Dipteryxin | 9.01 | 1.45 |
| 2-Dehydro-3-deoxy-L-arabinonate | 2.14 | 1.45 |
| D-Threose | 2.42 | 1.43 |
| ID | Name | Log2FC | ID | Name | Log2FC |
|---|---|---|---|---|---|
| WRKY | Streb.2G002460 | BZP23_ARATH | 1.9 | ||
| Streb.8G004280 | WRK21_ARATH | 2.0 | Streb.4G010840 | CPRF2_PETCR | 1.3 |
| Streb.11G007600 | WRK22_ARATH | 1.8 | Streb.7G020660 | GBF1_ARATH | 1.2 |
| Streb.1G002040 | WRK70_SOLLC | 1.8 | Streb.3G007530 | BZIP9_ARATH | −3.1 |
| Streb.3G012150 | WRKY6_ARATH | 1.6 | Streb.3G012510 | AI5L4_ARATH | −2.1 |
| Streb.4G015740 | WRKY4_ARATH | −2.5 | Streb.8G015740 | VIP1_ARATH | −1.0 |
| Streb.2G030570 | WRK51_ARATH | −1.4 | bHLH | ||
| Streb.4G005140 | WRK65_ARATH | −1.4 | Streb.Contig05035G000020 | ILR3_ARATH | 6.1 |
| Streb.2G051870 | WRK40_ARATH | −1.3 | Streb.2G020970 | BIM2_ARATH | 4.3 |
| NAC | Streb.1G023980 | IRO2_ORYSI | 3.9 | ||
| Streb.8G026410 | NAC83_ARATH | 9.2 | Streb.1G024080 | ORG3_ARATH | 3.8 |
| Streb.10G007870 | NTL9_ARATH | 1.9 | Streb.4G026850 | BH087_ARATH | 3.8 |
| Streb.5G033120 | NAC75_ARATH | 1.3 | Streb.10G018330 | BH128_ARATH | 3.2 |
| Streb.9G020010 | NAC73_ARATH | 1.3 | Streb.2G013370 | BH094_ARATH | 2.4 |
| Streb.8G033830 | NAC17_ARATH | 1.3 | Streb.2G028430 | RHL1_LOTJA | 1.4 |
| Streb.10G019010 | NAC43_ARATH | 1.2 | Streb.9G004040 | MYC2_ARATH | 1.3 |
| Streb.2G020770 | NAC2_ARATH | −3.0 | Streb.2G055730 | BIM1_ARATH | 1.3 |
| Streb.5G025450 | NAC6_SOYBN | −1.6 | Streb.11G016670 | ICE1_ARATH | 1.3 |
| Streb.7G009790 | NAC91_ARATH | −1.2 | Streb.11G003730 | BH030_ARATH | 1.1 |
| MYB | Streb.6G032180 | UNE10_ARATH | −2.5 | ||
| Streb.10G031370 | MY111_ARATH | 8.1 | Streb.7G020980 | BH025_ARATH | −1.9 |
| Streb.11G027310 | MYB4_ORYSJ | 4.0 | Streb.3G020800 | BH153_ARATH | −1.7 |
| Streb.11G030580 | KUA1_ARATH | 2.0 | Streb.4G022320 | PIF7_ARATH | −1.5 |
| Streb.6G002150 | MB101_ARATH | 1.9 | AP2/ERF | ||
| Streb.7G028220 | RVE8_ARATH | 1.8 | Streb.5G008600 | AP21_ORYSI | 4.2 |
| Streb.11G013850 | MYB90_ARATH | 1.7 | Streb.8G009050 | AP23_ORYSI | 3.3 |
| Streb.2G049910 | MYB06_ANTMA | 1.3 | Streb.11G011700 | ERF34_ARATH | 1.7 |
| Streb.4G021750 | MYBS1_ORYSI | 1.1 | Streb.4G000620 | ERF54_ARATH | 1.5 |
| Streb.2G045880 | LHY_PETHY R | 1.1 | Streb.5G004440 | EF118_ARATH | 1.5 |
| Streb.1G015820 | MYB44_ARATH | −1.9 | Streb.1G037650 | DREB3_ARATH | 1.4 |
| Streb.9G001240 | MYB38_MAIZE | −1.9 | Streb.9G000120 | ERF39_ARATH | 1.1 |
| Streb.2G021360 | MYB1_ACTCC | −1.9 | Streb.10G004210 | EF102_ARATH | 1.1 |
| Streb.9G010620 | MYB48_ARATH | −1.6 | Streb.Contig03456G000010 | ERF61_ARATH | −2.1 |
| Streb.9G019430 | MY102_ARATH | −1.2 | Streb.10G009110 | RAP23_ARATH | −1.5 |
| Streb.1G000660 | MYB17_ARATH | −1.0 | Streb.4G017620 | ERF08_ARATH | −1.5 |
| bZIP | Streb.4G017600 | AP2_ARATH | −1.2 | ||
| Streb.9G027550 | BZP44_ARATH | 6.1 | Streb.6G008530 | ERF3_TOBAC | −1.0 |
| Streb.10G008960 | BZP16_ARATH | 2.8 |
| ID | Name | log2FC | ID | Name | log2FC |
|---|---|---|---|---|---|
| Ion transport protein | ABC transporter | ||||
| Streb.3G013310 | CNGC2_ARATH | 3.1 | Streb.1G035320 | AB39G_ARATH | 4.2 |
| Streb.2G032360 | CNGC2_ARATH | 1.2 | Streb.3G009500 | AB10I_ARATH | 3 |
| Streb.7G022990 | CNGC1_ARATH | −1.2 | Streb.5G025360 | AB11G_ARATH | 2.4 |
| Sugar (and other) transporter | Streb.5G001490 | AB3C_ARATH | 1.8 | ||
| Streb.9G006200 | EDL16_ARATH | 2.1 | Streb.4G013640 | AB26B_ARATH | 1.4 |
| Streb.9G025500 | STP13_ARATH | 1.0 | Streb.1G016980 | AB5G_ARATH | 1.2 |
| Streb.10G002460 | PHT11_PETHY | −2.3 | Streb.3G008960 | AB3C_ARATH | 1.0 |
| Streb.2G008870 | EDL16_ARATH | −1.6 | Streb.9G030840 | ABCG1_PETHY | −2.2 |
| ZIP Zinc transporter | Streb.10G006460 | AB10C_ARATH | −1.7 | ||
| Streb.Contig03477G000010 | ZIP6_ARATH | 5.6 | Streb.10G027430 | AB20G_ARATH | −1.3 |
| Streb.6G017260 | ZIP2_ARATH | 5.7 | Triose-phosphate Transporter | ||
| Streb.1G047870 | ZIP6_ARATH | −2.6 | Streb.6G014120 | UGNT1_ARATH | 2.5 |
| Streb.10G018100 | IRT3_ARATH | −1.7 | Streb.5G032270 | GFT1_ARATH | 2.3 |
| CorA-like Mg2+ transporter protein | Streb.5G022800 | GPT1_ARATH | 1.6 | ||
| Streb.2G000140 | MRS23_ARATH | 6.6 | Streb.9G009400 | UUAT1_ARATH | 1.2 |
| Streb.9G031220 | MRS23_ARATH | 3.6 | Sugar efflux transporter for intercellular exchange | ||
| Streb.11G025820 | MRS24_ARATH | 1.3 | Streb.11G000040 | NOD3_MEDTR | 1.4 |
| Transmembrane amino acid transporter protein | Streb.1G032140 | SWET1_ARATH | 1.4 | ||
| Streb.2G034940 | AAP6_ARATH | 2.3 | Streb.2G031580 | SWT17_ARATH | 1.1 |
| Streb.9G013250 | LAX4_MEDTR | 1.1 | Streb.8G017430 | SWET3_ARATH | −1.1 |
| Streb.2G018370 | LHT1_ARATH | −1.9 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Wang, J.; Chen, M.; Meng, W.; Ding, A.; Chen, M.; Ding, R.; Tan, M.; Xiang, Z. Multi-Omics Analyses Uncover the Mechanism Underlying Polyploidization-Enhanced Steviol Glycosides Biosynthesis in Stevia rebaudiana. Plants 2024, 13, 2542. https://doi.org/10.3390/plants13182542
Liu J, Wang J, Chen M, Meng W, Ding A, Chen M, Ding R, Tan M, Xiang Z. Multi-Omics Analyses Uncover the Mechanism Underlying Polyploidization-Enhanced Steviol Glycosides Biosynthesis in Stevia rebaudiana. Plants. 2024; 13(18):2542. https://doi.org/10.3390/plants13182542
Chicago/Turabian StyleLiu, Juan, Jiaxue Wang, Mingjia Chen, Wenna Meng, Anping Ding, Miao Chen, Rongping Ding, Mingpu Tan, and Zengxu Xiang. 2024. "Multi-Omics Analyses Uncover the Mechanism Underlying Polyploidization-Enhanced Steviol Glycosides Biosynthesis in Stevia rebaudiana" Plants 13, no. 18: 2542. https://doi.org/10.3390/plants13182542