
Integration of Metabolomic and Transcriptomic Analyses Reveals the Molecular Mechanisms of Flower Color Formation in Prunus mume


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
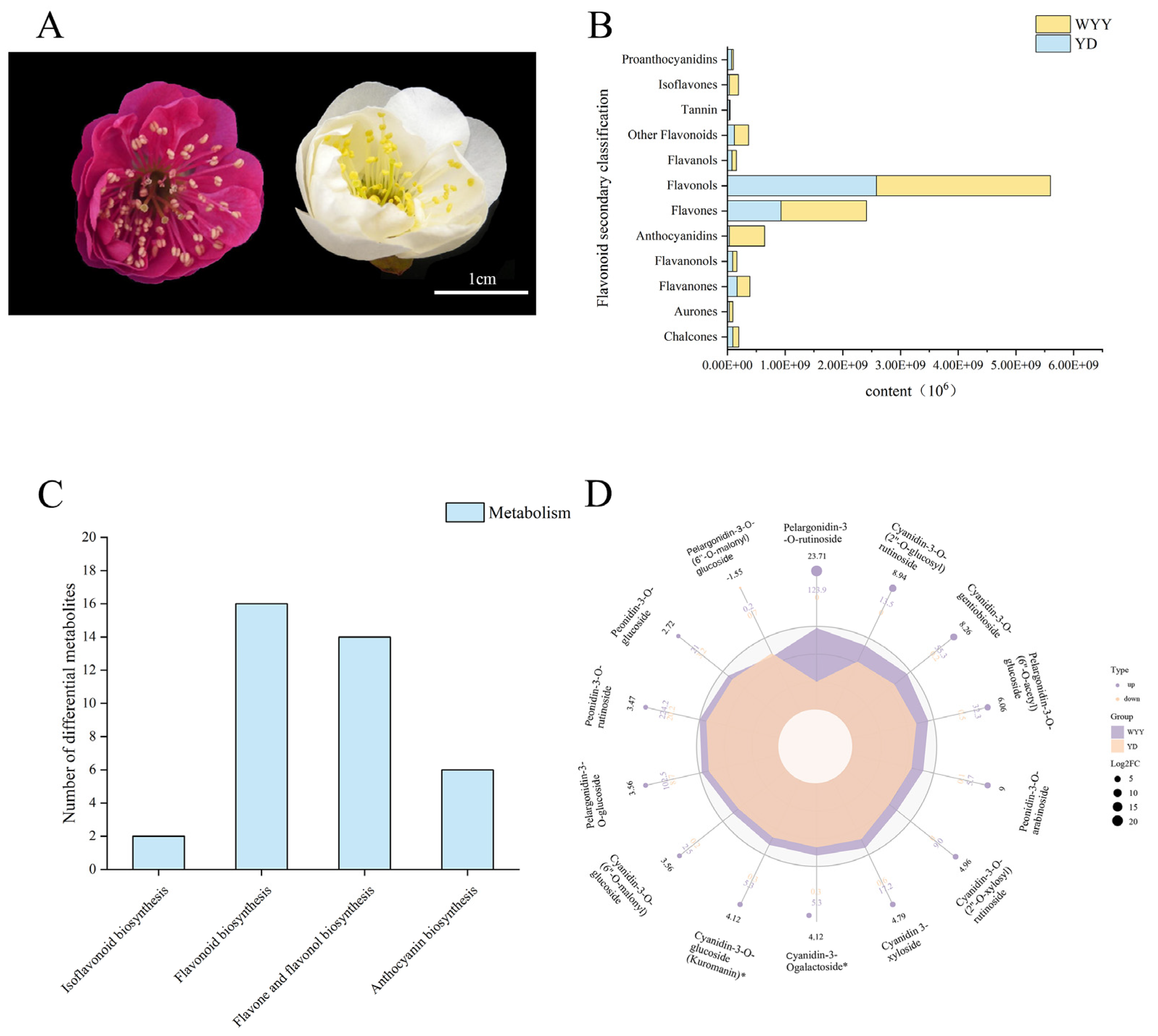
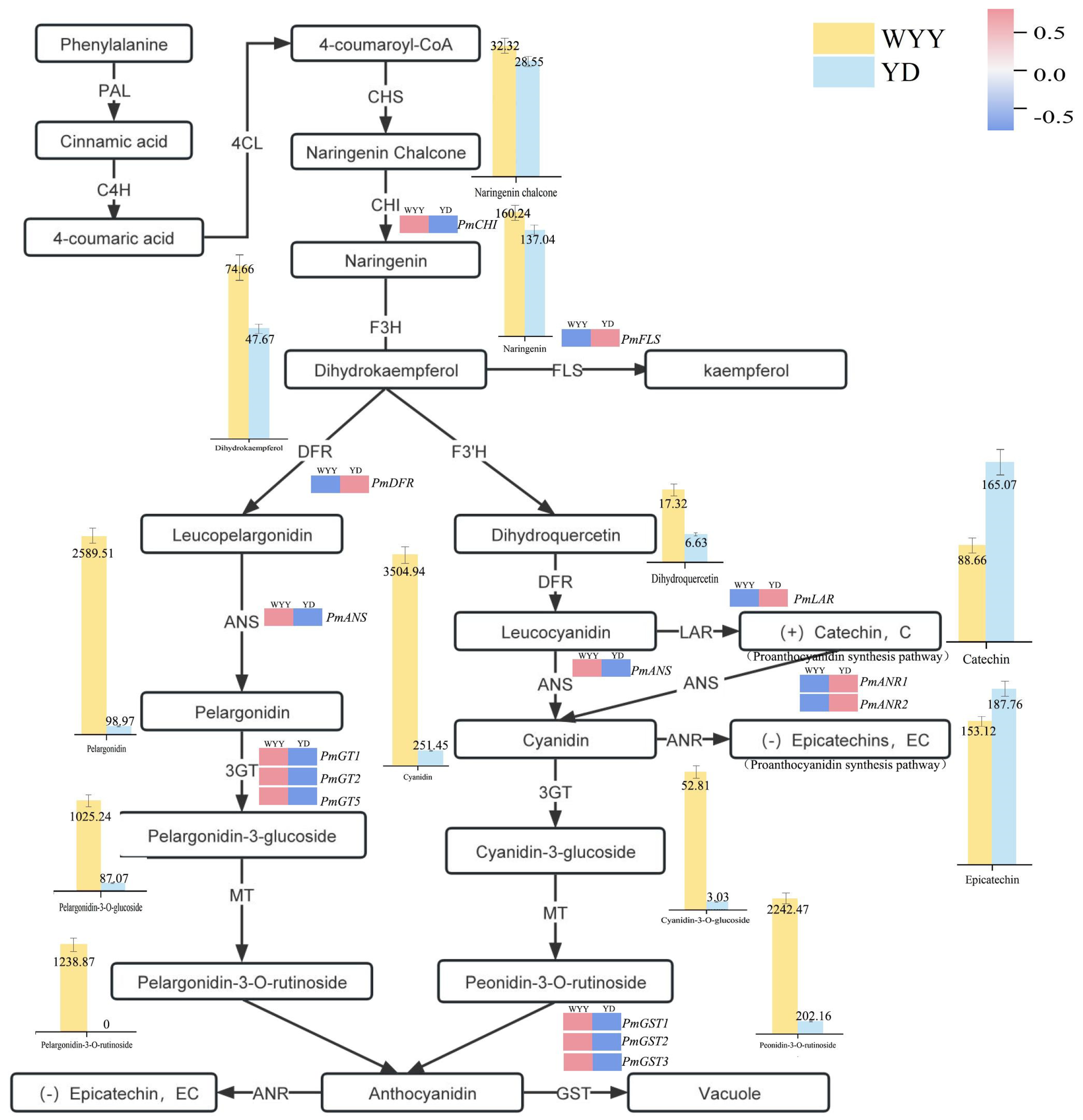
2.1. Identification of Flavonoids and Differentially Accumulated Metabolites between Purple–Red and White Flowers in P. mume
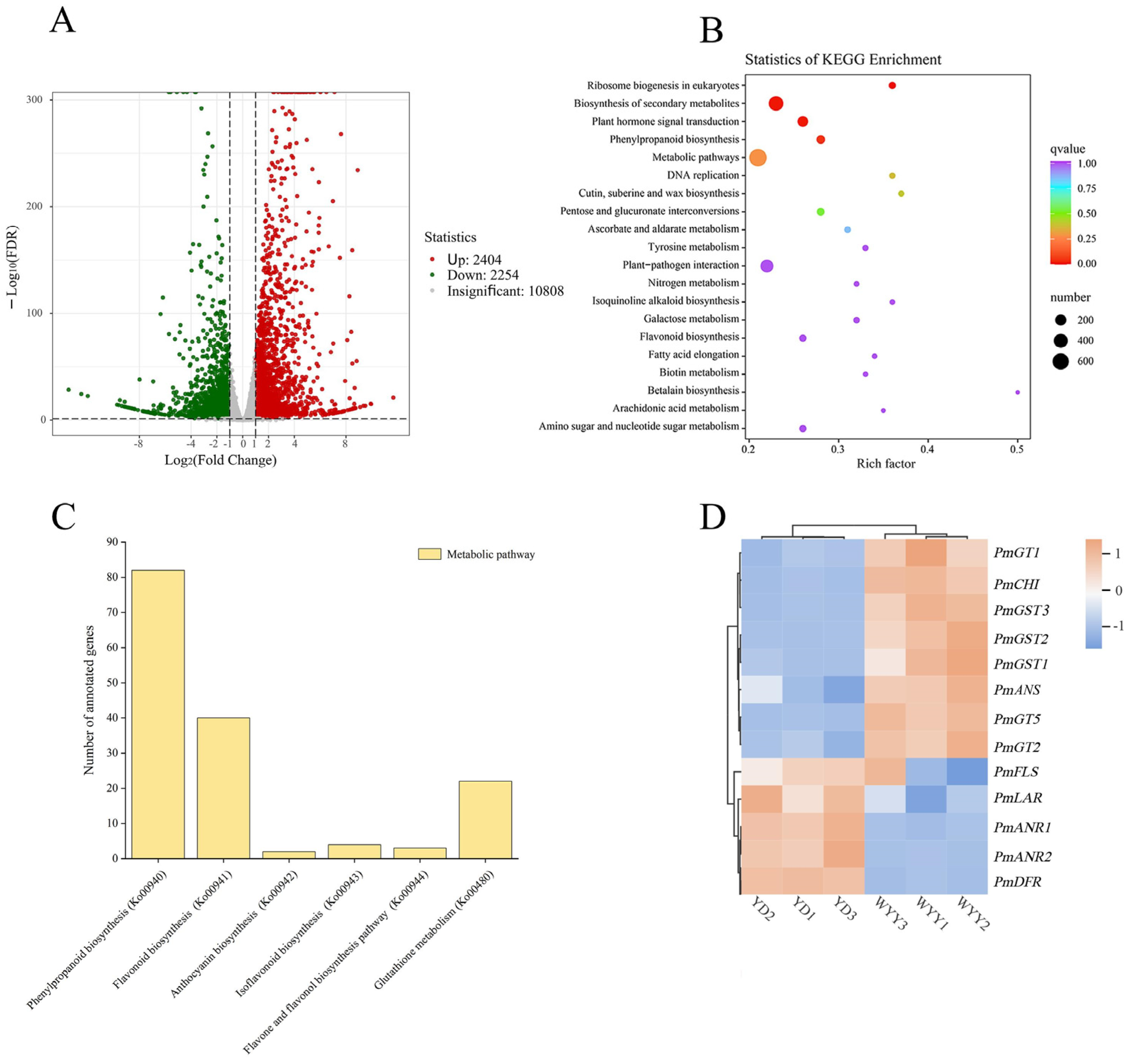
2.2. Transcriptome Sequencing and Analysis of DEGs
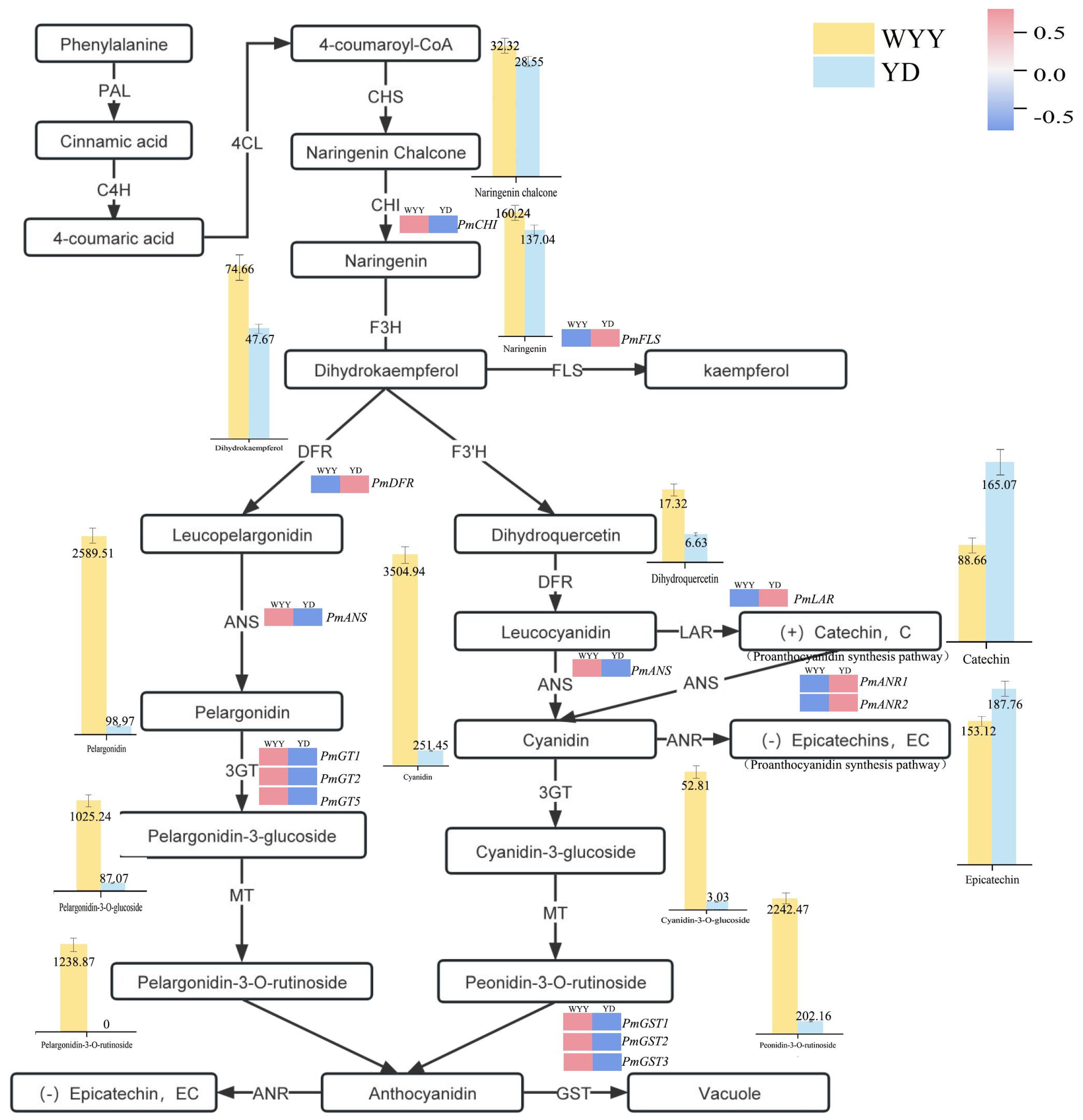
2.3. KEGG Pathway Enrichment Analysis and Identification of DEGs Associated with Anthocyanin Synthesis
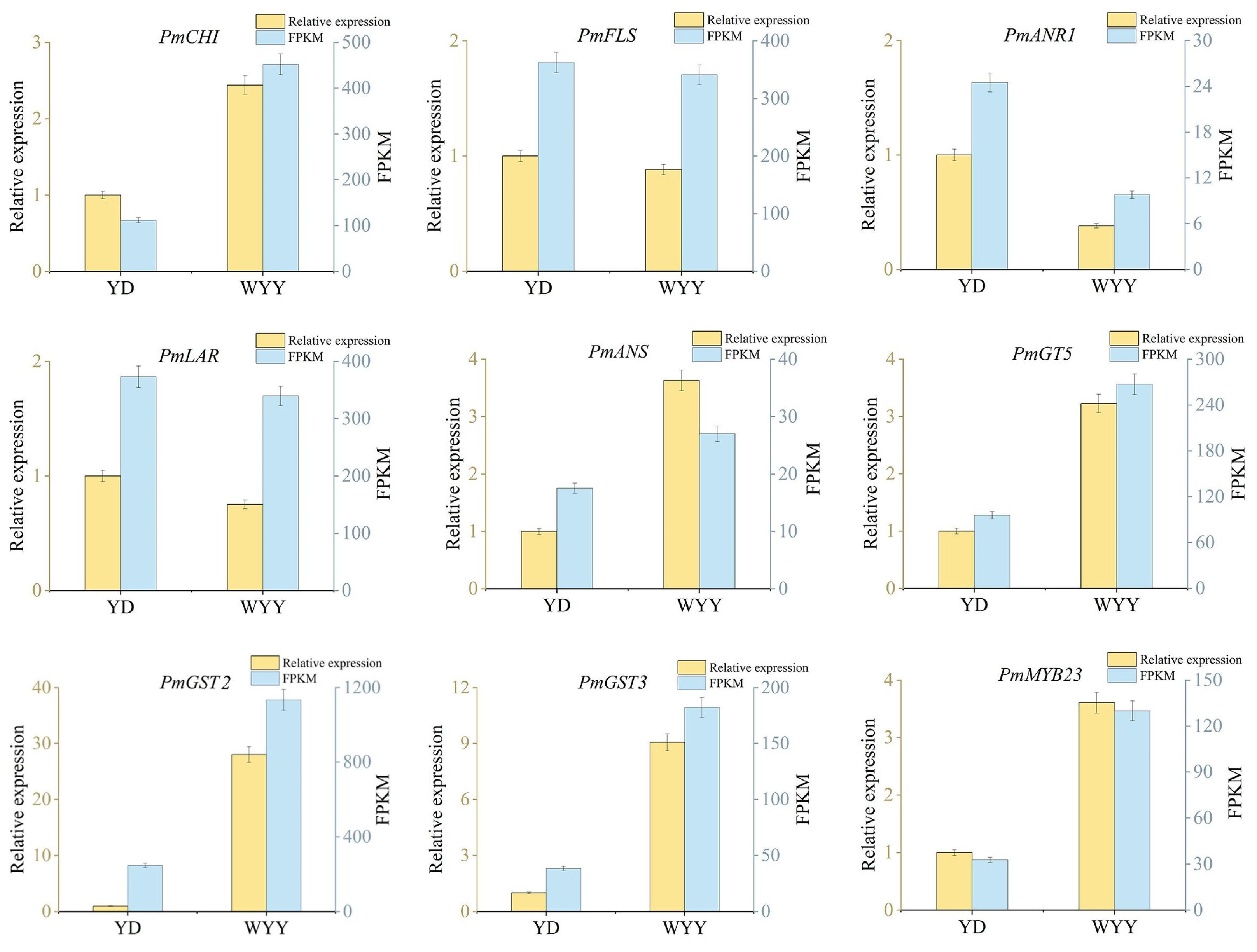
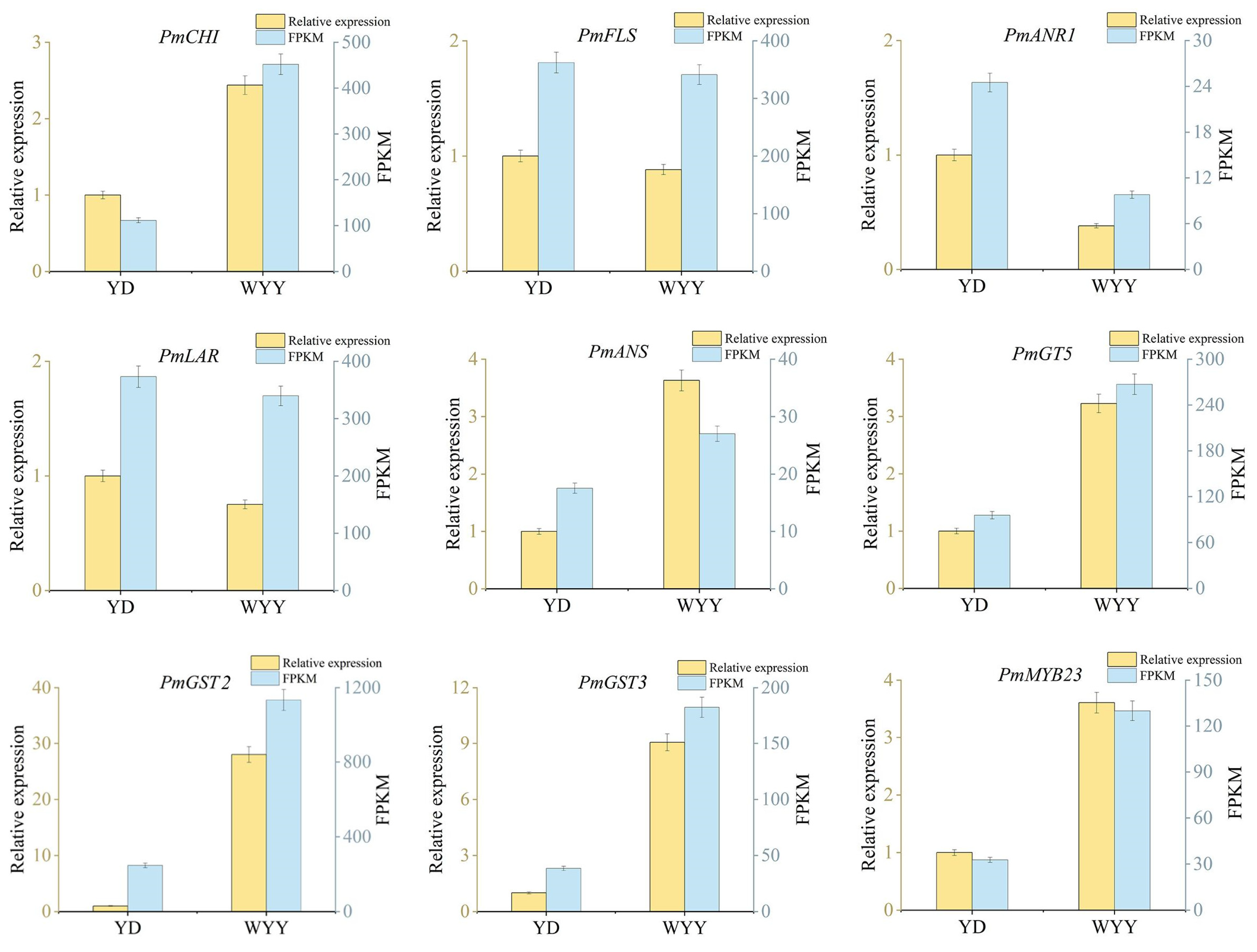
2.4. RT-qPCR Validation of Genes Related to Anthocyanin Synthesis
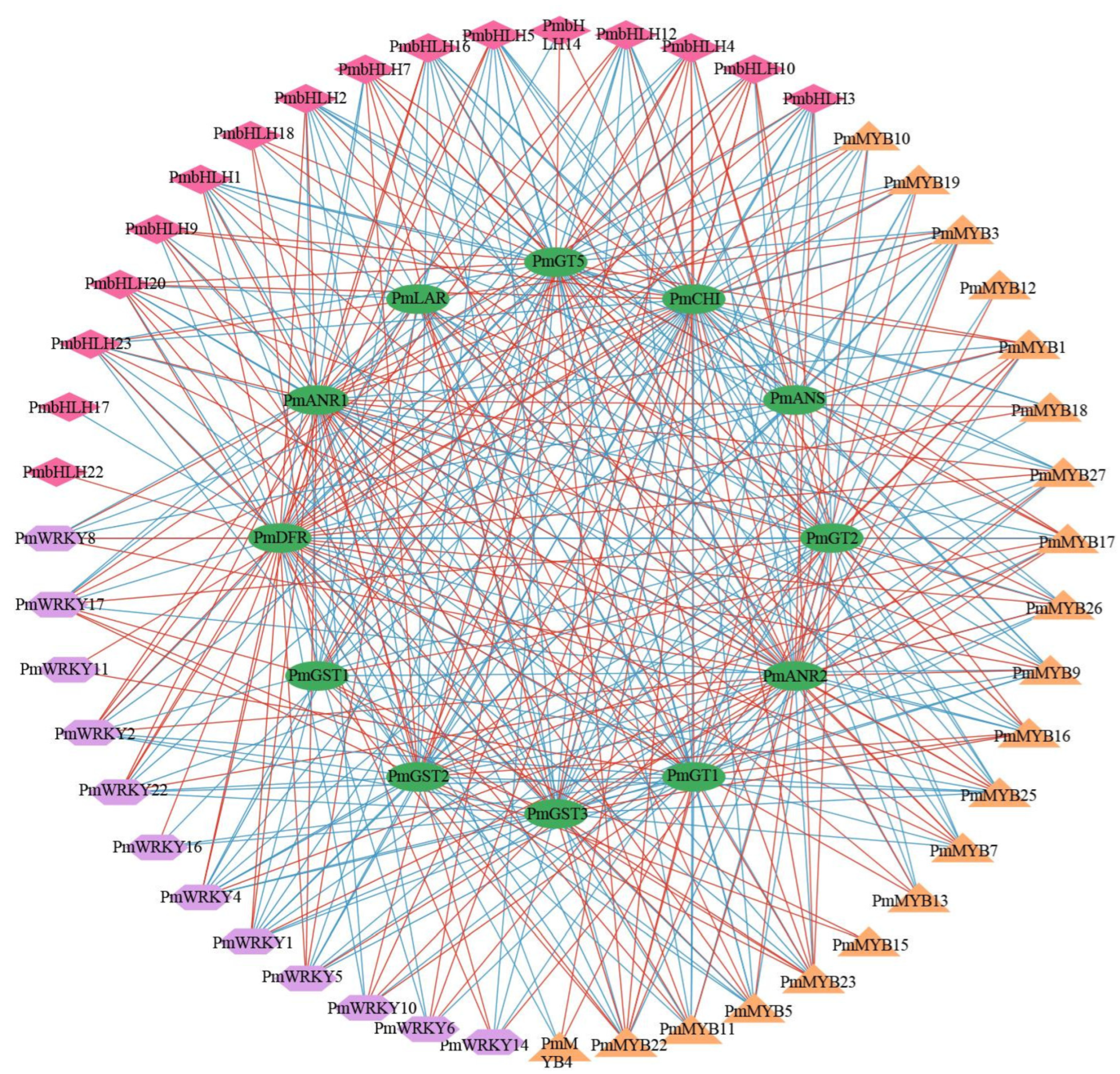
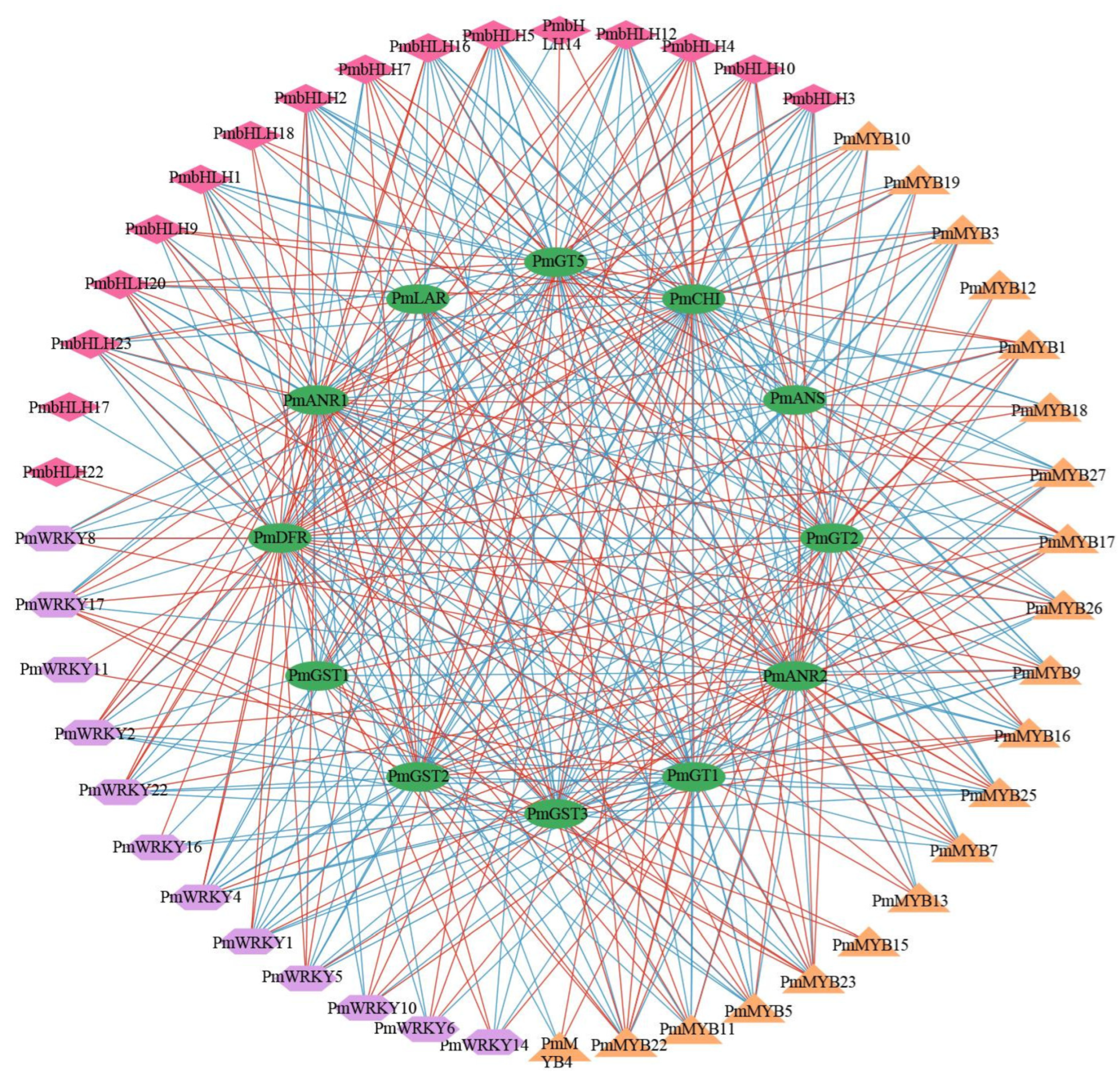
2.5. Transcription Factor Analysis
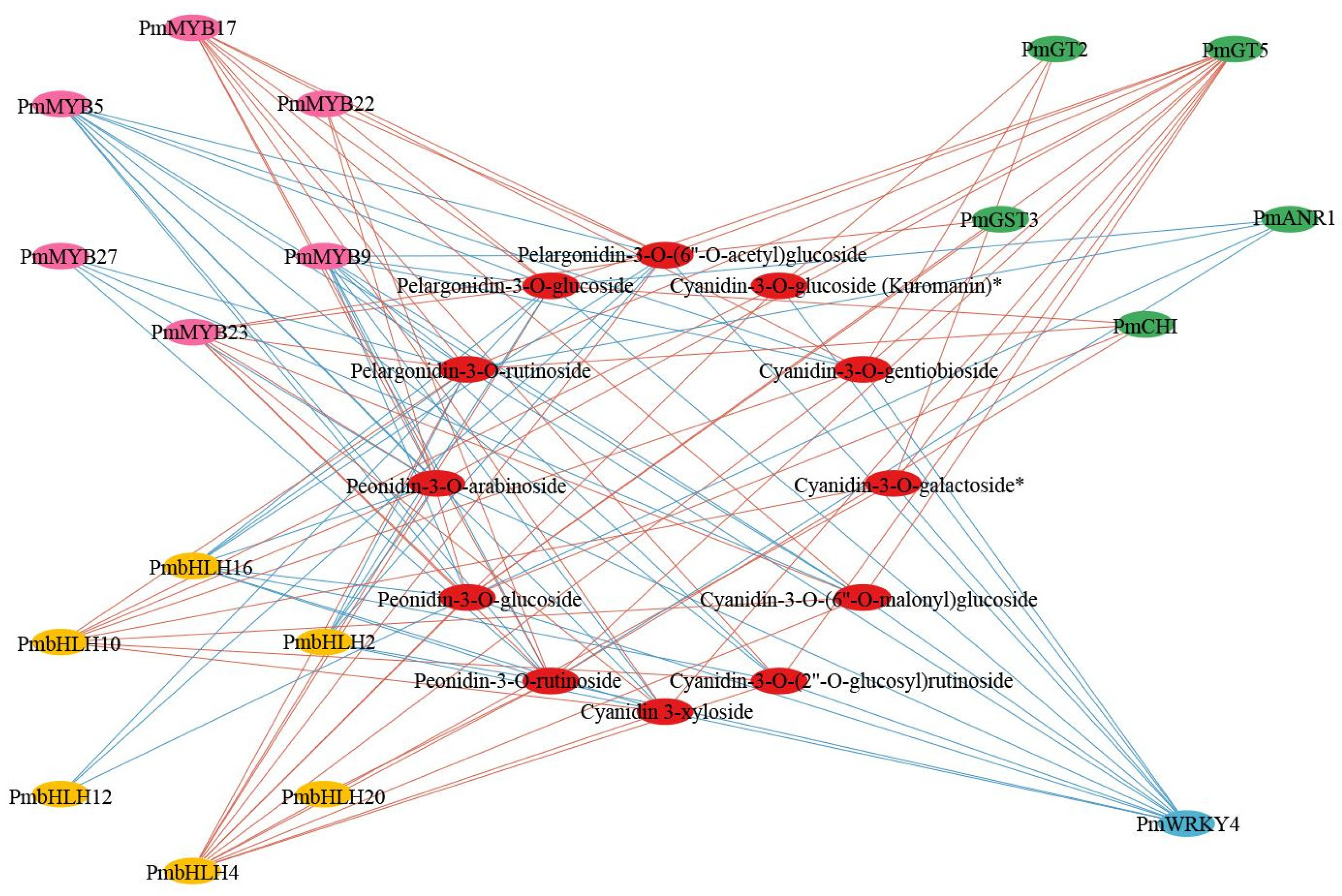
2.6. Construction of Regulatory Networks Associated with Anthocyanin Synthesis
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Metabolomic Analysis and Identification of DAMs
4.3. Transcriptome Analyses and Identification of DEGs
4.4. RT-qPCR Validation of Crucial Genes
4.5. Integrative Analysis of DAMs and DEGs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhao, D.; Tao, J. Recent advances on the development and regulation of flower color in ornamental plants. Front. Plant Sci. 2015, 6, 261. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.M.; Jibran, R.; Zhou, Y.; Albert, N.W.; Brummell, D.A.; Jordan, B.R.; Bowman, J.L.; Schwinn, K.E. The Evolution of Flavonoid Biosynthesis: A Bryophyte Perspective. Front. Plant Sci. 2020, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.M.; Chia, L.S.; Goh, N.K.; Chia, T.F.; Brouillard, R. Analysis and biological activities of anthocyanins. Phytochemistry 2003, 64, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Hichri, I.; Barrieu, F.; Bogs, J.; Kappel, C.; Delrot, S.; Lauvergeat, V. Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 2011, 62, 2465–2483. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, A.; Brockman, A.; Aguirre, L.; Campbell, A.; Bean, A.; Cantero, A.; Gonzalez, A. Advances in the MYB-bHLH-WD Repeat (MBW) Pigment Regulatory Model: Addition of a WRKY Factor and Co-option of an Anthocyanin MYB for Betalain Regulation. Plant Cell Physiol. 2017, 58, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Wang, J.; Lu, C.; Li, Y.; Kong, D.; Hong, Y.; Huang, H.; Dai, S. CcMYB6-1 and CcbHLH1, two novel transcription factors synergistically involved in regulating anthocyanin biosynthesis in cornflower. Plant Physiol. Biochem. 2020, 151, 271–283. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, S.; Peng, D.; Wu, Q.; Liao, X.; Xiang, K.; Wang, Z.; Tembrock, L.R.; Bendahmane, M.; Bao, M.; et al. Integrated multi-omic data and analyses reveal the pathways underlying key ornamental traits in carnation flowers. Plant Biotechnol. J. 2022, 20, 1182–1196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, W.; Sun, L.; Zhao, F.; Huang, B.; Yang, W.; Tao, Y.; Wang, J.; Yuan, Z.; Fan, G.; et al. The genome of Prunus mume. Nat. Commun. 2012, 3, 1318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, H.; Sun, L.; Fan, G.; Ye, M.; Jiang, L.; Liu, X.; Ma, K.; Shi, C.; Bao, F.; et al. The genetic architecture of floral traits in the woody plant Prunus mume. Nat. Commun. 2018, 9, 1702. [Google Scholar] [CrossRef]
- Chen, J. Chinese Prunus mume; China Hainan Publishing House: Haikou, China, 1996; pp. 12–58. [Google Scholar]
- Zhao, Y.; Guo, W.; Chen, J. Preliminary study on the categories and contents of the flower color pigments of Prunus mume Sieb et Zucc. J. Beijing For. Univ. 2004, 26, 68–73. [Google Scholar] [CrossRef]
- Zhang, Q.; Hao, R.; Xu, Z.; Yang, W.; Wang, J.; Cheng, T.; Pan, H.; Zhang, Q. Isolation and functional characterization of a R2R3-MYB regulator of Prunus mume anthocyanin biosynthetic pathway. PCTOC 2017, 131, 417–429. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, Y.; Zhang, C.; Qi, F.; Li, X.; Mu, S.; Peng, Z. Characterization of the floral transcriptome of Moso bamboo (Phyllostachys edulis) at different flowering developmental stages by transcriptome sequencing and RNA-seq analysis. PLoS ONE 2014, 9, e98910. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Nie, R.; Yang, N.; Cai, L.; Hu, Y.; Chen, S.; Cheng, X.; Wang, Z.; Chen, L. Integrated transcriptome and metabolome profiling of Camellia reticulata reveal mechanisms of flower color differentiation. Front. Genet. 2022, 13, 1059717. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zhou, H.; Ren, T.; Yu, E.R.; Feng, B.; Wang, J.; Zhang, C.; Zhou, C.; Li, Y. Integrated transcriptome and metabolome analysis revealed that HaMYB1 modulates anthocyanin accumulation to deepen sunflower flower color. Plant Cell Rep. 2024, 43, 74. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, H.; Liu, J.; Huang, T.; Zhang, X.; Xie, H.; Guo, Y.; Wang, Q.; Zhang, P.; Qin, P. Grain color formation and analysis of correlated genes by metabolome and transcriptome in different wheat lines at maturity. Front. Nutr. 2023, 10, 1112497. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.P.; Zhao, G.; Xie, Y.F.; Owusu, A.G.; Wu, Y.; Gao, J.S. Transcriptome and Metabolome Profiling Unveil Pigment Formation Variations in Brown Cotton Lines (Gossypium hirsutum L.). Int. J. Mol. Sci. 2023, 24, 5249. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Gong, R.; Zhang, C. Integrative analysis of transcriptome and metabolome reveals flavonoid biosynthesis regulation in Rhododendron pulchrum petals. BMC Plant Biol. 2022, 22, 401. [Google Scholar] [CrossRef]
- Feng, X.; Gao, G.; Yu, C.; Zhu, A.; Chen, J.; Chen, K.; Wang, X.; Abubakar, A.S.; Chen, P. Transcriptome and metabolome analysis reveals anthocyanin biosynthesis pathway associated with ramie (Boehmeria nivea (L.) Gaud.) leaf color formation. BMC Genom. 2021, 22, 684. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Devi, M.J.; Feng, X.; Li, Y.; Song, L.; Gao, H.; Cao, B. Transcriptome analysis reveals the fruit color variation in Ailanthus altissima. Physiol. Plant. 2023, 175, e13867. [Google Scholar] [CrossRef]
- Zhu, L.; Wen, J.; Ma, Q.; Yan, K.; Du, Y.; Chen, Z.; Lu, X.; Ren, J.; Wang, Y.; Li, S.; et al. Transcriptome profiling provides insights into leaf color changes in two Acer palmatum genotypes. BMC Plant Biol. 2022, 22, 589. [Google Scholar] [CrossRef]
- Parrish, S.B.; Paudel, D.; Deng, Z. Transcriptome analysis of Lantana camara flower petals reveals candidate anthocyanin biosynthesis genes mediating red flower color development. G3 2023, 14, jkad259. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Tang, D.; Liu, X.; Li, Z.; Xiang, Y.; Gao, K.; Li, H.; Yuan, L.; Huang, B.; Li, J. Transcriptome analysis reveals important regulatory genes and pathways for tuber color variation in Pinellia ternata (Thunb.) Breit. Protoplasma 2023, 2260, 1313–1325. [Google Scholar] [CrossRef]
- Guan, L.; Liu, J.; Wang, R.; Mu, Y.; Sun, T.; Wang, L.; Zhao, Y.; Zhu, N.; Ji, X.; Lu, Y.; et al. Metabolome and Transcriptome Analyses Reveal Flower Color Differentiation Mechanisms in Various Sophora japonica L. Petal Types. Biology 2023, 12, 1466. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Cai, C.; Mo, X.; Zhao, X.; Wu, L.; Liu, F.; Li, R.; Liu, C.; Chen, J.; Tian, M. Transcriptome and metabolome analysis reveals the effect of flavonoids on flower color variation in Dendrobium nobile Lindl. Front. Plant Sci. 2023, 14, 1220507. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Pires, N.; Dolan, L. Origin and diversification of basic-helix-loop-helix proteins in plants. Mol. Biol. Evol. 2010, 27, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liu, W.; Zhang, T.; Jiang, S.; Xu, H.; Wang, Y.; Zhang, Z.; Wang, C.; Chen, X. Transcriptomic Analysis of Red-Fleshed Apples Reveals the Novel Role of MdWRKY11 in Flavonoid and Anthocyanin Biosynthesis. J. Agric. Food Chem. 2018, 66, 7076–7086. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, J.; Hu, K.D.; Wei, S.W.; Sun, H.Y.; Hu, L.Y.; Han, Z.; Yao, G.F.; Zhang, H. PyWRKY26 and PybHLH3 cotargeted the PyMYB114 promoter to regulate anthocyanin biosynthesis and transport in red-skinned pears. Hortic. Res. 2020, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Qu, Y.; Sha, G.; Zhang, S.; Ma, Y.; Chen, M.; Zhai, R.; Yang, C.; Xu, L.; Wang, Z. PbWRKY75 promotes anthocyanin synthesis by activating PbDFR, PbUFGT, and PbMYB10b in pear. Physiol. Plant. 2021, 173, 1841–1849. [Google Scholar] [CrossRef]
- Zhu, J.; Guo, X.; Li, X.; Tang, D. Composition of flavonoids in the petals of freesia and prediction of four novel transcription factors involving in freesia lavonoid pathway. Front. Plant Sci. 2021, 12, 756300. [Google Scholar] [CrossRef]
- Bao, F.; Ding, A.; Cheng, T.; Wang, J.; Zhang, Q. Genome-wide analysis of members of the WRKY gene family and their cold stress esponse in Prunus mume. Genes 2019, 10, 911. [Google Scholar] [CrossRef] [PubMed]
- Yue, F.; Chen, Y.; Liu, B.; Gan, D.; Wang, H.; Wang, D. Identification of WD40 gene family in Prunus mume and its expression level under light illumination. PeerJ 2018, 6, e27106v1. [Google Scholar] [CrossRef]
- Ding, A.; Ding, A.; Li, P.; Wang, J.; Cheng, T.; Bao, F.; Zhang, Q. Genome-Wide Identification and Low-Temperature Expression Analysis of bHLH Genes in Prunus mume. Front. Genet. 2021, 12, 762135. [Google Scholar] [CrossRef] [PubMed]
- Sunil, L.; Shetty, N.P. Biosynthesis and regulation of anthocyanin pathway genes. Appl. Microbiol. Biotechnol 2022, 106, 1783–1798. [Google Scholar] [CrossRef] [PubMed]
- Saigo, T.; Wang, T.; Watanabe, M.; Tohge, T. Diversity of anthocyanin and proanthocyanin biosynthesis in land plants. Curr. Opin. Plant Biol. 2020, 55, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Marinova, K.; Pourcel, L.; Weder, B.; Schwarz, M.; Barron, D.; Routaboul, J.M.; Debeaujon, I.; Klein, M. The Arabidopsis MATE transporter TT12 acts as a vacuolar flavonoid/H+ -antiporter active in proanthocyanidin-accumulating cells of the seed coat. Plant Cell 2007, 19, 2023–2038. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Vimolmangkang, S.; Wei, G.; Zhou, H.; Korban, S.S.; Han, Y. Molecular characterization of genes encoding leucoanthocyanidin reductase involved in proanthocyanidin biosynthesis in apple. Front. Plant Sci. 2015, 6, 243. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Vimolmangkang, S.; Soria-Guerra, R.; Korban, S.S. Introduction of apple ANR genes into tobacco inhibits expression of both CHI and DFR genes in flowers, leading to loss of anthocyanin. J. Exp. Bot. 2012, 63, 2437–2447. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gong, Q.; Ni, X.; Zhou, Y.; Gao, Z. UFGT: The key enzyme associated with the petals variegation in japanese apricot. Front. Plant Sci. 2017, 8, 108. [Google Scholar] [CrossRef]
- Kitamura, S.; Shikazono, N.; Tanaka, A. TRANSPARENT TESTA 19 is involved in the accumulation of both anthocyanins and proanthocyanidins in Arabidopsis. Plant J. 2004, 37, 104–114. [Google Scholar] [CrossRef]
- Jiang, S.; Chen, M.; He, N.; Chen, X.; Wang, N.; Sun, Q.; Zhang, T.; Xu, H.; Fang, H.; Wang, Y.; et al. MdGSTF6, activated by MdMYB1, plays an essential role in anthocyanin accumulation in apple. Hortic. Res. 2019, 6, 40. [Google Scholar] [CrossRef]
- Chai, Q.; Wang, X.; Gao, M.; Zhao, X.; Chen, Y.; Zhang, C.; Jiang, H.; Wang, J.; Wang, Y.; Zheng, M.; et al. A glutathione S-transferase GhTT19 determines flower petal pigmentation via regulating anthocyanin accumulation in cotton. Plant Biotechnol. J. 2023, 21, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Dong, W.; Zhu, Y.; Allan, A.C.; Lin-Wang, K.; Xu, C. PpGST1, an anthocyanin-related glutathione S-transferase gene, is essential for fruit coloration in peach. Plant Biotechnol. J. 2020, 18, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Lai, B.; You, Y.; Zhang, L.; Wang, Q.; Chen, F.; Luo, G.; Du, L.; Wang, H. Identification and functional characterization of RsGST1, an anthocyanin-related glutathione S-transferase gene in radish. J. Plant Physiol. 2021, 263, 153468. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, Y.; Zhang, X.; Tian, F.; Yu, F.; Li, X.; Huang, D. Association Analysis of Transcriptome and Targeted Metabolites Identifies Key Genes Involved in Iris germanica Anthocyanin Biosynthesis. Int. J. Mol. Sci. 2023, 24, 16462. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.; Barbas, C. Gas chromatography-mass spectrometry (GCMS)-based metabolomics. Methods Mol. Biol. 2011, 708, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, R.; Song, Y.; He, J.; Sun, J.; Bai, J.; An, Z.; Dong, L.; Zhan, Q.; Abliz, Z. RRLC-MS/MS-based metabonomics combined with in-depth analysis of metabolic correlation network: Finding potential biomarkers for breast cancer. Analyst 2009, 134, 2003–2011. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Yang, X.; Wang, T.; Li, B.; Li, P.; Zhang, Q. Integration of Metabolomic and Transcriptomic Analyses Reveals the Molecular Mechanisms of Flower Color Formation in Prunus mume. Plants 2024, 13, 1077. https://doi.org/10.3390/plants13081077
Wang R, Yang X, Wang T, Li B, Li P, Zhang Q. Integration of Metabolomic and Transcriptomic Analyses Reveals the Molecular Mechanisms of Flower Color Formation in Prunus mume. Plants. 2024; 13(8):1077. https://doi.org/10.3390/plants13081077
Chicago/Turabian StyleWang, Ruyi, Xin Yang, Tao Wang, Baohui Li, Ping Li, and Qin Zhang. 2024. "Integration of Metabolomic and Transcriptomic Analyses Reveals the Molecular Mechanisms of Flower Color Formation in Prunus mume" Plants 13, no. 8: 1077. https://doi.org/10.3390/plants13081077