
Impact of Chromosomal Fusion and Transposable Elements on the Genomic Evolution and Genetic Diversity of Ilex Species


 , and
, and 
Abstract
1. Introduction
2. Results
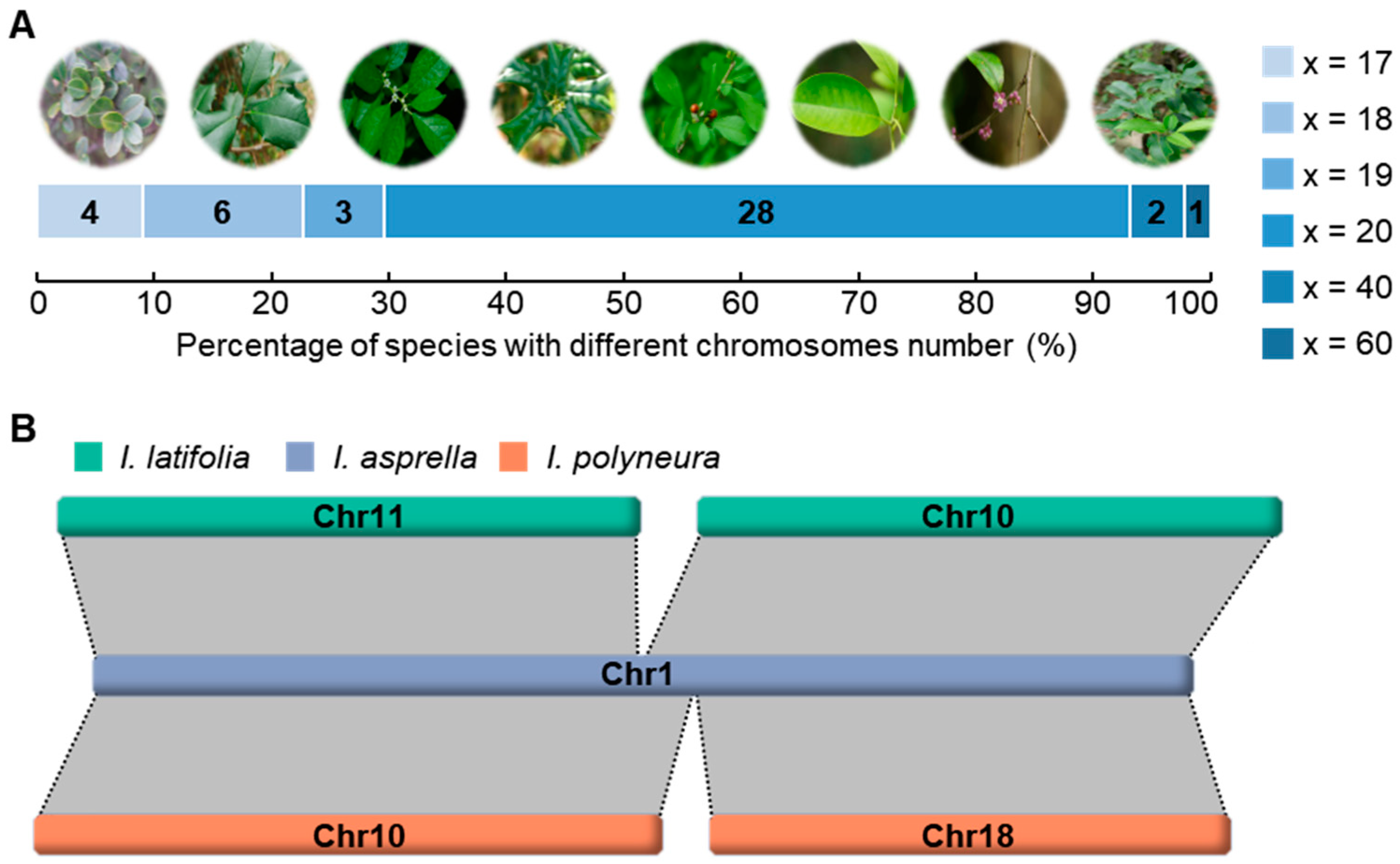
2.1. Karyotype Evolution Driven by Chromosome Fusion
2.2. Comparative Genome Reveals Slight Differences in Genes Annotation
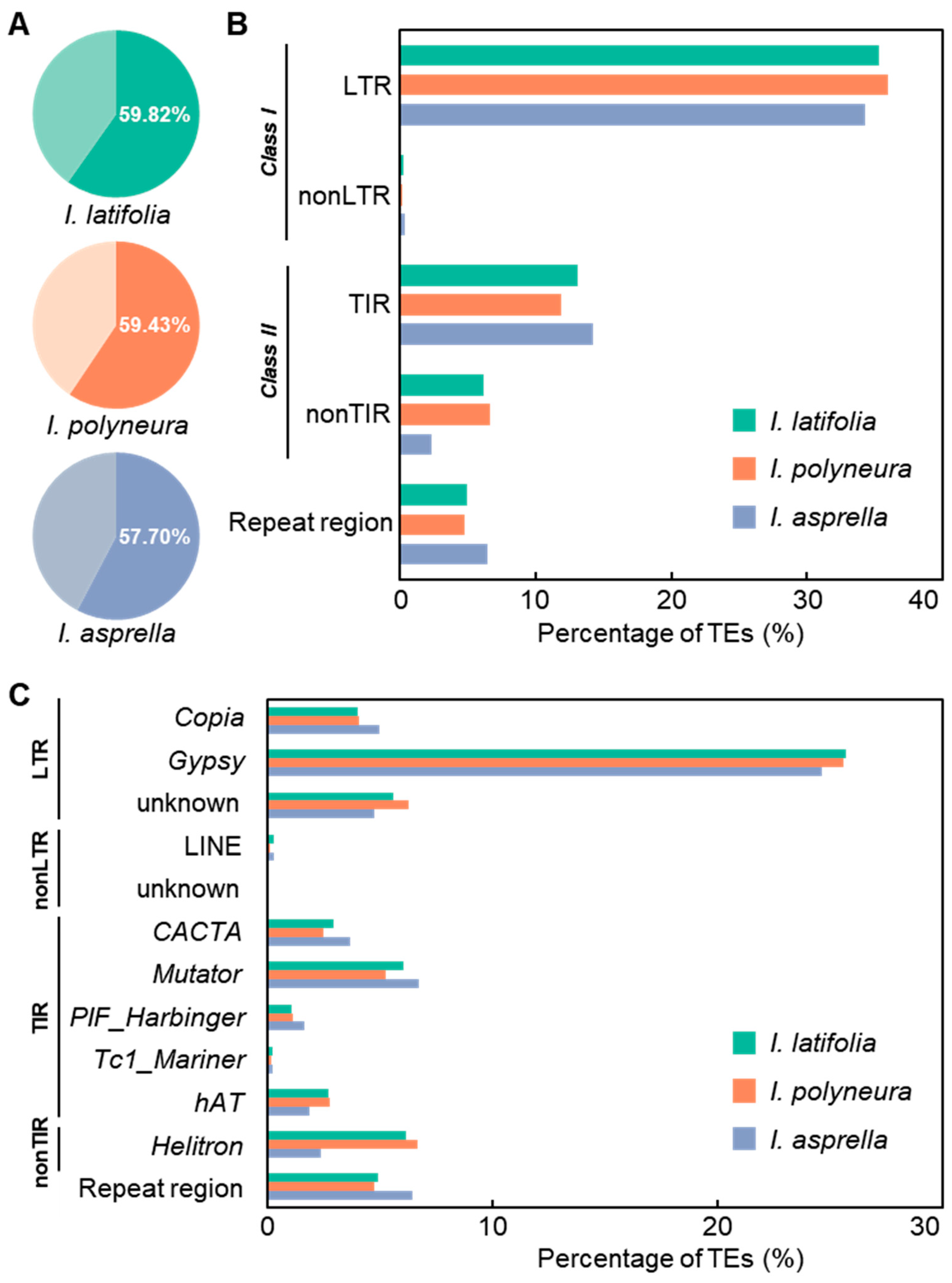
2.3. Identification of TEs in Three Ilex Species
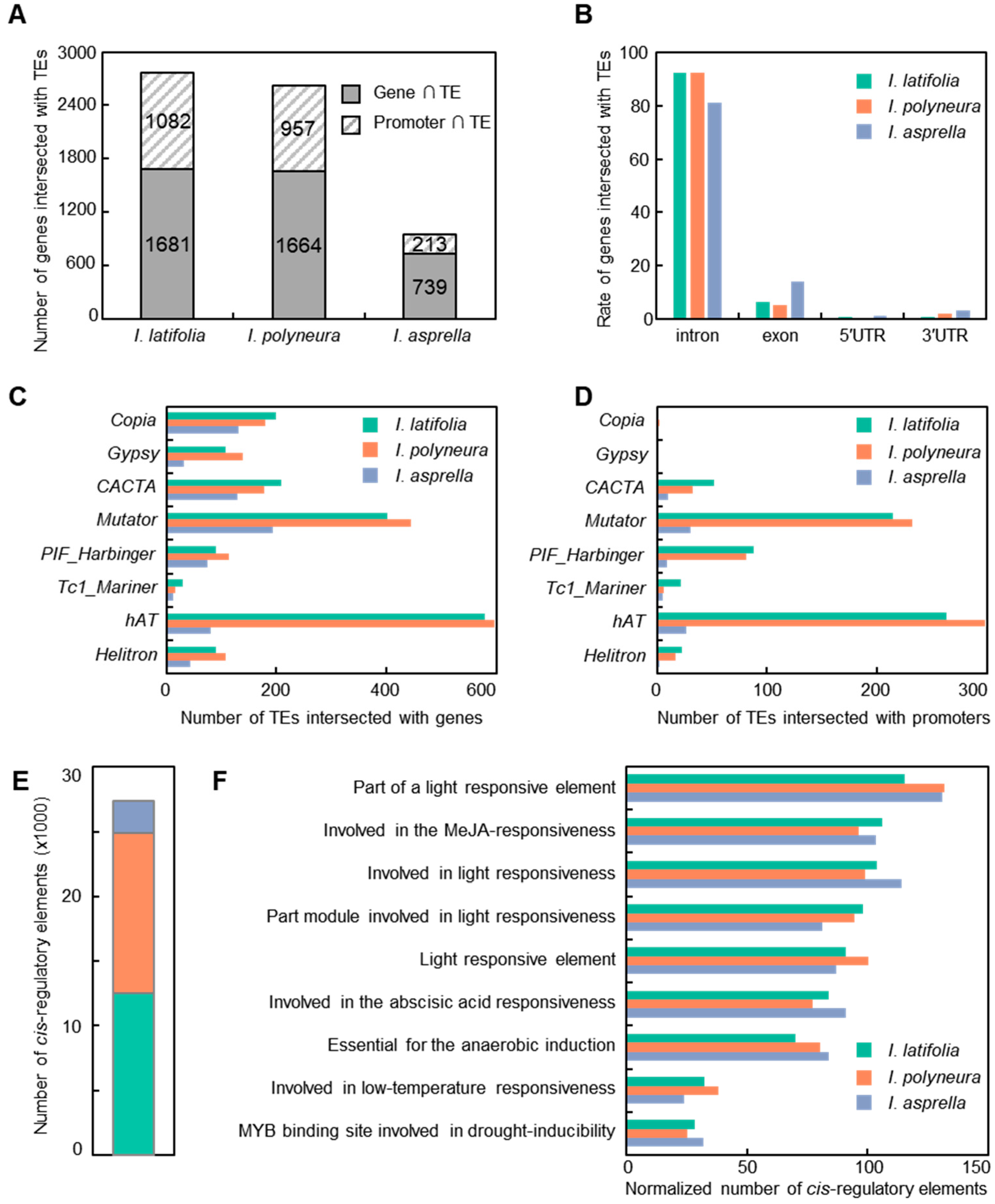
2.4. TEs Mediate Genetic Effects in Three Ilex Species
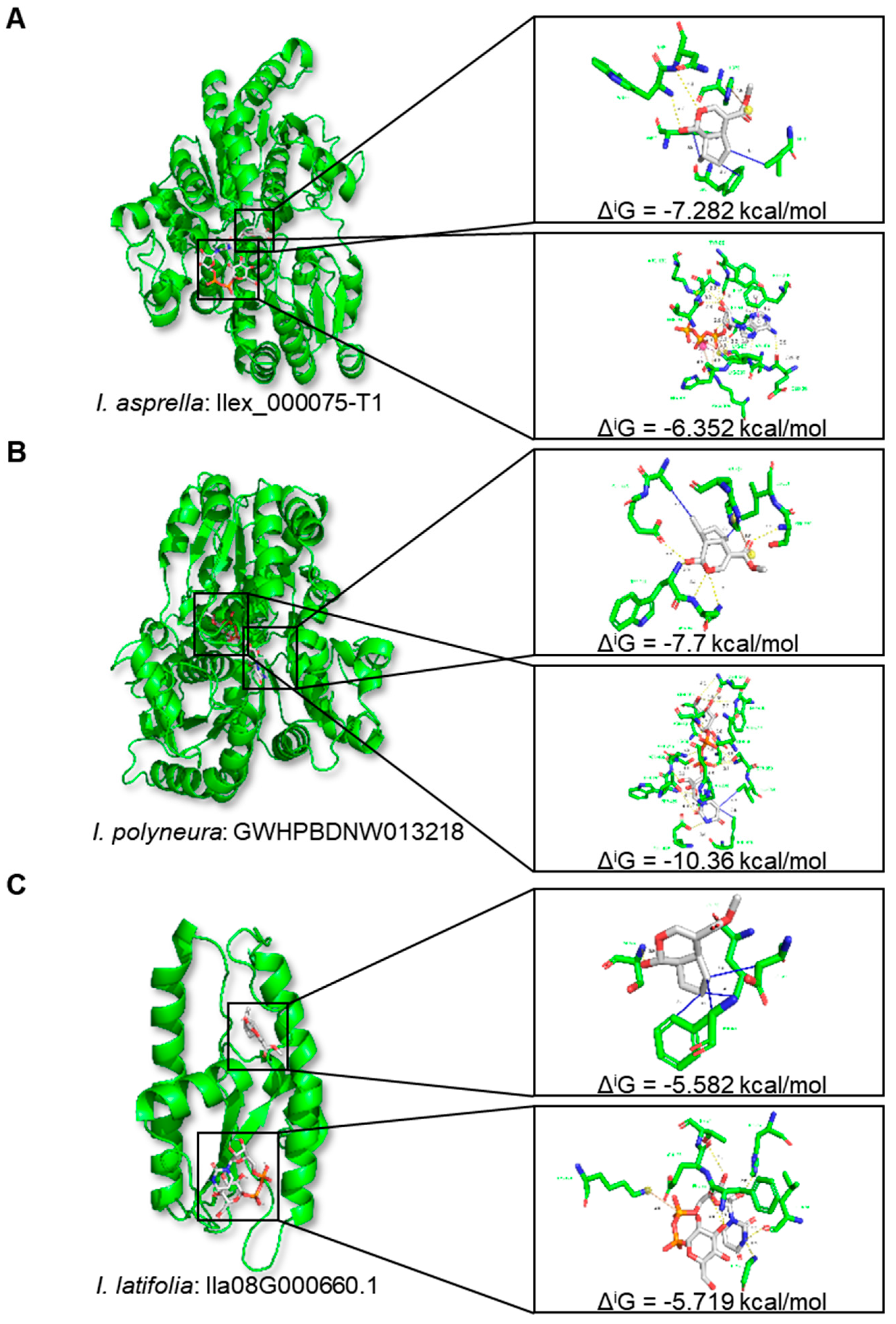
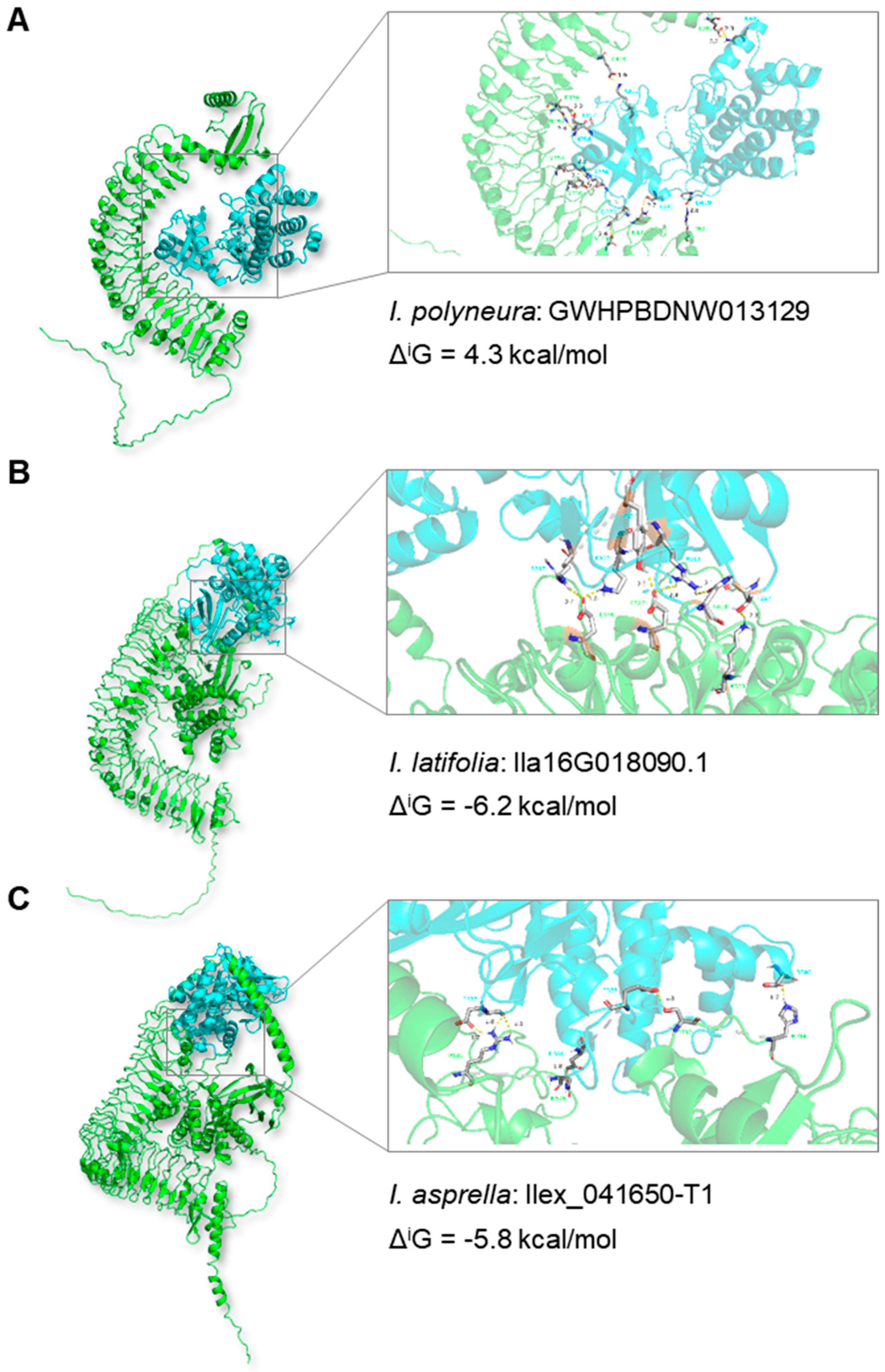
2.5. Impact of TE Insertions on Protein Structures: Two Case Studies
3. Discussion
4. Materials and Methods
4.1. Source of Genomes and Chromosome Counts
4.2. Collinearity-Based Chromosome Fusion Analysis
4.3. Identification of Transcription Factor Families
4.4. Identification of Transposable Elements
4.5. Distribution of Genes and TEs
4.6. Intersection of Genes and TEs
4.7. Extraction of Intron Positions in Genome
4.8. Identification of Cis-Regulatory Elements
4.9. Prediction of Protein Structure
4.10. Protein–Protein Interaction Simulation
4.11. Molecular Docking Simulation of Proteins and Small-Molecule Ligands
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feulner, P.G.D.; De-Kayne, R. Genome evolution, structural rearrangements and speciation. J. Evol. Biol. 2017, 30, 1488–1490. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.R.; Inouye, B.D.; Johnson, M.T.J.; Underwood, N.; Vellend, M. Ecological consequences of genetic diversity. Ecol. Lett. 2008, 11, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Reed, D.H.; Frankham, R. Correlation between fitness and genetic diversity. Conserv. Biol. 2003, 17, 230–237. [Google Scholar] [CrossRef]
- Otto, S.P.; Whitton, J. Polyploid incidence and evolution. Annu. Rev. Genet. 2000, 34, 401–437. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.A. The Role of Chromosomal Change in Plant Evolution; Oxford University Press: Oxford, UK, 2002; ISBN 9780197701928. [Google Scholar]
- Wessler, S.R. Transposable elements and the evolution of eukaryotic genomes. Proc. Natl. Acad. Sci. USA 2006, 103, 17600–17601. [Google Scholar] [CrossRef]
- Schubert, I.; Lysak, M.A. Interpretation of karyotype evolution should consider chromosome structural constraints. Trends Genet. 2011, 27, 207–216. [Google Scholar] [CrossRef]
- Weiss-Schneeweiss, H.; Schneeweiss, G.M. Karyotype Diversity and Evolutionary Trends in Angiosperms. In Plant Genome Diversity Volume 2: Physical Structure, Behaviour and Evolution of Plant Genomes; Greilhuber, J., Dolezel, J., Wendel, J.F., Eds.; Springer: Vienna, Austria, 2013; pp. 209–230. [Google Scholar]
- Miao, Y.; Luo, D.; Zhao, T.; Du, H.; Liu, Z.; Xu, Z.; Guo, L.; Chen, C.; Peng, S.; Li, J.X.; et al. Genome sequencing reveals chromosome fusion and extensive expansion of genes related to secondary metabolism in Artemisia argyi. Plant Biotechnol. J. 2022, 20, 1902–1915. [Google Scholar] [CrossRef]
- JW, I.J.; Baldini, A.; Ward, D.C.; Reeders, S.T.; Wells, R.A. Origin of human chromosome 2: An ancestral telomere-telomere fusion. Proc. Natl. Acad. Sci. USA 1991, 88, 9051–9055. [Google Scholar]
- Cicconardi, F.; Lewis, J.J.; Martin, S.H.; Reed, R.D.; Danko, C.G.; Montgomery, S.H. Chromosome fusion affects genetic diversity and evolutionary turnover of functional loci but consistently depends on chromosome size. Mol. Biol. Evol. 2021, 38, 4449–4462. [Google Scholar] [CrossRef]
- Xuan, Y.; Ma, B.; Li, D.; Tian, Y.; Zeng, Q.; He, N. Chromosome restructuring and number change during the evolution of Morus notabilis and Morus alba. Hortic. Res. 2022, 9, uhab030. [Google Scholar] [CrossRef]
- Vara, C.; Paytuví-Gallart, A.; Cuartero, Y.; Álvarez-González, L.; Marín-Gual, L.; Garcia, F.; Florit-Sabater, B.; Capilla, L.; Sanchéz-Guillén, R.A.; Sarrate, Z.; et al. The impact of chromosomal fusions on 3D genome folding and recombination in the germ line. Nat. Commun. 2021, 12, 2981. [Google Scholar] [CrossRef] [PubMed]
- Hauffe, H.C.; Searle, J.B. Chromosomal heterozygosity and fertility in house mice (Mus musculus domesticus) from Northern Italy. Genetics 1998, 150, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- de Vos, J.M.; Augustijnen, H.; Bätscher, L.; Lucek, K. Speciation through chromosomal fusion and fission in Lepidoptera. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190539. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Sun, X.; Cormack, B.P.; Boeke, J.D. Karyotype engineering by chromosome fusion leads to reproductive isolation in yeast. Nature 2018, 560, 392–396. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. USA 2012, 36, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.N.; Feschotte, C. A field guide to eukaryotic transposable elements. Annu. Rev. Genet. 2020, 54, 539–561. [Google Scholar] [CrossRef]
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Annu. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Makałowski, W.; Gotea, V.; Pande, A.; Makałowska, I. Transposable Elements: Classification, Identification, and Their Use As a Tool for Comparative Genomics; Anisimova, M., Ed.; Evolutionary Genomics: Humana, New York, NY, USA, 2019; Volume 1910, pp. 177–207. [Google Scholar]
- Klein, S.J.; O’Neill, R.J. Transposable elements: Genome innovation, chromosome diversity, and centromere conflict. Chromosome Res. 2018, 26, 5–23. [Google Scholar] [CrossRef]
- Stritt, C.; Wyler, M.; Gimmi, E.L.; Pippel, M.; Roulin, A.C. Diversity, dynamics and effects of long terminal repeat retrotransposons in the model grass Brachypodium distachyon. New Phytol. 2019, 227, 1736–1748. [Google Scholar] [CrossRef]
- Ramakrishnan, M.; Satish, L.; Sharma, A.; Kurungara Vinod, K.; Emamverdian, A.; Zhou, M.; Wei, Q. Transposable elements in plants: Recent advancements, tools and prospects. Plant Mol. Biol. Rep. 2022, 40, 628–645. [Google Scholar] [CrossRef]
- Hassan, A.H.; Mokhtar, M.M.; El Allali, A. Transposable elements: Multifunctional players in the plant genome. Front. Plant Sci. 2024, 14, 1330127. [Google Scholar] [CrossRef] [PubMed]
- Parisod, C.; Alix, K.; Just, J.; Petit, M.; Sarilar, V.; Mhiri, C.; Ainouche, M.; Chalhoub, B.; Grandbastien, M.A. Impact of transposable elements on the organization and function of allopolyploid genomes. New Phytol. 2010, 186, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Lockton, S.; Gaut, B.S. The contribution of Transposable Elements to expressed coding sequence in Arabidopsis thaliana. J. Mol. Evol. 2009, 68, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Gisby, J.S.; Catoni, M. The widespread nature of Pack-TYPE transposons reveals their importance for plant genome evolution. PLoS Genet. 2022, 18, e1010078. [Google Scholar] [CrossRef]
- Niu, X.-M.; Xu, Y.-C.; Li, Z.-W.; Bian, Y.-T.; Hou, X.-H.; Chen, J.-F.; Zou, Y.-P.; Jiang, J.; Wu, Q.; Ge, S.; et al. Transposable elements drive rapid phenotypic variation in Capsella rubella. Proc. Natl. Acad. Sci. USA 2019, 116, 6908–6913. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, J.; Han, X.; Li, J.; Gao, Y.; Richards, C.M.; Zhang, C.; Tian, Y.; Liu, G.; Gul, H.; et al. A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 2019, 10, 1494. [Google Scholar] [CrossRef]
- Chen, G.; Wang, R.; Jiang, Y.; Dong, X.; Xu, J.; Xu, Q.; Kan, Q.; Luo, Z.; Springer, N.M.; Li, Q. A novel active transposon creates allelic variation through altered translation rate to influence protein abundance. Nucleic Acids Res. 2023, 51, 595–609. [Google Scholar] [CrossRef]
- Cai, X.; Lin, R.; Liang, J.; King, G.J.; Wu, J.; Wang, X. Transposable element insertion: A hidden major source of domesticated phenotypic variation in Brassica rapa. Plant Biotechnol. J. 2022, 20, 1298–1310. [Google Scholar] [CrossRef]
- Li, X.; Dai, X.; He, H.; Lv, Y.; Yang, L.; He, W.; Liu, C.; Wei, H.; Liu, X.; Yuan, Q.; et al. A pan-TE map highlights transposable elements underlying domestication and agronomic traits in Asian rice. Natl. Sci. Rev. 2024, 11, nwae188. [Google Scholar] [CrossRef]
- Guo, Z.; Kuang, Z.; Tao, Y.; Wang, H.; Wan, M.; Hao, C.; Shen, F.; Yang, X.; Li, L.; Arkhipova, I. Miniature Inverted-repeat Transposable Elements drive rapid microRNA diversification in angiosperms. Mol. Biol. Evol. 2022, 39, msac224. [Google Scholar] [CrossRef] [PubMed]
- Loizeau, P.A.; Andrews, S.V.; Spichiger, S.; Aquifoliaceae, R. The families and genera of vascular plants. In Flowering Plants. Eudicots; Kubitzki, K., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; Volume 10, pp. 31–36. ISBN 978-3-319-93604-8. [Google Scholar]
- Yao, X.; Zhang, F.; Corlett, R.T. Utilization of the hollies (Ilex L. spp.): A review. Forests 2022, 13, f13010094. [Google Scholar] [CrossRef]
- Xu, K.W.; Wei, X.F.; Lin, C.X.; Zhang, M.; Zhang, Q.; Zhou, P.; Fang, Y.M.; Xue, J.Y.; Duan, Y.F. The chromosome-level holly (Ilex latifolia) genome reveals key enzymes in triterpenoid saponin biosynthesis and fruit color change. Front. Plant Sci. 2022, 13, 982323. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Lu, Z.; Song, Y.; Hu, X.; Corlett, R.T. A chromosome-scale genome assembly for the holly (Ilex polyneura) provides insights into genomic adaptations to elevation in southwest China. Hortic. Res. 2022, 9, uhab049. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.L.; Nong, W.; Wong, K.H.; Law, S.T.; So, W.L.; Chan, J.J.; Zhang, J.; Lau, T.D.; Hui, J.H.; Shaw, P.C. Chromosomal level genome of Ilex asprella and insight into antiviral triterpenoid pathway. Genomics 2022, 114, 110366. [Google Scholar] [CrossRef]
- Guo, Z.; Wei, J.; Xu, Z.; Lin, C.; Peng, Y.; Wang, Q.; Wang, D.; Yang, X.; Xu, K.W. HollyGTD: An integrated database for holly (Aquifoliaceae) genome and taxonomy. Front. Plant Sci. 2023, 14, 1220925. [Google Scholar] [CrossRef]
- Rice, A.; Glick, L.; Abadi, S.; Einhorn, M.; Kopelman, N.M.; Salman-Minkov, A.; Mayzel, J.; Chay, O.; Mayrose, I. The Chromosome Counts Database (CCDB)—A community resource of plant chromosome numbers. New Phytol. 2014, 206, 19–26. [Google Scholar] [CrossRef]
- Yang, Y.; Jiang, L.; Liu, E.D.; Liu, W.L.; Chen, L.; Kou, Y.X.; Fan, D.M.; Cheng, S.M.; Zhang, Z.Y.; Peng, H. Time to update the sectional classification of Ilex (Aquifoliaceae): New insights from Ilex phylogeny, morphology, and distribution. J. Syst. Evol. 2023, 61, 1036–1046. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids. Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Jin, J.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids. Res. 2019, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Rombauts, S.; Dehais, P.; Van Montagu, M.; Rouze, P. PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res. 1999, 27, 295–296. [Google Scholar] [CrossRef] [PubMed]
- Nagatoshi, M.; Terasaka, K.; Nagatsu, A.; Mizukami, H. Iridoid-specific Glucosyltransferase from Gardenia jasminoides. J. Biol. Chem. 2011, 286, 32866–32874. [Google Scholar] [CrossRef] [PubMed]
- Asada, K.; Salim, V.; Masada-Atsumi, S.; Edmunds, E.; Nagatoshi, M.; Terasaka, K.; Mizukami, H.; De Luca, V. A 7-Deoxyloganetic acid glucosyltransferase contributes a key step in secologanin biosynthesis in madagascar periwinkle. Plant Cell 2013, 25, 4123–4134. [Google Scholar] [CrossRef]
- Li, J.; Wen, J.; Lease, K.A.; Doke, J.T.; Tax, F.E.; Walker, J.C. BAK1, an Arabidopsis LRR Receptor-like Protein Kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell 2002, 110, 213–222. [Google Scholar] [CrossRef]
- Sun, Y.; Li, L.; Macho, A.P.; Han, Z.; Hu, Z.; Zipfel, C.; Zhou, J.-M.; Chai, J. Structural basis for flg22-induced activation of the Arabidopsis FLS2-BAK1 immune complex. Science 2013, 342, 624–628. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Kim, J.M.; Vanguri, S.; Boeke, J.D.; Gabriel, A.; Voytas, D.F. Transposable Elements and Genome Organization: A comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae senome sequence. Genome Res. 1998, 8, 464–478. [Google Scholar] [CrossRef]
- Flavell, R.B.; Bennett, M.D.; Smith, J.B.; Smith, D.B. Genome size and the proportion of repeated nucleotide sequence DNA in plants. Biochem. Genet. 1974, 12, 257–269. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chao, H.; Chen, L.; Craig, P.A.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB.org): Delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2023, 51, D488–D508. [Google Scholar] [CrossRef] [PubMed]
- Honorato, R.V.; Trellet, M.E.; Jiménez-García, B.; Schaarschmidt, J.J.; Giulini, M.; Reys, V.; Koukos, P.I.; Rodrigues, J.P.G.L.M.; Karaca, E.; van Zundert, G.C.P.; et al. The HADDOCK2.4 web server for integrative modeling of biomolecular complexes. Nat. Protoc. 2024. [Google Scholar] [CrossRef]
- Chrödinger, L.L.C. The PyMOL Molecular Graphics System, Version 3.0.0; PyMOL. Available online: https://pymol.org/ (accessed on 15 June 2024).
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids. Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, andpython bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Superfamily | I. latifolia | I. asprella | I. polyneura | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Count | Length (bp) | % | Count | Length (bp) | % | Count | Length (bp) | % | ||
| LTR | Copia | 38,219 | 30,700,831 | 4.01% | 51,998 | 35,494,928 | 4.96% | 35,857 | 29,629,464 | 4.08% |
| Gypsy | 145,246 | 196,792,508 | 25.69% | 170,245 | 176,221,772 | 24.63% | 146,734 | 186,042,503 | 25.59% | |
| unknown | 74,946 | 42,916,075 | 5.60% | 54,846 | 33,952,958 | 4.75% | 83,403 | 45,770,036 | 6.29% | |
| Non-LTR | LINE_element | 3491 | 2,077,187 | 0.27% | 3790 | 2,279,086 | 0.32% | 2605 | 1,113,338 | 0.15% |
| unknown | 570 | 167,545 | 0.02% | 0 | 0 | 0.00% | 0 | 0 | 0.00% | |
| TIR | CACTA | 69,302 | 22,641,095 | 2.96% | 79,124 | 26,401,225 | 3.69% | 58,667 | 18,315,040 | 2.52% |
| Mutator | 141,937 | 46,577,919 | 6.08% | 139,183 | 48,341,143 | 6.76% | 119,261 | 38,338,622 | 5.27% | |
| PIF_Harbinger | 26,367 | 8,491,650 | 1.11% | 37,818 | 11,904,229 | 1.66% | 26,525 | 8,109,161 | 1.12% | |
| Tc1_Mariner | 7196 | 1,968,434 | 0.26% | 6822 | 1,886,253 | 0.26% | 4588 | 1,248,614 | 0.17% | |
| hAT | 56,873 | 20,727,706 | 2.71% | 34,981 | 13,398,044 | 1.87% | 54,114 | 20,263,483 | 2.79% | |
| Non-TIR | Helitron | 139,768 | 47,218,541 | 6.16% | 52,976 | 16,905,447 | 2.36% | 143,665 | 48,573,149 | 6.68% |
| repeat_region | 125,490 | 37,903,467 | 4.95% | 165,145 | 45,991,681 | 6.43% | 123,449 | 34,683,080 | 4.77% | |
| Total | 829,405 | 458,182,958 | 59.82% | 796,928 | 412,776,766 | 57.70% | 798,868 | 432,086,490 | 59.43% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.; Wei, H.; Li, M.; Qiu, Y.; Li, L.; Xu, K.-W.; Guo, Z. Impact of Chromosomal Fusion and Transposable Elements on the Genomic Evolution and Genetic Diversity of Ilex Species. Plants 2024, 13, 2649. https://doi.org/10.3390/plants13182649
Xu Z, Wei H, Li M, Qiu Y, Li L, Xu K-W, Guo Z. Impact of Chromosomal Fusion and Transposable Elements on the Genomic Evolution and Genetic Diversity of Ilex Species. Plants. 2024; 13(18):2649. https://doi.org/10.3390/plants13182649
Chicago/Turabian StyleXu, Zhenxiu, Haikun Wei, Mingyue Li, Yingjie Qiu, Lei Li, Ke-Wang Xu, and Zhonglong Guo. 2024. "Impact of Chromosomal Fusion and Transposable Elements on the Genomic Evolution and Genetic Diversity of Ilex Species" Plants 13, no. 18: 2649. https://doi.org/10.3390/plants13182649
APA StyleXu, Z., Wei, H., Li, M., Qiu, Y., Li, L., Xu, K.-W., & Guo, Z. (2024). Impact of Chromosomal Fusion and Transposable Elements on the Genomic Evolution and Genetic Diversity of Ilex Species. Plants, 13(18), 2649. https://doi.org/10.3390/plants13182649