
Genome-Wide Identification of Osmanthus fragrans Histone Modification Genes and Analysis of Their Expression during the Flowering Process and under Azacytidine and Ethylene Treatments

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Identification and Characterization Analysis of HMs in the O. fragrans Genome
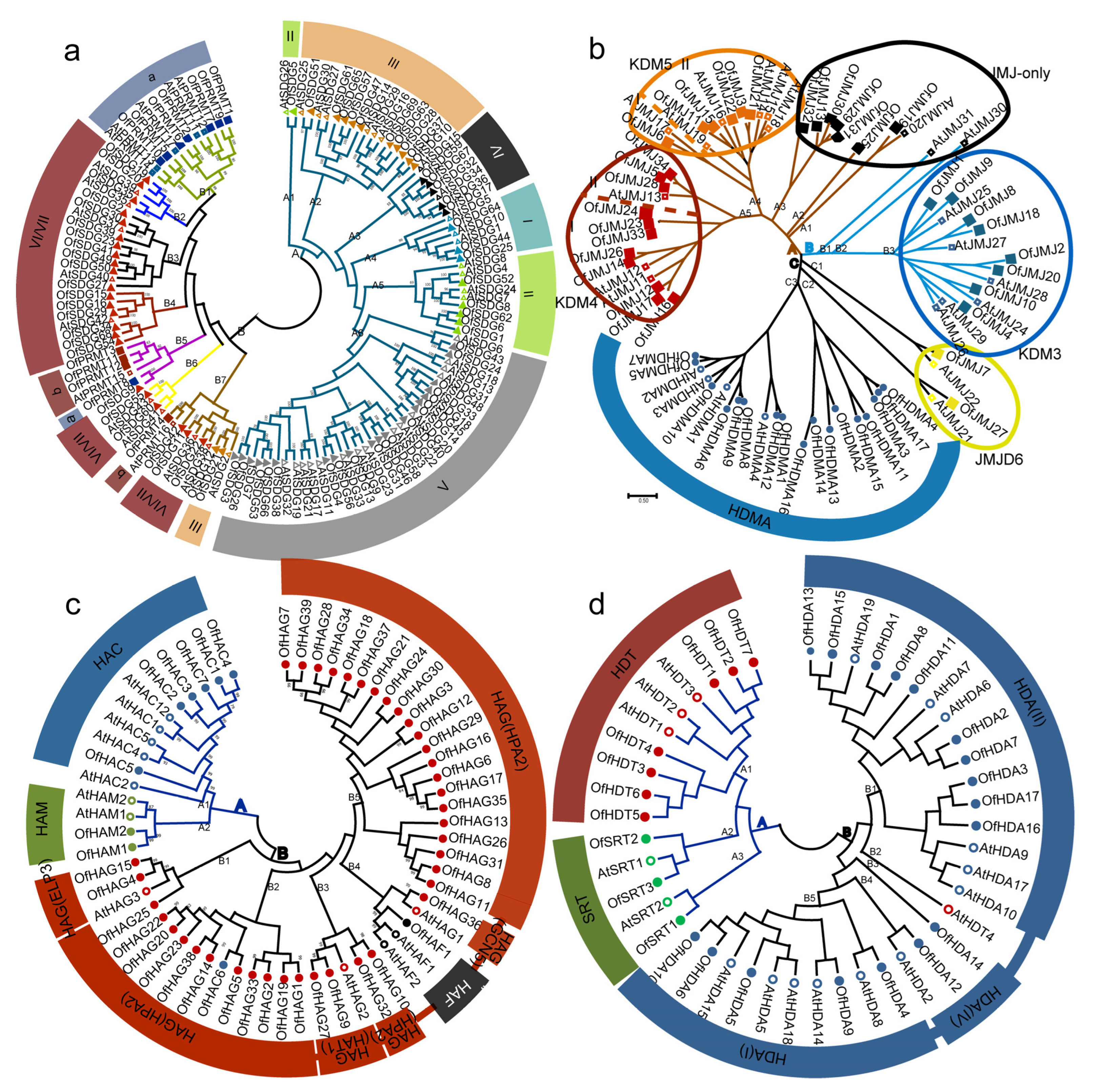
2.2. Phylogenetic Analysis of HMs between O. fragrans and Arabidopsis
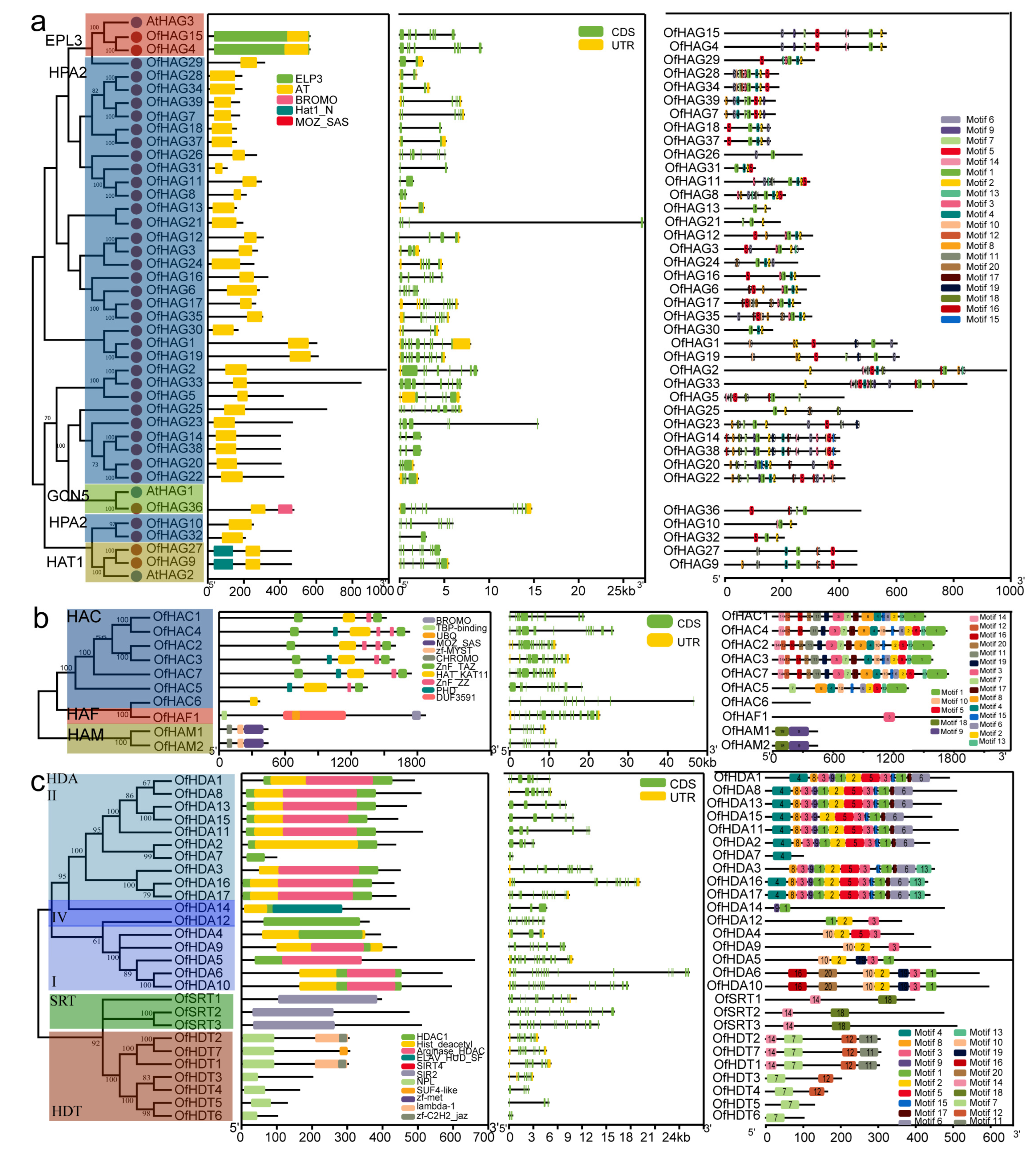
2.3. Gene Structure and Conserved Motif Analyses of OfHMs
2.4. Cis-Acting Element Analysis of OfHMs
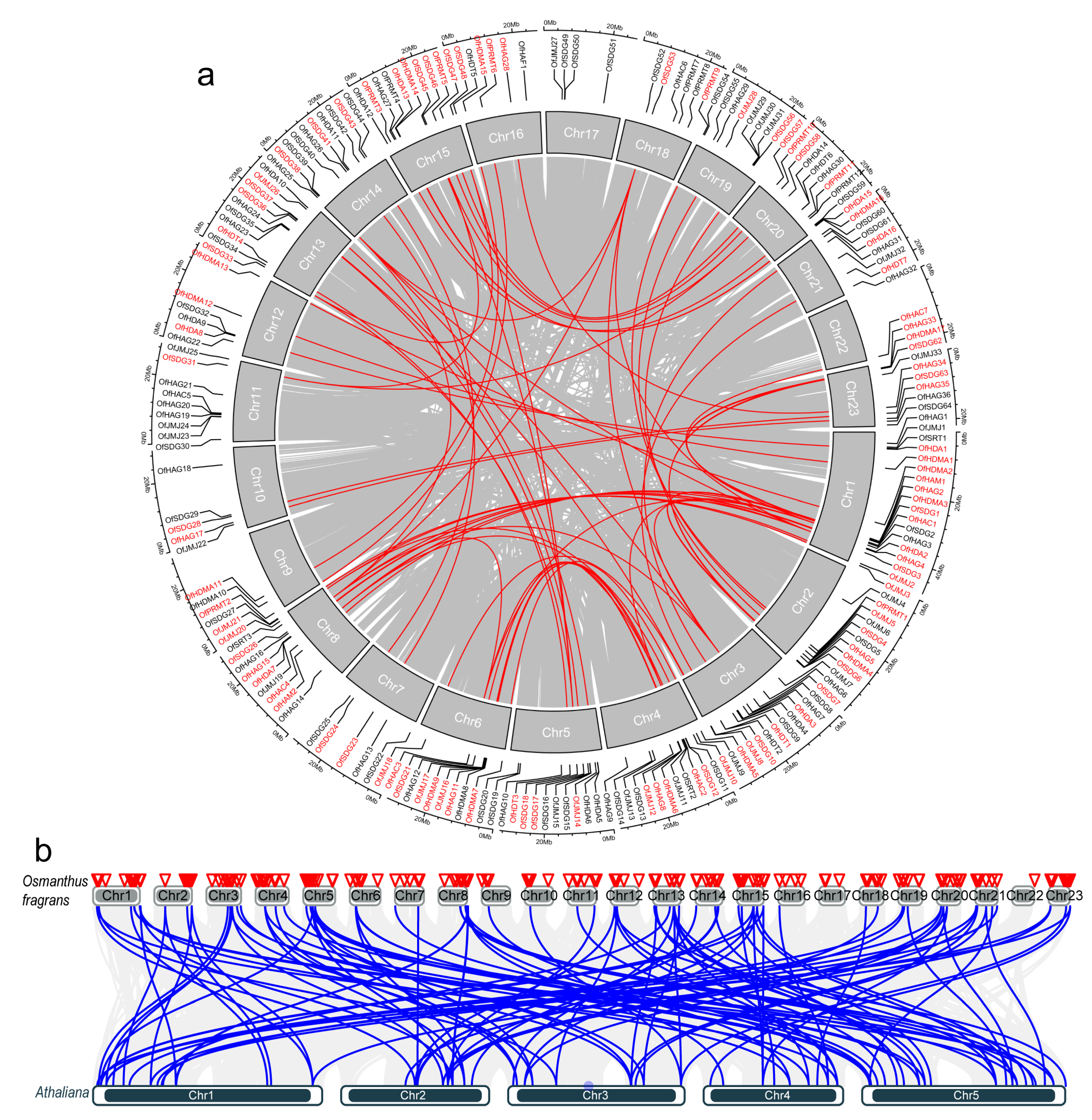
2.5. Chromosomal Distribution and Synteny Analysis of OfHMs
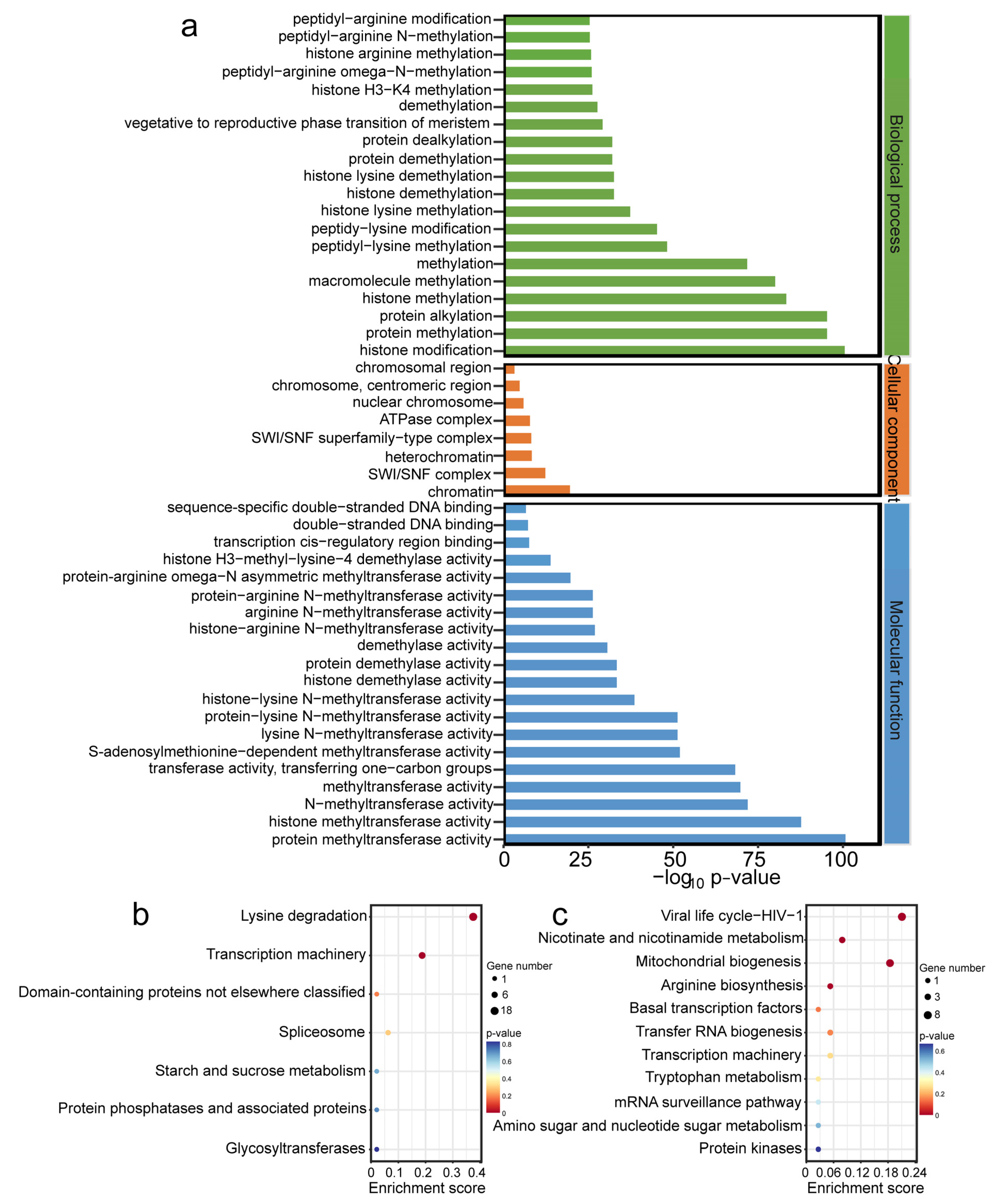
2.6. Functional Enrichment Analysis of OfHMs
2.7. Prediction of Interactions of OfHM Proteins
2.8. Expression Analysis of OfHMs in Different Tissues and Flowering Stages
2.9. Expression Analysis of OfHMs under Ethylene and 5′-Azacytidine (Aza) Treatment
2.10. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Analysis of OfHM Genes
3. Discussion
3.1. O. fragrans HM Genes in Comparison with Other Plant Species
3.2. Evolution and Amplification Analyses of OfHM Gene Family
3.3. OfHMs Are Involved in the Flowering Process and the Induction of Aza and Ethylene Responses
4. Materials and Methods
4.1. Identification and Chromosome Location of OfHM Gene Family
4.2. Physicochemical Characteristics Examination, Phylogenetic Tree Elaboration, and Gene Structure Research
4.3. Cis-Acting Regulatory Element Analysis of OfHM Genes
4.4. Tandem Duplication and Synteny Analyses
4.5. Protein–Protein Interaction Network Construction and Functional Enrichment Analysis of OfHM Genes
4.6. Plant Materials and Treatment
4.7. Identification of OfHM Expression Profiles Using High-Throughput Sequencing
4.8. qRT-PCR Analysis of OfHM Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pfluger, J.; Wagner, D. Histone modifications and dynamic regulation of genome accessibility in plants. Curr. Opin. Plant Biol. 2007, 10, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ahmad, B.; Liang, C.; Shi, X.; Sun, R.; Zhang, S.; Du, G. Bioinformatics and expression analysis of histone modification genes in grapevine predict their involvement in seed development, powdery mildew resistance, and hormonal signaling. BMC Plant Biol. 2020, 20, 412. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, J.; Liu, B.; Xu, Z.Y. Dynamic regulation of DNA methylation and histone modifications in response to abiotic stresses in plants. J. Integr. Plant Biol. 2022, 64, 23. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. David The language of covalent histone modifications. Nature 2000, 403, 41. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 307–318. [Google Scholar] [CrossRef]
- Lusser, A.; Kolle, D.; Loidl, P. Histone acetylation: Lessons from the plant kingdom. Trends Plant Sci. 2001, 6, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Müller, A.; Napoli, C.A.; Selinger, D.A.; Pikaard, C.S.; Richards, E.J.; Bender, J.; Mount, D.W.; Jorgensen, R.A. Analysis of histone acetyltransferase and histone deacetylase families of Arabidopsis thaliana suggests functional diversification of chromatin modification among multicellular eukaryotes. Nucleic Acids Res. 2002, 30, 5036–5055. [Google Scholar] [CrossRef]
- Fan, S.; Wang, J.; Lei, C.; Gao, C.; Yang, Y.; Li, Y.; An, N.; Zhang, D.; Han, M. Identification and characterization of histone modification gene family reveal their critical responses to flower induction in apple. BMC Plant Biol. 2018, 18, 173. [Google Scholar] [CrossRef]
- Verdone, L.; Caserta, M.; Mauro, E.D. Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. 2005, 83, 344–353. [Google Scholar] [CrossRef]
- Chen, Z.J.; Tian, L. Roles of dynamic and reversible histone acetylation in plant development and polyploidy. BBA-Gene Struct. Expr. 2007, 1769, 295–307. [Google Scholar] [CrossRef]
- Fu, W.; Wu, K.; Duan, J. Sequence and expression analysis of histone deacetylases in rice. Biochem. Biophys. Res. Commun. 2007, 356, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bernatavichute, Y.V.; Cokus, S.; Jacobsen, P.S.E. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 2009, 10, R62. [Google Scholar] [CrossRef] [PubMed]
- Peláez, I.M.; Kalogeropoulou, M.; Ferraro, A.; Voulgari, A.; Pankotai, T.; Boros, I.; Pintzas, A. Oncogenic RAS alters the global and gene-specific histone modification pattern during epithelial-mesenchymal transition in colorectal carcinoma cells. Int. J. Biochem. Cell Biol. 2010, 42, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Aiese Cigliano, R.; Sanseverino, W.; Cremona, G.; Ercolano, M.R.; Consiglio, F.M. Genome-wide analysis of histone modifiers in tomato: Gaining an insight into their developmental roles. BMC Genom. 2013, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Manjun, P.; Peiyuan, Y.; Xuncheng, L.; Caiqin, L.; Rui, X.; Jianguo, L.; Minglei, Z. Genome-wide identification of histone modifiers and their expression patterns during fruit abscission in litchi. Front. Plant Sci. 2017, 8, 639. [Google Scholar]
- Xu, J.; Xu, H.; Liu, Y.; Wang, X.; Xu, Q.; Deng, X. Genome-wide identification of sweet orange (Citrus sinensis) histone modification gene families and their expression analysis during the fruit development and fruit-blue mold infection process. Front. Plant Sci. 2015, 6, 607. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, M.; Zhang, W.; Zhao, J.; Zhang, J.; Wu, K.; Tian, L.; Duan, J. Histone acetyltransferases in rice (Oryza sativa L.): Phylogenetic analysis, subcellular localization and expression. BMC Plant Biol. 2012, 12, 145. [Google Scholar] [CrossRef]
- Gu, T.; Han, Y.; Huang, R.; McAvoy, R.J.; Li, Y. Identification and characterization of histone lysine methylation modifiers in Fragaria vesca. Sci. Rep. 2016, 6, 23581. [Google Scholar] [CrossRef]
- Alinsug, M.V.; Yu, C.; Wu, K. Phylogenetic analysis, subcellular localization, and expression patterns of RPD3/HDA1 family histone deacetylases in plants. BMC Plant Biol. 2009, 9, 37. [Google Scholar] [CrossRef]
- Thorstensen, T.; Grini, P.E.; Aalen, R.B. SET domain proteins in plant development. BBA-Gene Regul. Mech. 2011, 1809, 407–420. [Google Scholar] [CrossRef]
- Liu, X.C.; Yang, S.G.; Zhao, M.L.; Luo, M.; Yu, C.W.; Chen, C.Y.; Tai, R.; Wu, K. Transcriptional repression by histone deacetylases in plants. Mol. Plant. 2014, 7, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Mukherjee, I.; Thum, K.E.; Tanurdzic, M.; Katari, M.S.; Obertello, M.; Edwards, M.B.; McCombie, W.R.; Martienssen, R.A.; Coruzzi, G.M. The histone methyltransferase SDG8 mediates the epigenetic modification of light and carbon responsive genes in plants. Genome Biol. 2015, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Kikuchi, A.; Kamada, H. The Arabidopsis histone deacetylases HDA6 and HDA19 contribute to the repression of embryonic properties after germination. Plant Physiol. 2008, 146, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhang, H.; Xing, L.; Xu, S.; Liu, H.; Chong, K.; Xu, Y. Requirement of histone acetyltransferases HAM1 and HAM2 for epigenetic modification of FLC in regulating flowering in Arabidopsis. J. Plant Physiol. 2013, 170, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Liu, C.; Pei, Y.; Deng, X.; Niu, L.; Cao, X. Involvement of the histone acetyltransferase AtHAC1 in the regulation of flowering time via repression of FLOWERING LOCUS C in Arabidopsis. Plant Physiol. 2007, 143, 1660–1668. [Google Scholar] [CrossRef]
- Wu, K.; Zhang, L.; Zhou, C.; Yu, C.; Chaikam, V. HDA6 is required for jasmonate response, senescence and flowering in Arabidopsis. J. Exp. Bot. 2008, 59, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Latrasse, D.; Servet, C.; Zhou, D. Arabidopsis histone deacetylase HDA9 regulates flowering time through repression of AGL19. Biochem. Biophys. Res. Commun. 2013, 432, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.Q.; Chen, Q.; Lin, R.N.; Li, Y.Q.; Li, L.; Chen, S.; He, X.J. The HDA19 histone deacetylase complex is involved in the regulation of flowering time in a photoperiod-dependent manner. Plant J. 2019, 98, 448–464. [Google Scholar] [CrossRef]
- Yang, H.; Mo, H.; Fan, D.; Cao, Y.; Cui, S.; Ma, L. Overexpression of a histone H3K4 demethylase, JMJ15, accelerates flowering time in Arabidopsis. Plant Cell Rep. 2012, 31, 1297–1308. [Google Scholar] [CrossRef]
- Yang, H.; Han, Z.; Cao, Y.; Fan, D.; Li, H.; Mo, H.; Feng, Y.; Liu, L.; Wang, Z.; Yue, Y. A companion cell-dominant and developmentally regulated H3K4 demethylase controls flowering time in Arabidopsis via the repression of FLC expression. PLoS Genet. 2012, 8, e1002664. [Google Scholar] [CrossRef]
- Ning, Y.; Ma, Z.; Huang, H.; Mo, H.; Zhao, T.; Li, L.; Cai, T.; Chen, S.; Ma, L.; He, X. Two novel NAC transcription factors regulate gene expression and flowering time by associating with the histone demethylase JMJ14. Nucleic Acids Res. 2015, 43, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiu, L.; Huang, L.; Cao, J. BcJMJ30, the gene encoding jmjC domain-containing histone demethylase is associated with pollen development and fertilization in Brassica campestris ssp. chinensis. Plant Mol. Biol. Rep. 2011, 30, 529–538. [Google Scholar] [CrossRef]
- Berr, A.; Xu, L.; Gao, J.; Cognat, V.R.; Steinmetz, A. SET DOMAIN GROUP25 encodes a histone methyltransferase and is involved in FLOWERING LOCUS C activation and repression of flowering. Plant Physiol. 2009, 151, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.M.; Li, M.T.; Jin, W.W.; Li, S.; Zhang, S.Q.; Yu, L.J. Variations in the components of Osmanthus fragrans Lour. essential oil at different stages of flowering. Food Chem. 2009, 114, 233–236. [Google Scholar] [CrossRef]
- Wu, L.; Liu, J.; Huang, W.; Wang, Y.; Chen, Q.; Lu, B. Exploration of Osmanthus fragrans Lour.’s composition, nutraceutical functions and applications. Food Chem. 2021, 377, 131853. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Luan, F.; Bao, Y.; Peng, X.; Rao, Z.; Tang, Q.; Zeng, N. Traditional uses, phytochemical constituents and pharmacological properties of Osmanthus fragrans: A review. J. Ethnopharmacol. 2022, 293, 115273. [Google Scholar] [CrossRef]
- Zou, J.; Cai, X.; Wang, C. The spatial and temporal distribution of programmed cell death (PCD) during petal senescence of Osmanthus fragrans. Acta Hortic. 2017, 1185, 315–324. [Google Scholar] [CrossRef]
- Zou, J.J.; Zhou, Y.; Cai, X.; Wang, C.Y. Increase in DNA fragmentation and the role of ethylene and reactive oxygen species in petal senescence of Osmanthus fragrans. Postharvest Biol. Technol. 2014, 93, 97–105. [Google Scholar] [CrossRef]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef]
- Zou, J.J.; Cai, X.; Yang, J.; Zeng, X.; Liu, D.X.; Huang, S.; Chen, X.; Yang, Q.Y.; Wang, C.; Chen, H. DNA hypomethylation mediates flower opening and senescence in sweet osmanthus through auxin and ethylene responsive pathways. Postharvest Biol. Technol. 2023, 198, 112250. [Google Scholar] [CrossRef]
- Zheng, S.; Hu, H.; Ren, H.; Yang, Z.; Qiu, Q.; Qi, W.; Liu, X.; Chen, X.; Cui, X.; Li, S. The Arabidopsis H3K27me3 demethylase JUMONJI 13 is a temperature and photoperiod dependent flowering repressor. Nat. Commun. 2019, 10, 1033. [Google Scholar] [CrossRef]
- Kendall, M.; Colijn, C. Mapping phylogenetic trees to reveal distinct patterns of evolution. Mol. Biol. Evol. 2016, 33, 2735–2743. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, Z.; Vang, S.; Yu, J.; Wong, K.S.; Wang, J. Correlation between Ka/Ks and Ks is related to substitution model and evolutionary lineage. J. Mol. Evol. 2009, 68, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.C.; Sweetman, C.; Ford, C.M. Annotation of gene function in citrus using gene expression information and co-expression networks. BMC Plant Biol. 2014, 14, 186. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Bowman, M.J.; Vega, A.; Kim, H.R.; Mukherjee, A. Comparative transcriptome analysis provides key insights into gene expression pattern during the formation of nodule-like structures in Brachypodium. Funct. Integr. Genom. 2018, 18, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Ma, S.; Shen, D.; Fu, H.; Huang, J. Genome-wide identification of Gramineae histone modification genes and their potential roles in regulating wheat and maize growth and stress responses. BMC Plant Biol. 2021, 21, 543. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Gu, L.; Liu, C.; Lu, T.; Lu, F.; Lu, Z.; Cui, P.; Pei, Y.; Wang, B.; Hu, S.; et al. Arginine methylation mediated by the Arabidopsis homolog of PRMT5 is essential for proper pre-mRNA splicing. Proc. Natl. Acad. Sci. India Sect. B 2010, 107, 19114–19119. [Google Scholar] [CrossRef]
- Jiang, D.; Yang, W.; He, Y.; Amasino, R.M. Arabidopsis relatives of the human lysine-specific demethylase1 repress the expression of FWA and FLOWERING LOCUS C and thus promote the floral transition. Plant Cell 2007, 19, 2975–2987. [Google Scholar] [CrossRef]
- Li, C.; Huang, L.; Xu, C.; Zhao, Y.; Zhou, D.X. Altered levels of histone deacetylase OsHDT1 affect differential gene expression patterns in hybrid rice. PLoS ONE 2011, 6, e21789. [Google Scholar] [CrossRef]
- Zhong, X.; Zhang, H.; Zhao, Y.; Sun, Q.; Hu, Y.; Peng, H.; Zhou, D.X. The rice NAD+-dependent histone deacetylase OsSRT1 targets preferentially to stress- and metabolism-related genes and transposable elements. PLoS ONE 2013, 8, e66807. [Google Scholar] [CrossRef]
- Saleh, A.; Alvarez-Venegas, R.; Yilmaz, M.; Le, O.; Hou, G.; Sadder, M.; Al-Abdallat, A.; Xia, Y.; Lu, G.; Ladunga, I. The highly similar Arabidopsis homologs of trithorax ATX1 and ATX2 encode proteins with divergent biochemical functions. Plant Cell 2008, 20, 568–579. [Google Scholar] [CrossRef]
- Tamada, Y.; Yun, J.Y.; Woo, S.C.; Amasino, R.M. ARABIDOPSIS TRITHORAX-RELATED7 is required for methylation of lysine 4 of histone H3 and for transcriptional activation of FLOWERING LOCUS C. Plant Cell 2009, 21, 3257–3269. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.B.; Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2011, 331, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Tai, R.; Yu, C.W.; Yang, S.; Chen, C.Y.; Lin, W.D.; Schmidt, W.; Wu, K. Regulation of flowering time by the histone deacetylase HDA 5 in Arabidopsis. Plant J. 2015, 82, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Gan, E.S.; Xu, Y.; Wong, J.Y.; Geraldine Goh, J.; Sun, B.; Wee, W.Y.; Huang, J.; Ito, T. Jumonji demethylases moderate precocious flowering at elevated temperature via regulation of FLC in Arabidopsis. Nat. Commun. 2014, 5, 5098. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Choudhary, P.; Caruana, J.; Raina, R. JMJ27, an Arabidopsis H3K9 histone demethylase, modulates defense against Pseudomonas syringae and flowering time. Plant J. 2017, 91, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Hung, F.Y.; Lai, Y.C.; Wang, J.; Feng, Y.R.; Wu, K. The Arabidopsis histone demethylase JMJ28 regulates CONSTANS by interacting with FBH transcription factors. Plant Cell 2021, 33, 1196–1211. [Google Scholar] [CrossRef] [PubMed]
- Han, B.S. Flowering induction of Guzmania by ethylene. Sci. Hortic. 2006, 110, 104–108. [Google Scholar]
- Grbi, V.; Bleecker, A.B. Ethylene regulates the timing of leaf senescence in Arabidopsis. Plant J. 1995, 8, 595–602. [Google Scholar] [CrossRef]
- Wang, X.; Gao, J.; Gao, S.; Song, Y.; Yang, Z.; Kuai, B. The H3K27me3 demethylase REF6 promotes leaf senescence through directly activating major senescence regulatory and functional genes in Arabidopsis. PLoS Genet. 2019, 15, e1008068. [Google Scholar] [CrossRef]
- Yu, C.; Liu, X.; Luo, M.; Chen, C.; Lin, X.; Tian, G.; Lu, Q.; Cui, Y.; Wu, K. HISTONE DEACETYLASE6 interacts with FLOWERING LOCUS D and regulates flowering in Arabidopsis. Plant Physiol. 2011, 156, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zeng, X.; Yang, J.; Cai, X.; Shi, Y.; Zheng, R.; Wang, Z.; Liu, J.; Yi, X.; Xiao, S.; et al. Whole-genome resequencing of Osmanthus fragrans provides insights into flower color evolution. Hortic. Res. 2021, 8, 98. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; de Peer, Y.V.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Smyth, G.K. limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor, 1st ed.; Gentleman, R., Carey, V.J., Huber, W., Irizarry, R.A., Dudoit, S., Eds.; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR. Methods 2002, 25, 402–408. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, H.; Zhang, Y.; Chen, X.; Zeng, X.; Cai, X.; Li, Z.; Chen, H.; Yang, J.; Zou, J. Genome-Wide Identification of Osmanthus fragrans Histone Modification Genes and Analysis of Their Expression during the Flowering Process and under Azacytidine and Ethylene Treatments. Plants 2024, 13, 777. https://doi.org/10.3390/plants13060777
Xia H, Zhang Y, Chen X, Zeng X, Cai X, Li Z, Chen H, Yang J, Zou J. Genome-Wide Identification of Osmanthus fragrans Histone Modification Genes and Analysis of Their Expression during the Flowering Process and under Azacytidine and Ethylene Treatments. Plants. 2024; 13(6):777. https://doi.org/10.3390/plants13060777
Chicago/Turabian StyleXia, Hui, Yingting Zhang, Xiang Chen, Xiangling Zeng, Xuan Cai, Zeqing Li, Hongguo Chen, Jie Yang, and Jingjing Zou. 2024. "Genome-Wide Identification of Osmanthus fragrans Histone Modification Genes and Analysis of Their Expression during the Flowering Process and under Azacytidine and Ethylene Treatments" Plants 13, no. 6: 777. https://doi.org/10.3390/plants13060777