
Morphological and Transcriptomic Analyses Reveal the Involvement of Key Metabolic Pathways in Male Sterility in Chimonanthus praecox (L.) Genotypes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Morphological Characterization and Paraffin Section Analysis
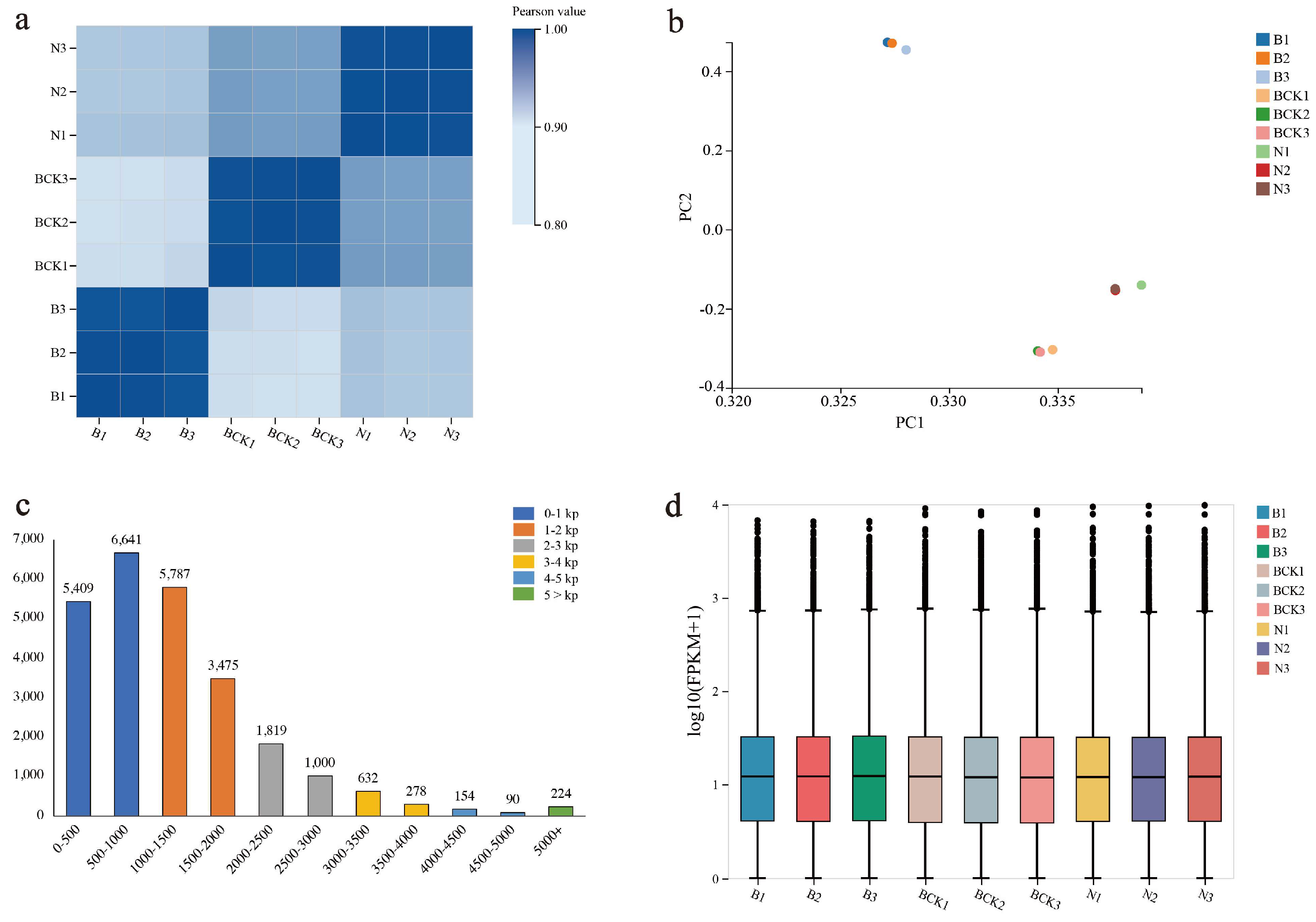
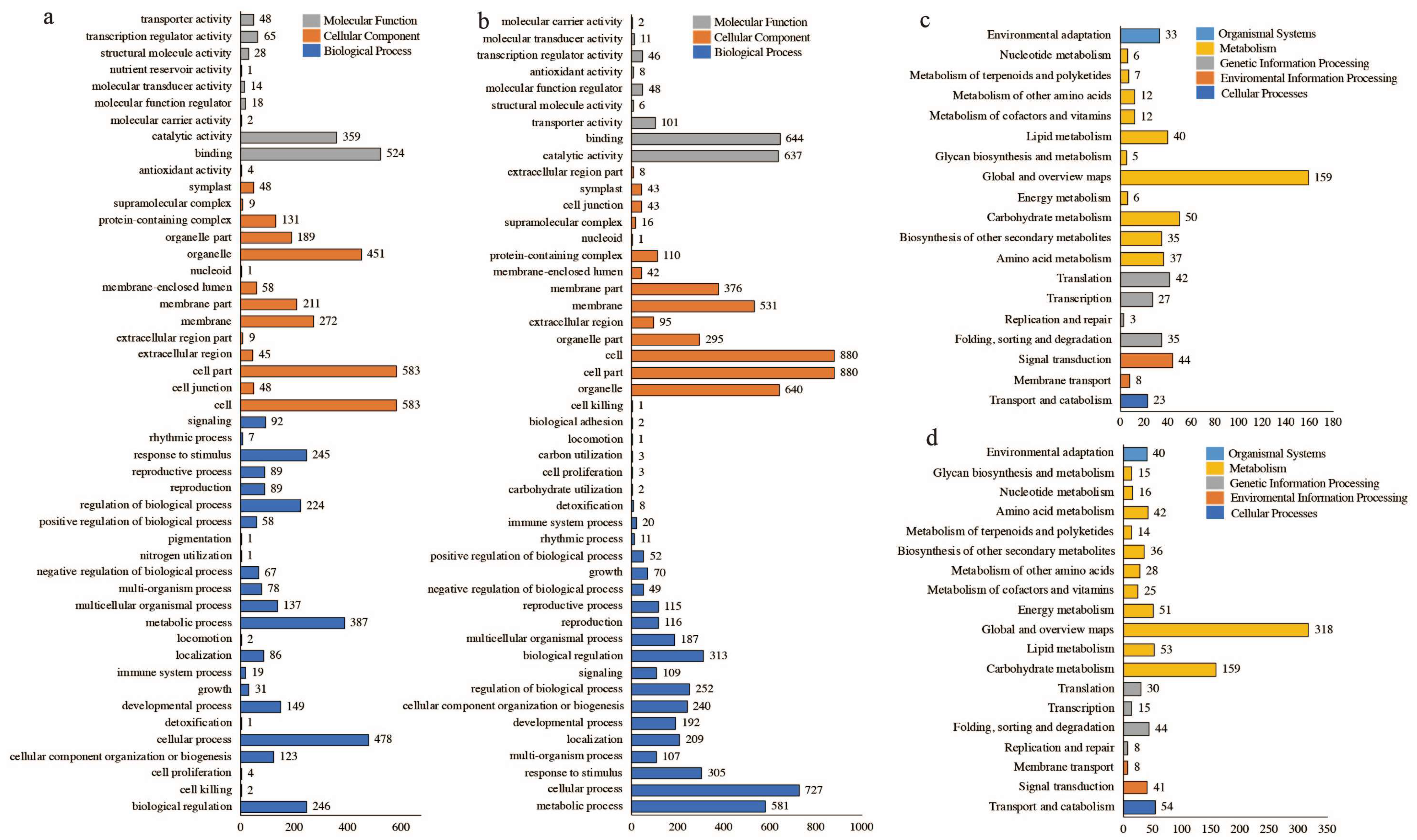
2.2. Overview of Transcriptome Data and Functional Annotation Information
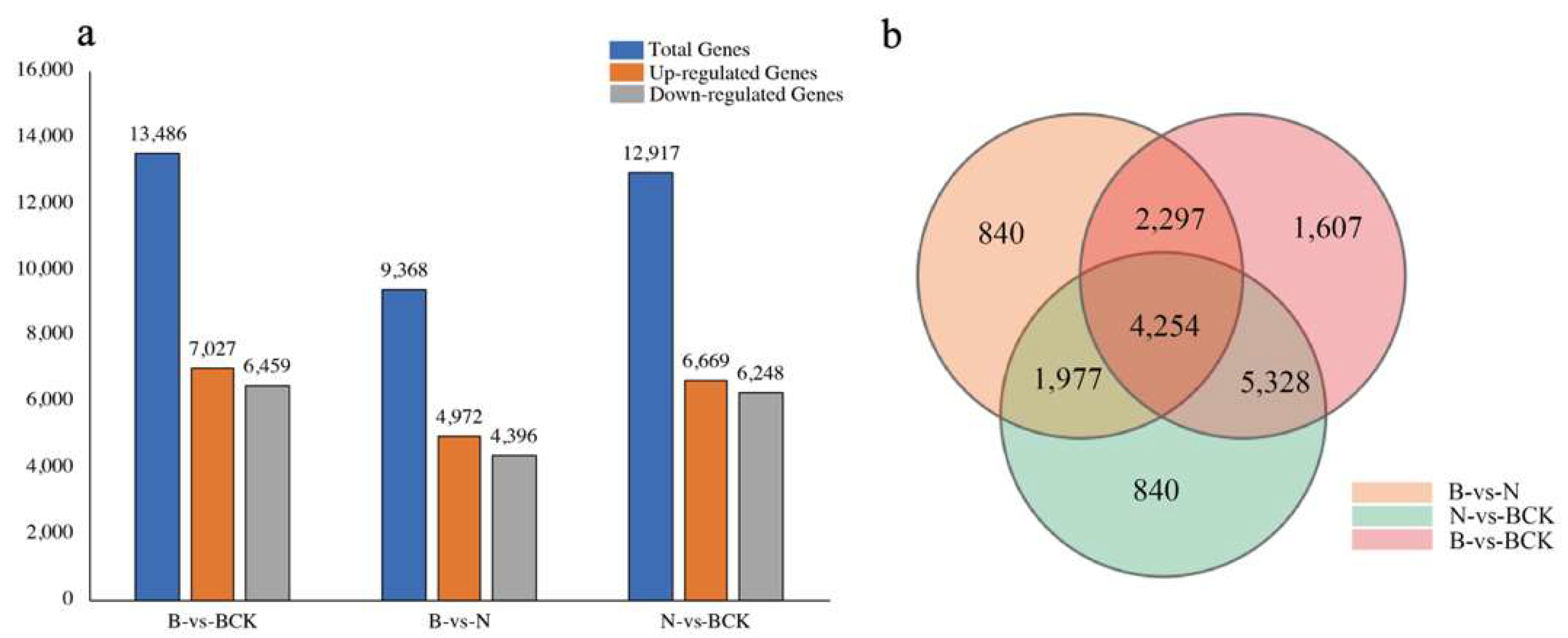
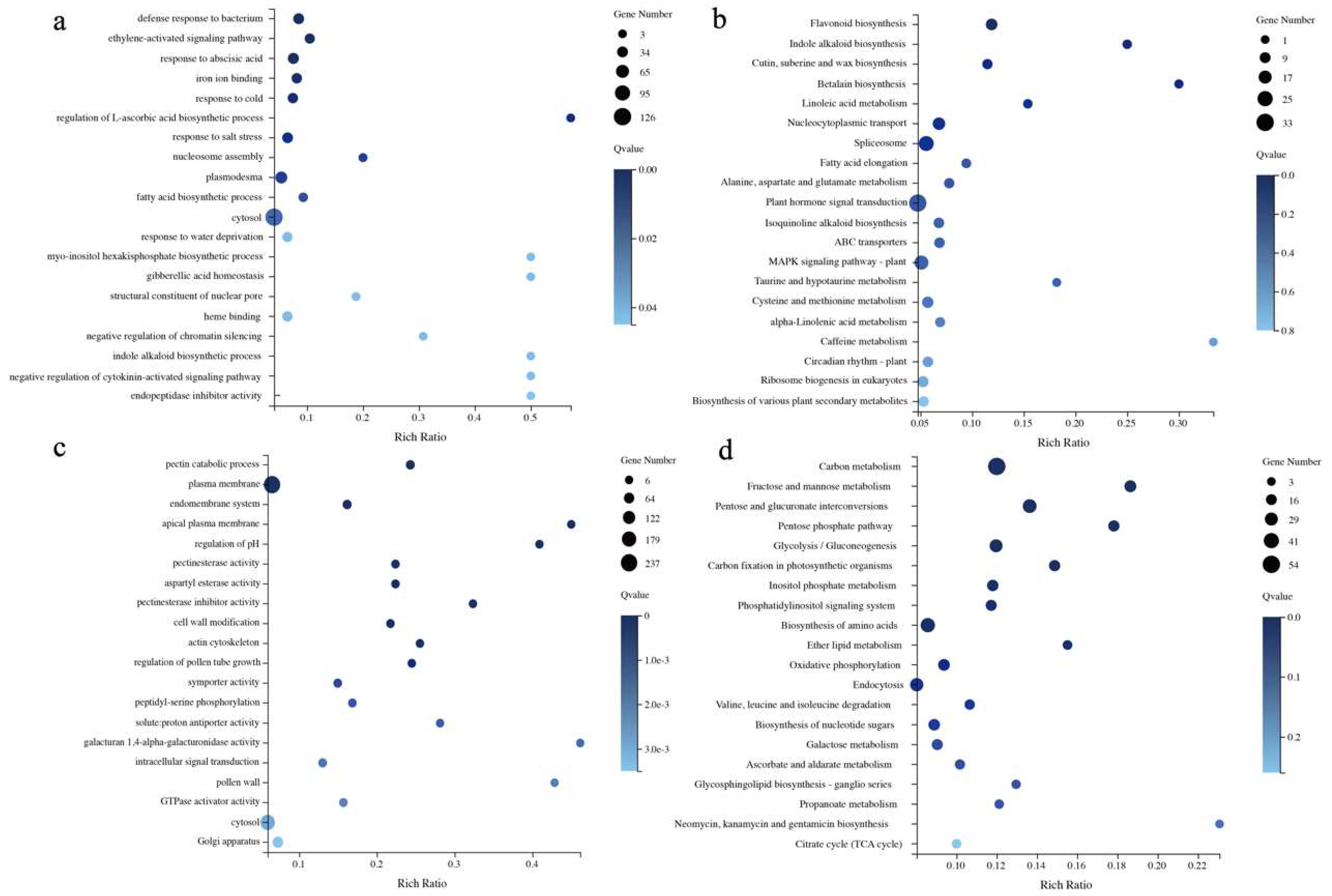
2.3. Analysis of Differentially Expressed Genes
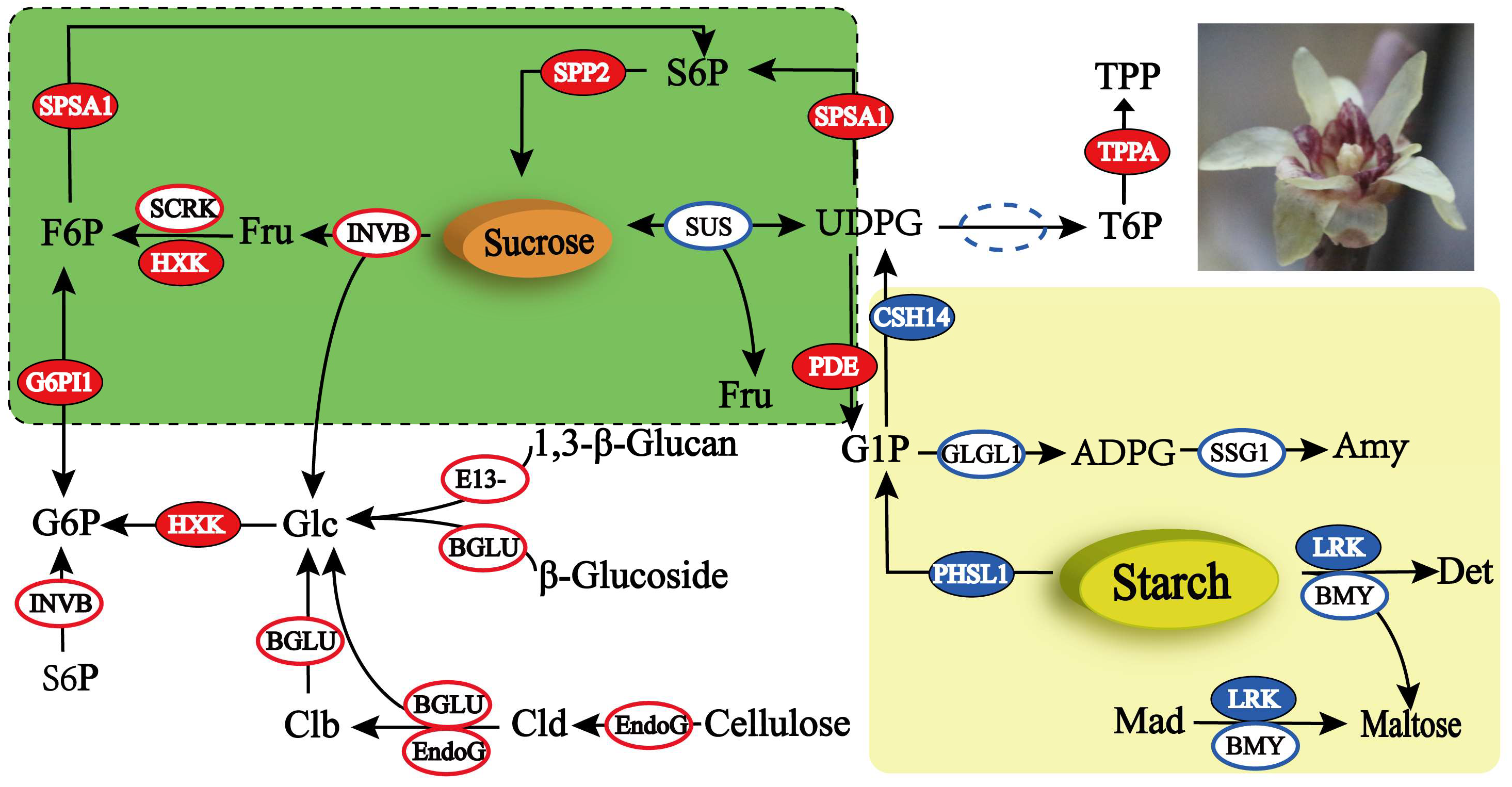
2.4. DEG Analysis of the Starch and Sucrose Metabolism Pathway
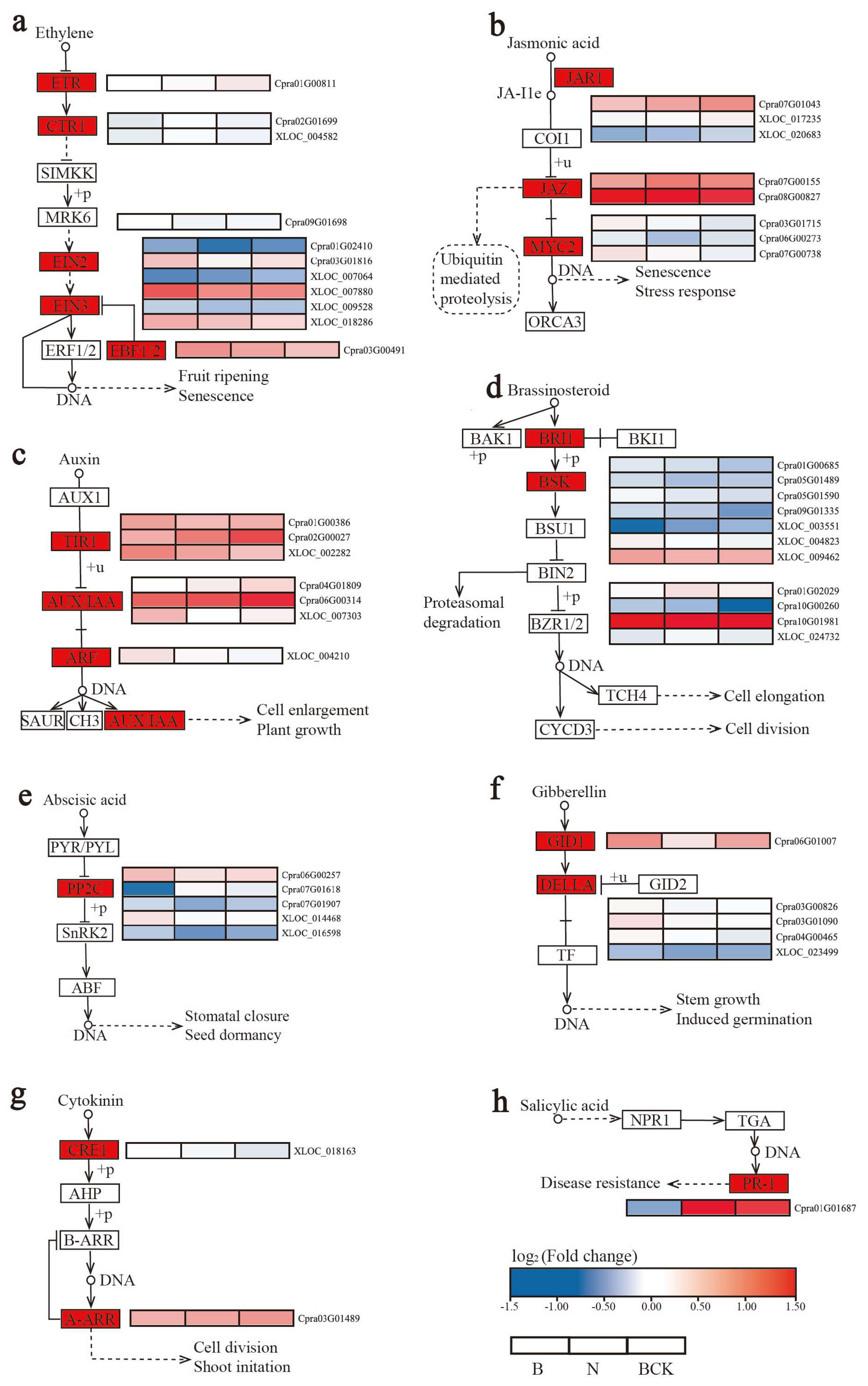
2.5. DEG Analysis of the Plant Hormone Signal Transduction Pathway
2.6. Verification of DEGs by qPCR
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Paraffin Sections
4.3. RNA Extraction, Library Construction, and Sequencing
4.4. Assembly, Functional Annotation, and DEG Analysis
4.5. Validation of DEGs Using Quantitative PCR (qPCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yang, J.; Dai, P.; Zhou, T.; Huang, Z.; Feng, L.; Su, H.; Liu, Z.; Zhao, G. Genetic diversity and structure of wintersweet (Chimonanthus praecox) revealed by EST-SSR markers. Sci. Hortic. 2013, 150, 1–10. [Google Scholar] [CrossRef]
- Shang, J.; Tian, J.; Cheng, H.; Yan, Q.; Chen, L. The chromosome-level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biol. 2020, 21, 200. [Google Scholar] [CrossRef] [PubMed]
- Sui, S.; Luo, J.; Ma, J.; Zhu, Q.; Lei, X.; Li, M. Generation and analysis of expressed sequence tags from Chimonanthus praecox (Wintersweet) flowers for discovering stress-responsive and floral development-related genes. Int. J. Genom. 2012, 2012, 134596. [Google Scholar]
- Chen, L. Research advances on Calycanthaceae. Chin. Landsc. Archit. 2012, 28, 49–53. [Google Scholar]
- Kitagawa, N.; Ninomiya, K.; Okugawa, S.; Motai, C.; Nakanishi, Y.; Yoshikawa, M.; Muraoka, O.; Morikawa, T. Quantitative determination of principal alkaloid and flavonoid constituents in wintersweet, the flower buds of Chimonanthus praecox. Nat. Prod. Commun. 2016, 11, 953–956. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, N.; Ma, X.; Yang, J.; Li, X.; Pang, W.; Lin, C. Chemical composition and in vitro antibacterial and antifungal activity of essential oils from different parts of Chimonanthus praecox (Linn.) Link. Chin. J. Mod. Appl. Pharm. 2019, 36, 901–905. [Google Scholar] [CrossRef]
- Chen, L.; Lu, D. Cultivar classification system of Chimonanthus praecox (L.) Link. J. Beijing For. Univ. 2001, 23, 107–108. [Google Scholar]
- Hedrick, P.W. Recent developments in conservation genetics. For. Ecol. Manag. 2004, 197, 3–19. [Google Scholar] [CrossRef]
- Li, F. Analysis of Genetic Diversity and Mating System in Natural Populations of Ormosia hosiei. Ph.D. Thesis, Chinese Academy of Forestry, Beijing, China, 2017. [Google Scholar]
- Audigeos, D.; Brousseau, L.; Traissac, S.; Scotti-Saintagne, C.; Scotti, I. Molecular divergence in tropical tree populations occupying environmental mosaics. J. Evol. Biol. 2013, 26, 529–544. [Google Scholar] [CrossRef]
- Berg, H. Factors influencing seed: Ovule ratios and reproductive success in four cleistogamous species: A comparison between two flower types. Plant Biol. 2003, 5, 194–202. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, G.; Zhang, S.; Chen, S.; Wang, Y.; Wen, P.; Ma, X.; Shi, Y.; Qi, R.; Yang, Y. Genomes of the banyan tree and pollinator wasp provide insights into fig-wasp coevolution. Cell 2020, 183, 875–889.e817. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Jia, H.; Wang, J.; Zhang, Z. Flowering and pollination patterns of Magnolia denudata with emphasis on anatomical changes in ovule and seed development. Flora-Morphol. Distrib. Funct. Ecol. Plants 2010, 205, 259–265. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, S.; Chen, L.; Liu, B.; Li, H. Research advances on dichogamy of Walnut. China Fruits 2018, 3, 65–71. [Google Scholar] [CrossRef]
- Li, L.; Lu, N.; Fan, B.; Zhao, Z. Effect of flowering time on floral sexual durations and phenotypic gender in dichogamous Aconitum gymnandrum. Biodivers. Sci. 2016, 24, 665–671. [Google Scholar] [CrossRef]
- Jin, X.; Xiao, C.; Zhang, J.; Zhao, J.; Xiao, Z.; Zhang, H.; Jin, Z. Comparison study of flowering process in three dichogamous Lauraceae species. J. Trop. Subtrop. Bot. 2021, 29, 162–170. [Google Scholar]
- Lloyd, D.G.; Webb, C. The avoidance of interference between the presentation of pollen and stigmas in angiosperms I. Dichogamy. N. Z. J. Bot. 1986, 24, 135–162. [Google Scholar] [CrossRef]
- Lv, C.; Liu, L.; Zhang, L.; Hu, Q.; Chen, M. Characteristics of pollination ecology for wintersweet (Chimonanthus praecox ( L. ) Link) after transplanted to Yantai. J. Ludong Univ. (Nat. Sci. Ed.) 2017, 33, 309–314. [Google Scholar]
- Worboys, S.J.; Jackes, B.R. Pollination processes in Idiospermum australiense (Calycanthaceae), an arborescent basal angiosperm of Australia’s tropical rain forests. Plant Syst. Evol. 2005, 251, 107–117. [Google Scholar] [CrossRef]
- Li, C. Revealing the Genetic Basis of Male Sterility Based on the Evolution of Citrus Cytonuclear. Ph.D. Thesis, Huazhong Agricultural University, Wuhan, China, 2023. [Google Scholar]
- Shen, Z.; Li, W.; Li, Y.; Liu, M.; Cao, H.; Provart, N.; Ding, X.; Sun, M.; Tang, Z.; Yue, C. The red flower wintersweet genome provides insights into the evolution of magnoliids and the molecular mechanism for tepal color development. Plant J. 2021, 108, 1662–1678. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, Q. Genetic diversity of germplasm resources of Chimonanthus praecox (L.) Link based on AFLP marker. Acta Ecol. Sin. 2007, 27, 4452–4459. [Google Scholar]
- Baránková, S.; Pascual-Díaz, J.P.; Sultana, N.; Alonso-Lifante, M.P.; Balant, M.; Barros, K.; D’Ambrosio, U.; Malinská, H.; Peska, V.; Lorenzo, I.P. Sex-chrom, a database on plant sex chromosomes. New Phytol. 2020, 227, 1594–1604. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, Q.; Xiao, Y.; Chen, X.; Chen, X.; Hu, X. On the theories of plant mating system and molecular evolution and their applications. Sci. Sin. (Vitae) 2023, 53, 50–63. [Google Scholar] [CrossRef]
- Singh, S.; Dey, S.; Bhatia, R.; Kumar, R.; Behera, T. Current understanding of male sterility systems in vegetable Brassicas and their exploitation in hybrid breeding. Plant Reprod. 2019, 32, 231–256. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Huang, W.; Huang, Q.; Qin, X.; Yu, C.; Wang, L.; Li, S.; Zhu, R.; Zhu, Y. Mitochondria and cytoplasmic male sterility in plants. Mitochondrion 2014, 19, 282–288. [Google Scholar] [CrossRef]
- Ramsey, M. Ant pollination of the perennial herb Blandfordia grandiflora (Liliaceae). Oikos 1995, 74, 265–272. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, H.; Hu, M.; Den, L.; Wang, Q.; Li, J.; Yuan, H. Application progress of Omics in the research of anther development I: Transcriptomics. Curr. Biotechnol. 2019, 9, 433–439. [Google Scholar] [CrossRef]
- Vedel, F.; Pla, M.; Vitart, V.; Gutierres, S.; Chétrit, P.; Paepe, R.d. Molecular basis of nuclear and cytoplasmic male sterility in higher plants. Plant Physiol. Biochem. 1994, 32, 601–618. [Google Scholar]
- Chen, H.; Qian, B.; Pei, X.; Li, Y.; Huang, C.; Liu, Y.; Yuan, C.; Yi, H.; Zeng, J.; Yi, B. Cytological and transcriptome analysis of cytoplasmic male sterility and maintainer line in glu-CMS tobacco. Acta Tabacaria Sin. 2023, 29, 64–70. [Google Scholar] [CrossRef]
- Herbert, S.W.; Walton, D.A.; Wallace, H.M. Pollen-parent affects fruit, nut and kernel development of Macadamia. Sci. Hortic. 2019, 244, 406–412. [Google Scholar] [CrossRef]
- Chase, C.D. Cytoplasmic male sterility: A window to the world of plant mitochondrial–nuclear interactions. Trends Genet. 2007, 23, 81–90. [Google Scholar] [CrossRef]
- Wei, B. Inheritance and Candidate Genes Identification for the Fertility Restorer of Cytoplasmic Male Sterility in Pepper. Ph.D. Thesis, Gansu Agricultural University, Lanzhou, China, 2017. [Google Scholar]
- He, C.; Liu, Z.; Xiong, X.; Zou, X.; Xiao, L. Cytologic observations on anther development of 9704A, a cytoplasmic male sterile line in Capsicum annum L. Acta Hortic. Sin. 2008, 4, 521–528. [Google Scholar] [CrossRef]
- Nie, M.; Wang, G.; Zhu, W. Cytology research on the anther abortion of three male sterility lines in Rapeseed (Brassica napus L.). Sci. Agric. Sin. 2007, 7, 1543–1549. [Google Scholar]
- Zhou, T.; Wei, Y.; Liang, W.; Guan, C.; Yang, S. Transcriptome analysis of gene expression characteristics at anther development stages of the male sterile in Wolfberry (Lycium barbarum). Genom. Appl. Biol. 2023, 3, 43. [Google Scholar]
- Kang, H.; Zhang, S.; Luo, T.; Chen, J.; Yang, W. Development mechanism of anther on hermaphrodite flower in Nyssa yunnanensis. J. West China For. Sci. 2019, 48, 63–69+75. [Google Scholar] [CrossRef]
- Wang, J.; Tang, X.; Yuan, L.; Chen, G.; Hou, J.; Yang, Y.; Huang, X.; Wang, C. Integrated analysis of transcriptome and proteome changes related to the male-sterile mutant MS7-2 in wucai (Brassica campestris L.). Sci. Hortic. 2023, 312, 111889. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Li, C.; Zhao, Z.; Liu, Y.; Liang, B.; Guan, S.; Lan, H.; Wang, J.; Lu, Y.; Cao, M. Comparative transcriptome analysis of isonuclear-alloplasmic lines unmask key transcription factor genes and metabolic pathways involved in sterility of maize CMS-C. PeerJ 2017, 5, e3408. [Google Scholar] [CrossRef]
- Li, F.; Chen, S.; Chen, F.; Teng, N.; Fang, W.; Zhang, F.; Deng, Y. Anther wall development, microsporogenesis and microgametogenesis in male fertile and sterile Chrysanthemum (Chrysanthemum morifolium Ramat., Asteraceae). Sci. Hortic. 2010, 126, 261–267. [Google Scholar] [CrossRef]
- Fan, W.; Liu, X.; Ma, X.; Yang, H.; Zhu, J.; Tang, J.; Yue, S.; Zheng, R. Mining of male sterility-related genes in Lycium barbarum using transcriptome data. Chin. Tradit. Herb. Drugs 2023, 54, 2226–2234. [Google Scholar]
- Fu, T.; Wang, Y.; Gao, H.; Zhuang, N. Expression and bioinformatics analysis of male sterility-related gene HbBPC7 in Hevea brasiliensis. Mol. Plant Breed. 2023. Available online: https://kns.cnki.net/kcms/detail/46.1068.S.20230510.1617.006.html (accessed on 11 May 2023).
- Zhang, X.; Feng, J.; Chen, J.; Fan, W.; Wang, L.; Yue, S.; Zhen, R. Comparative transcriptome analysis of male sterile anther in Wolfberry (Lycium barbarum L. ). Genom. Appl. Biol. 2022, 41, 1274–1285. [Google Scholar] [CrossRef]
- Yan, J.; Wang, J.; Zhao, Y.; Jia, X.; Guo, J.; Zhu, L. Transcriptome analysis of anthers of the maize CMS-S type sterile line. J. Hebei Agric. Univ. 2023, 46, 1–9. [Google Scholar] [CrossRef]
- Yu, W.; Miao, Y.; Mu, H.; Jia, X.; Yan, D.; Xu, B. Transcriptomic analysis on the mechanism of polen abortion in male sterile lines of Alfalfa. Acta Agrestia Sin. 2023, 31, 1702–1713. [Google Scholar]
- Kretschmer, M.; Croll, D.; Kronstad, J.W. Maize susceptibility to Ustilago maydis is influenced by genetic and chemical perturbation of carbohydrate allocation. Mol. Plant Pathol. 2017, 18, 1222–1237. [Google Scholar] [CrossRef]
- He, D. Transcriptome Analysis of Cytoplasmic Male Sterile Lines and Their Maitainers in Soybean. Master’s Thesis, Inner Mongolia University for Nationalities, Tongliao, China, 2020. [Google Scholar]
- Heng, S.; Chen, F.; Wei, C.; Li, X.; Yi, B.; Ma, C.; Tu, J.; Shen, J.; Fu, T.; Wen, J. Cytological and iTRAQ-based quantitative proteomic analyses of hau CMS in Brassica napus L. J. Proteom. 2019, 193, 230–238. [Google Scholar] [CrossRef]
- Sawhney, V.K.; Shukla, A. Male sterility in flowering plants: Are plant growth substances involved? Am. J. Bot. 1994, 81, 1640–1647. [Google Scholar] [CrossRef]
- Wang, H.; Wu, Z.; Han, Y. Relationships between endogenous hormone contents and cytoplasmic male sterility in sugarbeet. Sci. Agric. Sin. 2008, 41, 230–238. [Google Scholar]
- Wu, Z.; Hu, K.; Fu, J.; Qiao, A. Relationships between cytoplasmic male sterility and endogenous hormone content of pepper bud. J. South China Agric. Univ. 2010, 31, 1–4. [Google Scholar]
- Dubas, E.; Janowiak, F.; Krzewska, M.; Hura, T.; Żur, I. Endogenous ABA concentration and cytoplasmic membrane fluidity in microspores of oilseed rape (Brassica napus L.) genotypes differing in responsiveness to androgenesis induction. Plant Cell Rep. 2013, 32, 1465–1475. [Google Scholar] [CrossRef]
- Tianxia, G.; Zhanhai, D.; Jianping, Z. Studies on the changes of phytohormones during bud development stage in thermo-sensitivity genic male-sterile flax. Chin. J. Oil Crop Sci. 2007, 29, 248–253. [Google Scholar]
- Huang, S.; Zhou, X. Relationship between rice cytoplasmic male sterility and contents of GA_(1+ 4) and IAA. Acta Agric. Boreali-Sin. 1994, 9, 16–20. [Google Scholar]
- Duan, Q.; Wang, D.; Xu, Z.; Bai, S. Stamen development in Arabidopsis is arrested by organ-specific overexpression of a cucumber ethylene synthesis gene CsACO2. Planta 2008, 228, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Caldelari, D.; Wang, G.; Farmer, E.E.; Dong, X. Arabidopsis lox3 lox4 double mutants are male sterile and defective in global proliferative arrest. Plant Mol. Biol. 2011, 75, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Cecchetti, V.; Altamura, M.M.; Falasca, G.; Costantino, P.; Cardarelli, M. Auxin regulates Arabidopsis anther dehiscence, pollen maturation, and filament elongation. Plant Cell 2008, 20, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, gix120. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, F.; Song, A.; Zhao, Y.; Chen, X.; Gao, Y.; Wei, G.; Zhang, W.; Guan, Y.; Fu, J.; et al. The genome assembly of Chimonanthus praecox var. concolor and comparative genomic analysis highlight the genetic basis underlying conserved and variable floral traits of wintersweet. Ind. Crops Prod. 2023, 206, 117603. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5. [Google Scholar] [CrossRef]
- Kolde, R.; Kolde, M.R. Package ‘pheatmap’. R Package 2015, 1, 790. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- John, D.S.; Andrew, J.B.; Alan, D.; David, R.; Gregory, W. Qvalue: Q-Value Estimation for False Discovery Rate Control. R Package Version 2.26.0. Available online: http://github.com/jdstorey/qvalue (accessed on 2 September 2023).
- Chen, S.; Zhao, W.; Fu, X.; Guo, C.; Cai, Y.; Huang, S.; Chen, L.; Yang, N. Screening and verification of reference genes of wintersweet (Chimonanthus praecox L.) in real-time quantitative PCR analysis. Mol. Plant Breed. 2022. Available online: https://kns.cnki.net/kcms/detail/46.1068.S.20220810.1001.002.html (accessed on 11 August 2022).
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Wu, H.; Cao, Y.; Zheng, X.; Zhu, H.; Sui, S. Morphological and Transcriptomic Analyses Reveal the Involvement of Key Metabolic Pathways in Male Sterility in Chimonanthus praecox (L.) Genotypes. Plants 2024, 13, 2571. https://doi.org/10.3390/plants13182571
Liu B, Wu H, Cao Y, Zheng X, Zhu H, Sui S. Morphological and Transcriptomic Analyses Reveal the Involvement of Key Metabolic Pathways in Male Sterility in Chimonanthus praecox (L.) Genotypes. Plants. 2024; 13(18):2571. https://doi.org/10.3390/plants13182571
Chicago/Turabian StyleLiu, Bin, Huafeng Wu, Yinzhu Cao, Xiaowen Zheng, Haoxiang Zhu, and Shunzhao Sui. 2024. "Morphological and Transcriptomic Analyses Reveal the Involvement of Key Metabolic Pathways in Male Sterility in Chimonanthus praecox (L.) Genotypes" Plants 13, no. 18: 2571. https://doi.org/10.3390/plants13182571